
Abstract
There is a growing awareness that gut commensal metabolites play a major role in host physiology and indeed the pathophysiology of several illnesses. The composition of the microbiota largely determines the levels of tryptophan in the systemic circulation and hence, indirectly, the levels of serotonin in the brain. Some microbiota synthesize neurotransmitters directly, e.g., gamma-amino butyric acid, while modulating the synthesis of neurotransmitters, such as dopamine and norepinephrine, and brain-derived neurotropic factor (BDNF). The composition of the microbiota determines the levels and nature of tryptophan catabolites (TRYCATs) which in turn has profound effects on aryl hydrocarbon receptors, thereby influencing epithelial barrier integrity and the presence of an inflammatory or tolerogenic environment in the intestine and beyond. The composition of the microbiota also determines the levels and ratios of short chain fatty acids (SCFAs) such as butyrate and propionate. Butyrate is a key energy source for colonocytes. Dysbiosis leading to reduced levels of SCFAs, notably butyrate, therefore may have adverse effects on epithelial barrier integrity, energy homeostasis, and the T helper 17/regulatory/T cell balance. Moreover, dysbiosis leading to reduced butyrate levels may increase bacterial translocation into the systemic circulation. As examples, we describe the role of microbial metabolites in the pathophysiology of diabetes type 2 and autism.
Similar content being viewed by others

Introduction
The understanding of the importance of the gut microbiota and their metabolites in host physiology and pathology is advancing apace. Important, if not crucial, effects of gut commensal bacteria and their products have been revealed in both the intestinal and extraintestinal environments, including adipose tissues, the systemic immune system, the neuroendocrine system, and the central nervous system [1–3]. Qualitative and quantitative changes in a range of metabolites produced by gut microbiota appear to be involved in the development of a wide range of diseases including metabolic syndrome, obesity, inflammatory bowel disease, type 1 and type 2 diabetes mellitus, asthma autism, colon cancer, and major depression [1, 2, 4, 5].
One of the mechanisms by which abnormalities in the composition of the microbiota and their metabolites could provoke pathology is via adverse effects on the gut-brain axis. The gut-brain axis describes well-documented, bidirectional immune, hormonal, and neurally mediated routes of communication between the gut and the central nervous system aimed at maintaining or restoring the homeostatic performance of the gastrointestinal (GI) tract [6, 7]. There is now copious evidence demonstrating the importance of gut microbiota in influencing these interactions [8]. Accordingly, this and other evidence has led to the establishment of the microbiota-gut-brain axis in place of the well-established brain-gut-axis concept [9, 10].
The optimal gut-brain functioning of this axis is essential for maintaining GI and central nervous system homeostasis [7, 11]. Disorders in the microbiome-gut-brain axis are associated with the pathophysiology of a wide variety of diseases such as irritable bowel syndrome, depression, chronic fatigue syndrome, anxiety, and autism [9, 12, 13].
Dysbiosis refers to detrimental changes in the normal composition of the microbiota. Such a state may be caused by a range of dietary and environmental/genetic factors, increased intestinal permeability, or as a result of impaired communication between gut commensals and the antigen-presenting cells in the mucosal immune system. One common cause is increased physical or psychological stress, and in such circumstances, the resultant dysbiosis contributes to stressor-induced immune dysregulation in the gastrointestinal mucosa and systemic compartments of the immune system, leading to a systemic inflammation and other immune changes capable of affecting behavior and central nervous system function [9, 12, 14–17]. In addition to stress, high-fat and high sugar western-type diets, exceedingly common in many populations, are well-established promoters of dysbiosis and intestinal permeability [18, 19], while other aspects of unhealthy diets—such as emulsifiers in processed foods—could also potentially promote intestinal permeability and thus systemic inflammation.
One potential mechanism contributing to such inflammatory alterations is increased intestinal permeability and subsequent bacterial translocation or translocation of bacterial components such as the lipopolysaccharides (LPS) of the cell wall of Gram-negative bacteria into the systemic circulation [12, 20, 21]. However, another but certainly not mutually exclusive mechanism promoting illness involves increased neuroactive microbial metabolites such as tryptophan catabolites (TRYCATs); these are thought to play a role in the genesis of several neuroimmune diseases including major depression [12, 22, 23]. Other microbial metabolites are described as short chain fatty acids (SCFAs) and are also increasingly recognized as vital in human physiology and health. An insufficiency in SCFAs is a likely contributor to the development of several illnesses, including inflammatory bowel disease, obesity, metabolic syndrome, type 1 and 2 diabetes mellitus, autism, major depression, and colon cancer [1, 2, 4, 5]. Abnormalities in the concentrations in SCFAs have been described in these conditions [24–28]. This review aims to describe the mechanisms by which abnormalities in metabolite levels produced by gut microbiota as a result of dysbiosis may influence the genesis of disease, beginning with TRYCATs and a number of other neuroactive metabolites, before going on to focus on the specific effects of SCFAs.
Gut Dysbiosis and TRYCATs
Gut-Derived TRYCAT Formation and Immune Modulation
Commensals play crucial roles in metabolizing tryptophan and other amino acids such as tyrosine, with the subsequent production of tryptophan metabolites (e.g., indole-3-propionic acid). These are capable of modulating the immune system by various mechanisms, including shaping the differentiation of innate lymphoid cells or T cells, notably Th17 lymphocytes [29, 30]. Moreover, some species of Clostridia clusters show a high expression of indoleamine 2,3-dioxygenase (IDO), an enzyme that is induced by proinflammatory cytokines and induces the catabolism of tryptophan lowering plasma tryptophan and increasing TRYCAT levels [23]. TRYCATs also interact with the aryl hydrocarbon receptor, expressed on a number of immune or somatic cells, such as differentiating Th17 T cells [30]. The aryl hydrocarbon receptor is involved in the differentiation of Th17 and Tregs and the production of interleukin-22 (IL-22) by human Th22 cells. IL-22 regulates intestinal immune homeostasis and barrier function [29, 31]. This aryl hydrocarbon receptor (Arh) also regulates the activity of several classes of innate lymphoid cells, which play a major role in the formation and function of gut-associated lymphoid tissue (GALT) [31]. Hence, tryptophan catabolites and other aryl hydrocarbon receptor ligands can alter a number of innate and adaptive immune responses, including the outcomes of naive T cell differentiation [32].
IDO expression by dendritic cells causes accumulation of kynurenine, itself capable of upregulating aryl hydrocarbon receptors, resulting in a cascade of tolerogenic responses including the differentiation of Tregs [32, 33]. L-kynurenine, in particular, activates aryl hydrocarbon receptors in lymphoid tissue, resulting in IL-22 production by naive and activated T cells, leading to the development of Tregs [33–36]. Interestingly, agonism of the aryl hydrocarbon receptor is required to induce IDO expression [37], and once synthesized, kynurenine may activate the aryl hydrocarbon receptor for IDO induction in an autocrine manner, and form an aryl hydrocarbon receptor/kynurenine positive-feedback loop [38]. The aryl hydrocarbon receptor is also expressed by IL-22-producing innate lymphoid cells and intestinal intraepithelial lymphocytes [35, 39].
While tryptophan metabolism appears to play a pivotal role in T cell homeostasis, the uptake of tryptophan and other dietary amino acids by intestinal epithelial cells plays an indispensable role in antimicrobial peptide production and is thus a crucial factor in maintaining the homeostasis of the microbiota [40]. Aryl hydrocarbon receptors are also involved in the development of a beneficial phenomenon known as endotoxin tolerance, whereby tryptophan 1,2-dioxygenase (TDO) 2 expression, induced by lipopolysaccharides in a Toll-like receptor (TLR)4-dependent manner, results in the production of L-kynurenine and the activation of aryl hydrocarbon receptor in cells of the innate immune system. Subsequent challenge by LPS does not provoke an immune-inflammatory response by those cells in the absence of IDO [33, 37]. However, this defense clearly has its limits in an environment of increased intestinal permeability and translocation of LPS into the circulation, which is invariably accompanied by chronic immune activation, and systemic inflammation and an oxidative and nitrosative stress (O&NS) response [20, 21, 41, 42].
Moreover, elevated LPS and LPS-induced inflammatory cytokines provoke the synthesis of kynurenine 3-monooxygenase (KMO) which drives further catabolism of kynurenine toward neurotoxic molecules such as quinolinic acid [43]. Chronic immune activation and Th17 T cell differentiation can also downregulate tryptophan catabolism [44]. Other tryptophan catabolites such as tryptamine and N-dimethyl tryptamine, produced by the competitive pathways of tryptophan metabolism, inhibit IDO activation [45], while 6-formylindolol [3,2-b] (FICZ) actively enhances Th17 T cell development [46, 47]. Clearly dysbiosis-induced changes in the levels or relative proportions of TRYCATs, notably kynurenine, could be a source of systemic neuropathology. However, while TRYCATs are readily found in the systemic circulation, the same is true of microbial derived tryptophan [47, 48]. The presence of these and other neuroactive metabolites in the systemic circulation goes some way to explain the major role played by the microbiota in the development of the central nervous system (CNS) disorders leading to neuropathology [9, 12, 22, 23, 49, 50].
The Role of Microbial Derived TRYCATs in CNS Function
The composition of the microbiota affect, or possibly even control, circulating levels of tryptophan, which is an essential neurotransmitter at all levels of the gut-brain axis [49, 51, 52]. Tryptophan absorption from the gut into the systemic circulation regulates central glutamate and serotonin systems [53]. The alternative degradation of tryptophan, along the TRYCAT pathway, via the microbial manipulation of IDO and TDO levels, is a well-known pathway enabling neuromodulation as previously discussed [23, 54, 55]. It is also worth re-emphasizing that these pathways are the subject of immune and glucocorticosteroid regulation, respectively, which means that the performance of these pathways may change in an inflammatory environment [49, 51, 52]. Some commensal species can affect the concentration of circulating tryptophan via other means. For example, Bacteroides fragilis utilizes tryptophan as an energy source via the production of a tryptophanase enzyme [56]. Some other species are capable of increasing gut tryptophan levels by producing a range of synthase enzymes [57], and yet others display a capacity ex vivo for using tryptophan as a substrate for the synthesis of serotonin [58]. However, the predominant ability of the microbiota to regulate tryptophan metabolism along the TRYCAT pathway has the simultaneous effect of reducing the activity of the pathway involved in serotonin production and increasing the synthesis of quinolinic acid and kynurenic acid, as well as a range of other neuroactive metabolites [55, 59]. This is highly significant as serotonin is a crucial neurotransmitter at each signaling terminus of the gut-brain axis [60]. There are a range of neural processes in the gastrointestinal tract, which may be influenced by localized changes in serotonin concentrations provoking qualitative and quantitative changes in signals propagated along the scaffolding of the gut-brain axis, which in turn provokes changes in central nervous system neurotransmission [60, 61]. This is another route by which changes in the composition of the microbiota could compromise the bidirectional signaling between the brain and the gut [60, 61].
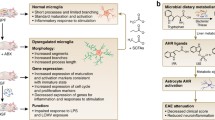
Microbiota also influence the levels of γ-aminobutyric acid (GABA), brain-derived neurotrophic factor (BDNF), dopamine, and noradrenaline, which are key players in the communication between the GI tract and the brain [1, 62]. GABA is metabolized from glutamate by a range of Lactobacillus and Bifidobacterium species [63, 64]. Moreover, a relationship between changed levels of GABA in the gut and in the brain has now been established [65]. Several commensal species have the capacity to modulate levels of BDNF in the gut with consequent changes in the levels and performance of this factor in the brain where BDNF supports neuronal survival, differentiation, and growth [1, 66, 67]. There is also some evidence that changes in the composition of the microbiota affect levels of the N-methyl-D-aspartate receptor (NMDA) subunits NR2A and NR2B and other proteins involved in synaptic function, such as postsynaptic density protein-95 and synaptophysin, which play a vital role in memory formation and retrieval [1, 67, 68]. While TRYCAT abnormalities have an acknowledged role in the pathogenesis and persistence of diseases such as major depression, chronic fatigue syndrome, and somatoform disorder [12, 22, 23], abnormalities in the levels of neurotoxic and immune-modulatory TRYCAT are also seen in a range of neurodegenerative and neuroimmune diseases, including multiple sclerosis [69]. Hence, given considerable evidence indicating the existence of increased intestinal permeability in patients with these illnesses [70], there would seem to be an urgent need for studies directed at the metagenomic analysis of the commensal population in such illnesses, which are currently sparse or even nonexistent in humans. A diagrammatic representation of the effects of microbial metabolites on the production of neuroactive molecules and on the performance of the immune system is provided by Fig. 1. We next turn to a consideration of microbiota metabolites, which have an established role in the regulation of human physiology and immune responses and are increasingly implicated in the pathophysiology of several illnesses. These are described as SCFAs and are the fermentative end products of insoluble fiber breakdown by anaerobic bacteria largely concentrated in the colon [48].

A diagrammatic representation of the effects of microbial metabolites on the production of neuroactive molecules and on the performance of the immune system
Short Chain Fatty Acids
Production and Mode of Action of SCFAs
Commensal bacteria inhabiting the intestinal tract gain energy primarily through the fermentation of nondigested dietary components and of host secretions, mainly mucin, generating SCFAs, including acetate, propionate, and butyrate. However, they differ greatly in their genetic capacity to synthesize enzymes needed to produce SCFAs [71, 72]. For example, acetate (C2) is the most abundant SCFAs produced by enteric and acetogenic (the process whereby acetate is produced from CO2 and an electron source) commensals [73], while Bacteroidetes and some Firmicutes species preferentially produce propionate [74]. Firmicutes include several species (Anaerostipes spp., Eubacterium spp., Roseburia spp., and particularly Faecalibacterium prausnitzii) identified as the dominant producers of butyrate and specialist degraders of indigestible polysaccharides [75, 76]. Butyrate production may depend on cross-feeding mechanisms that require the participation of enzymatic pathways encoded by different bacteria. For example, Bacteroides thetaiotaomicron can generate acetate via its wide array of glycoside hydrolases that can be further utilized by Eubacterium halli to generate butyrate [77]. This reveals the importance of bacterial interactions for ensuring ecosystem stability and defining the functional activity of the microbiota.
SCFAs ligate and activate a range of G protein-coupled receptors, which are involved in intracellular signal transduction and, consequently, cellular responses. For example, G protein-coupled receptor-41 and G protein-coupled receptor-43 are activated by almost all SCFAs [78] and are expressed at physiological levels on enteroendocrine cells and normal enterocytes [79–82]. G protein-coupled receptor-41 is also present on the cell membranes of other cells such as adipocytes, pancreatic cells, and enteric neuronal cells [83, 84]. Extraintestinal expression of G protein-coupled receptor-43 appears on some myeloid cells and all granulocytes [85, 86]. G protein-coupled receptor-109a is expressed by gut epithelial cells, dendritic cells, macrophages, and extraintestinal adipocytes and appears to be a specific receptor for butyrate and niacin [87, 88].
The activation of G protein-coupled receptors 41, 43, and 109a, on intestinal epithelial cells mediates a significant proportion of the functions of SCFAs [87, 89–91]. SCFAs ligation of these G protein-coupled receptors activates a range of signaling processes such as protein kinase A and ERK1/2 and many transcription factors, e.g., activating transcription factor 2 [92, 93]. Activation of this latter pathway is an indispensable element in the expression of pivotal immune and inflammatory cytokines and chemokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, and chemokine (C-X-C motif) ligand (CXCL)-1 and CXCL-2. SCFAs also activate G protein-coupled receptors 41 and 43 on specialized secretory epithelial cells leading to the synthesis and secretion of glucagon-like peptide (GLP)-1 [94].
SCFAs also modulate cell differentiation and proliferation and the secretion of hormones, including leptin and peptide YY [95, 96]. These actions, and others, enable SCFAs to regulate a wide range of regulatory effects on metabolic pathways and the immune response to infection or inflammation. The multiple metabolic effects of SCFAs are depicted in Fig. 2. We next turn to a consideration of their actions on the immune system. While these effects are varied, and at times somewhat contradictory, they can be usefully categorized into effects on the recruitment of leucocytes from the peripheral circulation to areas of inflammatory activity, effects on naive T cell differentiation, modulation of cytokine expression by antigen-presenting cells, and disruption of immune homeostasis by effects on epithelial barrier function. We will discuss each in turn.

Regulation of fatty acid metabolism. SCFAs increase hepatic and striated muscle AMPK activity, via an increase in the AMP/ATP ratio and/or activation of the GPR43-leptin pathway, leading to increased expression of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α expression and subsequent elevations of peroxisome proliferator-activated receptor (PPAR)α and farnesoid X receptor (FXR). The sum of these effects is enhanced fatty acid (FA) oxidation in both tissues and the decrease of a de novo hepatic fatty acid production. SCFAs stimulate PGC-1α and uncoupling protein (UCP)-1 activity in brown adipose tissue (BAT), thereby increasing FA oxidation and thermogenesis and fatty acid oxidation. The inhibition of lipolysis in adipose tissue mediated by SCFA engagement of GP43 likely involves suppression of the hormone-sensitive lipase (HSL), the activation of the Gi/o protein and the subsequent decrease in the levels of cAMP and protein kinase A (PKA). SCFA stimulation of adipose-specific GPR43 inhibits insulin signaling via suppression of Akt phosphorylation, subsequently inhibiting adipose tissue fat storage and promoting lipid and glucose metabolism in other tissues
Effects of SCFAs on Leucocyte Recruitment to Sites of Inflammation
Inflammatory damage to tissue leads to the recruitment of neutrophils and other leucocytes from the bloodstream and migration to the site of inflammation. This is a complex process, composed of several steps involving the coordinated activity of selectins and integrins, which enable neutrophil egress into tissue via human umbilical vein endothelial cells (HUVEC), and a range of chemokines, which summon and guide leucocytes to the sites of damage [74, 97]. SCFAs suppress the adherence of leucocytes to HUVEC and hence impede the translocation of neutrophils to sites of inflammation and hence exert an anti-inflammatory effect [74, 76] in a process involving nuclear factor (NF)-kB and peroxisome proliferator-activated receptor (PPAR)γ inhibition, and a subsequent downregulation in the expression of selectins and integrins [98]. SCFAs can also modulate the expression of chemokines and adhesion molecules on leucocytes and endothelial cells [76, 99]. SCFAs can stimulate or inhibit the recruitment of circulating lymphocytes to the sites of inflammation dependent on physiological conditions [3, 98, 100]. For example, butyrate inhibits the LPS-stimulated increase in CXCL-2 and macrophage chemoattractant protein-1 (MCP-1), leading to a decrease in leucocyte recruitment.
SCFAs exert a somewhat paradoxical effect on neutrophils. They modulate the synthesis and secretion of chemokines and adhesion molecule expression on the surface of neutrophils, in a similar manner to leucocytes, and thus inhibit the recruitment of these PMBCs to sites of inflammation [100]. However, SCFAs induce an increase in neutrophil chemotaxis, which is an effect dependent on G protein-coupled receptor-43 ligation [101, 102] and activation of ERK and p38 pathways [102]. There is, however, some evidence that the interaction of G protein-coupled receptor-43 and SCFAs may render neutrophils refractory to the chemoattractant effects of chemokines [3]. Butyrate, in particular, inhibits LPS-induced migration of macrophages via inhibition of histone deacetylase activity within these antigen-presenting cells [72]. Butyrate also modulates the synthesis and secretion of IL-8, MCP-1, and CXCL-1 by intestinal epithelial cells in response to peptidoglycan lipopolysaccharides and proinflammatory cytokines [103, 104]. Butyrate and propionate ameliorate the LPS-stimulated synthesis of CXCL-2 by neutrophils [98], while butyrate alone inhibits interferon (IFN)-γ-inducible protein-10 (IP-10). Both SCFAs inhibit the production of MCP-1 in the presence or absence of LPS [92].
Effects of SCFAs on T Cell Differentiation
Effects on Regulatory T Cell Production
Recent studies, utilizing metabolome analysis and other investigative techniques, have revealed that the number of colonic Tregs in steady-state conditions are heavily influenced or even determined by the luminal concentration of propionate, butyrate, and perhaps acetate, produced by commensal bacteria [105–107]. The precise mechanisms by which SCFAs promote the production of colonic Tregs are still a matter of debate and may well vary with the SCFAs involved. For example, colonic Tregs express G protein-coupled receptor-43, which has been implicated in ameliorating intestinal inflammation, and the function of this receptor is an essential element in colonic Treg differentiation mediated by propionate [108]. Butyrate, on the other hand, appears to mediate Treg differentiation by engagement with the butyrate-specific receptor G protein-coupled receptor-109a, which is extensively expressed on intestinal epithelial cells and antigen-presenting cells, but—crucially—not on lymphocytes [109]. Butyrate engagement with this receptor leads to the production of IL-10 and a range of metabolites, including retinoic acid, leading to the production of Tregs [109]. However, while the evidence that Treg production is mediated by butyrate appears robust, it must be remembered that T lymphocytes as a whole do not seem to express physiologically relevant levels of G protein-coupled receptors [110]. It is also worthy of note that the effect of propionate on Treg differentiation appears to be limited to colonic Tregs and not to the wider inducible Treg population [108]. Hence, either the effect of propionate on G protein-coupled receptor-43 is specific for colonic Tregs, or there is an alternative explanation for the observations. This alternative explanation may stem from the fact that short chain fatty acids, notably butyrate, also act as histone deacetylase inhibitors and promote the production or expansion of Tregs by epigenetic modification of forkhead box-P3 promoter and enhancer regions [105, 106]. Butyrate also potentiates Treg differentiation by intestinal dendritic cells by suppressing the transcription of genes involved in the response to LPS, leaving intestinal dendritic cells, TLR4 receptors, hyporesponsive to the presence of this commensal polysaccharide [106, 111]. Several uncertainties remain regarding the precise effects of SCFAs on the colonic Treg population, such as whether the provocative effects of SCFAs on Treg numbers are limited to increased naive CD4 T cell differentiation, or whether the effects include expansion of pre-existing resident Tregs [105, 106, 108]. It must be noted that, under certain physiological conditions, SCFAs can induce the production of effector Th1 and Th17 lymphocytes, and we now turn to a consideration of potential mechanisms enabling this phenomenon [110, 112].
SCFAs and Th1 and Th17 Differentiation
T cells can potentially be regulated by SCFAs via three different mechanisms. The first mechanism involves the activation of G protein-coupled receptors 41, 43, and 109a, which can modulate cell activation and differentiation. However, the weight of evidence indicates that T lymphocytes lack significant levels of these receptors, and hence, the current consensus is that SCFA receptors are unlikely to be an important mechanism enabling direct regulation of these lymphocytes by SCFAs. Another route is the regulation of cellular bioenergetic metabolism via the conversion of SCFAs to acetyl-CoA and integration into the tricarboxylic acid (Krebs cycle), providing an upsurge in energy production and an increase in adenosine triphosphate/adenosine diphosphate levels. This increase amplifies mTOR activation [113], which skews T cell differentiation into Th17 and Th1 T lymphocytes at the expense of regulatory FoxP3+ T cells [114]. Increased mTOR activity also provokes the differentiation of IL-10+ T cells [111]. Hence, the modulation of SCFAs of cellular energy metabolism and subsequent activation of mTOR accounts for the increased production of Th1, Th17 lymphocytes, and IL-10+ generating cells. Finally, the third route involved in T cell regulation involves histone deacetylase inhibitor activity, shared to various degrees by all SCFAs [112, 115, 116]. Such histone deacetylase inhibitor activity necessitates the internalization of SCFAs into lymphocytes and contact-based enzymatic inhibition, mainly of class I/II histone deacetylase, although some studies also suggest inhibition of Sirt-1 and other class III histone deacetylase [117]. SCFAs can directly induce the formation of IL-17, IFN-γ, and IL-10 secreting T lymphocytes cells via inhibition of histone deacetylase, leading to acetylation of p70 S6 kinase and subsequent phosphorylation of rS6 and increased mTOR activation [112]. This is of interest as this route to mTOR activation is quite distinct from the effects of SCFAs on energy metabolism discussed above.
In vitro evidence suggests that SCFAs can also affect T cell differentiation patterns indirectly via a broadly immunosuppressive or tolerogenic effect on antigen-presenting cells. For example, SCFAs inhibit the development of myeloid dendritic cells from their progenitors [118], as well their functional maturation [119, 120]. A key mechanism is the attenuation of histone deacetylase activity. Among the SCFAs, butyrate is one of the most potent, while acetate is one of the least potent inhibitors of histone deacetylase [121, 122]. This enzyme, together with the histone acetyltransferases (HAT), controls the degree of protein acetylation. By inhibiting the histone deacetylase activity, SCFAs increase the acetylation of histone and nonhistone proteins including different transcription factors, including NF-kB and p53 [123].
Effects of SCFAs on Cytokine Production by Antigen-Presenting Cells
SCFAs inhibit LPS or cytokine-stimulated production of nitric oxide (NO), IL-6, TNF-α and other inflammatory molecules in macrophages, monocytes, and PMBCs [110]. Butyrate and propionate suppress the synthesis of proinflammatory mediators, e.g., TNF-α and NO, in neutrophils via a mechanism involving the attenuation of NF-kB expression and/or activation [98]. The effect of propionate or butyrate on reactive oxygen species synthesis by neutrophils, on the other hand, remains controversial. Some authors have reported increased reactive oxygen species production by SCFAs [124, 125], while others have demonstrated inhibitory effects [126, 127]. Controversy also surrounds the possible impact of SCFAs on microglial production of reactive oxygen species, NO, prostaglandin-E2, etc. [128, 129]. However, the majority of studies demonstrate that these fatty acids ameliorate microglial activation via a mechanism once again involving histone deacetylase inhibitors [129, 130]. Accumulating evidence from animal models [130] also supports the view that SCFAs may be useful in reducing inflammatory responses in the central nervous system [131].
Effects of SCFAs on Gastrointestinal Epithelial Barrier Integrity
Butyrate-producing bacteria play a vital role in regulating the functions and integrity of the intestinal epithelial barrier [132–135], and hence, dysbiosis involving a reduction in butyrate producers could disrupt the immune homeostasis in the gastric mucosa with profound systemic effects [136]. SCFAs stimulate the secretion of glucagon-like peptides 1 and 2 via ligation of glucagon-like peptide-43 [137–139] leading to decreased epithelial permeability and increased mucosal antibacterial defenses [140]. Glucagon-like peptides 1 and 2 improve barrier function via a number of different mechanisms, including via the stimulation of L class enteroendocrine cells and the inhibition of apoptosis of intestinal epithelial cells [141]. These hormones also engage in a highly complex interaction with endocannabinoid receptors, whose actions are arguably the major players in regulating the permeability of the intestinal epithelium [142, 143]. When considered as a whole, the weight of evidence indicates that epithelial barrier integrity is governed by a complex cross talk between endocannabinoids, commensal LPS, SCFAs, and glucagon-like peptide hormones [142, 143]. It probably comes as no surprise to learn that changes in the commensal population, leading to a higher proportion of SCFA producers, increase the expression of glucagon-like peptides 1 and 2 and an increase in barrier integrity [144, 145]. On the other hand, dysbiosis leading to a loss of SCFA producers can result in adverse changes in the localization and distribution of two epithelial tight junction proteins, occludin and zonula occludens-1, leading to increased epithelial permeability in rodent models of type 2 diabetes [142, 145–147]. Increased epithelial permeability, linked to changes in the microbiota or other factors such as poor diet or stress, can lead to the translocation of LPS into the circulation and resulting systemic immune activation: this is a phenomenon often described as “metabolic endotoxemia” [145–148]. This phenomenon resulting from increased intestinal permeability is a triggering factor in the development of metabolic syndrome, insulin sensitivity, and type 2 diabetes (TD2) in humans [145, 146, 149]. Clearly, dysbiosis leading to a reduction in SCFAs can result in impaired barrier function leading to metabolic endotoxemia, loss of broadly anti-inflammatory effects, and metabolic dysregulation, notably in the domain of energy homeostasis. These tripartite effects have the potential to be a source of pathology in a host.
Effects of SCFAs on Energy Homeostasis
SCFAs manufactured by gut commensals, notably propionate and acetate, can be detected in hepatic, portal, and peripheral blood, suggesting that they have the ability to cross the gut-blood barrier [148, 150, 151]. This capacity for migration enables SCFAs to influence lipids, glucose, and cholesterol in a range of remote tissues [152–154] (Figs. 2 and 3). Positive benefits on satiety, cholesterol levels, and glucose metabolism have been recorded by increasing SCFA production in animals fed resistant starch [155, 156]. The positive effects of butyrate-producing bacteria on blood glucose and lipid metabolism are also evidenced by fecal transplantation studies [157, 158]. The positive effects on glucose are achieved largely by increases in glucagon-like peptide-1, which increases insulin secretion, lowering plasma glucose and preserving beta cell function [159, 160]. SCFAs also appear to affect epigenetic regulation of gene expression in type 2 diabetes via activation of G-coupled receptors and their capacity to act as histone deacetylation inhibitors [148, 161].

Regulation of glucose metabolism. Schematic representation of the likely pathways by which SCFAs regulate glucose metabolism. SCFAs can increase GLP-1 and PYY in the colon via engagement of GPR42 and GPR43. The latter molecule increases the uptake of glucose in muscle and adipose tissue, whereas GLP-1 decreases glucagon production and increases the production of insulin in the pancreas. In addition, SCFAs possess the capacity to decrease gluconeogenesis in the liver by increasing the phosphorylation and activity of AMPK
SCFAs ligate and regulate the balance between fatty acid oxidation and lipolysis, and increase fatty acid oxidation and decrease lipolysis in a mechanism involving increased activation and/or upregulation of PGC-1a peroxisome proliferator-activated receptors and leptin [95, 162, 163]. SCFAs also exert profound effects on cholesterol metabolism by inhibiting the activity of hydroxymethylglutaryl coenzyme A synthase, reducing hepatic glucogenesis [164, 165].
Raised levels of GLP-1 following engagement of SCFAs with free fatty acid receptors lead to elevated production of insulin in the pancreas and decreased levels of glucagon, while raised levels of pancreatic peptide (PYY) increase glucose uptake by striated muscle and adipose tissue [163, 166]. Gut commensals can also exert a regulatory influence on energy homeostasis via SCFA-induced increase in the activity of the sympathetic nervous system, which in turn may cause immune-inflammatory responses [167, 168]. All in all, it is clear that a complex interplay between the microbiota and SCFA production has a profound effect on energy homeostasis. We now move on to examine the potential role of impaired SCFA levels in the pathogenesis of complex illness where there is evidence of immune and metabolic dysregulation, using type 2 diabetes and autism spectrum disorders as exemplars.
Examples of How Dysbiosis May Play a Role in Human Disease
Type 2 Diabetes
Evidence for Dysbiosis in the Pathophysiology of Type 2 Diabetes
The altered ratio of Firmicutes/Bacteroides seen in patients with type 2 diabetes is associated with greater energy harvesting and the development of insulin resistance [169, 170]. Recently, a potential relationship between gut microbiota and TD2 pathophysiology has been suggested by two independent studies comparing metagenomes from TD2 and healthy controls. They have provided particularly strong evidence for the role of gut dysbiosis in the pathophysiology of the disease and TD2 subjects [171, 172]. These authors demonstrated an increase in Clostridium clostridioforme numbers and a decrease in the levels of Roseburia 272 in TD2 patients in both European and Chinese populations [171, 172]. Moreover, increased levels of Roseburia following fecal transplants from lean donors to patients with metabolic syndrome seemingly resulted in improved insulin sensitivity [173]. These positive effects appeared to be related to the presence of SCFAs, propionic and butyric acids [174], which ameliorate low-grade inflammation and stimulate the release of hormones such as leptin and adipokine in adipose tissue as discussed above [95, 175, 176]. These authors reported reduced counts of Prevotella and Atopobium and Clostridium coccoides group, but significantly elevated numbers of Lactobacillus spp. in patients with TD2. Such a microbiota profile was also associated with reduced production of butyrate and other SCFAs. Furthermore, this study reported significant increases in the number of commensals in the bloodstream of their diabetic patients compared to controls, indicating increased intestinal permeability and commensal translocation from the gut lumen into the systemic circulation [174]. Moreover, bacterial DNA (mostly belonging to the phylum Proteobacteria) has also been detected in the blood of subjects before diabetes onset and was higher in those who had abdominal adiposity suggesting that translocation could predict disease manifestation [177]. We now move on to consider this phenomenon and disease-specific consequences in a little more detail.
Increased Intestinal Permeability in Patients with Type 2 Diabetes
Several teams have reported a relationship between impaired GI permeability and the translocation of live bacteria or LPS fragments into the bloodstream with the development of TD2 [178–180]. Translocation of bacterial fragments or whole bacteria into the systemic circulation leads to chronic immune activation and low-grade inflammation, which is the hallmark of metabolic endotoxemia as discussed previously. Such metabolic toxemia is related to the development of the metabolic syndrome, insulin sensitivity, and ultimately TD2 [181–183]. Interestingly, the severity of low-grade inflammation, as evidenced by biomarkers including C-reactive protein, TNF-α, and IL-6, is considered as a predictor of the progression from metabolic syndrome to TD2 at least in high-risk patients [184–186].
Translocated LPS exert pathophysiological effects adversely affecting the function of several organs and tissues. For example, elevated levels of TNF-α, IL-1β, IL-6, and NF-kB, produced by LPS-induced activation of TLR2 and TLR4, are upregulated in the liver, as well as in adipose tissue and striated muscles, and make a major contribution to the development of increased insulin resistance [184, 187, 188]. LPS activation of TLR4 and the subsequent increased production of proinflammatory cytokines in macrophages and beta cells conspire to decrease the function, and ultimately the viability, of the latter cells; this is a critical element in the development of insulin resistance and TD2 [189, 190]. The Th17/Treg balance is disturbed in patients with TD2, with elevated numbers of circulating Th17 T cells and reduced numbers and function of Tregs [191, 192]. The direct adverse effects on glucose metabolism of elevated LPS in the systemic circulation have been reported in an interesting study [193]. These authors reported that LPS activation of TLR4 in myotubes leads to the creation of an inflammatory environment and phosphorylation of AKT and impaired glucose transport. LPS-mediated activation of TLR2 also leads to the phosphorylation and activation of AKT [194]. This is highly significant, given data demonstrating that blunted AKT activity is required for the persistent immunosuppressive properties of Tregs [195, 196]. Metabolic endotoxemia may also have a direct inhibitory effect on Treg function leading to the unrestrained differentiation of Th17 T cells. Engagement of TLR2 receptors on naive T cells and on the surface of Tregs drives their differentiation toward the Th17 phenotype, with a loss of immunosuppressive properties [197].
It is clear that the abnormalities discussed above could, at least in part, stem from dysbiosis leading to impoverished levels of SCFA production and increased intestinal permeability. However, TD2 is a disease with a strong genetic component, with interactive polymorphisms in HLA and vitamin D receptor (VDR) being a critical element in increased disease susceptibility [198]. It is, however, of interest that these authors reported VDR polymorphisms to be correlated with HLA-DRB1*04 and with metabolic and immunologic parameters [198] as reduced SCFAs would likely compound this association.
TRYCATs and SCFAs in Type 2 Diabetes
As discussed above, dysbiosis leading to decreased SCFA production and resultant increase in the translocation of LPS into the systemic circulation likely underpins the state of metabolic endotoxemia seen in patients with metabolic syndrome, insulin resistance, and TD2. This state is characterized by increased levels of proinflammatory cytokines, which upregulate the activity of IDO and hence the activation of the TRYCAT pathway seen in these patients [199]. Metabolic endotoxemia also plays a major role in the development of insulin resistance, metabolic syndrome, and TD2 via the activation and dysregulation of the TRYCAT pathway in the blood, immune cells, pancreas, and CNS; this produces a range of adverse effects on the synthesis, release, and performance of insulin [200]. The activation of this pathway in patients with TD2, coupled with low levels of pyridoxal-5′-phosphate, leads to chronically upregulated levels of kynurenic acid (KYNA) and xanthurenic acid and a number of other TRYCATs, all of which have a range of adverse effects on insulin production and function [201–204]. Briefly, LPS translocated into the systemic circulation provokes the synthesis of proinflammatory cytokines from antigen-presenting cells, resulting in the activation of IDO.
Elevated levels of these TRYCATs display pro-oxidative, neurotoxic, and apoptotic effects via upregulation of inducible NO synthase (iNOS), arachidonic acid, prostaglandins, phospholipase A2, and leukotriene cascades, each of which play a major role in the development of metabolic syndrome and insulin resistance (reviewed in [205]). Kynurenine may be alternatively metabolized to form KYNA and, ultimately, quinaldic acid (QA) in a reaction catalyzed by the action of kynurenine aminotransferase (KAT) or 3-hydroxy kynurenine (3-HK), catalyzed by KYN 3-monooxygenase [54]. 3-HK, in turn, may either be metabolized to produce quinolinic acid and, ultimately, nicotinamide, with the first step being catalyzed by kynureninase or metabolized to produce xanthurenic acid, and 8-hydroxyquinaldic acid (8-HQ), catalyzed by 3-HK-transaminase. IDO, KAT, and IMO activity is far higher in the periphery than in the brain, and hence, the activation of the TRYCAT pathway in the periphery has a major impact on the performance of this pathway in the CNS [206]. IDO activation in the periphery results in the production of kynurenine, 3-HK, and quinolinic acid, which can readily cross the blood brain barrier and be further metabolized in the CNS. KMO is relatively inactive in microglia and is readily saturated by chronically elevated levels of kynurenine entering the brain, which is the case in the presence of metabolic endotoxemia in the periphery. This inactivation results in increased production of KYNA by the relatively active KAT [206]. Levels of KYNA are elevated in patients with TD2 [206], which is pathologically significant as this TRYCAT can inhibit the conversion of proinsulin to insulin in the pancreas [201].
Many patients with TD2 also present with low levels of pyridoxal-5′-phosphate (P5P), an active form of vitamin B6 that acts as an essential cofactor for kynureninase [203, 204]. Hence, this enzyme is extremely sensitive to the depletion of P5P [200], and the upregulated TRYCAT pathway, in an environment of decreased P5P, leads to an increased availability of 3-HK for 3-HK-transaminase and subsequently increased production of xanthurenic acid and 8-HQ. This increases in the availability of kynurenine for KAT and thus increases the levels of KYNA and QA in the brain, blood, urine, and pancreatic islets [207–210]. Chronically elevated levels of these TRYCATs, notably xanthurenic acid, can have a number of deleterious effects on the production and function of insulin. Xanthurenic acid inhibits the release of insulin and induces apoptosis of pancreatic cells [211, 212]. This TRYCAT can also chelate insulin, which can reduce the activity of the hormone [203, 213].
The cause or causes of P5P depletion in patients with TD2 remain a matter of controversy, but it is fair to say that this phenomenon is not unique to people suffering from this illness as it also occurs in patients with inflammatory bowel disease, rheumatoid arthritis, cardiovascular disease, and other illnesses underpinned by the presence of chronic systemic inflammation [214]. Indeed, there is accumulating evidence that the depletion of this essential coenzyme is a consequence of chronic inflammation, which drives the catabolism of pyridoxol to pyridoxic acid and hydrolysis of P5P by alkaline phosphatase [215, 216].
Now we turn from a consideration of an illness where excessive levels of SCFA production might play a pathological role, to a group of illnesses where excessive SCFA production, notably propionate, may well underpin the symptoms of a disorder.
Autism Spectrum Disorder
Evidence for Dysbiosis in the Pathophysiology of Autism
Autism spectrum disorders are a heterogeneous set of neurobiological illnesses whose etiology cannot be explained by genetics alone [217, 218]. Idiopathic gastrointestinal abnormalities are a common comorbidity in children with autism spectrum disorder, suggesting that abnormalities in the microbiota and microbial metabolites may play a role in the pathophysiology of these illnesses [217, 219–221]. Indeed, numerous reports, utilizing both animal models and human epidemiological evidence, have linked disruptive changes in the composition in the microbiota and subsequent abnormalities in microbial metabolites leading to immune dyshomeostasis and changes in the gut-brain axis, with the development of autism spectrum disorder [217–219, 221–223].
Several authors have reported increased intestinal permeability and a range of gastrointestinal abnormalities, such as dysmotility, in some children with autism [224, 225]. A recent study by Emanuele and colleagues [226] reported the presence of LPS and elevated levels of proinflammatory cytokines in autism. In a similar vein, several research teams have reported the presence of gut dysbiosis, with an altered composition of the microbiota in individuals with autism spectrum disorder [223, 227–234]. Spore-bearing anaerobes and microaerophilic bacteria, mainly Clostridia, are elevated in many people with autism spectrum disorder [228, 235, 236]. Infectious pathobionts are also implicated in the etiology of autism spectrum disorders [236–238].
SCFAs and Autism
One animal model of autism spectrum disorders is based on administration of high levels of the SCFA propionate. We now focus on the consequences of such administration and highlight the mechanisms by which dysbiosis, leading to an overproduction of propionic acid and other SCFAs, could underpin many of the abnormalities seen in children with a diagnosis of autism spectrum disorder.
Propionate acid is a fermentation product of many bacteria, including Clostridia, Desulfovibrio, and Bacteroidetes, that are markedly overabundant in stool samples from patients with autism spectrum disorder [228, 238]. Importantly, this dysbiosis corresponds with excessively elevated propionic acid levels in the stool samples of autism spectrum disorder patients [221]. Other studies report alterations in Bacteroides and Prevotella species, but by far the most consistent finding is a major increase in the levels and diversity of Clostridia species, with one study reporting almost a 50-fold increase [232, 235, 238]. This gives further grounds for suspecting the presence of excessive SCFAs in at least some patients afforded this diagnosis. The study conducted by Kang and others cited above is of particular interest as they reported that the presence and severity of “autistic” symptoms was significantly correlated with lower levels of Coprococcus and Prevotella and several species of currently unclassified Veillonellaceae species [231]. These species are all propionate producers, and hence, the results of this study refute the suggestion that excessive propionate production is the cause of abnormalities seen in all children with an autism spectrum diagnosis. However, results of other studies do support the proposition that excessive SCFAs could partly explain the observed pathology in some of these illnesses. The possible mechanisms whereby propionic acid might cause these effects are not completely understood but we will now consider the mechanistic explanations as understood thus far.
Potential Mechanistic Explanations for Propionic Acid-Induced Pathology in Autism Spectrum Disorder
Many environmental toxins are implicated in the development of autism spectrum disorder, including propionic acid and other metabolic products of commensal bacteria [239–241]. These SCFAs readily cross the gut-blood barrier and gain access to the CNS, at least partly via uptake by monocarboxylate transporters in the gut lumen[242]. Excessive levels of SCFAs in the systemic circulation can lead to adverse metabolic and neurobiological effects, leading to mitochondrial and CNS pathology proposed to play a causative role in the pathophysiology of autism spectrum disorder [243]. It is also of interest that propionic acid has a number of direct actions on GI physiology very similar to the pattern of GI pathology seen in patients with autism spectrum disorder (reviewed in [218]). Propionic acid inhibits mitochondrial functions, via the production of the cytotoxic propionyl CoA, and the subsequent sequestration of carnitine impaired membrane stability and levels of cardiolipin 51 [239, 244, 245]. It is of interest that intraventricular administration of propionic acid results in an abnormal acylcarnitine profile and relative carnitine deficiency seen in some children with autism spectrum disorder [246–248]. Carnitine plays a key role in β-oxidation of fatty acids and hence makes a major contribution to energy production; thus, depleted levels could be a source of relative mitochondrial dysfunction [249].
The capability of circulating propionic acid and other SCFAs to directly influence the brain via the assistance of monocarboxylate transporters in the blood brain barrier(BBB) is perhaps underappreciated [250, 251]. Propionic acid is internalized by microglia, astrocytes and, to a lesser degree, neurons [251, 252], which also express monocarboxylate transporting receptors [253, 254]. Once internalized, these SCFAs appear to be a major energy source involved in many aspects of cellular metabolism, particularly during postnatal brain development [251, 252, 255].
Perhaps most compellingly, animal models of autism spectrum disorder based on pulsed infusions of propionic acid and other SCFAs have demonstrated that such administration produces neurocognitive defects, abnormal motor movements, impaired social interactions, and other symptoms reminiscent of autism spectrum disorder [217, 239–241]. Moreover, the brain tissue of rats treated with propionic acid shows neurochemical changes reminiscent of autism, including glutathione depletion, neuroinflammation, oxidative stress, and altered phospholipid/acylcarnitine profiles [217, 239–241]. The presence of SCFAs in the CNS also affects lipid metabolism [256], the release and synthesis of neurotransmitters [257], oxidative stress-related mitochondrial functions [258], and intracellular pH.
Propionic acid can also modulate serotonin, dopamine, and glutamate systems in a manner similar to that observed in autism spectrum disorder [259, 260], largely by potentiating the release of intracellular calcium [261]. In general, SCFAs may increase dopamine synthesis via the induction of tyrosine hydroxylase in the sympathetic ganglia and the adrenals, leading to neuroendocrine abnormalities such as the cardiovascular instability and increased sympathetic tone seen in many children with a diagnosis of autism spectrum disorder [262, 263]. It must be stressed, however, that levels of propionate achieved by the mode of administration in such animal models may exceed those which can occur in children with autism spectrum disorder, and thus, extreme caution must be exercised in extrapolating such data. However, there is evidence that increased intestinal permeability or apoptosis of intestinal epithelial cells may allow for increased SCFA levels in the systemic circulation and consequently the CNS [264, 265]. In any event, SCFAs are unlikely to be the only metabolites ultimately derived from the microbiota involved in the pathogenesis of these illnesses, as abnormal TRYCAT levels are also implicated [266].
Summary and Conclusions
Tryptophan metabolites and IDO produced by microbial metabolites can shape the immune response by engaging aryl hydrocarbon receptors and promoting the differentiation of Th17, or regulatory T cells and IL-22 production by Th22 T cells, which in turn regulates epithelial barrier function. Elevated levels of IDO produced by a range of gut commensals increase the levels of kynurenine, which can also engage aryl hydrocarbon receptors. This induces the differentiation of Treg cells and increases the levels of IDO, leading to profound immunosuppression. Increased levels of IDO in the periphery can transverse the BBB leading to the production of toxic metabolites such as quinolinic acid. The composition of the microbiota is largely responsible for the levels of tryptophan in the systemic circulation and influences the levels of other neuroactive substances in the intestine, blood, and brain.
The levels of systemic tryptophan influence the performance of the serotoninergic and glutamatergic systems in the CNS and the production of neurotoxic molecules by the TRYCAT pathway in astrocytes and microglia. Microbiota also influence the levels of GABA, BDNF, dopamine, noradrenaline, and serotonin in the gut, the systemic circulation, and in the CNS. The presence of these and other neuroactive metabolites in the systemic circulation goes some way to explain the major role played by the microbiota in the functioning and development of the central nervous system and their potential to disrupt the brain functions leading to neuropathology.
Microbially generated SCFAs activate G protein-coupled receptors 41, 43, and 109a, on intestinal epithelial cells, which mediate many of their functions including the activation of intracellular signaling and the expression of pivotal immune and inflammatory cytokines and chemokines. SCFAs also activate G protein-coupled receptors 41 and 43 on specialized secretory epithelial cells, leading to the synthesis and secretion of GLP-1, which plays a major role in regulating epithelial barrier integrity. SCFAs also exert profound effects on chemokine production and neutrophil recruitment to sites of inflammation. Their capacity to promote the differentiation of Tregs, IL-17 producing Th17 T cells, and the pattern of cytokine production by antigen-presenting cells is mediated by their capacity to act as histone deacetylase inhibitors or activators of mTOR signaling.
SCFAs, notably butyrate, stimulate the secretion of glucagon-like peptides 1 and 2 via ligation of glucagon-like peptide-43, leading to decreased epithelial permeability and increased mucosal antibacterial defenses. On the other hand, dysbiosis leading to a loss of butyrate or propionate producers can result in adverse changes in the localization and distribution of two epithelial tight junction proteins, occludin and zonula occludens-1, leading to increased epithelial permeability and translocation of bacterial LPS into the systemic circulation.
References
Heijtz RD, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H et al (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 108:3047–3052
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R et al (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328:228–231
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS et al (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461:1282–1286
Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C (2009) Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One 4:e6026
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G et al (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107:14691–14696
De Palma G, Collins S, Bercik P, Verdu E (2014) The microbiota-gut-brain axis in gastrointestinal disorders: stressed bugs, stressed brain or both? J Physiol 592:2989–2997
Burokas A, Moloney R, Dinan T, Cryan J (2015) Microbiota regulation of the mammalian gut-brain axis. Adv Appl Microbiol 9:1–62
Carabotti M, Scirocco A, Maselli M, Severia C (2015) The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 28:203–209
Maes M, Kubera M, Leunis JC (2008) The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett 29:117–124
Stilling R, Dinan T, Cryan J (2013) Microbial genes, brain & behaviour—epigenetic regulation of the gut-brain axis. Genes Brain Behav 13:69–86
Grenham S, Clarke G, Cryan JF, Dinan TG (2011) Brain–gut–microbe communication in health and disease. Front Physiol 2:1–15. doi:10.3389/fphys.2011.00094
Maes M, Mihaylova I, Ruyter M, Kubera M, Bosmans E (2007) The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): relevance for depression—and other conditions involving inflammation. Neuro Endocrinol Lett 28:826–831
Borre Y, Moloney R, Clarke G, Dinan T, Cryan J (2014) The impact of microbiota on brain and behavior: mechanisms & therapeutic potential. Adv Exp Med Biol 817:373–403
Gur T, Worly B, Bailey M (2015) Stress and the commensal microbiota: importance in parturition and infant neurodevelopment. Front Psychiatry 6:5
Galley J, Bailey M (2014) Impact of stressor exposure on the interplay between commensal microbiota and host inflammation. Gut Microbes 5:390–396
Bailey M (2012) The contributing role of the intestinal microbiota in stressor-induced increases in susceptibility to enteric infection and systemic immunomodulation. Horm Behav 62:286–294
Bailey M, Dowd S, Galley J, Hufnagle A, Allen R, Lyte M (2011) Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun 25:397–407
Chassaing B, Koren O, Goodrich JK et al (2015) Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519:92–96
Murphy EA, Velazquez KT, Herbert KM (2015) Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr Opin Clin Nutr Metab Care 18:515–520
Morris G, Maes M (2012) A neuro-immune model of myalgic encephalomyelitis/chronic fatigue syndrome. Metabolic Brain Dis 28:523–540
Morris G, Berk M, Galecki P, Walder K, Maes M (2016) The neuro-immune pathophysiology of central and peripheral fatigue in systemic immune-inflammatory and neuro-immune diseases. Mol Neurobiol 53(2):1195–1219
Maes M, Leonard B, Myint A, Kubera M, Verkerk R (2011) The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog NeuroPsychopharmacol Biol Psychiatry 35:702–721
Maes M, Rief W (2012) Diagnostic classifications in depression and somatization should include biomarkers, such as disorders in the tryptophan catabolite (TRYCAT) pathway. Psychiatry Res 196:243–249
Huda-Faujan N, Abdulamir AS, Fatimah AB, Anas OM, Shuhaimi M, Yazid AM, Loong YY (2010) The impact of the level of the intestinal short chain fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J 4:53–58
Vernia P, Caprilli R, Latella G, Barbetti F, Magliocca FM, Cittadini M (1988) Fecal lactate and ulcerative colitis. Gastroenterology 95:1564–1568
Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, Clarke SF, O’Toole PW et al (2010) Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59:1635–1642
McIntyre A, Gibson PR, Young GP (1993) Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut 34:386–391
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, Hardt PD (2010) Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18:190–195
Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L (2012) The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36:92–104
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109
Mjösberg J, Bernink J, Peters C, Spits H (2012) Transcriptional control of innate lymphoid cells. Eur J Immunol 42:1916–1923
Julliard W, Fechner J, Mezrich J (2014) The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Front Immunol 5:458
Mezrich J, Fechner J, Zhang X, Johnson B, Burlingham W, Bradfield C (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L et al (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203
Qiu J, Guo X, Chen Z, He L, Sonnenberg G, Artis D, Fu Y, Zhou L (2013) Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39:386–399
Fallarino F, Grohmann U, Puccetti P (2012) Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur J Immunol 42:1932–1937
Nguyen N, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci 107:19961–19966
Nguyen N, Nakahama T, Le D, Van Son L, Chu H, Kishimoto T (2014) Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol 5:551
Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M (2011) Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147:629–640. doi:10.1016/j.cell.2011.09.025
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T et al (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487:477–481
Maes M, Leunis JC, Geffard M, Berk M (2014) Evidence for the existence of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) with and without abdominal discomfort (irritable bowel) syndrome. Neuro Endocrinol Lett 35:445–453
Moylan S, Berk M, Dean OM, Samuni Y, Williams LJ, O’Neil A, Hayley AC, Pasco JA et al (2014) Oxidative & nitrosative stress in depression: why so much stress? Neurosci Biobehav Rev 45:46–62
Connor T, Starr N, O’Sullivan J, Harkin A (2008) Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-γ? Neurosci Lett 441:29–34
Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P (2008) IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J Immunol 180:5157–5162
Tourino M, de Oliveira E, Bellé L, Knebel F, Albuquerque R, Dörr F, Okada S, Migliorini S et al (2013) Tryptamine and dimethyltryptamine inhibit indoleamine 2,3 dioxygenase and increase the tumor-reactive effect of peripheral blood mononuclear cells. Cell Biochem Funct 31:361–364
Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B (2008) Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med 206:43–49
Stephens G, Wang Q, Swerdlow B, Bhat G, Kolbeck R, Fung M (2013) Kynurenine 3-monooxygenase mediates inhibition of Th17 differentiation via catabolism of endogenous aryl hydrocarbon receptor ligands. Eur J Immunol 43:1727–1734
Dinan T, Borre Y, Cryan J (2014) Genomics of schizophrenia: time to consider the gut microbiome? Mol Psychiatry 19:1252–1257
Clarke G, Grenham S, Fitzgerald P, Moloney R, Shanahan F, Dinan T, Cryan J (2012) Su1992 regulation of serotonergic neurotransmission and behaviour by the brain-gut-microbiome axis. Gastroenterology 142:S–555
Forsythe P, Kunze W, Bienenstock J (2012) On communication between gut microbes and the brain. Curr Opin Gastroenterol 28:557–562
Clarke G, Stilling R, Kennedy P, Stanton C, Cryan J, Dinan T (2014) Minireview: Gut microbiota: the neglected endocrine organ. Mol Endocrinol 28:1221–1238
Bercik P, Verdu EF, Foster JA et al (2010) Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 139:2102–2112.e1
Barry S, Clarke G, Scully P, Dinan T (2008) Kynurenine pathway in psychosis: evidence of increased tryptophan degradation. J Psychopharmacol 23:287–294
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477
Stone TW, Stoy N, Darlington LG (2013) An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci 34:136–143
Hsiao EY (2013) Immune dysregulation in autism spectrum disorder. Int Rev Neurobiol 113:269–302
Yanofsky C (2007) RNA-based regulation of genes of tryptophan synthesis and degradation, in bacteria. RNA 13:1141–1154
Shishov VA, Kirovskaia TA, Kudrin VS, Oleskin AV (2009) Amine neuromediators, their precursors, and oxidation products in the culture of Escherichia coli K-12 [in Russian]. Prikl Biokhim Mikrobiol 45:550–554
Mawe G, Hoffman J (2013) Serotonin signalling in the gut—functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol 10:564–564
O’Mahony S, Clarke G, Borre Y, Dinan T, Cryan J (2015) Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 277:32–48
Dash S, Clarke G, Berk M, Jacka F (2015) The gut microbiome and diet in psychiatry. Curr Opin Psychiatry 28:1–6
Keightley P, Koloski N, Talley N (2015) Pathways in gut-brain communication: evidence for distinct gut-to-brain and brain-to-gut syndromes. Aust N Z J Psychiatry 49:207–214
Zhang Y, Song L, Gao Q, Yu S, Li L, Gao N (2012) The two-step biotransformation of monosodium glutamate to GABA by Lactobacillus brevis growing and resting cells. Appl Microbiol Biotechnol 94:1619–1627
Barrett E, Ross R, O’Toole P, Fitzgerald G, Stanton C (2012) γ-Aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol 113:411–417
Bravo JA, Julio-Pieper M, Forsythe P, Kunze W, Dinan TG, Bienenstock J et al (2012) Communication between gastrointestinal bacteria and the nervous system. Curr Opin Pharmacol 12:667–672. doi:10.1016/j.coph.2012.09.010
Matsumoto M, Kibe R, Ooga T, Aiba Y, Sawaki E, Koga Y, Benno Y (2013) Cerebral low-molecular metabolites influenced by intestinal microbiota: a pilot study. Front Syst Neurosci 7:9
Neufeld KM, Kang N, Bienenstock J, Foster JA (2011) Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil 23(3):255–264. e119
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y (2004) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558:263–275
Markelov V, Trushin M (2007) Multiple sclerosis and neurochemical disturbances. Pak J Med Sci 23:145–149
Lucas K, Morris G, Anderson G, Maes M (2015) The Toll-like receptor radical cycle pathway: a new drug target in immune-related chronic fatigue. CNS Neurol Disord Drug Targets 14:838–854
Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M et al (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422:173–176
Maa C, Chang MY, Hsieh MY, Chen YJ, Yang CJ, Chen ZC, Li YK, Yen CK et al (2010) Butyrate reduced lipopolysaccharide-mediated macrophage migration by suppression Maa of Src enhancement and focal adhesion kinase activity. J Nutr Biochem 21:1186–1192
Owen KA, Pixley FJ, Thomas KS, Vicente-Manzanares M, Ray BJ, Horwitz AF, Parsons JT, Beggs HE et al (2007) Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J Cell Biol 179:1275–1287
Zapolska-Downar D, Naruszewicz M (2009) Propionate reduces the cytokine-induced VCAM-1 and ICAM-1 expression by inhibiting nuclear factor-kappa B (NF-kappaB) activation. J Physiol Pharmacol 60:123–131
Nilsson NE, Kotarsky K, Owman C, Olde B (2003) Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acid. Biochem Biophys Res Commun 303:1047–1052
Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M (2004) Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: the role of NF-kappaB and PPARalpha. J Nutr Biochem 15:220–228
Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C et al (2009) Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A 106:5859–5864
Bocker U, Nebe T, Herweck F, Holt L, Panja A, Jobin C, Rossol S, Sartor RB et al (2003) Butyrate modulates intestinal epithelial cell-mediated neutrophil migration. Clin Exp Immunol 131:53–60
Fusunyan RD, Quinn JJ, Fujimoto M, MacDermott RP, Sanderson IR (1999) Butyrate switches the pattern of chemokine secretion by intestinal epithelial cells through histone acetylation. Mol Med 5:631–640
Inatomi O, Andoh A, Kitamura K, Yasui H, Zhang Z, Fujiyama Y (2005) Butyrate blocks interferon-gamma-inducible protein-10 release in human intestinal subepithelial myofibroblasts. J Gastroenterol 40:483–489
Menzel T, Luhrs H, Zirlik S, Schauber J, Kudlich T, Gerke T, Gostner A, Neumann M et al (2004) Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm Bowel Dis 10:122–128
Bohmig GA, Krieger PM, Saemann MD, Wenhardt C, Pohanka E, Zlabinger GJ (1997) n-Butyrate downregulates the stimulatory function of peripheral blood-derived antigen-presenting cells: a potential mechanism for modulating T-cell responses by short-chain fatty acids. Immunology 92:234–243
Dianzani C, Cavalli R, Zara GP, Gallicchio M, Lombardi G, Gasco MR, Panzanelli P, Fantozzi R (2006) Cholesteryl butyrate solid lipid nanoparticles inhibit adhesion of human neutrophils to endothelial cells. Br J Pharmacol 148:648–656
Allport JR, Ding HT, Ager A, Steeber DA, Tedder TF, Luscinskas FW (1997) L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J Immunol 158:4365–4372
Griffin WS (2006) Inflammation and neurodegenerative diseases. Am J Clin Nutr 83:470S–474S
Boyle JJ (2005) Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol 3:63–68
Chakravortty D, Koide N, Kato Y, Sugiyama T, Mu MM, Yoshida T, Yokochi T (2000) The inhibitory action of butyrate on lipopolysaccharide-induced nitric oxide production in RAW 264.7 murine macrophage cells. J Endotoxin Res 6:243–247
Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH (2013) Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145:396–406. doi:10.1053/j.gastro.2013.04.056
Perez R, Stevenson F, Johnson J, Morgan M, Erickson K, Hubbard NE, Morand L, Rudich S et al (1998) Sodium butyrate upregulates Kupffer cell PGE2 production and modulates immune function. J Surg Res 78:1–6
Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 19:587–593
Glozak MA, Sengupta N, Zhang X, Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23
Cox MA, Jackson J, Stanton M, Rojas-Triana A, Bober L, Laverty M, Yang X, Zhu F et al (2009) Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J Gastroenterol 15:5549–5557
Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S (2009) Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med 15:633–640
Sakata D, Yao C, Narumiya S (2010) Prostaglandin E2, an immunoactivator. J Pharmacol Sci 112:1–5
Zaibi MS, Stocker CJ, O’Dowd J, Davies A, Bellahcene M, Cawthorne MA, Brown AJ, Smith DM et al (2010) Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett 584:2381–2386
Plaisancie P, Dumoulin V, Chayvialle JA, Cuber JC (1996) Luminal peptide YY-releasing factors in the isolated vascularly perfused rat colon. J Endocrinol 151:421–429
Luster AD, Alon R, von Andrian UH (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 6:1182–1190
Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R (2011) Suppressive effect of short chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem 22:849–855
Miller SJ, Zaloga GP, Hoggatt AM, Labarrere C, Faulk WP (2005) Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition 21:740–748
Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R (2009) Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci 117:331–338
Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F, Raabe B, Chalaris A et al (2009) G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 183:7514–7522
Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly YM, Stephens L, Hawkins PT et al (2011) SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One 6, e21205
Blais M, Seidman EG, Asselin C (2007) Dual effect of butyrate on IL-1beta-mediated intestinal epithelial cell inflammatory response. DNA Cell Biol 26:133–147
Leung CH, Lam W, Ma DL, Gullen EA, Cheng YC (2009) Butyrate mediates nucleotide-binding and oligomerisation domain (NOD) 2-dependent mucosal immune responses against peptidoglycan. Eur J Immunol 39:3529–3537
Furusawa Y, Obata Y, Fukuda S, Endo T, Nakato G, Takahashi D, Nakanishi Y, Uetake C et al (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504:446–450
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross J et al (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455
Hoeppli R, Wu D, Cook L, Levings M (2015) The environment of regulatory T cell biology: cytokines, metabolites, and the microbiome. Front Immunol 6:61
Smith P, Howitt M, Panikov N, Michaud M, Gallini C, Bohlooly-Y M, Glickman J, Garrett W (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad P et al (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40:128–139
Kim C, Park J, Kim M (2014) Gut microbiota-derived short-chain fatty acids, T cells, and inflammation. Immune Netw 14:277
Chang P, Hao L, Offermanns S, Medzhitov R (2014) The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci 111:2247–2252
Park J, Kim M, Kang S, Jannasch A, Cooper B, Patterson J, Kim C (2014) Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol 8:80–93
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105
Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC et al (2009) The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30:832–844
Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA (2002) The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr 132:1012–1017
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Yu X, Shahir AM, Sha J, Feng Z, Eapen B, Nithianantham S, Das B, Karn J et al (2014) Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J Virol 88:4466–4479
Singh N, Thangaraju M, Prasad PD, Martin PM, Lambert NA, Boettger T, Offermanns S, Ganapathy V (2010) Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem 285:27601–27608
Berndt BE, Zhang M, Owyang SY, Cole TS, Wang TW, Luther J, Veniaminova NA, Merchant JL et al (2012) Butyrate increases IL-23 production by stimulated dendritic cells. Am J Physiol Gastrointest Liver Physiol 303:G1384–G1392
Frikeche J, Simon T, Brissot E, Gregoire M, Gaugler B, Mohty M (2012) Impact of valproic acid on dendritic cells function. Immunobiology 217:704–710
Mace TA, King SA, Ameen Z, Elnaggar O, Young G, Riedl KM, Schwartz SJ, Clinton SK et al (2014) Bioactive compounds or metabolites from black raspberries modulate T lymphocyte proliferation, myeloid cell differentiation and Jak/STAT signaling. Cancer Immunol Immunother 63:889–900
Wang A, Gu Z, Heid B, Akers RM, Jiang H (2009) Identification and characterization of the bovine G protein-coupled receptor GPR41 and GPR43 genes. J Dairy Sci 92:2696–2705
Tazoe H, Otomo Y, Karaki S, Kato I, Fukami Y, Terasaki M, Kuwahara A (2009) Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed Res 30:149–156
Stringer RE, Hart CA, Edwards SW (1996) Sodium butyrate delays neutrophil apoptosis: role of protein biosynthesis in neutrophil survival. Br J Haematol 92:169–175
Nakao S, Moriya Y, Furuyama S, Niederman R, Sugiya H (1998) Propionic acid stimulates superoxide generation in human neutrophils. Cell Biol Int 22:331–337
Liu Q, Shimoyama T, Suzuki K, Umeda T, Nakaji S, Sugawara K (2001) Effect of sodium butyrate on reactive oxygen species generation by human neutrophils. Scand J Gastroenterol 36:744–750
Sandoval A, Trivinos F, Sanhueza A, Carretta D, Hidalgo MA, Hancke JL, Burgos RA (2007) Propionate induces pH(i) changes through calcium flux, ERK1/2, p38, and PKC in bovine neutrophils. Vet Immunol Immunopathol 115:286–298. doi:10.1016/j.vetimm.2006.11
Huuskonen J, Suuronen T, Nuutinen T, Kyrylenko S, Salminen A (2004) Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br J Pharmacol 141:874–880
Park JS, Woo MS, Kim SY, Kim WK, Kim HS (2005) Repression of interferon-gamma-induced inducible nitric oxide synthase (iNOS) gene expression in microglia by sodium butyrate is mediated through specific inhibition of ERK signaling pathways. J Neuroimmunol 168:56–64
Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, Lu RB, Gean PW et al (2007) Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 149:203–212
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM (2007) Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther 321:892–901
Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A et al (2014) Intestinal permeability—a new target for disease prevention and therapy. BMC Gastroenterol 14:189. doi:10.1186/s12876-014-0189-7
Ferreira T, Leonel A, Melo M, Santos R, Cara D, Cardoso V, Correia M, Alvarez-Leite J (2012) Oral supplementation of butyrate reduces mucositis and intestinal permeability associated with 5-fluorouracil administration. Lipids 47:669–678
Canani R (2011) Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol 17:1519
Peng L, Li Z, Green R, Holzman I, Lin J (2009) Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 139:1619–1625
Kelly C, Zheng L, Campbell E, Saeedi B, Scholz C, Bayless A, Wilson K, Glover L et al (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17:662–671
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J et al (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61:364–371. doi:10.2337/db11-1019
Hadjiyanni I, Li K, Drucker D (2009) Glucagon-like peptide-2 reduces intestinal permeability but does not modify the onset of type 1 diabetes in the nonobese diabetic mouse. Endocrinol 150:592–599
Psichas A, Sleeth ML, Murphy KG, Brooks L, Bewick GA, Hanyaloglu AC, Ghatei MA, Bloom SR et al (2015) The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes (Lond) 39:424–429. doi:10.1038/ijo.2014.153
Kaji I, Karaki S, Kuwahara A (2014) Short-chain fatty acid receptor and its contribution to glucagon-like peptide-1 release. Digestion 89:31–36. doi:10.1159/000356211
Brubaker PL, Drucker DJ (2004) Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinol 145:2653–2659
Geurts L, Lazarevic V, Derrien M, Everard A, Van Roye M, Knauf C, Valet P, Girard M et al (2011) Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front Microbiol 2:149
Muccioli GG, Naslain D, Backhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:392
Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, Possemiers S, Van Holle A et al (2011) Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60:2775–2786
Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D et al (2009) Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP‐2‐driven improvement of gut permeability. Gut 58:1091–1103
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57:1470–1481
Cani P (2012) Crosstalk between the gut microbiota and the endocannabinoid system: impact on the gut barrier function and the adipose tissue. Clin Microbiol Infect 18:50–53
Puddu A, Sanguineti R, Montecucco F, Viviani G (2014) Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators Inflamm 2014:1–9
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28:1221–1227
den Besten G, van Eunen K, Groen A, Venema K, Reijngoud D, Bakker B (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54:2325–2340
Fushimi T, Suruga K, Oshima Y, Fukiharu M, Tsukamoto Y, Goda T (2006) Dietary acetic acid reduces serum cholesterol and triacylglycerols in rats fed a cholesterol-rich diet. Br J Nutr 95:916–924
Demigné C, Morand C, Levrat MA, Besson C, Moundras C, Rémésy C (1995) Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Br J Nutr 74:209–219
Todesco T, Rao AV, Bosello O, Jenkins DJ (1991) Propionate lowers blood glucose and alters lipid metabolism in healthy subjects. Am J Clin Nutr 54:860–865
Topping DL, Clifton PM (2001) Short-chain fatty acids and human colonic function: roles of resistant starch and non-starch polysaccharides. Physiol Rev 81:1031–1064
Stoddart N, Smith J, Milligan G (2008) International union of pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev 60(no. 4):405–417
Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT et al (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterol 143:e913–e917
Udayappan SD, Hartstra AV, Dallinga-Thie GM, Nieuwdorp M (2014) Intestinal microbiota and faecal transplantation as treatment modality for insulin resistance and type 2 diabetes mellitus. Clin Exper Immunol 177:24–29
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439
Ross SA, Ekoé J (2010) Incretin agents in type 2 diabetes. Can Fam Physician 56:639–648
Remely M, Aumueller E, Merold C, Dworzak S, Hippe B, Zanner J, Pointner A, Brath H et al (2014) Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene 537:85–92
Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen J, Tian H, Li Y (2008) Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinol 149:4519–4526
Yamashita H, Fujisawa K, Ito E, Idei S, Kawaguchi N, Kimoto M, Hiemori M, Tsuji H (2007) Improvement of obesity and glucose tolerance by acetate in type 2 diabetic Otsuka Long-Evans Tokushima fatty (OLETF) rats. Biosci Biotechnol Biochem 71:1236–124
Wright RS, Anderson JW, Bridges SR (1990) Propionate inhibits hepatocyte lipid synthesis. Proc Soc Exp Biol Med 195:26–29
Boey D, Lin S, Karl T, Baldock P, Lee N, Enriquez R, Couzens M, Slack K et al (2006) Peptide YY ablation in mice leads to the development of hyperinsulinaemia and obesity. Diabetologia 49:1360–1370
Freeland KR, Wolever TM (2010) Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br J Nutr 103:460–466
Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A et al (2011) Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci 108:8030–8035
Inoue D, Kimura I, Wakabayashi M, Tsumoto H, Ozawa K, Hara T, Takei Y, Hirasawa A et al (2012) Short-chain fatty acid receptor GPR41-mediated activation of sympathetic neurons involves synapsin 2b phosphorylation. FEBS Lett 586:1547–1554
Tilg H, Kaser A (2011) Gut microbiome, obesity, and metabolic dysfunction. The J Clin Invest 121:2126–2132
Tremaroli V, Bäckhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249
Karlsson F, Tremaroli V, Nookaew I, Bergström G, Behre C, Fagerberg B, Nielsen J, Bäckhed F (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498:99–103
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W et al (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60
Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte R, Bartelsman J, Dallinga–Thie G, Ackermans M et al (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterol 143:913–916.e7
Sato J, Kanazawa A, Ikeda F, Yoshihara T, Goto H, Abe H, Komiya K, Kawaguchi M et al (2014) Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care 37(8):2343–2350
Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM, Yanagisawa M (2004) Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc Natl Acad Sci 101:1045–1050. doi:10.1073/pnas.2637002100
Al-Lahham SH et al (2010) Regulation of adipokine production in human adipose tissue by propionic acid. Eur J Clin Invest 40:401–407
Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, Lepage P, Klopp C et al (2011) Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia 54:3055–3061
Dixon A, Valsamakis G, Hanif M, Field A, Boutsiadis A, Harte A, McTernan P, Barnett A et al (2008) Effect of the orlistat on serum endotoxin lipopolysaccharide and adipocytokines in South Asian individuals with impaired glucose tolerance. Int J Clin Prac 62:1124–1129
Pussinen P, Havulinna A, Lehto M, Sundvall J, Salomaa V (2011) Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care 34:392–397
Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán L, Smirnova N, Bergé M et al (2011) Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3:559–572
Diamant M, Blaak E, de Vos W (2010) Do nutrient-gut-microbiota interactions play a role in human obesity, insulin resistance and type 2 diabetes? Obesity Rev 12:272–281
Everard A, Cani P (2013) Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol 27:73–83
Musso G, Gambino R, Cassader M (2010) Gut microbiota as a regulator of energy homeostasis and ectopic fat deposition: mechanisms and implications for metabolic disorders. Curr Opin Lipidol 21:76–83
Shoelson S (2006) Inflammation and insulin resistance. J Clin Invest 116:1793–1801
Dandona P (2005) Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 111:1448–1454
Shoelson S, Herrero L, Naaz A (2007) Obesity, inflammation, and insulin resistance. Gastroenterol 132:2169–2180
Pickup JC, Crook MA (1998) Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41:1241–1248. doi:10.1007/s001250051058
Chung S, Lapoint K, Martinez K, Kennedy A, Boysen Sandberg M, McIntosh MK (2006) Preadipocytes mediate lipopolysaccharide-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinol 147:5340–5351. doi:10.1210/en.2006-0536
Cucak H, Mayer C, Tonnesen M, Thomsen L, Grunnet L, Rosendahl A (2014) Macrophage contact dependent and independent TLR4 mechanisms induce β-cell dysfunction and apoptosis in a mouse model of type 2 diabetes. PLoS One 9:e90685
Garay-Malpartida H, Mourão R, Mantovani M, Santos I, Sogayar M, Goldberg A (2011) Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol 12:18
Zhang C, Xiao C, Wang P, Xu W, Zhang A, Li Q, Xu X (2014) The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Human Immunol 75:289–296
Zeng C, Shi X, Zhang B, Liu H, Zhang L, Ding W, Zhao Y (2011) The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med 90:175–186
Liang H, Hussey S, Sanchez-Avila A, Tantiwong P, Musi N (2013) Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS One 8:e63983
van Maren WW, Jacobs JF, de Vries IJ, Nierkens S, Adema GJ (2008) Toll-like receptor signalling on Tregs: to suppress or not to suppress? Immunol 124:445–452. doi:10.1111/j.1365-2567.2008.02871
Doan H, Bowen K, Evers B (2009) QS296. Toll-like receptor 4 activation in human colorectal cancer cells induces PI3K/AKT signaling. J Surgical Res 151:295
Basu S, Hubbard B, Shevach E (2014) Foxp3-mediated inhibition of Akt inhibits Glut1 (glucose transporter 1) expression in human T regulatory cells. J Leukoc Biol 97:279–283
Nyirenda M, Sanvito L, Darlington P, O’Brien K, Zhang G, Constantinescu C, Bar-Or A, Gran B (2011) TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol 187:2278–2290
Al-Daghri N, Al-Attas O, Alokail M, Alkharfy K, Draz H, Agliardi C, Mohammed A, Guerini F et al (2012) Vitamin D receptor gene polymorphisms and HLA DRB1*04 cosegregation in Saudi type 2 diabetes patients. J Immunol 188:1325–1332
Mangge H, Summers K, Meinitzer A, Zelzer S, Almer G, Prassl R, Schnedl W, Reininghaus E et al (2013) Obesity-related dysregulation of the tryptophan-kynurenine metabolism: role of age and parameters of the metabolic syndrome. Obesity 22:195–201
Oxenkrug G (2013) Insulin resistance and dysregulation of tryptophan–kynurenine and kynurenine–nicotinamide adenine dinucleotide metabolic pathways. Mol Neurobiol 48:294–301
Oxenkrug G, Ratner R, Summergrad P (2013) Kynurenines and vitamin B6: link between diabetes and depression. J Bioinform Diabetes 1:1–10
Oxenkrug G (2015) Increased plasma levels of xanthurenic and kynurenic acids in type 2 diabetes. Mol Neurobiol 52:805–810
Reginaldo C, Jacques P, Scott T, Oxenkrug G, Selhub J, Paul L (2015) Xanthurenic acid is associated with higher insulin resistance and higher odds of diabetes. FASEB 29:919.20
Nix W, Zirwes R, Bangert V, Kaiser R, Schilling M, Hostalek U, Obeid R (2015) Vitamin B status in patients with type 2 diabetes mellitus with and without incipient nephropathy. Diabetes Res Clin Pract 107:157–165
Oxenkrug G (2010) Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann N Y Acad Sci 1199:1–14
Allison D, Ditor D (2014) The common inflammatory etiology of depression and cognitive impairment: a therapeutic target. J Neuroinflamm 11:151
Midttun O, Ulvik A, Pedersen E, Ebbing M, Bleie O et al (2011) Low plasma vitamin B-6 status affects metabolism through the kynurenine pathway in cardiovascular patients with systemic inflammation. J Nutr 141:611–617
Kimoto M, Ogawa T, Tokushima Sasaoka K (1991) Accumulation of 3-hydroxy-L-kynurenine sulfate and ethanolamine in urine of the rat injected with 1-aminoproline. J Exp Med 38:37–44
Rogers KS, Evangelista SJ (1985) 3-Hydroxykynurenine, 3-hydroxyanthranilic acid, and o-aminophenol inhibit leucine-stimulated insulin release from rat pancreatic islets. Proc Soc Exp Biol Med 178:275–278
Sarkar SA, Wong R, Hackl SI, Moua O, Gill RC et al (2007) Induction of indoleamine 2,3-dioxygenase by interferon-gamma in human islets. Diabetes 56:72–79
Meyramov G, Korchin V, Kocheryzkina N (1984) Diabetogenic activity of xanturenic acid determined by its chelating properties? Acta Vitaminol Enzymol 6:221–228
Ikeda S, Kotake Y (1986) Urinary excretion of xanthurenic acid and zinc in diabetes: (3). Occurrence of xanthurenic acid-Zn2+ complex in urine of diabetic patients and of experimentally-diabetic rats. Ital J Biochem 35:232–241
Kotaki Y, Ueda T, Mori T, Igaki S, Hattori M (1975) Abnormal tryptophan metabolism and experimental diabetes by xanthurenic acid (XA). Acta Vitaminol Enzymol 29:236–239
Sakakeeny L, Roubenoff R, Obin M, Fontes J, Benjamin E, Bujanover Y, Jacques P, Selhub J (2012) Plasma pyridoxal-5-phosphate is inversely associated with systemic markers of inflammation in a population of U.S. adults. J Nutr 142:1280–1285
Ulvik A, Midttun O, Pedersen E, Eussen S, Nygard O, Ueland P (2014) Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am J Clin Nutr 100:250–255
Vasilaki A, McMillan D, Kinsella J, Duncan A, O’Reilly D, Talwar D (2008) Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr 88:140–146
Rosenfeld C (2015) Microbiome disturbances and autism spectrum disorders. Drug Metab Dispos 43:1557–1571
MacFabe D (2012) Short-chain fatty acid fermentation products of the gut microbiome implications in autism spectrum disorders. Microb Ecol Health Dis 23. doi: 10.3402/mehd.v23i0.19260
Mayer E, Padua D, Tillisch K (2014) Altered brain-gut axis in autism: comorbidity or causative mechanisms? Bioessays 36:933–939
Mayer E, Savidge T, Shulman R (2014) Brain-gut microbiome interactions and functional bowel disorders. Gastroenterol 146:1500–1512
Wang L, Conlon M, Christophersen C, Sorich M, Angley M (2014) Gastrointestinal microbiota and metabolite biomarkers in children with autism spectrum disorders. Biomark Med 8:331–344
Krajmalnik-Brown R, Lozupone C, Kang D, Adams J (2015) Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microbial Ecology in Health & Disease 26
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA et al (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155:1451–1463. doi:10.1016/j.cell.2013.11.024
Boukthir S, Matoussi N, Belhadj A, Mammou S, Dlala SB, Helayem M, Rocchiccioli F, Bouzaidis Abdennebi M (2010) Abnormal intestinal permeability in children with autism. Tunis Med 88:685–686
de Magistris L, Familiari V, Pascotto A, Sapone A, Frolli A, Iardino P, Carteni M, De Rosa M et al (2010) Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J Pediatr Gastroenterol Nutr 51:418–424
Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, di Nemi S, Politi P (2010) Low-grade endotoxemia in patients with severe autism. Neurosci Lett 471:162–165
Adams JB, Johansen LJ, Powell LD, Quig D, Rubin RA (2011) Gastrointestinal flora and gastrointestinal status in children with autism—comparisons to typical children and correlation with autism severity. BMC Gastroenterol 11:22
Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, Youn E, Summanen PH et al (2010) Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16:444–453
Finegold SM, Downes J, Summanen PH (2012) Microbiology of regressive autism. Anaerobe 18:260–262
Gondalia SV, Palombo EA, Knowles SR, Cox SB, Meyer D, Austin DW (2012) Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res 5:419–427
Kang DW, Park JG, Ilhan ZE, Wallstrom G, Labaer J, Adams JB, Krajmalnik-Brown R (2013) Reduced incidence of and other fermenters in intestinal microflora of autistic children. PLoS One 8, e68322
Parracho HM, Bingham MO, Gibson GR, McCartney AL (2005) Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J Med Microbiol 54:987–991
Williams BL et al (2011) Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One 6, e24585
Williams BL, Hornig M, Parekh T, Lipkin WI (2012) Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. MBio 3. doi: 10.1128/mBio.00261-11. Print 2012.
Song Y, Liu C, Finegold SM (2004) Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol 70:6459–6465
Finegold SM (2011) Desulfovibrio species are potentially important in regressive autism. Med Hypotheses 77:270–274
Finegold S (2011) State of the art; microbiology in health and disease. Intestinal bacterial flora in autism. Anaerobe 17:367–368
Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen ML, Bolte E et al (2002) Gastrointestinal microflora studies in late-onset autism. Clin Infect Dis 35:S6–S16
Thomas RH, Meeking MM, Mepham JR, Tichenoff L, Possmayer F, Liu S et al (2012) The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: further development of a rodent model of autism spectrum disorders. J Neuroinflamm 9:153
Shultz SR, MacFabe DF, Ossenkopp KP, Scratch S, Whelan J, Taylor R et al (2008) Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: implications for an animal model of autism. Neuropharmacology 54:901–911
MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F et al (2007) Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res 176:149–169
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T et al (2007) Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem 100:1469–1479
Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13:68
Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP et al (2006) Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J 398:107–112
Brass EP (1992) Interaction of carnitine and propionate with pyruvate oxidation by hepatocytes from clofibrate-treated rats: importance of coenzyme A availability. J Nutr 122:234–240
Clarke JT, Clark-Taylor BE (2004) Is autism a disorder of fatty acid metabolism? Possible dysfunction of mitochondrial beta oxidation by long chain acyl-CoA dehydrogenase. Med Hypotheses 62:970–975
Filipek P, Juranek J, Nguyen M, Cummings C, Gargus J (2004) Relative carnitine deficiency in autism. J Autism Dev Disord 34:615–623
Frye R (2012) Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. N A J Med Sci 5:141–147
Jones LL, McDonald DA, Borum PR (2010) Acylcarnitines: role in brain. Prog Lipid Res 49:61–75
Kekuda R, Manoharan P, Baseler W, Sundaram U (2013) Monocarboxylate 4 mediated butyrate transport in a rat intestinal epithelial cell line. Dig Dis Sci 58:660–667
Maurer M (2004) Correlation between local monocarboxylate transporter 1 (MCT1) and glucose transporter 1 (GLUT1) densities in the adult rat brain. Neurosci Lett 355:105–108
Peinado A, Yuste R, Katz LC (1993) Extensive dye coupling between rat neocortical neurons during the period of circuit formation. Neuron 10:103–114
Pierre K, Pellerin L (2005) Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem 94:1–14
Pellerin L (2005) How astrocytes feed hungry neurons. Mol Neurobiol 32:059–072
Rafiki A, Boulland J, Halestrap A, Ottersen O, Bergersen L (2003) Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neurosci 122:677–688
Hara H, Haga S, Aoyama Y, Kiriyama S (1999) Short-chain fatty acids suppress cholesterol synthesis in rat liver and intestine. J Nutr 129:942–948
DeCastro M, Nankova B, Shah P, Patel P, Mally P, Mishra R, La Gamma E (2005) Short chain fatty acids regulate tyrosine hydroxylase gene expression through a cAMP-dependent signaling pathway. Mol Brain Res 142:28–38
Wajner M, Latini A, Wyse AT, Dutra-Filho CS (2004) The role of oxidative damage in the neuropathology of organic acidurias: insights from animal studies. J Inherit Metab Dis 27:427–448
Neuhaus E, Beauchaine TP, Bernier R (2010) Neurobiological correlates of social functioning in autism. Clin Psychol Rev 30:733–748
El-Ansary AK, Ben BA, Kotb M (2012) Etiology of autistic features: the persisting neurotoxic effects of propionic acid. J Neuroinflammation 9:74
Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB (2003) Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci 6:1139–1140
Shah P, Nankova BB, Parab S, La Gamma EF (2006) Short chain fatty acids induce TH gene expression via ERK-dependent phosphorylation of CREB protein. Brain Res 1107:13–23
Ming X, Julu PO, Brimacombe M, Connor S, Daniels ML (2005) Reduced cardiac parasympathetic activity in children with autism. Brain Dev 27:509–516
Liu Z, Li N, Neu J (2005) Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr 94:386–393
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science 336:1262–1267
Anderson G, Maes M (2014) Redox regulation and the autistic spectrum: role of tryptophan catabolites, immuno-inflammation, autoimmunity and the amygdala. Curr Neuropharmacol 12:148–167
Acknowledgments
MB is supported by a NHMRC Senior Principal Research Fellowship 1059660.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
MB has received grants from the NIH, Simons Autism Foundation, Stanley Medical Research Foundation, NHMRC, and CRC for Mental Health; he has been a paid consultant for Astra Zeneca, Eli Lilly, Glaxo SmithKline, Janssen Cilag, Lundbeck, and Pfizer and a paid speaker for Astra Zeneca, Eli Lilly, Glaxo SmithKline, Lundbeck, and Merck. FNJ has received grants from the Brain and Behaviour Research Institute, NHMRC, Australian Rotary Health, and the Meat and Livestock Board and has been a paid speaker for Sanofi-Synthelabo, Janssen Cilag, Servier, and Health Ed. The other authors have no conflicts to declare.
Rights and permissions
About this article

Cite this article
Morris, G., Berk, M., Carvalho, A. et al. The Role of the Microbial Metabolites Including Tryptophan Catabolites and Short Chain Fatty Acids in the Pathophysiology of Immune-Inflammatory and Neuroimmune Disease. Mol Neurobiol 54, 4432–4451 (2017). https://doi.org/10.1007/s12035-016-0004-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0004-2