
Abstract
Background
Allogeneic mesenchymal stromal cells (MSCs) have been used extensively in various clinical trials. Nevertheless, there are concerns about their efficacy, attributed mainly to the heterogeneity of the applied populations. Therefore, producing a consistent population of MSCs is crucial to improve their therapeutic efficacy. This study presents a good manufacturing practice (GMP)-compatible and cost-effective protocol for manufacturing, banking, and lot-release of a homogeneous population of human bone marrow-derived clonal MSCs (cMSCs).
Methods
Here, cMSCs were isolated based on the subfractionation culturing method. Afterward, isolated clones that could reproduce up to passage three were stored as the seed stock. To select proliferative clones, we used an innovative, cost-effective screening strategy based on lengthy serial passaging. Finally, the selected clones re-cultured from the seed stock to establish the following four-tired cell banking system: initial, master, working, and end of product cell banks (ICB, MCB, WCB, and EoPCB).
Results
Through a rigorous screening strategy, three clones were selected from a total of 21 clones that were stored during the clonal isolation process. The selected clones met the identity, quality, and safety assessments criteria. The validated clones were stored in the four-tiered cell bank system under GMP conditions, and certificates of analysis were provided for the three-individual ready-to-release batches. Finally, a stability study validated the EoPCB, release, and transport process of the frozen final products.
Conclusion
Collectively, this study presents a technical and translational overview of a GMP-compatible cMSCs manufacturing technology that could lead to the development of similar products for potential therapeutic applications.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stromal cells (MSCs) have been explored in a broad range of preclinical to clinical studies and are considered as advanced therapy medicinal products (ATMPs) [1]. Until June 2021, MSCs have been used in 1114 ongoing or completed clinical trials for various diseases, including skeletal diseases, autoimmune and inflammatory disorders, cancers, and COVID-19 (https://www.clinicaltrials.gov). This extensive application of MSCs is mainly attributed to their proven safety and efficacy [2,3,4,5]. Actually, MSCs are promising candidates for allogeneic cell therapy because of their intrinsic features such as low immunogenicity, trophic effects, and unique immunomodulation properties, manifest as intelligent pro-inflammatory or anti-inflammatory effects in response to environmental signals [6, 7].
Despite the large amount of evidence supporting the safety and efficacy of MSCs in research and clinical trials (well discussed in [8]), there are still controversial results about the efficacy of these cells that is mainly due to the heterogeneity of the applied cell populations [9]. It seems that producing a pure, high-quality MSC population can eliminate the inconsistencies between studies and facilitate the achievement of robust, reliable, and reproducible outcomes for this highly potential therapeutic products. This dearth in cultured MSCs can be partly associated with the heterogeneity of initial MSCs, donor-to-donor variations, and the diversity of initial sources from which MSCs are derived [9,10,11]. However, MSC heterogeneity is an innate property that is associated to the niche and also to the isolation and culture methods [12, 13]. Furthermore, it is necessary to provide a good manufacturing practice (GMP) platform to ensure the purity and safety of final cell therapy products (CTPs) and meet the regulatory requirements for clinical applications. Establishing a tiered cell bank system is the final step to support the proper translation of CTPs into medicine by assuring standardization of the entire cell manufacturing process, accredited identity, function and safety assessments, and transparent sharing of standard operating procedures data.
Different approaches have been proposed for the production of a relatively high homogeneous population of MSCs like the purification of sub-population of MSCs by primary surface markers such as NGFR, STRO-1, BRIGHT/VCAM-1, MCAM/CD146, and CD34 [9, 14,15,16,17], and enrichment of MSCs by the subfractionation culturing method (SCM) [18, 19]. The first approach has some technical issues, such as the lack of a comprehensive agreement on specific surface markers [20]. However, the SCM method appears to be suited for production of a homogeneous MSC population by manufacturing single colony-forming units (CFUs) to produce clonal MSCs (cMSCs). However, although SCM is considered as an efficient and reproducible isolation technique, it should be improved in terms of biological and industrial applications before it can be successfully applied to the global development of clinical-grade cMSCs. The main challenge of this approach is the need to spend time and resources on expanding, characterizing, and biobanking numerous clones, while only a limited proportion of these clones eventually reach the final product.
Here, we implemented and optimized the manufacturing procedure of a homogeneous population of human bone marrow (BM)-derived cMSCs. Our minimal serial culturing method to screen clones is beneficial in cost management and commercialization of the final cell product. In order to ensure the accuracy of this clone screening method, we extensively compared the different cMSC batches with the heterogeneous MSC (hMSCs) counterpart in terms of identity, quality, and safety. Our GMP policy and quality assurance standards ensured that donor screening, clonal isolation, optimal clone selection, and establishment of initial, master, working, and end of product cell banks (ICB, MCB, WCB, and EoPCB, respectively), in addition to all related quality control (QC) procedures stringently met the Iran Food and Drug Administration (IFDA) regulatory guidelines. These guidelines, basically, follow the Pharmaceutical Inspection Co-operation Scheme principles. At the end of the manufacturing process, we released 40 packs of our final drug product (DP) for phase I/II clinical trials for COVID-19 patients (NCT04366063).
Methods
Ethical Approval
BM sample was collected from a healthy volunteer after receipt of written informed consent, which was obtained according to the Iran Food and Drug Administration guidelines and approval of the National Ethics Committee’s license for the use of an intended human biological sample for manufacturing and therapeutic goals (Approval ID: IR.ACECR.ROYAN.REC.1397.289).
Donor Selection and Evaluation
Eligible, healthy volunteers for BM donation were carefully chosen according to the inclusion criteria listed in Supplementary Table 1. Briefly, donor competency referred to general health through asking the volunteers’ medical history, physical examination, serological testing to insure the medical health and the absence of viral infections, chest X-ray, and electrocardiogram (EKG). From three voluntrees, we finally chose a 31-year-old healthy male donor who met all the inclusion criteria for BM donation.
Aspiration and Transport of Bone Marrow
BM was aspirated from the iliac crest of the healthy donor under sterile conditions in an operating room according to a procedure by Emadedin et al. [21]. The BM was harvested in a total volume of 40 ml. The aspirate was put in a sterile heparinized 50 ml tube and placed in a cool box (4 °C to 8 °C) for transport within 12 h to the GMP clean room of the manufacturing center with a temperature data logger.
Isolation and Expansion of Heterogeneous Mesenchymal Stromal Cells (hMSCs)
We used two approaches to isolate and expand MSCs from the aspirated BM sample in order to obtain the hMSC and cMSC lines. For hMSCs, the BM mononuclear cells were separated from the whole BM sample by density gradient centrifugation as previously described [22]. In brief, Ficoll®-Paque (GE Healthcare, USA) was used at a density of 1.077 g/ml, and the separated mononuclear cells were cultured in 175 cm2 ventilated flasks (BD Biosciences, France) at a density of 0.5 × 106 cells/cm2 in isolation culture medium. The isolation medium consisted of minimal essential medium Eagle, alpha formulation (alpha-MEM medium) supplemented with 20% defined fetal bovine serum (FBS; HyClone, USA), 1% MEM non-essential amino acids (MEM NEAAs), and 1% GlutaMAX (Supplementary Table 2). After one day, the floating cells were removed by changing the medium, and the adherent cells were sub-cultured when they were more than 70% confluent. An expansion culture medium was used for serial passaging. This medium consisted of an α-MEM base medium supplemented with 5% human platelet lysate (hPL) and the same additives as the isolation medium (Supplementary Table 3).
Isolation and Expansion of Clonal Mesenchymal Stromal Cells (cMSCs) and Establishment of Seed Stock Cell Bank
We used the SCM technique reported by Yi et al. to produce the cMSCs [18, 19]. Briefly, every 2.5 ml of BM aspirate from the iliac crest of a healthy donor was directly cultured in an isolation medium in a 100-mm tissue culture dish. The supernatant, which contained suspended cells, was daily transferred to the new dishes until day (D) 5 (Supplementary Fig. 1). Dishes were named based on the day of the transfer (D1-D5) and dishes from D2 to D5 were kept until colonies appeared (after about two weeks). The separate well-grown colonies were detached enzymatically with TrypLE (Thermo Fisher, USA) using cloning cylinders, and the cell suspension was transferred to 35-mm tissue culture dishes. In order to save all individual clones of the MSCs that had high CFU activity, we chose the clones that expanded and reached confluence over five to six days in each of three subsequent passages (from the well of 6-well plate to a T25 culture flask, and then to a T75 culture flask). These clones were candidate for cryopreservation as seed stock cell bank at passage 3 (P3). Before cryopreservation, about 120–150 × 103 cells from each clone was cultured in a T25 culture flask (P4; 5 × 103 cells/cm2 seeding density) in order to find the best clone(s). The rest of the cells from each clone at P3 were cryopreserved in at least three cryovials with a density of 0.5 ± 0.1 × 106 cells/500 μl freezing medium and stored in vapor phase of liquid nitrogen (LN) as seed stock cell bank. The cryopreservation of stationary-phase cells at seed stock was done in cryogenic vials (Greiner bio-one, Germany) in combination with a freezing medium comprising 90% FBS plus 10% dimethyl sulfoxide (DMSO; USP grade, Sigma-Aldrich, Germany). The nomenclature of each clone was done based on the D# C# combination, where D represents the name of the dishes (D2-D5) and C represents the colony number in each dish (for example, D2C3 indicates the third colony from dish D2).
Screening of Proliferative Clonal Mesenchymal Stromal cells
For selecting the best clones that had extensive serial passage ability, the P4 cells from each clone were cultured up to P15 in expansion culture medium. In order to save materials and time, a minimum number of cells were cultured in each passage at 5 × 103 cells/cm2 seeding density. The cells from each clones which exhibited acceptable proliferation rate was retained for the next round of passaging. Each clone could reach to P15 with conventional passage ability (confluence during 5–6 days) and spindle-shaped morphology were candidate for the further assessments. These analyses included the investigation of growth kinetics and doubling time, cellular senescence assay, and immunophenotyping. The cells from each clone could pass these assessments, were selected for establishment of the intended cell biobanks. The process of screening the best cMSCs was done in GLP laboratory due to reduce the cost of manufacturing.
Process of Manufacturing the Four-Tiered Cell Banks
The manufacturing process was divided into the ICB, MCB, WCB, and EoPCB steps. All manufacturing process were done in GMP condition. The biobank production was started from the seed stock of each selected clone. For this, one cryopreserved unit from the seed stock of each clone (P3) was thawed and passaged separately. At P6, the cells were cryopreserved as the ICB with the density of 0.5 ± 0.1 × 106 cells/0.5 ml freezing medium in each cryovial. After obtaining QC approval for ICB, including fibroblast-like cell morphology, viability, cell count, sterility, and mycoplasma, the ICB cells were used for MCB production.
In the next step, a cryopreserved unit from the ICB of each clone was separately thawed and expanded for three sequential passages in order to produce the MCB cells. At MCB step, the cells in P10 were cryopreserved with the density of 5 ± 0.5 × 106 cells/2 ml freezing medium for each cryovial. The quality and efficacy of the MCB were assessed by precise control of fibroblast-like cell morphology, cell count, viability, DT, and immunophenotyping for MSC markers and immune markers (CD80, CD86, and HLA-DR). Sterility tests (aerobic, anaerobic, and fungi) and mycoplasma assessments were conducted before and after the MCB storage. The presence of endotoxin, a panel of virus tests similar to that performed for the donor (Supplementary Table 1), in vivo transplantation to evaluate tumorigenesis, and karyotype analysis were conducted at the MCB stage. Potency assessments that included the differentiation potential of the three lineages, induction of angiogenesis, and inhibition of lymphocyte proliferation were also assessed in addition to the acquisition of a dedicated identity by determining short tandem repeats (STR) and HLA typing.
For WCB establishment, one vial from the MCB for each clone was thawed and after two sequential passages, the P13 cells were cryopreserved at a density of 10 ± 0.5 × 106 cells/4 ml freezing medium per cryovial. The assessments in this stage included the evaluation of cell morphology, viability, cell counts, sterility, mycoplasma, and immunophenotyping.
The cryopreservation of stationary-phase cell at ICB, MCB, and WCB steps was done in cryogenic vials (cat no. 123263 for ICB and MCB and 127,277 for WCB, Greiner bio-one, Frickenhausen, Germany) in combination with a freezing medium comprising 90% FBS plus 10% dimethyl sulfoxide.
In order to develop EoPCB, based on the number of cells needed as DP, one to several vials from WCB were thawed and after one sequential passage, the P15 cells were cryopreserved as EoPCB. For cryopreservation of cells at this step, cell suspensions were frozen in freezing bags (Pall Medical, USA) with the serum-free freezing medium specified in this article. The used freezing medium for the final product cryopreservation included injectable normal saline (Samen Pharmaceutical Company, Iran) with 10% pharmaceutical grade human serum albumin (HSA; Kedrion Biopharma, Italy) and 10% DMSO. Assessments of morphology, viability, cell count, sterility, mycoplasma, endotoxin, karyotyping and array comparative genomic hybridization (CGH) analysis were performed at this stage.
Mycoplasma, Bacterial and Fungal Culture Tests
The Mycoplasma Genesig® Standard Kit was used to detect 36 mycoplasma species by quantitative real-time PCR (RT-qPCR) according to the manufacturer’s instructions (Primerdesign, USA). For this purpose, cell supernatant and about 1 × 106 cells were used to identify possible mycoplasma contamination. The sampling process for the detection of mycoplasma was done both during the passaging and the cryopreservation of the cells. For monitoring mycoplasma detection during serial passaging, a schedule for mycoplasma testing was performed at least every two weeks. In each stage cell banking (ICB, MCB, WCB, EoPCB), before- and after freezing samples were used for sterility/mycoplasma inspection. Once each cell bank was stablished, some vials were thawed and tested for sterility/mycoplasma. We evaluated cell sterility by testing the cell supernatant for a wide range of microbes that included aerobic and anaerobic microorganisms, and fungi with a BD BACTEC™ system based on the company’s instructions (BD, USA).
Measurement of Endotoxin
The test was achieved by Gel Clot method LAL ENDOSAFE® (Charles River Endosafe, USA) with 0.125EU/ ml sensitivity as a single test according to the manufacturer’s instruction. Based on the presence or absence of a gel clot, the result is considered positive or negative, respectively. The endotoxin inspection was evaluated at EoPCB step for before/after freezing samples.
Investigation of Growth Kinetics and Doubling Time
Growth kinetics was determined by culturing and performing daily counts of MSCs at three serial passages (P7 to P9 for cMSCs and P2 to P4 for hMSCs) during six days with an initial seeding density of 5 × 103 cells/cm2 in 24-well plates. Cell counting was done both manually using Neubauer hemocytometer and automatically by NucleoCounter® NC-200™ (Chemometec, Denmark) cell counter. Doubling time was calculated during the logarithmic growth phase (days 2–4) with this formula: doubling time (h) = h × ln (2)/ln (C2/C1), where: C is the number of cells at each time of collection and ln is a Napierian logarithm [23].
Cellular Senescence Assay
Assessment of cellular senescence or biological aging was performed with a lysosomal senescence-associated beta-galactosidase (SA-β-GAL) activity assay and a Cellular Senescence Activity Assay Kit (Enzo Life Science, USA) according to the manufacturer’s instructions. Briefly, P10 and P15 cMSCs and P3 hMSCs were cultured for three days at a primary seeding density of 6 × 103 cells/cm2 in six-well plates. The cell lysate was prepared and incubated with SA-β-GAL substrate for one hour at 37 °C. Fluorescence was read with a plate reader (Synergy™ H4 Hybrid Multi-Mode Microplate Reader, BioTek®) at 360 nm (excitation)/465 nm (emission), and the results were recorded in relative fluorescence units.
Differentiation Potential Analysis
cMSCs and hMSCs were cultured and differentiated with StemPro osteogenesis, adipogenesis, and chondrogenesis differentiation kits (Thermo Fisher, USA) according to the manufacturer’s instructions. Briefly, MSCs were cultured and after reaching to 70–80% confluency, specific-differentiation media was added for 21 days. The degree of differentiation towards osteogenic, chondrogenic, and adipogenic lineages was determined by tissue-specific staining and gene expression. For staining, adipogenic and osteogenic differentiated cells were stained with oil red O (Sigma-Aldrich) and alizarin red S (Sigma-Aldrich) stains. Chondrogenic differentiated cells were processed as paraffin-embedded blocks. The blocks were sectioned into 5–6 µm thick sections, and the sections were transferred onto glass slides. After removal of the paraffin, the slides were stained with Alcian blue (Sigma-Aldrich). A phase-contrast CKX41 microscope (Olympus, Japan) connected to a DP72 camera (Olympus) was used for image acquisition. The gene expression level of lineage markers was examined by RT-qPCR. The evaluated genes were osteogenic-related markers including collagen type 1 (COL I), parathyroid hormone (PTH), and osterix (OSX), adipogenic-related genes including lipoprotein lipase (LPL), proliferator-activated receptor gamma (PPARg), and adiponectin (ADIPOQ), and chondrogenic-related genes COL I, COL II, and COL X.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR) Analysis
Briefly, an RNeasy Micro Kit (Qiagen, Germany) was used to extract total RNA from the three independent replicates of both cMSCs and hMSCs differentiated cells. Evaluation of purity and concentration of the extracted RNA was done at A260 nm/A280 nm using a Biowave II spectrophotometer (Biochrom, UK). The quality and integrity of the total RNA were verified by electrophoresis. According to the manufacturer’s instructions, a total amount of 2 µg of total RNA was converted into cDNA using a QuantiTect Reverse Transcription Kit (Qiagen, Germany). RT-qPCR was carried out using SYBR Green master mix (Qiagen). The reactions were carried out in duplicate, and RT-qPCR amplification was performed according to the following program: stage 1 at 95 °C for 10 min and stage 2 (40 cycles) at 95 °C for 10 s and 60 °C for 60 s. The target gene expression levels were normalized against the reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and compared with the cells’ undifferentiated states. The relative quantification of gene expression was calculated using the ΔΔCt method. Supplementary Table 1 lists the primers used in this study.
Immunomodulation Assay: Lymphocyte Proliferation Assay
The immunomodulatory properties of the cMSCs were compared to hMSCs by running the lymphocyte proliferation assay assay with peripheral blood mononuclear cells (PBMCs). First, both cMSCs and hMSCs were treated with 10 µg/ml mitomycin C (Sigma-Aldrich) for 2 h, after which the cells were washed and dissociated. The resultant cell suspension of either cMSCs or hMSCs was used to prepare a cell suspension of two ratios (1:2 and 1:10) for co-culturing with PBMCs. We added 250 µl of each prepared cell ratio to each well of a 96-well plate. The 96-well plate was put in the incubator (37 ˚C, 5% CO2, and 80% humidity) for 24 h. The next day, peripheral blood was collected from a random healthy donor, and the PBMCs were separated according to a conventional method using Ficoll®-Paque (GE Healthcare). The PBMCs were marked with 5 mM CellTrace™ (CFSE Cell Proliferation Kit; Thermo Fisher) for 5 min, and 105 PMBC was added to the wells that contained the test (with MSCs) and control groups (without MSCs). PBMCs were induced to proliferate by using 10 µg/ml phytohemagglutinin, PHA-P (Sigma-Aldrich). Cells were incubated for 72 h, and we determined the PBMC proliferation rate using a BD FACSCalibur™ (BD Bioscience, USA) flow cytometer. Data were analyzed using Flowing software 2.5.1 and compared to the non-co-cultured state.
Flow Cytometry Analysis
For immunophenotyping, the expression levels of the CD73, CD90, CD105, CD14, CD20, CD34, and CD45 cell surface markers were detected using a human MSC phenotyping kit (MACS Miltenyi Biotec, Germany). We determined immunogenicity with flow cytometry analysis of antibodies against the B7 family, CD80 (BD Biosciences), CD86 (BD Biosciences), and HLA-DR (DAKO, Denmark) family. Data acquisition and analysis were performed with a BD FACSCalibur™ and Flowing software 2.5.1, respectively.
Karyotyping and Array Comparative Genomic Hybridization Analysis
Karyotype assessments were performed as previously described by Fathi et al. [24]. For doing array CGH analysis, cMSCs at P15 were examined as its instruction (Agilent Technologies, USA).
Tube Formation and Scratch Assays
An angiogenesis starter kit (Thermo Fisher) was used to evaluate the ability of the cMSCs and hMSCs to induce angiogenesis. Briefly, 48-h conditioned media of both the cMSCs and hMSCs cultures were used to evaluate the induction of tube formation by Mitomycin C (10 μg/ml) inactivated human umbilical vein endothelial cells (HUVECs). The HUVECs were seeded at a density of 6 × 103 cells per well in Geltrex pre-coated 96-well plates and incubated at 37 °C for 24 h. Subsequently, each well was photographed under a phase-contrast microscope (magnifications: 40x, 100x). The observed nodes and branching points were counted in order to quantify angiogenesis, and the total tube length was measured by ImageJ software version 1.47 (National Institutes of Health, USA). All experiments were conducted independently and at least three times.
The scratch assay was performed in a 24-well plate to evaluate the induction potential of migration by cMSCs. The supernatant of cMSCs and hMSCs were collected in both the resting and primed conditions with Interferon gamma (IFNγ, R&D Systems, USA). As a prerequisite step, 4 × 103 cells of both cMSCs and hMSCs were separately seeded in each well of a 24-well plate (two wells for each resting and primed state). On day three of the culture, the primed wells under consideration were treated with 10 ng/ml IFNγ. The culture media was removed about 18 h after priming, and the cells were washed with PBS buffer (without Ca2+ and Mg2+; Thermo Fisher). Fresh culture media was added, and after 48 h, the conditioned media was collected for the assay.
Further, mitomycin C-treated HUVECs were cultured, and a scratch area was created using a sterile 2 µL pipette tip on the cell monolayers. The cells were subsequently treated with a 48-h conditioned medium. The closure area was photographed by inverted phase-contrast microscopy (Olympus) and an installed digital camera (Olympus) at 12, 24, and 48 h after generation of the scratch area. ImageJ software was used to calculate the average closure area. This experiment was conducted in triplicate.
Tumorigenicity Assessment
Around 5 × 106 cells of P15 cMSCs and the same amount of human embryonic stem cells (hESCs), as the positive control cells, were harvested at their logarithm phase, centrifuged, and mixed with 50 µl ice-cold Matrigel (Sigma-Aldrich). The cells were injected into the flanks of six-week-old male nude mice at the subcutaneous root. The injected mice were followed for three months. Mice in the groups with teratoma formation were sacrificed when the tumors were 25 to 50 mm in diameter. The tumors were surgically removed, fixed, embedded in paraffin, and sectioned into 60 µm thick sections for hematoxylin–eosin staining. The stained sections were photographed by a bright field microscope.
Statistical Analysis
All data are presented as mean ± SD. Data were analyzed with GraphPad Prism, version 6.0. One-way analysis of variance (ANOVA) following Tukey’s post hoc was used to determine statistically significant differences between values of cMSCs and hMSCs in senescence, doubling time (DT), and immunosuppression of lymphocyte assays. Immunogenicity, angiogenesis, and scratch assay data were assessed by two-way ANOVA following Tukey’s post hoc. The level of significance was set at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Culture of bone marrow-derived colony forming units to isolate mesenchymal stromal cells and establish the seed stock cell bank
SCM approach allowed us to obtain 27 single CFU-derived colonies from the supernatants of a BM aspirate culture. From these, the cells from 21 clones could reach to P3 with an acceptable proliferation rate and a fibroblastic spindle-shaped morphology. Two separate processes were performed on the cells derived from these clones. First, a small number of cells from each clone was passaged again in order to find the best clone(s), mainly based on the long-term passage ability, during the serial passaging (see the next result). The culture dishes of these cells were transferred from the GMP clean room to the GLP laboratory to reduce the cost of the manufacturing. Second, the remaining cells of each clone were cryopreserved at P3 as the seed stock cell bank. The term ‘seed stock’ is designated to the cryopreserved stocks of cMSCs established in the early passages. After selecting the best clone(s) in the GLP laboratory, the seed stock of the selected clone(s) would be considered as the starting materials for the establishment of ICB, MCB, WCB, and EoPCB under GMP-condition for the further applications in clinical trials.
Screening of Clonal Mesenchymal Stromal Cells with the Capability of Lengthy Serial Passaging Under Good Laboratory Practice Conditions
During clone selection process we found that among 21 clones, five reached P10 and were expanded for further analysis (Fig. 1A). These clones underwent additional passaging until P15 (Fig. 1A, B). Assessment of SA-β-GAL activity between these cMSCs and their P3 hMSCs counterparts revealed that all five derived cMSCs, either at P10 or P15, were significantly less senescent than the P3 hMSCs (Fig. 1C). Flow cytometry data showed that the specific mesenchymal surface markers (CD37, CD90, and CD105) were expressed more than 95% in all five cMSCs and concomitant with decreased expressions of non-specific markers (CD14, CD20, CD34, and CD45) (Fig. 1D). Interestingly, expressions of the CD markers at P15 of the five cMSCs were more uniform than hMSCs at P3 (Fig. 1E). Evaluation of the growth curve during three subsequent passages and the daily cell counts revealed that the logarithmic phase of these five cMSCs began on day two and continued up to day 5 (Fig. 1F). However, calculating DT showed that cMSCs derived from the D2C4, D5C1, and D5C3 clones were similar to young hMSCs (50 h; p > 0.05), whereas the DT was over 70 h and proliferation was slower than the hMSCs (p > 0.05) in cMSCs derived from the D2C3 and D5C2 clones (Fig. 1G). Finally, we chose three cMSCs derived from the D2C4, D5C1, and D5C3 clones for further evaluation based on the proliferation ability.

Clone selection based on long-term proliferation ability and MSC-related criteria. A Bars represent long-term serial passaging of the different clones derived from 2 ml of aspirated BM. As shown in the chart, 5 out of 21 clones reached P15 (*). B Cell morphology of selected cMSCs at P15 and their counterpart hMSCs at P5. C Senescence was evaluated by measuring senescence-associated β-galactosidase (SA-β-GAL) activity and expressed as relative fluorescence units (RFU) of the same number of cultured cells. Data are expressed as mean ± SD of six replicates. Statistically significant difference compared with the hMSCs group. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. D Quantitative flow cytometry data show the positive expressions of CD73, CD90, and CD105 and negative expressions of CD14, CD20, CD34, and CD45. Mean ± SD, n = 3. E Histogram view of specific MSC markers (solid line) and their isotype controls (shaded line). F Growth curves were calculated by counting cultured cells with the same seeding count during six days. Each line represents a passage number (three serial passages) with three replicates per day (n = 9). G DT was generated using cell counts at days 2 and 4 as the growth curve’s logarithm phase. Results are expressed as mean ± SD of nine replicates. Statistically significant difference compared with the hMSCs group. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. DT: Doubling time, MSC: Mesenchymal stromal cells; SCM: Subfractionation culturing method, cMSCs: Clonal mesenchymal stromal cells, BM: Bone marrow, hMSCs: Heterogeneous mesenchymal stromal cells
The Selected Proliferative Clones Meet the Criteria for Mesenchymal Stromal Cell Potential
We comprehensively characterized the selected cMSCs and compared them with the hMSCs. Lineage differentiation potential of three P15 selected cMSCs were compared with P3 hMSCs after culturing in differentiation media for 21 days. The osteogenic potential was visualized by alizarin red staining which show that the mineralized nodules accumulated after 21 days in ECM around of differentiated cells in three cMSCs as well as hMSCs (Fig. 2A). Also, RT-qPCR analysis confirmed that the expression level of Col I, PTH and OSX genes up-regulated within 3 weeks in both groups in comparison to their undifferentiated counterparts, although, slight differences were seen between the clones (Fig. 2B). Also, Oil red O staining in adipocytes differentiated cells revealed that the cytoplasmic oil droplets accumulated in all three cMSCs and hMSCs (Fig. 2A). RT-qPCR analysis of LPL, PPARg and ADIPOQ indicated a high expression level of genes in both groups in comparison to their undifferentiated counterparts (Fig. 2B). The ability of cells to chondrogenic differentiation was measured by Alcian blue staining which indicating sulphated proteoglycans areas on the cross-sections in three cMSCs and hMSCs (Fig. 2A). Analysis of genes involved in chondrogenesis, Col I, COL II and COL X, showed that both groups expressed chondrogenic-related genes and COL X showed significantly upregulation versus undifferentiated MSCs (Fig. 2B).

Full characterization of passage 15 (P15) selected clones. A Differentiation assessment of cMSCs and hMSCs using alizarin red S, oil red O, and Alcian blue, respectively, for osteogenic, adipogenic, and chondrogenic differentiation (bar: 100 µm). B Expressions of mesenchymal specific differentiation genes evaluated by RT-qPCR. Data are compared to the undifferentiated state. Each bar represents three individual replicates. Mean ± SD, n = 6. P-values less than 0.05 were considered significant (*P < 0.05; **P < 0.01, ***P < 0.001, ****P < 0.0001). C In vitro immunomodulation of P15 cMSCs and P5 hMSCs on healthy donor PBMCs under allogeneic conditions. Co-culture effect of MSCs on the suppression percentage of PBMCs. PBMCs were stimulated with PHA. Data were normalized with the stimulated PBMCs in the absence of MSCs. Each bar shows proliferation suppression of 1 × 105 CFSE-labeled PBMCs at ratios of 1:2 and 1:10 (MSCs:PBMCs). Mean ± SD, n = 2. P-values less than 0.05 were considered significant (***p < 0.001). D Scratch wound migration assay induced by 48-h secretome of cMSCs (P15) and hMSCs (P3) in resting and primed conditions. Migration was assessed by measuring the scratched area at 0, 12, 24, and 48 h after starting the test on mitomycin C-treated HUVECs. The bar chart shows the quantitative assessment of the wound area. P-values lower than 0.05 were considered significant (*p < 0.05; **p < 0.01; ***p < 0.001; N.S.: Non-significant). E In vitro angiogenesis stimulated by 48-h secretome of cMSCs (P15) and hMSCs (P3) (Scale bar: 100 µm). The bar chart shows the quantitative assessment of angiogenesis by ImageJ software. Each bar represents three replicates. F Histogram and bar charts for flow cytometry analysis of immunogenicity markers (HLA-DR, CD80, and CD86) in cMSCs, hMSCs, and moDCs (positive control). Data are from three replicates. P-values lower than 0.05 were considered significant (**p < 0.01; ***p < 0.001). G Karyotype of cMSCs (P15) through the G-banding method. cMSCs: Clonal mesenchymal stromal cells, hMSCs: Heterogeneous mesenchymal stromal cells, PBMCs: Peripheral blood mononuclear cells, PHP: Phytohemagglutinin PHA-P, HUVECs: Human umbilical vein endothelial cells, moDCs: Monocyte-derived dendritic cells
Furthermore, inhibition of lymphocyte proliferation was studied in order to evaluate the immunomodulation potential. The three P15 cMSCs and the P3 hMSCs showed almost more than 80% ability to inhibit immune cell proliferation compared to the untreated group (Fig. 2C). The migration ability of P15 cMSCs was compared with P3 hMSCs. The MSCs were first committed to anti-inflammatory condition by IFNγ treatment (primed state), and their secretome was compared with the un-committed condition (resting state). Scratch assay analysis showed that the secretome of 48 h-primed cMSCs significantly promoted migration of mitomycin C-treated HUVECs to decrease the closure area compared to hMSCs (p < 0.001; Fig. 2D). The secretome of cMSCs was also used to induce in vitro angiogenesis of HUVECs with an angiogenesis starter kit. Different angiogenesis parameters that included the numbers of nodes, junctions, and branches, in addition to the total tube length and total branching length, showed that secretome in both the resting and primed conditions could quantitatively support tube formation. There were no noticeable differences between cMSCs and hMSCs in this assessment (Fig. 2E).
It has been reported that prolonged cultivation of MSCs might harm their immunomodulatory ability [25]. A comparison of P15 cMSCs and P3 hMSCs showed low-level expressions (< 5%) of co-stimulatory factors CD80 and CD86 (Fig. 2F). The average expression of HLA-DR was less than 5% in the D2C4 and D5C1 clones and about 10% in the D5C3 clone and in hMSCs (Fig. 2F). There were no numerical or structural chromosomal aberrations observed in the P15 cMSCs after classical karyotyping (Fig. 2G). Overall, based on the evaluated criteria, the selected cMSCs were chosen to establish the cell bank system and guarantee the availability of high-quality, authentic human MSCs for clinical applications.
Establishment of Four-Tiered Clonal Mesenchymal Cell Banks Under Good Manufacturing Practice Conditions
The roadmap for manufacturing of ICB, MCB, WCB, and EoPCB steps and the corresponding QC has been exhibited in Fig. 3. The biobank production was started separately from the seed stock of the selected D2C4, D5C1, and D5C clones. As indicated in Table 1, the numbers of cryopreserved vials for each clone at ICB step were: 30 vials (D2C4), 36 (D5C1), and 36 (D5C3). This number of cells in the ICB provides a suitable downstream resource for each cMSC line to create future large biobanks without the need for new donor recruitment and a lengthy process for optimal clone selection. After obtaining a QC approval for morphology, viability, cell count, sterility, and mycoplasma, we could use the ICB cells for MCB production (Fig. 3).

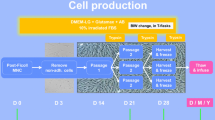
Process flow diagram for GMP-grade manufacturing of cMSCs and the necessary QC assessments. Schema indicated the master production schedule for BM sampling, the manufacturing steps, and biobanking concerning the passage numbers and QC tests. The approximate timescale for the main production stages from BM sampling to seed stock establishment as well as clone selection process is written in red. The timescale for establishment of the four tiered cell bank (written in blue) is started from the seed stock of the selected clones. The minimum time needed for production and QC assessments were considered for estimating the timescale. GMP: Good manufacturing practice; BM: Bone marrow; cMSCs: Clonal mesenchymal stromal cells; QC: Quality control; ICB: Initial cell bank, MCB: Master cell bank, WCB: Working cell bank, EoPCB: End of product cell bank, IPQC: In-process quality control, GLP: Good laboratory practice
In the MCB step, a total of 65 vials were cryopreserved for the D2C4 clone, 80 for the D5C1 clone, and 80 for the D5C3 clone (Table 1). A complete QC characterization of safety, efficacy, and quality was conducted based on the illustrated flowchart due to the importance of MCB as the initial stage for manufacturing the large biobank. Supplementary Fig. 2 shows the certificate of analysis, for the MCB of one clone, D5C1.
Because the number of cells used in each MSC-related clinical trial can differ by tens of millions per kilogram of body weight, we decided to add another tier, WCB, before the EoPCB. The WCB could ensure fast, reliable access to the required number of adequate cells in the EoPCB per requirements. In the WCB, we cryopreserved 70 vials for D2C4, 80 for D5C1, and 75 for D5C3 (Table 1). We assessed the minimum necessary qualifications at this stage (Fig. 3).
In order to develop EoPCB, we first needed to develop a formulation for serum- and xeno-free cryopreservation medium as the excipients of DP and determine the number of cells as the active pharmaceutical ingredient of DP (Fig. 4A). First, several GMP-compatible freezing media, which included sodium lactate with 10% human serum albumin (HSA), Plasma-Lyte A with 10% HSA, and normal saline with 10% HSA (all media plus 10% DMSO) were considered for storage of 2.5 ± 0.5 × 106 cells (Fig. 4B). After one month of preservation in the LN vapor phase, there was no significant difference observed in terms of cell count, viability, and fibroblast-like morphology among the cells cryopreserved with the above-mentioned media and the common research-grade freezing medium (90% FBS + 10% DMSO (Fig. 4B). Furthermore, cell-specific immunophenotyping of cMSCs (D5C1) was in the acceptable range for all of the freezing media (Fig. 4C). Therefore, we selected a freezing medium that contained normal saline with 10% HSA and 10% DMSO to store cells at EoPCB.

Development of the final product manufacturing process. A Flowchart of the final product manufacturing process development in which the number of packaged cells is determined for the target disease. B The table compares cell count and viability between several types of freezing media compatible with the therapeutic targets and the research-grade freezing medium that contains FBS. Morphology of the thawed cells, which were frozen in various clinical-grade media or research-grade freezing medium. C Immunophenotyping of thawed cells was performed using the MSC surface marker panels. MSC: Mesenchymal stromal cell; FBS: Fetal bovine serum
In order to determine the number of cells in DP, we selected about 75 × 106 cells/cryobag for future studies that needed 1 × 106 MSCs per kilogram of body weight for transplantation, which was based on the average adult human weight in Iran [26]. We used these packages to evaluate the quality of storage, stability, release, and transport of DP with almost rigorous cell numbers per package. After thawing three vials from WCB and one sequential passage, 9 cryobags for D2C4, 10 for D5C1, and 9 for D5C3 that each contained 75 ± 5 × 106 cells per 15 ml of freezing medium (5 × 106 cells/ml) were cryopreserved as EoPCB (Table 1). In addition to assessment of the minimum necessary qualifications, karyotyping and array CGH were also performed at this stage and confirmed the safety of the EoPCB cells (Supplementary Fig. 3). We followed the stability of these EoPCB cells after 12 and 18 months of storage in the LN vapor phase. Our results showed that the cell count, viability, and immunophenotyping were within the normal ranges compared to earlier time points (Table 2). Overall, the rigorous evaluations performed during the selection of reproducible cMSCs ensured that the selected cMSCs could establish four-tier cell banks within GMP-compliant conditions.
Release and Transport of Drug Product
Frozen CTP should be transported in high-quality containers that maintain a low temperature using either an LN-dry shipper or electrically driven cooling devices. Due to the limitations in the transport of CTPs with the mentioned devices that should be returned from the clinic to the cell processing laboratory, we evaluated the transport of DP with dry ice. First, we examined cell viability after removing frozen cells from the vapor phase of LN and storage in a -80 °C freezer (acceptable temperature range: -70 to -80 °C) for one week. Our validation study showed that the cell viability was ≥ 90% up to 96 h (Table 3). Next, we examined the maintenance of a temperature range (-70 to -80 °C) in a Styrofoam box that contained two, 1.5 kg dry ice bags placed at the bottom and top of the chamber. The sample and data sensor logger were placed in the middle (Supplementary Fig. 4). The data showed that the container could maintain the acceptable temperature range for up to 15 h (Table 4). Assessment of the cells after transport confirmed the integrity of the packaging in terms of leakage and breakage. Also, analysis of these cells after thawing showed that they had acceptable viability and sterility. These results could ensure that establishment of EoPCB and the transport process were designed entirely safe for transport of the cell therapy DP to the clinic.
Ethical approval was successfully obtained to release our final product in a clinical trial phase I/II for COVID-19 patients (NCT04366063). According to the previous studies published for COVID-19, a package of 108 cells/20 ml was considered for each patient [27]. We manufactured 40 cryobags containing 108 cells/20 ml freezing medium from D5C1 cMSCs for this clinical trial. The results are being studied (unpublished data).
Discussion
The isolation and manufacturing of human BM-cMSCs with SCM is an optimal method for establishing an allogeneic homogeneous cell bank under GMP conditions [18]. However, the isolation of numerous clones and the need for their simultaneous expansion to choose the best proliferative clones which meet the criteria for defining MSCs is costly and time-consuming. In this study, by placing a seed stock step in the process of manufacturing cMSCs, we could store all expanded clones at an early passage, P3. After the process of proliferative clone selection, only the selected clones were considered as the starting material for establishment of the GMP-compatible cell banks from the seed stock. By this strategy, we could start the process of establishment of the extensive qualified tiered cell banks from the three selected clones among the 21 isolated ones that had been preserved in the seed stock.
Our initial expectation was that the capabilities of the manufactured clones were different from each other. Although the strategy proposed by the others is to classify clones based on differences in their potential and assigning them to different indications [28, 29], our selected clones showed only minor differences in terms of differentiation and migration potential. We believe that, due to the strict restrictions we imposed during clone screening, similar clones were selected. However, further studies are needed to ensure functional differences between these clones. For example, it is shown that a line of cMSCs that highly express brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) could significantly reduce the fibrotic scar formation in a rat spinal cord injury model in comparison to hMSCs. This approach suggests a new attitude to select tissue-specific cMSCs to target specific tissues or diseases [30].
We applied SCM for establishment of the CFU-derived cMSCs in order to obtain homogeneous population; however, it is also believed that cMSCs display intracolony heterogeneity (reviewed in [31]). This heterogeneity may result from multiple progenies within a putative clone or even from single cell-derived one. It seems that asynchronous cell division, and the senescence that follows, drive intracolony heterogeneity [32]. The slow-dividing senescent MSCs are recognizable by their large spread cytoplasm from the fast-dividing slender spindle shaped cells [32, 33]. Although we could not claim that the offspring of each clone are molecularly and functionally homogenous, the three established cMSCs at P15 indicated high homogeneity in terms of displaying the fibroblast-like morphology. Moreover, this was quite noticeable to us that the cell proliferation rate was different among the BM-isolated clones and some clones exhibited the significant high proliferation ability expandable up to P15. Further analysis of secretome, proteome, and transcriptome of BM-isolated cMSCs may reveal more details about the characteristics of these clones. Moreover, these extensive analyses can provide a better understanding about cMSC identity through the comparative assessment with MSCs derived from the other important sources such as adipose and Wharton’s jelly of the human umbilical cord. For example, it is shown that Wharton’s jelly-derived MSCs overexpress genes involved in neurotrophic support and their secretome could induce maturation of neuroblastoma cells to a greater extent than hMSCs [34]. Also, the secretome analysis of Wharton’s jelly-derived MSCs exhibited more diverse composition than hMSCs suggesting greater therapeutic potential [35].
We performed all the QC tests during clone selection at P15 and compared cMSCs with hMSCs at P3-P5. We found that cMSCs at P15 could pass all capabilities needed to use these lines as CTPs. It has been previously shown that cMSCs have a high proliferation ability and are expandable up to P16 without significantly compromising its characteristics [19]. Increasing the number of passages could greatly increase the capacity of the number of cells in the EoPCB, which was of paramount importance to develop an allogeneic cell bank. Since expansion of hMSCs usually more than five passages lead to senescence [33], the use of proliferative cMSCs can overcome this challenge. On the other hand, the higher number of passage raises a major concern about the possibility of transformation and chromosome abnormality of the cells. Nevertheless, karyotyping and array CGH analysis at the process of clone selection and also the repeating of these assessment at the process of biobanking, especially in the step of EoPCB, confirmed the safety of cMSCs at the higher passages.
In order to comprehend the quantitative potential of our allogeneic biobank that can be virtually manufactured via GMP and based on the SCM, we have to interpret the practical data and present its estimated numbers. As shown in Table 1 and Fig. 5, by thawing and expansion all of the units in ICB, MCB, and WCB, we estimate between 430,000 to 760,000 cryobags that contain 75 ± 5 × 106 cells could be obtained. Thus, with only one BM sample, the use of SCM for the establishment of CFU-derived cMSCs, and through the strategy applied in this study that included the establishment of seed stock, selecting the best proliferative clones, and four-step cell banking, we could organize an allogenic MSC banking system that has a production capacity of nearly 1,880,000 cryobags with 75 ± 5 × 106 cells, as an example. Of course, it should be noted that the conventional two-dimensional monolayer condition in tissue culture flasks that we used in this study does not fit the large-scale industrial culture of MSCs. Nonetheless, scale-up of suspension culture system, such as culture on microcarriers in stirred-tank bioreactors, offers more advantages for use in such commercial processes [36,37,38]. However, the use of such an approach raises concerns about the main characteristics of MSCs, such as immunomodulatory properties and trophic effects, that requires precisely evaluation to ensure the efficacy of these cells [39]

Schematic layout of the biobanking system for the CFU-derived MSC lines. Schematic layout of the biobanking system and development of the cells for the COVID-19 clinical trial as an empirical example that can be extended to other clinical trials. CFU: Colony-forming unit; MSC: Mesenchymal stromal cell
For therapeutic applications, cMSCs must be produced without animal-derived components (xeno-free) following GMP standards. In addition, the excipients should be identified and examined precisely with the validated tests. At the beginning of the production process (direct BM culture), we had to use FBS to facilitate cell adhesion and the formation of early colonies. However, during the rest of the production, we replaced FBS with hPL. The use of hPL instead of FBS not only maintains and improves cell quality it also eliminates the risk of immune reactions and transmission of animal pathogens [40].
One of the significant challenges of using the cells as DP is their independence to the cell processing center. It needs a freezing medium that could be injected into the patient directly or just by dilution in physiological serum. We chose a freezing medium that contained 10% HSA and 10% DMSO in normal saline. The limit for DMSO administration that is recommended by the USA FDA-approved cord blood hematopoietic progenitor cell therapy is one gram (~ 1.1 ml) per kilogram of body weight per day (Package Insert – Ducord; https://www.fda.gov/media/84567/). Thus, the EoPCB-developed cryobags which contained 15 ml freezing medium (including 1.5 ml DMSO), can be injected directly into the patient. However, it is recommended to decrease the concentration of DMSO to less than 3.5% [41] which it can be done simply by diluting the cell product solution with normal saline during injection.
An important matter not mentioned in this study pertains to the preclinical toxicity studies for the established cMSC lines, which we are preparing in a separate report. Altogether, this study presents a technical and translational overview of cMSCs manufacturing technology under GMP conditions that could be used to guide the development of similar production processes with therapeutic goals.
Conclusion
We developed a fully defined roadmap for the isolation and selection of proliferative cMSCs by taking advantage of the SCM. The designed seed stock step for reserving all clones at early passages allowed us to store all of the isolated clones at early passages as an initial source for establishing a high capacity four-tired cell bank system for allogeneic cell therapy under GMP conditions. These studies would be beneficial for the application of MSCs in various clinical trials.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- BM:
-
Bone marrow
- CFUs:
-
Colony-forming units
- cMSCs:
-
Clonal MSCs
- CTPs:
-
Cell therapy products
- DP:
-
Drug product
- DT:
-
Doubling time
- EoPCB:
-
End of product cell bank
- GLP:
-
Good laboratory practice
- GMP:
-
Good manufacturing practice
- hPL:
-
Human platelet lysate
- HUVECs:
-
Human umbilical vein endothelial cells
- ICB:
-
Initial cell bank
- IFNγ:
-
Interferon gamma
- MCB:
-
Master cell bank
- QC:
-
Quality control
- RT-qPCR:
-
Quantitative reverse transcription polymerase chain reaction
- SCM:
-
Subfractionation culturing method
- WCB:
-
Working cell bank
References
Regulation, E., No. 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending directive 2001/83/EC and regulation (EC) no 726/2004. Journal of European Union, (324), 121–137.
Barnhoorn, M. C., et al. (2020). Long-term Evaluation of Allogeneic Bone Marrow-derived Mesenchymal Stromal Cell Therapy for Crohn’s Disease Perianal Fistulas. Journal of Crohn’s & Colitis, 14(1), 64–70.
Khalifeh Soltani, S., et al. (2019). Safety and efficacy of allogenic placental mesenchymal stem cells for treating knee osteoarthritis: A pilot study. Cytotherapy, 21(1), 54–63.
Levy, M. L., et al. (2019). Phase I/II Study of Safety and Preliminary Efficacy of Intravenous Allogeneic Mesenchymal Stem Cells in Chronic Stroke. Stroke, 50(10), 2835–2841.
Matthay, M. A., et al. (2019). Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): A randomised phase 2a safety trial. The Lancet Respiratory Medicine, 7(2), 154–162.
Gao, F., et al. (2016). Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death and Disease, 7, e2062.
Uccelli, A., Pistoia, V., & Moretta, L. (2007). Mesenchymal stem cells: A new strategy for immunosuppression? Trends in Immunology, 28(5), 219–226.
Moll, G., M.J. Hoogduijn, and J.A. Ankrum. (2020). Editorial: Safety, Efficacy and Mechanisms of Action of Mesenchymal Stem Cell Therapies. Frontiers in Immunology, 11(243).
Levy, O., et al., Shattering barriers toward clinically meaningful MSC therapies. Sci Adv, 2020. 6(30): p. eaba6884.
Wilson, A., Webster, A., & Genever, P. (2019). Nomenclature and heterogeneity: Consequences for the use of mesenchymal stem cells in regenerative medicine. Regenerative Medicine, 14(6), 595–611.
Phinney, D. G. (2012). Functional heterogeneity of mesenchymal stem cells: Implications for cell therapy. Journal of Cellular Biochemistry, 113(9), 2806–2812.
Costa, L. A., et al. (2021). Functional heterogeneity of mesenchymal stem cells from natural niches to culture conditions: Implications for further clinical uses. Cellular and Molecular Life Sciences, 78(2), 447–467.
Glenn, J. D., & Whartenby, K. A. (2014). Mesenchymal stem cells: Emerging mechanisms of immunomodulation and therapy. World of Journal Stem Cells, 6(5), 526–539.
Quirici, N., et al. (2002). Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Experimental Hematology, 30(7), 783–791.
Zhou, B. O., et al. (2014). Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell, 15(2), 154–168.
Gronthos, S., et al. (2003). Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. Journal of Cell Science, 116(9), 1827–1835.
Sacchetti, B., et al. (2007). Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell, 131(2), 324–336.
Yi, T., et al. (2015). Manufacture of Clinical-Grade Human Clonal Mesenchymal Stem Cell Products from Single Colony Forming Unit-Derived Colonies Based on the Subfractionation Culturing Method. Tissue Engineering. Part C, Methods, 21(12), 1251–1262.
Song, S. U., et al. (2008). Variations of clonal marrow stem cell lines established from human bone marrow in surface epitopes, differentiation potential, gene expression, and cytokine secretion. Stem Cells Devision, 17(3), 451–461.
Lee, A. S., et al. (2013). Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nature Medicine, 19(8), 998–1004.
Emadedin, M., et al. (2012). Intra-articular injection of autologous mesenchymal stem cells in six patients with knee osteoarthritis. Archives of Iranian Medicine, 15(7), 422–428.
Böyum, A. (1968). Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scandinavian Journal of Clinical and Laboratury Investigation Supplement, 97, 77–89.
Roth, V., Doubling time computing. Avaible from: http://www.doubling-time.com/compute. php, 2006.
Fathi, A., et al. (2009). Comparative proteome and transcriptome analyses of embryonic stem cells during embryoid body-based differentiation. Proteomics, 9(21), 4859–4870.
Ankrum, J. A., Ong, J. F., & Karp, J. M. (2014). Mesenchymal stem cells: Immune evasive, not immune privileged. Nature Biotechnology, 32(3), 252–260.
Ahranjani, S. A., et al. (2012). Waist Circumference, Weight, and Body Mass Index of Iranians based on National Non-Communicable Disease Risk Factors Surveillance. Iranian Journal of Public Health, 41(4), 35–45.
Rajarshi, K., Chatterjee, A., & Ray, S. (2020). Combating COVID-19 with mesenchymal stem cell therapy. Biotechnology Reports Amsterdam, 26, e00467.
Yin, J. Q., Zhu, J., & Ankrum, J. A. (2019). Manufacturing of primed mesenchymal stromal cells for therapy. Nat Biomed Eng, 3(2), 90–104.
Gronthos, S., et al. (2003). Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. Journal of Cell Science, 116(Pt 9), 1827–1835.
Kim, M., et al. (2018). Transplantation of human bone marrow-derived clonal mesenchymal stem cells reduces fibrotic scar formation in a rat spinal cord injury model. Journal of Tissue Engineering and Regenerative Medicine, 12(2), e1034–e1045.
Rennerfeldt, D. A., & Van Vliet, K. J. (2016). Concise Review: When Colonies Are Not Clones: Evidence and Implications of Intracolony Heterogeneity in Mesenchymal Stem Cells. Stem Cells, 34(5), 1135–1141.
Rennerfeldt, D.A., et al., (2019). Emergent heterogeneity in putative mesenchymal stem cell colonies: Single-cell time lapsed analysis. PLoS One, 14(4), e0213452.
Liu, J., et al., (2020). Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Frontiers in Cell and Developmental Biology, 8(258).
Donders, R., et al. (2018). Human Wharton’s Jelly-derived stem cells display a distinct immunomodulatory and proregenerative transcriptional signature compared to bone marrow-derived stem cells. Stem Cells and Development, 27(2), 65–84.
Shin, S., et al. (2021). Comparative Proteomic Analysis of the Mesenchymal Stem Cells Secretome from Adipose, Bone Marrow, Placenta and Wharton’s Jelly. International Journal of Molecular Sciences, 22(2), 845.
Abbasalizadeh, S., et al. (2017). Allogeneic cell therapy manufacturing: Process development technologies and facility design options. Expert Opinion on Biological Therapy, 17(10), 1201–1219.
Hanga, M. P., et al. (2021). Expansion of human mesenchymal stem/stromal cells on temporary liquid microcarriers. Journal of Chemical Technology & Biotechnology, 96(4), 930–940.
Rafiq, Q. A., et al. (2013). A quantitative approach for understanding small-scale human mesenchymal stem cell culture - implications for large-scale bioprocess development. Biotechnology Journal, 8(4), 459–471.
Cherian, D.S., et al. (2020). Biological Considerations in Scaling Up Therapeutic Cell Manufacturing. Frontiers in Pharmacology, 11(654).
Hemeda, H., Giebel, B., & Wagner, W. (2014). Evaluation of human platelet lysate versus fetal bovine serum for culture of mesenchymal stromal cells. Cytotherapy, 16(2), 170–180.
Windrum, P., et al. (2005). Variation in dimethyl sulfoxide use in stem cell transplantation: A survey of EBMT centres. Bone Marrow Transplantation, 36(7), 601–603.
Acknowledgements
The authors sincerely appreciate the efforts by their collaborators at Royan ATMP Technology Development Center and the Department of Stem Cells and Developmental Biology at Royan Institute. Special gratitude goes to the volunteer who donated primary materials for the study.
Funding
This work was supported by grants from Royesh Venture Capital Fund to HB. The funding source had no responsibilities in the study design, data collection, analysis, interpretation, manuscript writing, and the decision to submit this paper for publication.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: MP, SNH, HB. Analysis and interpretation of data: MP, FA, NF, NH, LT, MG, AS, EH. drafting or revising the manuscript: MP, SNH, EH, MD, HB. All authors approved the final article.
Corresponding authors
Ethics declarations
Conflicts of Interests
The authors have no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pakzad, M., Hassani, S.N., Abbasi, F. et al. A Roadmap for the Production of a GMP-Compatible Cell Bank of Allogeneic Bone Marrow-Derived Clonal Mesenchymal Stromal Cells for Cell Therapy Applications. Stem Cell Rev and Rep 18, 2279–2295 (2022). https://doi.org/10.1007/s12015-022-10351-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-022-10351-x