
Abstract
Neuroinflammation in the cardiovascular center plays a critical role in the progression of hypertensive heart disease. And microglial autophagy is involved in the regulation of neuroinflammation. Cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor, senses mitochondrial DNA (mtDNA) and regulates autophagy. The detailed mechanisms of central cGAS affects neuroinflammatory response in hypertensive heart disease via regulating autophagy remain unknown. Angiotensin II (Ang II, 1.5 mg·kg−1·12 h−1, 2 weeks) was intraperitoneally injected to induce hypertension in mice. The cGAS-STING pathway was activated in the paraventricular nucleus (PVN) of Ang II-induced hypertensive mice. The contractile dysfunction of heart was alleviated in Ang II-induced hypertensive cGAS−/− mice. To observe the central effects of cGAS on regulating hypertensive heart disease, the RU.521 (a cGAS inhibitor) was intracisternally infused in hypertensive mice. Intracisternal infusion of the RU.521-alleviated myocardial interstitial fibrosis, cardiomyocyte hypertrophy, and the contractile dysfunction in Ang II-induced hypertensive mice. Intracisternal infusion of RU.521 attenuated the microglial activation, neuroinflammation, sympathetic/parasympathetic activity ratio, and lowered blood pressure. The autophagic flux in the PVN cells was blocked, while intracisternal infusion of RU.521 alleviated this effect in the Ang II-induced hypertensive mice. In vitro, it was found that cGAS-STING activation-induced autophagic flux blockage, while when the impaired autophagic flux was facilitated by rapamycin, an autophagy inducer, the microglial M1 polarization was decreased correspondingly. In conclusion, cGAS induces the inflammatory phenotype of microglia via impairing autophagic flux, thereby participating in neuroinflammation, which leads to sympathetic overactivation in hypertension and further caused hypertensive myocardial injury.
Similar content being viewed by others


Introduction
With social health problems accumulating in modern society, the prevalence of hypertension continues to increase [1, 2]. Hypertension is an independent risk factor for cardiovascular events including myocardial ischemia, cerebral stroke, and heart failure [3]. Long-term exposure to the hemodynamic stress imposed by hypertension, along with the combination of other factors, including comorbidities (e.g., obesity, diabetes mellitus, and chronic kidney disease) eventually leads to left ventricular (LV) dysfunction and heart failure [4,5,6]. Therefore, the updated pathophysiological mechanisms of LV dysfunction and heart failure are required to control hypertension-related heart diseases.
It has been demonstrated that the sympathetic overactivity is an indispensable pathogenesis of hypertension and hypertension-induced end target organ injury [7]. Overactive local renin angiotensin system (RAS), oxidative stress, and neuroinflammation in paraventricular nucleus (PVN) and rostral ventrolateral medulla (RVLM) are implicated as major factors in sympathetic overactivity driving, thereby increased blood pressure (BP) and cardiac remodeling in hypertension [8, 9]. Angiotensin II (Ang II), one of the main components of RAS, is not only generated in the systemic circulation but also synthesized locally in tissues like vessel, heart, kidney and brain which increases the BP by multiple mechanisms, such as constricting resistance vessels, stimulating synthesis and release of aldosterone, and enhancing sympathetic outflow from brain [10, 11].
It was reported that Ang II triggers the release of proinflammatory cytokines (PICs) and the production of reactive oxygen species (ROS) in microglia and astrocytes [12]. Our previous studies found that activated proinflammatory/classic (M1) immunophenotypes microglia were increased in the RVLM of stress-induced hypertensive rats, which mediated the secretion of TNF-α, IL-1β, and other PICs, and we have confirmed that minocycline, a microglial M1 polarization inhibitor, reduced sympathetic hyperactivation and systolic BP in stress-induced hypertensive rats [13, 14].
Mitophagy is particularly important for cellular homeostasis in health and disease, and it is the autophagic response that specifically targets damaged mitochondria. Autophagosome-lysosome fusion and autolysosome acidification contribute to the late phase in the autophagic process which is essential to maintain functional autophagic flux and cellular homeostasis. Our recent study showed that stressed microglia existed the mitophagic flux blockage resulted from the lysosomal dysfunction [13, 15]. The increasing number of injured mitochondria induced the activation of NLRP3 (NLR family pyrin domain-containing 3) inflammasome, which led to increased release of PICs including IL-1β and IL-18 from microglia [15, 16]. It is confirmed that the blockage of mitophagic flux leads to increased ROS, which changes the permeability of mitochondrial membrane potential and promotes mitochondria decoupling to produce more ROS [17]. Excessively increasing ROS leads to the increasing number of injured mitochondria and promotes the release of mitochondrial DNA (mtDNA) into cytoplasm [18].
Cyclic GMP-AMP synthase (cGAS) is considered as one of the cytoplasmic DNA sensors [19,20,21]. It detects and binds DNA from multiple sources including exogenous (e.g., DNA viruses, retroviruses, intracellular bacteria) and endogenous sources (e.g., damaged mitochondria). After binding DNA, cGAS transforms ATP and GTP to the second messenger 2′-3′-cyclic GMP-AMP (2′-3′-cGAMP), which in turn activates the stimulator of interferon genes (STING) to induce the expression of type I interferons (IFN-1) and PICs via two main pathways. One is phosphorylating the transcription factor interferon regulatory factor 3 (IRF3) by TANK-binding kinase 1 (TBK1) [22, 23]; the other one is activating transcription factors nuclear factor κB (NF-κB) [20, 24]. In addition, it was reported cGAS induced the occurrence of mitophagy and degradation of injured mitochondria in the liver ischemia reperfusion mice [25]. Related studies suggest that under the stimulation of double stranded (ds) DNA, the direct interaction between cGAS and the Beclin-1 autophagy protein not only enhances the autophagy-mediated degradation of cytosolic DNAs but also suppresses cGAMP synthesis to halt IFN production stimulation [26, 27]. Recent study found that STING, upon binding cGAMP, activates autophagy through a mechanism that is independent of TBK1-IRF3 activation and interferon induction [28].
Series studies have suggested that cGAS, activated by mtDNA, not only plays a key role in the release of PICs but also mediates autophagy of injured mitochondria. However, the specific mechanism of central cGAS regulating microglial autophagy and neuroinflammation in hypertensive heart disease remains unclear. The aim of our present study is to explore the effects of cGAS on neuroinflammation and sympathetic activation in hypertension mice via regulating microglia autophagy.
Materials and Methods
Drugs and Reagents
Ang II (P1085), RU.521 (S6841), and SR-717 (S0853) were purchased from Selleckchem (Houston, TX, USA); rapamycin (S293790) was purchased from Aladdin (Shanghai, China); the flowing antibodies, LC3I/II (4108), p62/SQSTM1 (sequestosome 1) (23214), CD86 (91882S), and β-actin (3700), were purchased from Cell Signaling Technology (Boston, MA, USA); cGAS (sc-515777) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA); STING (19851-1-AP) and cGAS (26416-1-AP) were purchased from Proteintech (Chicago, IL, USA); Iba-1 (GB12105) was purchased from Servicebio (Wuhan, Hubei, China); OX42 (ab216355), donkey anti-rabbit IgG H&L (Alexa Fluor 647) (ab150075), donkey anti-rabbit IgG H&L (Alexa Fluor 488) (ab150073), and donkey anti-rabbit IgG H&L (HRP) (ab6802) were purchased from Abcam (Cambridge, MA, USA). AAV9-mCherry-eGFP-LC3 (HB-AP2100001) was purchased from Hanbio Co. Ltd. (Shanghai, China); Masson’s trichrome staining kit and wheat germ agglutinin were provided by Serivebio (Wuhan, Hubei, China).
Experiment Design
Experiment 1
To explore the role of cGAS in hypertension in vivo, the cGAS knockout (cGAS−/−) mice and C57BL/6 wild-type (WT) mice weighing (20 ± 5) g were divided into four groups: (1) wild-type sham group (CTRL), (2) wild-type + Ang II injection group (HTN), (3) cGAS−/− sham group (cGAS−/− CTRL), and (4) cGAS−/− + Ang II injection group (cGAS−/− HTN). In the HTN group, each mouse was intraperitoneally injected with angiotensin II (Ang II) (1.5 mg·kg−1·12 h−1) for 14 days [29]. The sham group was intraperitoneally injected with equivalent normal saline for 14 days. The BP was recorded and cardiac function was assessed on the 14th day of the experiment. To evaluate the pathological alterations, cardiac tissues were collected on the 28th day when the mice were sacrificed.
Experiment 2
To investigate the effects of central cGAS on microglial activation, RU.521 (a cGAS inhibitor) was infused into the cerebrellomedullary cistern of mice. The C57BL/6 wild-type (WT) mice weighing (20 ± 5) g were randomly divided into three groups: (1) control group (CTRL), (2) Ang II injection group (HTN), (3) Ang II injection + RU.521 intracisternal infusion group (HTN+RU.521). Hypertension mice was induced using the same method of experiment 1. RU.521 (0.4 mg/kg) was infused into cerebellomedullary cistern on the 17th day of the experiment. At the same time, the mice in HTN group were intracisternally infused with the vehicle. The BP and heart rate variability (HRV) were recorded, and cardiac function was assessed (the cardiac function was measured every week, which was attached in Fig. S1). Cardiac tissues were collected to evaluate the pathological alterations, and brain tissues were collected to measure cGAS, STING, LC3II/LCI, and p62 expressions in the PVN of mice on the 28th day when the experiment was terminated.
Experiment 3
In vitro, the cultured BV2 cells were assigned to five groups: (1) vehicle control group (CTRL), (2) Ang II group, (3) Ang II + rapamycin group, (4) Ang II + SR-717 group, (5) Ang II + SR-717 + rapamycin group. For drug treatment, cells were treated with 100 nmol/L Ang II for 12 h [12]. SR-717 (10 μmol/L, 12 h) was used to interfere cGAS pathway [30]. Furthermore, rapamycin (an autophagic inducer, 1 nmol/L, 12 h) [31] was used to activate autophagy pathway. Six individual repeats were arranged for each experiment group.
For the abovementioned experiments, each cohort (n=6 per group) was used to perform immunofluorescent staining. The mCherry-eGFP-LC3 adenovirus were transfected into microglia in vitro to further evaluate autophagic flux. The experiment design is shown in Fig. 1a.
Animal Preparation
The C57BL/6 wild-type male mice and cGAS−/− mice were used in our study and were obtained from Animal Laboratory Center of Fudan University. The genotyping identification of cGAS−/− mice was attached in supplementary data (Fig. S2). The mice were exposed to a 12-h light/dark cycle, with temperature controlled at standard 24 °C. Food and water were given ad libitum. The body weight of mice was monitored in experiment 2 (Fig. S3).
Cerebellomedullary (CM) Cistern Infusion
The mice were anesthetized with 1.5% isoflurane in a 30% O2 gas mixture delivered by a specially made facemask during the surgery. The mice were then placed in the prone position. The nape of the neck was incised at the midline to expose sagittal suture and midline of the nape, which are two important anatomic landmarks during the puncture. The needle then went through the muscles and ligaments in the midline into the gap between the occipital and the atlas vertebra, then 8 μl solution was slowly infused into the CM cistern, and the needle was withdrawn at the end of infusion. The incision was then stitched. We used Evans Blue dye to verify the successfully infusion [32] (Fig. S4). DMSO was used to dissolve the RU.521 to maximum solubility and then, artificial cerebrospinal fluid was used to dilute the solution to the final concentration.
Echocardiographic Assessment
To access the cardiac function, mice were anesthetized by isoflurane (3% for induction and 1.5% for maintenance) and maintained at a body temperature of 37 °C. M-mode images were acquired with a Vevo 3100 high-frequency ultrasound system (VisualSonics, Toronto, ON, Canada) and a 30-MHz imaging transducer. The echocardiographic parameters measured were left ventricular ejection fraction, left ventricular fractional shortening, left ventricular end-systolic volume, left ventricular end-diastolic volume, left ventricular internal dimension-systole, and left ventricular internal dimension-diastole [33, 34]. For the low heart rate which was associated with low cardiac function during the anesthesia, when the heart rate of mice was founded lower than 200 per minute during the anesthesia, the final monitoring of functional index would be stopped and the mice would be sacrificed for subsequent histological observation. One mouse of HTN (wild type + Ang II injection group) and one mouse of cGAS−/− CTRL (cGAS−/− sham group) in experiment 1 and three mice of CTRL (wild-type sham group) and one mouse of HTN (wild type + Ang II infusion group) in experiment 2 were excluded from the measurement.
Blood Pressure (BP) Recording
The noninvasive tail-cuff method (CODA; Kent Scientific, Torrington, Connecticut, USA) was used to record the BP of mice without anesthesia as our previous study [15].
Heart Rate Variability (HRV) Recording
Mice were anesthetized by isoflurane (3% for induction and 1.5% for maintenance). The heart rate was monitored and input into a PowerLab (AD Instruments, Australia) data-acquisition system and was saved in a computer using the LabChart 7 software. The 5-min electrocardiogram segments were recorded and analyzed by the HRV module of LabChart 7 software [35].
Cell Culture
Mouse BV2 microglia cells were cultured in MEM medium with 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 95% air CO2 and 5%. The medium was changed every 2 days. When the cells reached 80% confluence, they were split with 0.25% trypsin and sub-cultured for further passages as our previous study [36].
mCherry-eGFP-LC3 Adenovirus Transfection
To evaluate autophagic flux, the adeno-associated virus (AAV9) (HB-AP2100001) delivered the reporter fusion protein, tandem fluorescent-tagged LC3, and mCherry-eGFP-LC3 into the BV2 cells in the MOI of 10. The GFP signals were quenched in the lysosomal acidic conditions, whereas the mCherry signal was stable in acidic conditions. Therefore, yellow dots only exhibited in autophagosomes, while red puncta only exhibited in lysosomes [31]. These signals were then recorded and analyzed using ImageJ software.
Immunofluorescent Staining
The mouse was anesthetized and fixed in a supine position, then its left ventricle was perfused with 200 mL of 0.01 M PBS (PH 7.4), followed by 200 mL of 4% paraformaldehyde in 0.1 mol/L PB. The brain was taken out and then fixed in 4% paraformaldehyde for 24 h; then, it was placed overnight in 20% and 30% sucrose at 4 °C, respectively. The PVN is located 0.58~1.22 mm caudal from bregma, 0.25 mm lateral from sagittal suture, and 4.75 mm ventral from dura. The 30-μm coronal sections containing the PVN were cut using a freezing microtome (Leica Biosystems, Germany). The sections of the PVN were then washed in PBS and incubated with 0.3% Triton X-100 for 15 min, followed by incubation with staining blocking buffer for 15 min. The sections were then incubated with mice polyclonal antibody to Iba-1 and/or rabbit polyclonal antibody to cGAS (26416-1-AP) and STING (19851-1-AP) overnight at 4 °C. The goat anti-rabbit IgG H&L and goat anti-mouse IgG H&L were used as secondary antibody. The fluorescent signal was observed under a Fluorview FV300 laser scanning confocal microscope (Olympus, Tokyo, Japan). The microscopic observation and evaluation of slides were conducted by Yijun Huang in the author list, who is blinded to the experimental group of the slide and did not participate in tissue processing and staining. And the total number of Iba-1 co-staining with cGAS, STING, LC3, and p62 was also counted by a pathologist blinded to the treatment from the Department of Physiology and Pathophysiology in Fudan University.
Microglial Skeleton Analysis
Skeleton analysis method was used to analyze the morphology of microglia in immunofluorescent images of fixed brain tissues[37]. For microglia morphology analysis, the Iba-1 positive channel was enhanced to visualize all microglia processes. To eliminate single-pixel background fluorescence, noise de-speckling was applied. The image was then converted to a binary and then skeletonized using ImageJ. To collect the data including the number of endpoints per frame and process length, the AnalyzeSkeleton plugin was applied to skeletonized images. In addition, cell number per frame was also demonstrated in the data. Microglia morphology is often demonstrated by the number of endpoints per cell and the length of cell processes. Based on the data collected using the AnalyzeSkeleton, the number of endpoints per cell, the length of cell processes, and the cell body size could be calculated and then be analyzed using GraphPad Prism 8 software.
Western Blot Assay
The tissue homogenated from the PVN was subjected to western blot analysis. Protein samples (20 μg each) were subjected to SDS-PAGE in 8–12% gradient gel (Beyotime Biotechnology, Shanghai, China) and were transferred to PVDF membrane. cGAS (sc-515777) (1:500), STING (19851-1-AP) (1:1000), LC3I/II (1:1000), and p62 (1:1000) were measured, while β-actin (1:1000) was developed as a loading control. This was followed by incubation with horseradish peroxidase-conjugated donkey anti-rabbit IgG. ECL detection reagents (180-5001, Tanon Science & Technology, Shanghai, China) was used to assess the amount of detected protein, and the immunostaining band was revealed and quantitated by a fully automatic chemiluminescence image analysis system (Tanon-5200; Tanon Science & Technology, Shanghai, China).
Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from microglia using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. The mRNAs of IL-1β, TNF-α, IL-6, and IFN-β were analyzed by RT-PCR. cDNA was transcribed from 1 mg of total RNA using reverse transcription system (Promega, Madison, WI). SYBR-Green-based real-time quantitative RT-PCR was performed using the iCycler (Bio-Rad Laboratories Inc, Hercules, CA). The quantification of gene expression was performed using an ABI PRISM 7500 sequence detection system (Applied Biosystems Life Technologies, Foster City, CA) with SYBR Green (TransGen Biotech Co., Ltd.). Primers used in this study were provided by Sangon Biotech (Shanghai, China). The sequences of primers were designed using Primer Express 2.0 and are listed in Table 1.
Histological Staining
All mice were perfused as mentioned above, and 4% paraformaldehyde was used to fix the heart tissues. The tissues were cut into cross-sectional slices of 15 μm thick. The Masson’s staining was used in heart sections in order to reveal the degree of cardiac fibrosis. The degree of cardiac hypertrophy was evaluated by wheat germ agglutinin (WGA) staining. The microscopic observation and evaluation of slides was conducted by by Yijun Huang in the author list, who is blinded to the experimental group of the slide and did not participate in tissue processing and staining.
Statistical Analysis
Data were shown as mean ± standard error of measurement (SEM). For comparison between groups, one-way or two-way ANOVA was used to determine differences. For experiments that included two groups of samples, Student’s unpaired t-test was used. Statistical significance was set at P<0.05. Statistical analysis and graphing were performed by using GraphPad Prism 8 software.
Results
Cardiac Contractile Dysfunction and Cardiac Injury Were Alleviated in Ang II-Induced Hypertensive cGAS−/− Mice
In this study, intraperitoneal injection with Ang II for 14 days was used to induced hypertension in mice. The BP was monitored using the noninvasive tail-cuff method. Ang II intraperitoneal injection induced an increase in systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean blood pressure (MBP) compared with those of vehicle control group (CTRL). However, cGAS−/− mice injected with Ang II did not show an increase in the BP (Fig. 1b). Echocardiography was used to assess cardiac function of the mice. Compared with the vehicle control group (CTRL), Ang II-induced hypertensive mice (HTN) showed decreased left ventricular (LV) ejection fraction and LV fractional shortening, whereas cGAS−/− mice injected with Ang II (cGAS−/− HTN) did not show cardiac contractile dysfunction (Fig. 1c–e). The myocardial tissue was collected on the 28th day when the experiment was terminated, and the Masson’s trichrome staining and the WGA staining were used to evaluate the pathological alterations of cardiac fibrosis and cardiomyocyte hypertrophy, respectively. The images exhibited an increase in average collagen volume and cardiomyocyte cross section area in HTN group compared with CTRL group, whereas the myocardial interstitial fibrosis (Fig. 1g and i) and cardiomyocyte hypertrophy were alleviated in mice of cGAS−/− HTN group (Fig.1f–h). These results indicated that cGAS knockout lowered BP, alleviated cardiac contractile dysfunction and cardiac injury of Ang II-induced hypertensive mice. We have also explored that knocking out of cGAS in wild-type sham mice did not significantly affect the morphology of cardiomyocyte and myocardial interstitial fibrosis (Fig. S5a–S5e).

Cardiac contractile dysfunction and cardiac injury were alleviated in Ang II-induced hypertensive cGAS−/− mice. a The experiment design in vivo. b Average blood pressure of mice was measured using the non-invasive tail-cuff method after 2-week Ang II infusion. CTRL (wild type sham group, n = 6), HTN (wild type + Ang II injection group, n = 5), cGAS−/− CTRL (cGAS−/− sham group, n = 5), and cGAS−/− HTN (cGAS−/− + Ang II injection group, n = 6). SBP, systolic blood press; DBP, diastolic blood pressure; MBP, mean blood pressure. c The representative M-mode echocardiograms of left ventricle for mice after 2-week Ang II intraperitoneal injection. d Left ventricle ejection fraction. e Left ventricle fractional shortening. f The representative photomicrographs of Masson’s trichrome-stained heart sections in low-power field. Scale bar, 1 mm. g The representative photomicrographs of Masson’s trichrome-stained heart sections in high-power field. Scale bar, 200 μm. h The representative photomicrographs of WGA-stained heart sections. Scale bar, 50 μm. i The quantitative data of the cardiac fibrosis area (blue). j The quantitative data of the cardiomyocyte cross sectional area. The data are presented as mean ± SEM. *p < 0.05; ns, no significant
Microglial Activation Increased in the PVN of Ang II-Induced Hypertensive Mice, Accompanied by the Activation of cGAS-STING Pathway
The expression of OX42, a microglia marker, was increased in the PVN of Ang II-induced hypertensive mice (HTN) compared with those of vehicle control group (CTRL) using immunofluorescent staining examination (Fig. 2a–c). To further evaluate the activation of microglia, the morphology of the microglia was analyzed using skeleton analysis method. Results showed that the endpoints and the process length of microglia were significantly shorter in the PVN of the HTN group compared with the CTRL group, indicating microglia were activated in the HTN group (Fig. 2d and e). The cell body size was significantly bigger in the HTN group compared with the CTRL group, indicating microglia was activated and appeared amoeba-like morphology in the HTN group (Fig. 2f). It was reported that cGAS, a cytosolic DNA sensor, can be activated by mtDNA and regulates autophagy via STING pathway [28, 38]. The electron micrographs showed swollen mitochondria and fragmented mitochondrial crest in microglia of the PVN in the HTN group (Fig. S6). The quantitative analysis of the densities of cGAS and STING immunoblotting bands demonstrated that the expressions of cGAS and STING were increased in the PVN of mice with HTN group (Fig. 2g–i). The immunofluorescence analysis for cGAS or STING co-staining with microglia marker Iba-1 showed that immunofluorescence densities of cGAS and STING were increased (Fig. 3a–c). The colocalization of cGAS and Iba-1 and STING and Iba-1 of the PVN are shown in z-stacks in Fig. S7a and S7b. These results suggested that cGAS-STING pathway in the microglia was activated in the PVN of Ang II-induced hypertensive mice.

Activation of microglia, cGAS, and STING were increased in the PVN of Ang II-induced hypertensive mice. a The representative images of immunostained microglia for OX42 (CD11b/c) in the PVN of mice from CTRL (wild-type sham group) and HTN (wild type + Ang II infusion group). Scale bar, 100 μm or 25 μm. b The number of OX42 positive cells in the images of PVN. c The quantitative data on the mean densitometry of OX42 immuno-positivity. d The number of endpoints per microglia in the images of PVN stained with OX42. e The average length of microglia processes in the images of PVN stained with OX42. f The body size of microglia in the images of PVN stained with OX42. g, h, and i The representative immunoblotting bands and densitometry quantitation of cGAS and STING expression in the PVN of CTRL and HTN group mice were shown. Data are presented as mean ± SEM. n=6, *P < 0.05.

Intracisternal infusion of RU.521 alleviated the PVN cGAS and STING upregulation of Ang II-induced hypertensive mice. a and b The double immunofluorescent staining showed colocalization of cGAS and Iba-1, STING, and Iba-1 in the PVN of CTRL (wild-type sham group), HTN (wild-type + Ang II infusion group), and HTN + RU.521 (wild-type + Ang II infusion + RU.521 intracisternal infusion group), which were analyzed by confocal fluorescence microscopy. The colocalization dots of cGAS and Iba-1 and STING and Iba-1 were pointed with yellow arrows. Scale bar, 100 μm or 20 μm. c The quantitative analysis of fluorescence intensity of cGAS, STING staining, and levels of colocalization of cGAS and Iba-1, STING, and Iba-1 in microglia of PVN from different groups. The total number of Iba-1 co-staining with cGAS or STING was counted unilaterally in squares measuring 0.2 × 0.2 mm in size. Counting was performed by a pathologist blinded to the treatment.d, e, and f The representative immunoblotting bands and densitometry quantitation of cGAS and STING expression in the PVN. Data are presented as mean ± SEM. n=6, *P < 0.05
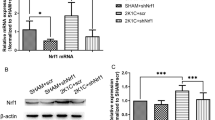
Autophagic Flux Was Blocked in the PVN of Ang II-Induced Hypertensive Mice, while This Effect Was Reversed by RU.521 (a cGAS Inhibitor) Intracisternal Infusion
Our previous studies showed that autophagic flux blockage contributed to microglial activation in the RVLM of stress-induced hypertensive mice [15]. Whether the activation of cGAS-STING pathway regulated the autophagic flux and thus mediated the activation of central microglia remained to be further studied. During the process of autophagy, LC3I can be lipidated to LC3II which is then recruited to autophagosomal membranes, and autophagic substrate p62 is degraded within the mature autolysosome. LC3II increment is taken as a marker of early-phase and p62 decrement is taken as a marker of late-phase autophagy [39, 40]. The immunofluorescent staining and immunoblotting were used to detect the expression of LC3 and p62 in the microglia. The colocalization of LC3 and Iba-1 and p62 and Iba-1 of the PVN are shown in z-stacks in Fig. S7c and S7d. The quantitative analysis showed increased colocalization of LC3 and Iba-1, p62, and Iba-1 in the PVN of the HTN group (Fig. 4g). The results showed that the autophagic marker LC3II/I ratio and the autophagic substrate p62 were increased (Fig.4a–c), which suggested a blockage of late-phase autophagic flux in the PVN of Ang II-induced hypertensive mice (HTN group). In order to further explore whether the autophagic flux was regulated by central cGAS, RU.521 (a cGAS inhibitor) was intracisternally infused to inhibit the cGAS-STING pathway in central nerve system of mice in HTN group. The immunoblotting bands showed that intracisternal infusion of RU.521 in hypertensive mice decreased the expression of cGAS and STING in the PVN (Fig.3d–f). At the same time, we found that intracisternal infusion of RU.521 overtly decreased LC3II/I ratio and the level of p62 expression (Fig.4a–f), which indicated that the inhibition of cGAS in the PVN facilitated resumption of autophagic flux in Ang II-induced hypertensive mice.

Intracisternal infusion of RU.521 alleviated the PVN autophagic flux blockage in of Ang II-induced hypertensive mice. a, b, and c The representative immunoblotting bands and densitometry quantitation of LC3 and p62 expression in the PVN of different groups. d and e The immunofluorescent staining showed expression of Iba-1, LC3, and p62, which were analyzed by confocal fluorescence microscopy. The colocalization dots of LC3 and Iba-1 and p62 and Iba-1 were pointed with yellow arrows. Scale bar, 100 μm or 20 μm. f Quantitative analysis of fluorescence intensity of LC3 and p62 from different groups. g The quantitative analysis the of the levels of colocalization of LC3 and Iba-1, p62, and Iba-1 in microglia of PVN from different groups. h The number of endpoints per microglia, the average length of microglia processes and the body size of microglia in the images of PVN stained with Iba-1. Data are presented as mean ± SEM. n=6, *P < 0.05
Intracisternal Infusion of RU.521 (a cGAS Inhibitor)-Attenuated Microglial Activation and Neuroinflammation, Lowered Sympathetic/Parasympathetic Activity Ratio and BP in Ang II-Induced Hypertensive Mice
The immunofluorescence staining showed that intracisternal infusion of RU.521 attenuated microglial activation (Fig. 4h). The quantitative real-time PCR assay was performed to test the mRNA levels of pro-inflammatory cytokines. The results showed that the IFN-β, IL-1β, IL-6, and TNF-α expressions were upregulated in the PVN of Ang II-induced hypertensive mice (HTN group), which was decreased by intracisternal infusion of RU.521 (HTN + RU.521 group) (Fig. 5a). In general, short recordings of HRV (i.e., less than 1 h) show two primary patterns of oscillation which are separated into frequency bands from ≈7 to 25 s (0.04–0.15 Hz; low frequency, or LF) and 2.5 to ≈7 s (0.15–0.4 Hz; high frequency, or HF). LF and HF frequency bands are widely used to quantify sympathetic and parasympathetic modulations, respectively [41, 42]. To evaluate the imbalance of sympathetic tone versus parasympathetic tone, we conducted the experiment to record the heart rate variability. The results showed an upregulation of the ratio of LF versus HF (LF/HF) in mice of HTN group, which suggested sympathetic overactivation. Intracisternal infusion of RU.521 lowered the sympathetic activity and BP in hypertensive mice (Fig. 5b and c).

Intracisternal infusion of RU.521 alleviated neuroinflammation, and lowered sympathetic/parasympathetic activity ratio and blood pressure. a The mRNA expression of IFN-β, IL-1β, IL-6, and TNF-α in the PVN of mice from CTRL, HTN, and HTN + RU.521 group. b The heart rate variability was analyzed by the LabChart 7 software. LF, low frequency; HF, the high frequency. c Average blood pressure of mice was measured using the non-invasive tail-cuff method 11 days after RU.521 intracisternal infusion. SBP, systolic blood press; DBP, diastolic blood pressure; MBP, mean blood pressure. CTRL (wild type sham group, n = 3), HTN (wild type + Ang II infusion group, n = 5), HTN + RU.521 (wild type + Ang II infusion + RU.521 intracisternal infusion group, n = 6). Data are presented as mean ± SEM, *p < 0.05
cGAS-STING Pathway Activation-Mediated Impairment of Autophagic Flux, which Contributed to M1 Polarization of Microglia In Vitro
BV2 murine microglial cells were used to explore the effects of activated cGAS pathway on autophagic flux blockage and microglial activation in vitro. The double immunofluorescent staining was used to observe the activation of cGAS pathway, the autophagic flux blockage, and microglial activation. The expression of microglial M1 phenotype marker (CD86) was upregulated in Ang II-treated microglia. The expression of CD86 was decreased in microglia co-treated Ang II and autophagy inducer (1 nmol/L rapamycin, 12 h), which suggested that the impairment of autophagic flux contributed to the M1 polarization. We next investigated the effects of exogenous cGAS agonist (10 μmol/L SR-717, 12 h) on autophagic flux blockage and microglial activation in vitro. The results showed SR-717 exacerbated M1 polarization of Ang II-activated microglia, while the cotreatment with rapamycin decreased M1 polarization (Fig. 6a and b). The mCherry-eGFP-LC3 adenovirus was transfected into microglia in vitro to further evaluate autophagic flux. It is well established that GFP fluorescence is quenched in autolysosomes due to the presence of acidic autolysosomes. Therefore, yellow dots only exhibited in autophagosomes, while red puncta only exhibited in lysosomes. The increase of mCherry + GFP+ (yellow dots) indicated the failed fusion of autophagosomes and lysosomes, which are taken as a marker of autophagic flux blockage. The mCherry + GFP+ (yellow) dots were increased in Ang II-activated cells, while rapamycin alleviated this effect. An excessive increase of mCherry + GFP+ dots was observed in Ang II-activated microglia treated with SR-717, which was reversed by the cotreatment of rapamycin (Fig. 6c and d). These results indicated that the activation of cGAS-STING pathway contributed to impaired autophagic flux in microglia, which triggered M1 polarization of microglia in vitro.

Impaired autophagic flux induced by cGAS-STING activation contributed to microglial M1 polarization. a The representative images showing the co-staining of M1 polarization marker (CD86) and microglial marker (OX42). Scale bar, 20 μm. b The ratio of CD86 immuno-positivity cells versus OX42 immuno-positivity cells. The ratio was analyzed by ImageJ. c and d LC3 dots were visualized under fluorescent confocal microscope and quantified after mCherry-eGFP-LC3 adeno-associated virus transfected to the BV2 cells. The number of mCherry + GFP + dots per cell was counted by a pathologist blinded to the treatment. Scale bar, 10 μm. Data are presented as mean ± SEM, n=6, *p < 0.05
Intracisternal Infusion of RU.521 (a cGAS Inhibitor) Alleviated the Myocardial Interstitial Fibrosis and Cardiomyocyte Hypertrophy in Ang II-Induced Hypertensive Mice
The present study had shown that intracisternal infusion of RU.521 had ameliorative effects on PVN microglial activation, neuroinflammation, and upregulation of sympathetic modulation. We then investigated whether these effects could ameliorate the cardiac pathological injury of hypertensive mice. The heart weight/body weight was increased in Ang II-treated hypertensive mice, while RU.521 intracisternal infusion alleviated these effects (Fig. 7a and b). The Masson’s trichrome staining and the WGA staining indicated that intracisternal infusion of RU.521 reduced average collagen volume and cardiomyocyte cross section area of hypertensive mice (Fig. 7c–g). These results indicated that intracisternal infusion of RU.521 alleviated the myocardial interstitial fibrosis and cardiomyocyte hypertrophy of Ang II-induced hypertensive mice.

Intracisternal infusion of RU.521-alleviated cardiac injury in Ang II-induced hypertensive mice. aThe representative images of whole hearts of mice from CTRL, HTN, and HTN + RU.521 group. b The ratio of heart weight/body weight. cThe representative photomicrographs of the Masson’s trichrome-stained heart sections in low-power field. Scale bar, 1 mm. d The representative photomicrographs of the Masson’s trichrome-stained heart sections in high-power field. Scale bar, 200 μm. e The quantitative data of the cardiac fibrosis area (blue). f Representative photomicrographs of the WGA-stained heart sections. Scale bar, 50 μm. g The quantitative data of the cardiomyocyte cross sectional area. Data are presented as mean ± SEM, n=6, *p < 0.05
Intracisternal Infusion of RU.521 (a cGAS Inhibitor)-Alleviated Cardiac Contractile Dysfunction in Ang II-Induced Hypertensive Mice
Echocardiography was performed after intracisternal infusion of RU.521 to assess cardiac functions of mice. Compared with control group (CTRL group), left ventricular (LV) ejection fraction (Fig. 8a and b) and LV fractional shortening (Fig.8a and c) were decreased in Ang II-induced hypertensive mice (HTN group). Furthermore, LV end-systolic volume (Fig. 8d), LV end-diastolic volume (Fig. 8e), LV internal dimension-systole (Fig. 8f), and LV internal dimension-diastole (Fig. 8g) were increased in mice of HTN group. These data indicated cardiac contractile dysfunction in Ang II-treated mice. In contrast, we did not observe cardiac contractile dysfunction in Ang II-induced hypertensive mice intracisternally infused of RU.521 (HTN + RU.521 group) (Fig. 8a–g). These results indicated that intracisternal infusion of RU.521 played a protective role in cardiac contractile dysfunction of Ang II-induced hypertensive mice.

Intracisternal infusion of RU.521-alleviated cardiac contractile dysfunction in Ang II-induced hypertensive mice. a The representative M-mode echocardiograms of left ventricle for mice 11 days after intracisternal infusion of RU.521. b Left ventricle ejection fraction. c Left ventricle fractional shortening. d Left ventricle end-systolic volume. e Left ventricle end-diastolic volume. f Left ventricle internal dimension-systole. g Left ventricle internal dimension-diastole. CTRL (wild-type sham group, n = 3), HTN (wild type + Ang II infusion group, n = 5), HTN + RU.521 (wild type + Ang II infusion + RU.521 intracisternal infusion group, n = 6). Data are presented as mean ± SEM. *p < 0.05
Discussion
It is hard to prevent the pathological progress of hypertension and hypertension-induced cardiac remodeling [43]. Evidence indicates that sympathetic nervous system plays essential roles in the development of hypertension and hypertensive cardiac contractile dysfunction [44]. In our present study, we found that centrally inhibited the cGAS-STING pathway alleviated the contractile dysfunction, cardiomyocyte hypertrophy, and myocardial interstitial fibrosis in Ang II-induced hypertensive mice. Moreover, in vitro, cGAS-STING pathway activation mediated the impairment of autophagic flux, which contributed to the M1 polarization of microglia. However, when the impaired autophagic flux was facilitated by rapamycin, an autophagy inducer, the microglial M1 polarization was decreased correspondingly, in vitro. This study implied that inhibition of cGAS-STING pathway alleviated microglial activation, neuroinflammation, and sympathetic activation via regulating autophagy.
The renin-angiotensin system (RAS) is one of the major systems affecting cardiac output and peripheral resistance [45]. Ang II, one of the main components of RAS, increases BP by multiple mechanisms, such as constricting resistance vessels, stimulating synthesis and release of aldosterone, and enhancing sympathetic outflow from brain [11]. Besides generated in the circulation, Ang II can also be synthesized locally in tissues like the vessel, heart, and brain [10]. Gao et al. intraperitoneally injected adult C57 mice with Ang II for 2 weeks in order to explore the potential mechanism of hypertension-associated cognitive decline and anxiety-like behavior [29]. Using the same method, our results showed that the SBP was increased in this animal model. Moreover, myocardial interstitial fibrosis, cardiomyocyte hypertrophy, and cardiac contractile dysfunction were observed in those mice. In animal models and patients with heart failure, the cardiac RAS is activated and local Ang II generation is enhanced, which contributes cardiomyocyte hypertrophy [46].
Under hypertension settings, it is reported that prolonged Ang II release results in increased permeability of the blood-brain barrier (BBB), giving circulating Ang II access to important sympathoexcitatory brain centers such as the PVN and RVLM [16]. In addition, central RAS can also affect sympathoexcitatory brain centers during hypertension [44]. It is reported that Ang II stimulates Toll-like receptor 4 (TLR4) via Ang II type 1 receptor (AT1R) and induces the activation of microglia [47]. Ang II activates NADPH-oxidase complex via binding to AT1R and results in ROS generation, which contributes to shifting the microglia from M0/resting to M1/proinflammatory microglial phenotypes [48]. Our previous study showed that the local prorenin, one of the components of the brain RAS, also activated the microglia, thereby participated in the development of hypertension via regulating mitophagy [16].
Our present results showed that cGAS-STING pathway participated in microglial activation via regulating autophagy. To further investigate its detail mechanisms, the Ang II-stimulated BV2 cells were co-treated with SR-717 (a cGAS agonist) and rapamycin (an autophagic inducer). We found that when facilitated the autophagic flux, the microglial activation mediated by cGAS-STING was attenuated. It is reported that cGAMP, produced by cGAS, binds to STING and translocates to ER-Golgi intermediate compartment (ERGIC) to serve as a membrane source for LC3, an autophagic protein lipidation [49]. Chen et al. reported that cGAMP induced LC3 lipidation, a key step in autophagosome biogenesis, via a pathway dependent on WIPI2 and ATG5 but independent of the ULK and VPS34-beclin kinase complexes [28]. Similar to what Chen et al. reported, our results showed that when cGAS was activated in the HTN group, the LCII/I ratio increased. Whereas, instead of autophagosome formation, we mainly focus on how cGAS affects autophagic flux. Our results showed increased expression of p62 in the PVN and increased mCherry + GFP+ dots in microglia when cGAS was activated. These results were unable to prove cGAS effects on regulating autophagosome biogenesis but were adequate to confirm the blockage of autophagic flux. The mechanistic target of rapamycin (mTOR) plays a crucial role in regulating autophagy and can be inhibited by rapamycin [50]. The activation of mTORC1 inhibited autophagy via phosphorylating multiple autophagy-related proteins [51]. mTORC1 can phosphorylate the transcription factor EB (TFEB), a main regulator of lysosomal and autophagic gene expression, and inhibit its nuclear localization [52]. Proper lysosome function is essential for autophagy completion, especially for the late phase. Therefore, rapamycin was used to activate late-phase autophagy. Our results showed that cGAS blocked p62 degradation and can be reversed by rapamycin, which indicated that cGAS blocked the late-phase autophagic flux. The detailed mechanisms need to be further investigated.
Our previous study showed that the accumulation of injured mitochondria induced activation of NLRP3 inflammasome in microglia and resulted in increased microglia release of PICs including IL-1β and IL-18 [15, 16]. Our updated study further confirmed that impaired autophagic flux induced the accumulation of injured mitochondria, which led to proinflammatory state in microglia of stress-induced hypertensive mice [15]. Our present study showed autophagic flux blockage in the PVN of Ang II-induced hypertensive mice, accompanied by microglial activation. The relationship between autophagic flux blockage and microglial activation remains unclear. It was noted that impaired Beclin1-driven autophagy induces microglial activation and neuroinflammation via regulating NLRP3 degradation [53]. Moreover, TNF-α inhibits autophagic flux and promotes microglial M1 polarization via activating AKT/mTOR signaling [54].
It has been demonstrated that sympathetic overactivity participates in the development of the essential hypertension and pathophysiology of the complex cardiometabolic alterations called “end-organ damage” [7]. Prolonged sympathetic activation contributes to the development of myocardial cell injury, cardiac fibrosis, and left ventricular dysfunction. Neuroinflammation in cardiovascular regulatory centers is implicated as main factors in augmenting sympathetic activity in hypertension and other cardiovascular abnormalities [8]. Overactive local RAS and neuroinflammation in PVN and RVLM are key factors in regulating sympathetic activity [55].
HRV has been of great value in the attempt to develop noninvasive sensors that apply to clinical use [56]. Therefore, HRV measurement was used to assess the imbalance of sympathetic tone versus parasympathetic tone. Frequency-domain analysis of HRV includes LF, HF, and LF/HF. It is commonly accepted that the HF peak and power can always be recognized as a valid index of vagal activity, while LF component of HRV is mediated by both cardiac vagal and sympathetic activity [57]. It is reported that the ratio of LF/HF can be considered as an index of sympathovagal imbalance [52]. The recording of renal sympathetic nerve activity (RSNA) is demonstrated to be a more reliable way to reflect sympathetic nerve activation. Due to the technical difficulty and limitations of recording RSNA in mice reported by other researches, we choose the HRV analysis to reflex the sympathetic activity indirectly. We found that intracisternal infusion of RU.521 attenuated the microglial activation, neuroinflammation, sympathetic/parasympathetic activity ratio, and lowered the BP in Ang II-induced hypertensive mice.
Limitations
First, intracisternal infusion of RU.521 was used instead of PVN microinjection because intracisternal infusion has much more possibility of potential clinical application. After intracisternal infusion, significant molecular changes were observed in the PVN of different groups mice, which supported our conclusion. Second, we have not specifically interfered microglia in vivo; thus, we cannot exclude functions of other cells and the interactions between microglia and the rest types of cells.
Conclusions
Collectively, we demonstrated that inhibition of cGAS in the PVN attenuates hypertensive heart injury via facilitating microglia autophagic flux (Fig. 9). Antagonizing the activation of central cGAS-STING signaling pathway might be a new strategy against hypertension and its target organ injury.

Schematic illustrating cGAS mediating Ang II-induced PVN neuroinflammation via impairing autophagic flux in microglia. cGAS induces the M1 polarization of microglia via impairing autophagic flux, thereby participating in neuroinflammation, which leads to sympathetic overactivation in hypertension and hypertensive heart injury
Data Availability
Data will be made available upon reasonable request.
Abbreviations
- PVN:
-
hypothalamic paraventricular nucleus
- cGAS:
-
cytosolic DNA sensor cyclic GMP-AMP synthase
- mtDNA:
-
mitochondrial DNA
- Ang II:
-
angiotensin II
- BP:
-
blood pressure
- HRV:
-
heart rate variability
- WGA:
-
wheat germ agglutinin
- STING:
-
stimulator of interferon genes
- LC3:
-
microtubule-associated protein light chain 3
- p62:
-
SQSTM1 (sequestosome 1)
- LV:
-
left ventricular
- RAS:
-
renin angiotensin system
- RVLM:
-
rostral ventrolateral medulla
- ROS:
-
reactive oxygen species
- NLRP3:
-
the nucleotide-binding oligomerization domain (NOD)-like receptor containing pyrin domain 3
- PICs:
-
proinflammatory cytokines
- IL-1β:
-
interleukin-1β
- IL-18:
-
interleukin-18
- ROS:
-
reactive oxygen species
- ATP:
-
adenosine triphosphatase
- GTP:
-
guanosine triphosphate
- cGAMP:
-
2′-3′-cyclic GMP-AMP
- IL-6:
-
interleukin-6
- TNF-α:
-
tumor necrosis factor α
- IRF3:
-
interferon regulatory factor 3
- TBK1:
-
TANK-binding kinase 1
- NF-κB:
-
nuclear factor-kappa B
- RT-PCR:
-
quantitative real-time polymerase chain reaction
- PBS:
-
phosphate buffer saline
- PB:
-
phosphate buffer
- SDS-PAGE:
-
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PVDF:
-
polyvinylidene fluoride
- ECL:
-
electrochemiluminescence
- SBP:
-
systolic blood pressure
- DBP:
-
diastolic blood pressure
- MBP:
-
mean blood pressure
- IFN-β:
-
interferon β
- LF:
-
low frequency
- HF:
-
high frequency
- ACE2:
-
angiotensin-converting enzyme 2
- AT1:
-
angiotensin II receptor type 1
- mTOR:
-
mammalian target of rapamycin
- WIPI2:
-
WD repeat domain, phosphoinositide interacting 2
- ATG5:
-
autophagy related 5
- ULK:
-
Unc-51-like autophagy activating kinase
- VPS34:
-
PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3
References
Kelly T, Yang W, Chen CS, Reynolds K, He J (2008) Global burden of obesity in 2005 and projections to 2030. Int J Obes 32(9):1431–1437. https://doi.org/10.1038/ijo.2008.102
Liu M-Y, Li N, Li WA, Khan H (2017) Association between psychosocial stress and hypertension: a systematic review and meta-analysis. Neurol Res 39(6):573–580. https://doi.org/10.1080/01616412.2017.1317904
Fuchs FD, Whelton PK (2020) High blood pressure and cardiovascular disease. Hypertension 75(2):285–292. https://doi.org/10.1161/hypertensionaha.119.14240
Drazner MH (2011) The progression of hypertensive heart disease. Circulation 123(3):327–334. https://doi.org/10.1161/CIRCULATIONAHA.108.845792
Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, Chen J, He J (2016) Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation 134(6):441–450. https://doi.org/10.1161/CIRCULATIONAHA.115.018912
Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, Alexander L, Estep K, Hassen Abate K, Akinyemiju TF, Ali R, Alvis-Guzman N, Azzopardi P, Banerjee A, Bärnighausen T, Basu A, Bekele T, Bennett DA, Biadgilign S et al (2017) Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. JAMA 317(2):165. https://doi.org/10.1001/jama.2016.19043
Grassi G, Ram VS (2016) Evidence for a critical role of the sympathetic nervous system in hypertension. J Am Soc Hypertens 10(5):457–466. https://doi.org/10.1016/j.jash.2016.02.015
Haspula D, Clark MA (2018) Neuroinflammation and sympathetic overactivity: Mechanisms and implications in hypertension. Auton Neurosci 210:10–17. https://doi.org/10.1016/j.autneu.2018.01.002
Li T, Chen Y, Gua C, Wu B (2018) Elevated oxidative stress and inflammation in hypothalamic paraventricular nucleus are associated with sympathetic excitation and hypertension in rats exposed to chronic intermittent hypoxia. Front Physiol 9:840. https://doi.org/10.3389/fphys.2018.00840
Te Riet L, van Esch JHM, Roks AJM, van den Meiracker AH, Danser AHJ (2015) Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res 116(6):960–975. https://doi.org/10.1161/CIRCRESAHA.116.303587
Oparil S, Zaman MA, Calhoun DA (2003) Pathogenesis of hypertension. Ann Intern Med 139(9):761–776
Cui C, Xu P, Li G, Qiao Y, Han W, Geng C, Liao D, Yang M, Chen D, Jiang P (2019) Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: Role of renin-angiotensin system. Redox Biol 26:101295. https://doi.org/10.1016/j.redox.2019.101295
Du D, Hu L, Wu J, Wu Q, Cheng W, Guo Y, Guan R, Wang Y, Chen X, Yan X, Zhu D, Wang J, Zhang S, Guo Y, Xia C (2017) Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J Neuroinflammation 14(1). https://doi.org/10.1186/s12974-017-0942-2
Hu L, Zhang S, Wen H, Liu T, Cai J, Du D, Zhu D, Chen F, Xia C (2019) Melatonin decreases M1 polarization via attenuating mitochondrial oxidative damage depending on UCP2 pathway in prorenin-treated microglia. PLoS One 14(2):e0212138. https://doi.org/10.1371/journal.pone.0212138
Zhang S, Hu L, Jiang J, Li H, Wu Q, Ooi K, Wang J, Feng Y, Zhu D, Xia C (2020) HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation 17(1). https://doi.org/10.1186/s12974-019-1673-3
Hu L, Zhang S, Ooi K, Wu X, Wu J, Cai J, Sun Y, Wang J, Zhu D, Chen F, Xia C (2020) Microglia-derived NLRP3 activation mediates the pressor effect of prorenin in the rostral ventrolateral medulla of stress-induced hypertensive rats. Neurosci Bull 36(5):475–492. https://doi.org/10.1007/s12264-020-00484-9
Jia G, Meng Z, Liu C, Ma X, Gao J, Liu J, Guo R, Yan Z, Christopher T, Lopez B, Liu W, Dai H, Lau WB, Jiao X, Zhao J, Wang ZX, Cao J, Wang Y (2020) Nicotine induces cardiac toxicity through blocking mitophagic clearance in young adult rat. Life Sci 257:118084. https://doi.org/10.1016/j.lfs.2020.118084
Tsutsui H, Kinugawa S, Matsushima S (2011) Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301(6):H2181–H2190. https://doi.org/10.1152/ajpheart.00554.2011
Gao D, Wu J, Wu Y-T, Du F, Aroh C, Yan N, Sun L, Chen ZJ (2013) Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science (New York, NY) 341(6148):903–906. https://doi.org/10.1126/science.1240933
Cai X, Chiu Y-H, Chen ZJ (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54(2):289–296. https://doi.org/10.1016/j.molcel.2014.03.040
Hopfner K-P, Hornung V (2020) Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21(9):501–521. https://doi.org/10.1038/s41580-020-0244-x
Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17(10):1142–1149. https://doi.org/10.1038/ni.3558
Xia P, Wang S, Gao P, Gao G, Fan Z (2016) DNA sensor cGAS-mediated immune recognition. Protein Cell 7(11):777–791. https://doi.org/10.1007/s13238-016-0320-3
Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V (2013) Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503(7477):530–534. https://doi.org/10.1038/nature12640
Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, Li T, Loughran PA, Griepentrog JE, Huang H, Scott MJ, Huang F, Billiar TR (2018) cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. American Journal of Physiology-Gastrointestinal and Liver. Physiology 314(6):G655–G667. https://doi.org/10.1152/ajpgi.00326.2017
Liang Q, Seo GJ, Choi YJ, Kwak M-J, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner K-P, Ha T, Oh B-H, Jung JU (2014) Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15(2):228–238. https://doi.org/10.1016/j.chom.2014.01.009
Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS (2015) The cytosolic sensor cGAS detects mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17(6):811–819. https://doi.org/10.1016/j.chom.2015.05.004
Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, Chen ZJ (2019) Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567(7747):262–266. https://doi.org/10.1038/s41586-019-1006-9
Gao N, Wang H, Xu X, Yang Z, Zhang T (2021) Angiotensin II induces cognitive decline and anxiety-like behavior via disturbing pattern of theta-gamma oscillations. Brain Res Bull 174:84–91. https://doi.org/10.1016/j.brainresbull.2021.06.002
Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, Vernier W, Ali SH, Kissai M, Lazar DC, Nguyen N, Pereira LE, Benish B, Woods AK, Joseph SB, Chu A, Johnson KA, Sander PN, Martínez-Peña F et al (2020) Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369(6506):993–999. https://doi.org/10.1126/science.abb4255
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140(3):313–326. https://doi.org/10.1016/j.cell.2010.01.028
Xu D, Zhou J, Mei H, Li H, Sun W, Xu H (2021) Impediment of cerebrospinal fluid drainage through glymphatic system in glioma. Front Oncol 11:790821. https://doi.org/10.3389/fonc.2021.790821
Chen Y, Zeng M, Zhang Y, Guo H, Ding W, Sun T (2021) Nlrp3 deficiency alleviates angiotensin II-induced cardiomyopathy by inhibiting mitochondrial dysfunction. Oxidative Med Cell Longev 2021:1–10. https://doi.org/10.1155/2021/6679100
Yang M, Wang S, Fu S, Wu NN, Xu X, Sun S, Zhang Y, Ren J (2021) Deletion of the E3 ubiquitin ligase, Parkin, exacerbates chronic alcohol intake-induced cardiomyopathy through an Ambra1-dependent mechanism. Br J Pharmacol 178(4):964–982. https://doi.org/10.1111/bph.15340
Ye T, Liu X, Qu C, Zhang C, Fo Y, Guo Y, Chen X, Shi S, Yang B (2019) Chronic inhibition of the sigma-1 receptor exacerbates atrial fibrillation susceptibility in rats by promoting atrial remodeling. Life Sci 235:116837. https://doi.org/10.1016/j.lfs.2019.116837
Ooi K, Hu L, Feng Y, Han C, Ren X, Qian X, Huang H, Chen S, Shi Q, Lin H, Wang J, Zhu D, Wang R, Xia C (2021) Sigma-1 receptor activation suppresses microglia M1 polarization via regulating endoplasmic reticulum-mitochondria contact and mitochondrial functions in stress-induced hypertension rats. Mol Neurobiol 58(12):6625–6646. https://doi.org/10.1007/s12035-021-02488-6
Morrison HW, Filosa JA (2013) A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:4. https://doi.org/10.1186/1742-2094-10-4
Lautrup S, Lou G, Aman Y, Nilsen H, Tao J, Fang EF (2019) Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem Int 129:104469. https://doi.org/10.1016/j.neuint.2019.104469
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117(Pt 13):2805–2812
Katsuragi Y, Ichimura Y, Komatsu M (2015) p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282(24):4672–4678. https://doi.org/10.1111/febs.13540
Akselrod S, Gordon D, Ubel FA, Shannon DC, Berger AC, Cohen RJ (1981) Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science (New York, NY) 213(4504):220–222
Wang Y, Hu H, Yin J, Shi Y, Tan J, Zheng L, Wang C, Li X, Xue M, Liu J, Wang Y, Li Y, Li X, Liu F, Liu Q, Yan S (2019) TLR4 participates in sympathetic hyperactivity post-MI in the PVN by regulating NF-κB pathway and ROS production. Redox Biol 24:101186. https://doi.org/10.1016/j.redox.2019.101186
Tomek J, Bub G (2017) Hypertension-induced remodelling: on the interactions of cardiac risk factors. J Physiol 595(12):4027–4036. https://doi.org/10.1113/JP273043
Jackson L, Eldahshan W, Fagan SC, Ergul A (2018) Within the brain: the renin angiotensin system. Int J Mol Sci 19(3):876. https://doi.org/10.3390/ijms19030876
Saxena T, Ali AO, Saxena M (2018) Pathophysiology of essential hypertension: an update. Expert Rev Cardiovasc Ther 16(12):879–887. https://doi.org/10.1080/14779072.2018.1540301
Wollert KC, Drexler H (1999) The renin-angiotensin system and experimental heart failure. Cardiovasc Res 43(4):838–849. https://doi.org/10.1016/s0008-6363(99)00145-5
Mowry FE, Peaden SC, Stern JE, Biancardi VC (2021) TLR4 and AT1R mediate blood-brain barrier disruption, neuroinflammation, and autonomic dysfunction in spontaneously hypertensive rats. Pharmacol Res 174:105877. https://doi.org/10.1016/j.phrs.2021.105877
Labandeira-Garcia JL, Rodríguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ (2017) Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci 9:129. https://doi.org/10.3389/fnagi.2017.00129
Yang J, Tang X, Nandakumar KS, Cheng K (2019) Autophagy induced by STING, an unnoticed and primordial function of cGAS. Cell Mol Immunol 16(8):683–684. https://doi.org/10.1038/s41423-019-0240-2
Lamming DW, Bar-Peled L (2019) Lysosome: The metabolic signaling hub. Traffic 20(1):27–38. https://doi.org/10.1111/tra.12617
Kim YC, Guan KL (2015) mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 125(1):25–32. https://doi.org/10.1172/jci73939
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31(5):1095–1108. https://doi.org/10.1038/emboj.2012.32
Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M (2019) Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J 38(4). https://doi.org/10.15252/embj.201899430
Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, Yang YP, Zhao Z, Hu LF, Liu CF (2018) A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci 10:378. https://doi.org/10.3389/fnagi.2018.00378
Agarwal D, Welsch MA, Keller JN, Francis J (2011) Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res Cardiol 106(6):1069–1085. https://doi.org/10.1007/s00395-011-0231-7
Valenza G, Citi L, Saul JP (1985) Barbieri R (2018) Measures of sympathetic and parasympathetic autonomic outflow from heartbeat dynamics. J Appl Physiol 125(1):19–39. https://doi.org/10.1152/japplphysiol.00842.2017
Reyes del Paso GA, Langewitz W, Mulder LJ, van Roon A, Duschek S (2013) The utility of low frequency heart rate variability as an index of sympathetic cardiac tone: a review with emphasis on a reanalysis of previous studies. Psychophysiology 50(5):477–487. https://doi.org/10.1111/psyp.12027
Acknowledgements
We thank Dr. Dapeng Yan and his research team (Department of Immunology, School of Basic Medical Sciences and Shanghai Public Health Clinical Center, Key Laboratory of Medical Molecular Virology of MOE/MOH, Fudan University, Shanghai, 200032, China) for their kindly gifted cGAS−/− mice from Animal Laboratory Center of Fudan University. We thank for Dr. Limin Lu (Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, China) for his technical support in Western blot assay.
Funding
This work was supported by the Chinese National Natural Science Foundation no. 82171803 and no. 81770423 to CMX; the Hui-Chun Chin and Tsung-Dao Lee Chinese Undergraduate Research Endowment (CURE) no. 2193101011001 to CZH.
Author information
Authors and Affiliations
Contributions
CMX, CZH, and XYQ conceived and designed the study. CMX and STZ coordinated the study. CZH, XYQ, and XRR carried out the animal experiments. LH carried out the analysis and interpretation of RT-PCR and the cell experiment. CZH and JYL carried out the western blot assay. CZH, XYQ, XRR, RHH, and HL worked on tissue processing and staining. YJH conducted the microscopic observation and evaluation of slides. CZH and XYQ wrote and prepared the manuscript. CMX and KO revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval
All experimental protocols were approved by the Fudan University Animal Care Committee, and all operations in this study were conformed to the guidelines of the Institutional Ethics Committee.
Consent to Participate
The manuscript does not contain data from any individual person.
Consent for Publication
Not applicable.
Conflict of Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.







Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Han, C., Qian, X., Ren, X. et al. Inhibition of cGAS in Paraventricular Nucleus Attenuates Hypertensive Heart Injury Via Regulating Microglial Autophagy. Mol Neurobiol 59, 7006–7024 (2022). https://doi.org/10.1007/s12035-022-02994-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02994-1