
Abstract
Excess greenhouse gas emissions, primarily carbon dioxide (CO2), have caused major environmental concerns worldwide. The electroreduction of CO2 into valuable chemicals using renewable energy is an ecofriendly approach to achieve carbon neutrality. In this regard, copper (Cu) has attracted considerable attention as the only known metallic catalyst available for converting CO2 to high-value multicarbon (C2+) products. The production of C2+ involves complicated C–C coupling steps and thus imposes high demands on intermediate regulation. In this review, we discuss multiple strategies for modulating intermediates to facilitate C2+ formation on Cu-based catalysts. Furthermore, several sophisticated in situ characterization techniques are outlined for elucidating the mechanism of C–C coupling. Lastly, the challenges and future directions of CO2 electroreduction to C2+ are envisioned.
Similar content being viewed by others


Introduction
Massive carbon dioxide (CO2) emissions arising from industrial activities have caused a substantial increase in the atmospheric CO2 concentration, and the ensuing global warming has resulted in increasingly frequent environmental disasters such as starvation, habitat loss, species extinction, and sea-level rise [1, 2]. To tackle these problems, carbon capture, utilization, and storage have been conceived as pathways to peak carbon emissions and eventually reach a carbon–neutral society before 2060 [3]. Electrochemical CO2 reduction reaction (CO2RR) powered by renewable electricity provides a sustainable solution for converting waste CO2 emissions into useful feedstocks and thereby realizing a net negative carbon footprint and long-term storage of the intermittent renewable electricity in chemical bonds [4, 5]. In the past three decades, significant progress has been achieved in using CO2RR to generate diverse products, including CO [6,7,8,9], HCOOH [10, 11], CH4 [12,13,14], C2H4 [15,16,17], C2H5OH [18,19,20], and n-C3H7OH [21,22,23]. Compared to monocarbon (C1) products, multicarbon (C2+) products are more desirable because of their higher energy density, richer chemical structure, and more versatile applications [24].
Since CO2 is a thermodynamically stable molecule with strong C=O bonds (750 kJ/ mol) [25], converting CO2 to C2+ products is extremely difficult. Copper (Cu) is the only known metallic electrocatalyst capable of generating C2+ products in considerable amounts [26,27,28]. However, its selectivity and the activity of the C2+ products are drastically limited by the sluggish kinetics of the C–C coupling step (i.e., the bifurcation for the generation of C2+ and C1 products), as well as because of competition with H–H formation [29]. In the multielectron and multiproton transfer process of CO2RR, the adsorption energy and coverage of key intermediates, such as *H, *COOH, *OCHO, and *CO, are commonly considered to determine the reaction routes and activity of CO2RR [30, 31]. For example, the first proton-coupled transfer reaction will generate *OOCH or *COOH intermediates. *OOCH is the intermediate for formate production, while *COOH is the intermediate for the formation of CO gas or *CO, which is a key intermediate for C–C coupling [32, 33]. Theoretical calculations indicate that the binding energy of *H weakens with increasing *CO coverages, and this in turn inhibits the activity of the competing hydrogen evolution reaction (HER) [31]. Therefore, controlling the intermediate adsorption is critical for inhibiting the formation of H2 and C1 products but facilitating the formation of adsorbed CO dimers (e.g., *CO–CO or *CO–CHO). In addition, Cu moderately binds with most intermediates, resulting in multiple reaction pathways and thus yielding a mixture of numerous products [26]. To improve the selectivity and activity of C2+ products, a variety of strategies such as grain boundary exposure [34,35,36], heteroatom doping [37, 38], and local microenvironment regulation [39, 40] were developed to manipulate the adsorption state of intermediates and facilitate their deep reduction.
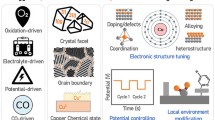
This review focuses on integrated strategies for regulating the adsorption state of intermediates and their influence on C2+ production for Cu-based electrocatalysts (Fig. 1). Moreover, we discuss the vital role of in situ characterization techniques for examining CO2RR intermediates. Finally, we highlight the challenges and perspectives associated with the electroreduction of CO2 to C2+ products. This review can provide a better understanding of the principles of catalyst design with the aim of achieving an overall improvement for the CO2-to-C2+ catalytic systems.

Schematic illustration of the intermediate manipulation strategies for the electrochemical CO2 reduction reaction (CO2RR) toward multicarbon products
Proposed Mechanisms for CO2RR Toward C2+ Products
Since Hori et al. [41, 42] first discovered that Cu is capable of electroreducing CO2 to C2+ products, Cu-based catalysts have been recognized as the best electrocatalysts for C2+ production during the past three decades. To investigate the origin of the deep reduction activity of Cu, the binding energies of the adsorbed CO (*CO) and hydrogen (*H) were used as a descriptor to predict the CO2RR product distribution of different metal catalysts [26, 43]. In the case of metals such as Au and Ag that bind weakly to *CO and *H, *CO prefers direct desorption to form CO. Metals such as Pt and Fe have high binding strength with *CO, but their favorable adsorption of *H facilitates HER. Unlike in the case of those metals, the adsorption energy of *CO and *H on Cu is neither too strong to poison its active sites nor too weak to be immediately desorbed. This moderate intermediate binding energy of Cu enables continuous C–C coupling and multistep hydrogenation, which is essential for the deep reduction of CO2 beyond CO.
Different reaction routes have been proposed for C2+ formation (Fig. 2). *CO is generally recognized as a key reaction intermediate in the electroreduction of CO2 to C2+ products [29, 44, 45]. The dimerization of *CO [46, 47] and coupling of *CO with protonated *CO (*CHO or *COH) [48, 49] are widely accepted possible routes for C–C coupling. In the *CO dimerization mechanism, C–C coupling is generally regarded as the rate-determining step (RDS) for C2+ production and has been widely applied in theoretical calculations [50, 51]. In this case, two *CO intermediates on the Cu surface are coupled to form *OCCO, and this process is accompanied by an electron transfer [52]. However, density functional theory (DFT) simulations suggest that the RDS of C2+ products may be changed from *CO dimerization to *CO–*CHO or *CO–*COH coupling with the increase in the local *CO coverage at higher negative potentials [53]. In the mechanism of *CO coupling with hydrogenated *CO, *CO hydrogenation is considered the RDS for C2+ formation on Cu (111) and (100) surfaces [54]. The competition between *CO and proton sources (i.e., *H, H+, or adsorbed H2O) on the active sites of Cu will lead to a shift in product distribution [55, 56]. After the C–C coupling step, the *COCHO or *COCOH is subsequently reduced to *CHCOH, which is a common precursor of C2H4 and C2H5OH [57, 58]. *CHCOH deoxygenation to *CCH or hydrogenation to *CHCHOH can generate C2H4 and C2H5OH, respectively [20].

Possible reaction routes for the electrochemical CO2RR toward multicarbon products
In addition, the formation of C3 products on Cu has been observed. Some rationally designed Cu catalysts attained a Faradaic efficiency (FE) of up to 15.4% for n-propanol (the major C3 product) in CO2RR (33% in CO reduction reaction (CORR)) [21, 59]. However, the reaction pathway to C3 products, which inevitably requires two successive C–C coupling steps, has not been well-studied yet because of the lack of sophisticated characterization techniques for *C2+ intermediates. As an approximate solution, electroreducing C2/C3 compounds toward those C3 products that may share the reaction pathway as the electrochemical CO2RR toward the same products is employed for a preliminary mechanistic exploration. Hori et al. [60] found that the electroreduction of propionaldehyde produces large amounts of n-propanol, suggesting that propionaldehyde is a potential intermediate for the n-propanol product. Through electrochemical differential mass spectrometry, Bell’s group [61] observed that the relative abundance of ethanol increased at the expense of propionaldehyde at more cathodic potentials. This observation indicates that ethanol and propionaldehyde share the same intermediate (*CH3CHO). To identify the intermediates for the second C–C coupling step, Zhang et al. [62] performed a co-electroreduction of isotopically labeled CO and acetaldehyde and found that only a minor fraction (up to 36%) of the n-propanol product originates from the cross-coupling between CO and acetaldehyde. The adsorbed methylcarbonyl (*OCH2CH3 or *CHOHCH3) is considered a possible intermediate for the second C–C coupling step, in which the reaction pathways bifurcate toward C2 or C3 products. Therefore, a reasonable speculation for the C3 pathway is that a *C2 intermediate undergoes intermolecular C–C coupling with a neighboring *CO intermediate, and this is followed by proton or electron transfer to form propionaldehyde. The key to C3 production is to stabilize *C2 intermediates and optimize the *CO coverage [22, 63].
In conclusion, the C–C coupling step is essential for the conversion of CO2 to C2+ products. This step is regulated by the adsorption states of the intermediates on Cu. With regard to the initial intermediate for C2+ generation, the surface coverage and adsorption duration of *CO affect the probability of C–C coupling. Thereafter, the adsorption states of the subsequent intermediates determine the energy barrier of C–C coupling and the followed reaction routes. Thus, manipulating intermediates by rational Cu-based catalyst design is a promising way to produce high-value C2+ products efficiently.
Strategies of Intermediate Manipulation for Promoting C2+ Production
In the following section, we will summarize the principal strategies of manipulating intermediates, including the surface structural effects, introduction of additional elements, chemical state effects, electric field effects, substrate effects, confinement effects, and local microenvironment effects for promoting the deep conversion of CO2 to C2+ products.
Surface Structural Effects
Crystal Facets
The crystal facet is one of the critical structural parameters for Cu. The adsorption strength of intermediates has a high sensitivity to the crystal facets. Hence, the distributions of CO2RR products on various crystal facets are different. In general, Cu (111) facets have an appropriate ratio of *CO to *H, and thus, these facets favor CH4 production. Unlike Cu (111), Cu (100) facets are more favorable for *CO adsorption, thereby promoting C–C coupling to generate C2H4 (Fig. 3a, b) [64]. DFT calculations demonstrate that *CO dimerization on Cu (100) exhibits the lowest energy among the low-index Cu facets [49]. Therefore, the selective exposure of Cu (100) facet is an important strategy to improve C2+ selectivity [65]. Wang’s group [23] selectively exposed Cu (100) facets through the metal-ion battery cycling method, achieving a sixfold improvement in the ratio of C2+ and C1 products compared with the polished Cu foil. Furthermore, the C2+/CH4 value was plotted as a function of crystal orientation, illustrating that high-index facets can further improve the selectivity for C2+ products (Fig. 3c) [42]. For example, Cu (711) is thermodynamically favored for C–C coupling via cross-coupling of the *CO and *COH intermediates based on DFT calculations [66]. Similarly, Cu (751) has higher activity and selectivity for C–C coupling compared to low-index facets [67]. Changes in the Cu facets also affect the activity and selectivity of CO2RR. High-purity 4H Cu and heterogeneous 4H/ face-centered cubic (fcc) Cu synthesized on the template of 4H and 4H/fcc Au exhibit higher overall activity and catalytic selectivity than fcc Cu, indicating the high dependence of electrocatalytic behaviors on crystal facets (Fig. 3d) [68].

Reproduced with permission from Ref. [64]. Copyright 2020 American Chemical Society. c Variation in C2+/CH4 with the angle of the crystal orientation with reference to Cu (100). Reproduced with permission from Ref. [42]. Copyright 2003 Elsevier. d Faradic efficiencies (FEs) for producing C2H4 on different catalysts. Reproduced with permission from Ref. [68]. Copyright 2020 American Chemical Society. e Wulff construction clusters of Cu with the adsorption of CO2RR or hydrogen evolution reaction (HER) intermediates. Reproduced with permission from Ref. [80]. Copyright 2020 Springer Nature. f Transmission electron microscopy (TEM) image of an electrodeposited Cu TEM grid after electrocatalysis in the electrolyte to which ethylenediamine tetramethylenephosphonic acid (EDTMPA) was added. Inset shows the corresponding electron diffraction pattern of the selected area. g Grazing incidence X-ray diffraction (GI-XRD) patterns of polycrystalline Cu electrodes before and after electrocatalysis in the electrolytes with and without EDTMPA. Reproduced with permission from Ref. [39]. Copyright 2022 Springer Nature
Improvement of C2+ selectivity by crystal facets. a X-ray diffraction patterns of Cu octahedra (Cuoh), Cu cubes (Cucub), and Cu spheres (Cusph). b Selectivity of CO2RR products on Cu with different facet exposures.
Although the C2+ activity and selectivity can be effectively improved by controlling the crystal facet, Cu usually suffers from dynamical reconstruction during the CO2RR process. This reconstruction causes changes in its original crystal facets and catalytic performance [69, 70]. Besides, note that the active Cu surface may also undergo an irreversible evolution even after the removal of the applied potential [71, 72]. This susceptibility of Cu reconstruction makes it difficult to identify the active sites under realistic conditions [73]. To probe the actual active sites on Cu and to obtain fundamental evidence on structure–activity relationships, numerous in situ characterization techniques have been developed under controlled conditions during CO2RR [74,75,76]. Through operando electrochemical scanning tunneling microscopy (EC-STM), Kim et al. [77, 78] found that a polycrystalline Cu electrode held at a fixed negative potential undergoes stepwise surface reconstruction in both alkaline and neutral electrolytes, first transforming to Cu (111) and then to Cu (100). The resulting Cu (100) surface remains stable, without further surface transformations, during the subsequent tests. In situ grazing incidence X-ray diffraction (GI-XRD) also revealed the reconstruction of polycrystalline Cu toward the (100) facet in the presence of CO2 [71]. The degree of reconstruction increases as the applied potential becomes more negative, and the reconstructed facets are partially preserved in the subsequent anodic scanning step. The in situ characterizations described earlier indicate that the dynamic reconstruction of Cu catalysts is mainly driven by the cathode potential, but the influence of adsorbates cannot be ruled out.
Recently, the adsorption of intermediates was shown to affect the formation of preferred crystal facets with high C2+ selectivity during the CO2RR. Wang et al. [79] reported a self-selective method to stabilize the crystal surface with the strongest binding to the target intermediates because the adsorption of reactants tends to reduce the relative surface energies of these surfaces. Sargent’s group [80] performed Cu electrodeposition in the presence of CO2RR intermediates and achieved a 70% increase in the ratio of Cu (100) facets to the total surface area compared to Cu electrodeposited in the presence of HER intermediates (Fig. 3e). This Cu catalyst has a high FE of 90% for the total C2+ products at current densities higher than 580 mA/cm2, and the FE of C2H4 remains constant over 65 h of electrolysis. In addition to the reaction intermediates, electrolyte additives also affect the crystal facet reconstruction on Cu catalysts. Our recent work demonstrated that ethylenediamine tetramethylenephosphonic acid (EDTMPA) molecules preferentially adsorb on Cu (110) during the CO2RR, inducing the selective generation of Cu (110) facets with an intrinsically high *CO binding strength (Fig. 3f, g) [39]. These studies demonstrate that the adsorption of intermediates or electrolyte additives on the catalyst surface can induce the formation of specific crystal facets with high activity; these facets are beneficial for the adsorption and conversion of intermediates.
Unsaturated Coordination Atoms
After obtaining an in-depth understanding of the crystallographic dependence of the product distribution for Cu, the high proportion of unsaturated coordination atoms was also considered to contribute to the high catalytic activity of the high-index crystalline facets for C2+ production [67, 81]. CO adsorption energies on low-index Cu facets (i.e., Cu (111), Cu (100), and Cu (110)) and several regular stepped and kinked facets (i.e., Cu (211), Cu (221), and Cu (532)) were examined by thermal desorption spectroscopy. The results reveal that the high-index crystalline surfaces with a lower coordination number (CN) of surface atoms have higher CO adsorption energies than the low-index ones [82]. In addition to the sites on step edges and joints, the adatoms on the surface also tend to have a higher degree of unsaturation and bind strongly with CO. Theoretical calculations are usually used to reveal the role of adparticles with low CN and surface clusters on the pristine Cu surface in concentrating *CO and facilitating the formation of CO dimers (Fig. 4a–c) [83]. In the presence of Cu adparticles, the reaction barriers between *CO and *C2 intermediates (*OCCOH or *CCH2) are dramatically reduced, enabling the selective electrosynthesis of n-propanol. DFT calculations revealed that the highly undercoordinated sites (CN < 5.9) promote C2H5OH production; moderate coordination sites (5.9 < CN < 7.5) are beneficial for C2H4 production; and high coordination sites (CN > 7.5) have a strong hydrogen adsorption energy [84]. Therefore, the CN of surface catalytic active sites is closely correlated with CO2RR product selectivity.

Reproduced with permission from Ref. [83]. Copyright 2018 Springer Nature. d Images of the computationally produced electropolished Cu surface (left) and the surface after Ar plasma bombardment (right). e Predicted distribution of CO adsorption energies. Reproduced with permission from Ref. [86]. Copyright 2020 American Chemical Society. f A correlation plot between the FE of C2H4 (FEC2H4) and partial current density of C2H4 (jC2H4) values with the crystallite sizes. g A correlation plot between the charges of the CO adsorption peaks with FEC2H4 and jC2H4. Reproduced with permission from Ref. [88]. Copyright 2016 American Chemical Society. h Population of surface atoms with a specific CN as a function of particle diameter. Reproduced with permission from Ref. [89]. Copyright 2014 American Chemical Society
Improvement of C2+ selectivity by unsaturated coordination atoms. a Demonstration of various low coordination number (CN) Cu sites with different numbers of Cu adatoms (ADs) added on various Cu facets. b The adsorption energy of CO. c The reaction energies of *CO dimerization.
The CN of the atoms on the catalyst surface can be adjusted not only by crystal surface control, but also by manipulating the macroscopic morphology. Nanostructuring of the catalyst allows for increasing the specific surface area and exposing more unsaturated coordination atoms, thereby resulting in a low CN catalyst. For example, a high density of undercoordinated Cu was formed on the surface of the Cu foil treated by anodic halogenation and subsequent electroreduction processes, resulting in a selective conversion of CO2 to C2+ products with a FE of 72% [85]. Machine learning was applied to predict the *CO binding energy of 10,433 surface atoms on a rough Cu model, and the results show that a high percentage of undercoordinated Cu sites preferentially bind to *CO (Fig. 4d, e) [86]. These results illustrate that the undercoordinated sites formed by the nanostructuring of catalysts greatly increase the activity and selectivity of C2+ products.
In addition, the size of nanocatalysts also influences the concentration of unsaturated atoms. Consider the example of a Cu nanocrystal cube, for which the percentage of low coordination atoms on corners and edges decreases with the increasing size of the cube; therefore, the surface structure of the nanocrystal cube is closer to that of a single crystal [87]. Cu2O-derived Cu particles were selected to investigate the correlation between the statistical microcrystal size and selectivity of CO2 electroreduction to C2H4 [88]. With a decrease in the microcrystal size from 41 to 18 nm, the selectivity of C2H4 increased linearly from 10 to 43% (Fig. 4f). The cyclic voltammetric analysis of Cu particles was performed in CO- and N2-saturated electrolytes, and the results revealed a linear correlation between the adsorption charge and the selectivity of C2H4. This implies that smaller particles have more sites available for CO adsorption (Fig. 4g). However, hydrocarbon selectivity is sharply inhibited when the nanoparticles are smaller than 5 nm [89]. This is because the stronger binding of *CO and *H on atoms with CN < 8 largely reduces the surface mobility of intermediates, which results in a lower probability of subsequent CO hydrogenation to form hydrocarbons (Fig. 4h). Rong et al. [90] synthesized Cu catalysts with a size gradient from single atoms to nanoclusters on a graphdiyne substrate by an alkyne-bond-directed site-trapping method. Surprisingly, the increased size remarkably improved the selectivity of CORR, showing a high C2+ FE of 91.2% at 312 mA/cm2 for 1–1.5 nm Cu nanoclusters with a large number of low CN atoms. These results indicate that Cu nanoparticles of size between 5 and 18 nm with moderately unsaturated coordination can exhibit suitable *CO and *H binding energies for C2+ production. Thus, this result highlights the practicality of unsaturated coordination atoms.
Grain Boundaries
Li et al. [91] first discovered that the reduction of Cu oxides greatly increases the catalytic performance compared to the performance of pure Cu. This increase is attributed to the abundant grain boundary (GB) structure on the oxide-derived Cu (OD-Cu). Electroreduction and H2 annealing reduction were performed to reduce Cu2O to Cu, and similar networks of interconnected nanocrystals with distinct GBs between the nanocrystals were obtained [92]. To investigate the effect of numerous GB structures in OD-Cu on CO2RR and CORR performances, a series of intensive mechanistic explorations and discussions were performed by Kanan’s group [34,35,36, 93]. The CO temperature-programmed desorption results reveal that the binding of CO on OD-Cu is stronger than that on a Cu foil, hence improving the catalytic performance of CORR for OD-Cu (Fig. 5a, b) [35]. The direct correlation between CORR activity and GB density was further quantified to establish a design principle for solid catalysts [93]. The bulk electrolysis of Cu nanoparticles with different GB densities reveals that the specific activity of CO reduction depends linearly on the ratio of GB surface terminations (Fig. 5c). Finally, GB terminations on the electrode surface are more active for CO2RR than for HER; this finding was confirmed by scanning electrochemical cell microscopy and electron backscatter diffraction studies (Fig. 5d, e) [36]. The surface-terminating dislocations accumulated at the GBs modify the density of undercoordination sites, selectively increasing the activity of CO2RR [34]. In addition, some research groups reported that GB structures have an excellent electrocatalytic performance for CO2 reduction to C2+ products [94,95,96,97,98,99,100]. This performance is attributed to the facilitated CO2 activation [94, 95], increased *CO adsorption energy [96, 97], and reduced C–C coupling barriers [98, 99].

Reproduced with permission from Ref. [35]. Copyright 2015 American Chemical Society. c Specific activity for CO reduction versus the grain boundary (GB) surface density at − 0.5 V vs. a reversible hydrogen electrode (RHE). Reproduced with permission from Ref. [93]. Copyright 2016 American Chemical Society. d Electron backscatter diffraction orientation map of the tested sample with GBs. Inset text and paths indicate the locations where line scans were collected. e Line scan generated from individual constant potential electrolysis across the GB at 1 atm Ar or CO2. Reproduced with permission from Ref. [36]. Copyright 2017 the American Association for the Advancement of Science. f CO vibrational frequency (νCO) observed during chronoamperometric scans. Reproduced with permission from Ref. [102]. Copyright 2020 the Royal Society of Chemistry. g Physical proximity of the optimal C1 and C2 sites that facilitate the coupling of C1–C2 into C3 products. Reproduced with permission from Ref. [103]. Copyright 2019 Springer Nature. h The FE versus the size of the atomic-scale interspace (atomic-dS) measured at − 0.9 V versus RHE. Reproduced with permission from Ref. [48]. Copyright 2020 Wiley–VCH
Improvement of C2+ selectivity by grain boundaries. a, b CO temperature-programmed desorption profiles of a polycrystalline Cu and b oxide-derived Cu (OD-Cu) under air oxidation at 500 °C (i.e., OD-Cu–500).
Because *CO with a low vibrational frequency has been observed on the surface, fragmented Cu is considered to be active for rapid CO dimerization (Fig. 5f) [101, 102]. Moreover, the highly fragmented Cu also assists in clustering the binding sites of the C1 and C2 intermediates, thereby facilitating further coupling of these intermediates (Fig. 5g) [94, 103]. Adjusting the atomic-level spacing (atomic-dS) between Cu particles is another efficient approach to achieve the highly active and selective generation of C2+ products [48]. Metallic Cu-based catalysts with different particle spacings were constructed by lithiation, delithiation, and electroreduction of CuOx particles. The spacing range was confirmed by examining three-dimensional tomographs obtained using a Cs-corrected scanning transmission electron microscope. Theoretical and experimental results show that a spacing of 5–6 Å maximizes the binding energy of the intermediates involved in C–C bond formation, achieving a FE of ~ 80% for C2+ products (Fig. 5h). These results further confirm the essential effect of GBs on C2+ activity and selectivity.
Vacancies
Vacancy engineering enables the alteration of the surface electron structure of catalysts, thereby facilitating CO2 activation and intermediate adsorption to generate C2+ products. For example, Cu surface vacancies with a Cu2S core increase the energy barrier of the ethylene pathway but leave the ethanol pathway virtually uninfluenced; thus, a selective conversion of CO2 to polyalcohol is achieved (Fig. 6a–e) [104]. To accurately regulate the percentage of vacancies on Cu-based catalysts, the lithium electrochemical tuning method was proposed to remove anions while preserving the nanostructure of the electrode. In this method, double S vacancies are formed on the hexagonal CuS (100) surface, and the density of the S vacancies can be regulated by controlling the number of charging–discharging cycles (Fig. 6f) [21]. The unique double S vacancy structure provides efficient electrocatalytic active sites to stabilize *CO and *OCCO dimers simultaneously, and these sites facilitate the CO–OCCO coupling to form C3 species with an FE of 15.4% toward n-propanol in H-cells (Fig. 6g). In conclusion, the local electron-rich environment at the vacancy sites favors electron transfer to the CO2RR intermediates, thereby facilitating the coupling of the intermediates to generate C2+ products. However, the stability of the S vacancies at cathodic potentials needs to be determined in future studies by in situ characterization.

Reproduced with permission from Ref. [104]. Copyright 2018 Springer Nature. f Mechanism of n-propanol formation on adjacent CuSx with a double-sulfur vacancy (CuSx-DSV). g FEn-PrOH and FEn-PrOH/FEC1+C2+C3 of various catalysts. Reproduced with permission from Ref. [21]. Copyright 2021 Springer Nature
Modulation of intermediate adsorption by vacancy. a–d: a Atomic models and b the reaction Gibbs free energy diagram from the adsorbed *C2H3O intermediate to ethylene and ethanol for pristine Cu, c Cu with Cu vacancy and d Cu with Cu vacancy and subsurface S slab models. e The FE of alcohols and alkenes on various catalysts, where V denotes vacancy.
Additional Element Introduction
Heteroatom Doping
Heteroatom doping has been demonstrated as an efficient way to increase active site exposure. Because of the lattice mismatch between the dopant and Cu, the strain generated at the interface adjusts the electronic structure and the intermediate adsorption strength for CO2RR. Doping by p-block elements with higher electronegativity than Cu will induce the presence of oxidation states without a phase change [105,106,107]. In addition, the dopants with strong oxygen affinity facilitate the breaking of C–O bonds in *OCHCH2; this condition thermodynamically favors the generation of ethylene and ethane but inhibits the formation of ethanol [108]. Zhou et al. [37] found that B doping can tune the local electronic structure of Cu via the transfer of electrons from Cu to B; thus, B doping can regulate the active site ratios (Cuδ+ to Cu0). B doping also persistently stabilizes Cuδ+ to facilitate the adsorption and dimerization of CO, and thus, there is a high tendency for C2 formation (Fig. 7a–c). To further improve the long-term stability of C2+ production, Zn atoms were introduced in the B-doped Cu [109, 110]. Recently, F-doped Cu was demonstrated to promote water activation, and the resulting local enrichment of *H facilitates the hydrogenation of *CO to *CHO, which in turn lowers the C–C coupling energy barrier and promotes the formation of C2+ products [111]. Similarly, Cu-based catalysts derived from metal–organic framework (MOF) with different organic linkers were used to reveal the mechanism for C2+ generation [112]. The C2/C1 product ratios can be adjusted from 0.6 to 3.8 following the order NH2− < OH− < bare < F− < 2F−, suggesting that the increased dissociation of H2O by strongly electronegative groups in organic linkers favors C2 production.

Reproduced with permission from Ref. [37]. Copyright 2018 Springer Nature. d Reaction barriers for C1–C1 and C1–C2 coupling on M-doped Cu systems calculated based on the density functional theory (DFT). Reproduced with permission from Ref. [59]. Copyright 2019 Springer Nature. e Calculated water dissociation reaction energies and hydrogen adsorption energies on various surfaces. f Product distributions of various hydroxides/oxide-modified Cu/PTFE electrodes, along with the corresponding C2H5OH/C2H4 ratio. Reproduced with permission from Ref. [19]. Copyright 2019 Springer Nature
Modulation of intermediate adsorption by doping. a CO adsorption energy as a function of the oxidation state of Cu. b The FE of C2 and C1 for different oxidation states of Cu on B-doped Cu (Cu(B)). c The partial current density of C2 at different potentials on Cu(B)-2, oxidized nano-Cu (Cu(C)), and pristine Cu (Cu(H)).
Since the electrosynthesis of C3 products depends greatly on the presence of both C1 and C2 intermediates, metal doping strategies were proposed to achieve the simultaneous stabilization of these intermediates. Among metal-doped Cu candidates, Ag-doped Cu favors both C1−C1 coupling and C1−C2 coupling. This Ag-doped Cu has the highest FE (33%) for the reduction of CO to n-propanol [59]. The facilitation of multiple C−C coupling is attributed to the strain and ligand effects induced by Ag doping, which result in an energetic asymmetry of the adjacent Cu atoms (Fig. 7d). Compressive strain induced by doping with Ag atoms in the Cu lattice also shifts the valence band density of Cu to a deeper level [113]. This electronic structure lowers the binding energies of H and O compared to the binding energy of CO, and thus, the HER is selectively inhibited, and the generation of carbonyl-containing C2+ products, accompanied by decreased generation of hydrocarbons, is facilitated.
Given the important role of hydrogenation reactions in multicarbon alcohol production [114, 115], heteroatom doping is also proposed to modulate the *H on the surface of catalysts for facilitating the hydrogenation of intermediates after C−C coupling. Theoretical calculations revealed that Pt- or Pd-doped Cu exhibits the optimal H binding energy, which promotes the hydrogenation of C2 intermediates to generate C2H5OH [116]. The results show that the CuPd and CuPt catalysts have an alcohol-to-ethylene FE ratio that is twice that of bare Cu. In addition, hydroxide- and oxide-doped Cu catalysts stabilized at reduction potential can also activate water and tune the surface *H coverage, thereby regulating the ethanol and ethylene reaction pathways (Fig. 7e) [19]. The increased *H is only involved in the branched reaction toward ethanol, accelerating the conversion of *HCCOH to *HCCHOH (a key intermediate for ethanol generation). Among all the hydroxide- and oxide-doped Cu catalysts, Ce(OH)x/Cu/PTFE exhibits the maximum FE of 43% for ethanol generation at a current operating density of 300 mA/cm2 (Fig. 7f). In conclusion, doping affects the adsorption of different intermediates, and thus, it is thus beneficial to break the linear adsorption relationship of intermediates for the selective generation of certain C2+ products.
Cu-Based Alloys
Alloying is another effective strategy to modulate the electronic and geometrical structures of catalysts for inhibiting HER competition and promoting CO2RR activity [117]. The electronic structure of the catalyst is directly correlated with the binding strength of intermediates, and the geometrical structure affects the local distribution of certain intermediates at active sites. Several Cu-based alloys were extensively studied from the viewpoint of C2+ production [118, 119]. For example, He’s group [120] used E-beam evaporation to fabricate thin films of CuAg to precisely regulate the stoichiometric ratio of the two elements. Thus, they built a good model to reveal the real reaction mechanisms during the CO2RR process. The operando synchrotron radiation–Fourier transform infrared spectroscopy (SR-FTIR) demonstrated that the CuAg bimetal greatly inhibits the formation of O−C−O intermediates and increases the coverage of *CO and *OCCO intermediates, thus promoting the C2+ production (Fig. 8a). Moreover, Cu supported on amorphous CuTi alloys (a-CuTi@Cu) can electroreduce CO2 to C2−4 products, such as ethanol, propanol, and n-butanol (Fig. 8b) [121]. Theoretical simulations and in situ characterization demonstrate that the subsurface Ti atoms increase the electron density of the surficial Cu sites with improved adsorption ability of *CO intermediates. Thus, the energy barriers for the dimerization or trimerization of *CO are reduced. The function of the interface in Cu alloys in promoting C−C coupling and C2+ formation was also confirmed by theoretical calculations [122].

Reproduced with permission from Ref. [120]. Copyright 2022 American Chemical Society. b FE for various electrocatalysts at − 0.8 V versus RHE. Reproduced with permission from Ref. [121]. Copyright 2021 Wiley–VCH. c A two-dimensional activity volcano plot for CO2RR. d A two-dimensional selectivity volcano plot for CO2RR. Reproduced with permission from Ref. [123]. Copyright 2020 Springer Nature
Modulation of intermediate adsorption by Cu-based alloys. a Operando synchrotron radiation–Fourier transform infrared (SR-FTIR) spectroscopy during CO2RR.
To expedite the discovery of Cu-based catalysts with high C2+ selectivity, Sargent’s group [123] developed a machine learning-accelerated high-throughput DFT framework for material selection. They selected 228,969 adsorption sites from 244 different Cu-containing intermetallic compound crystals to train a machine learning model. The framework was subjected to approximately 4000 DFT simulations for CO adsorption energy calculations, and the Cu-Al alloy exhibited the highest abundance of adsorption sites and site types with near-optimal CO adsorption energies. These properties created a favorable Cu coordination environment for C–C dimerization (Fig. 8c, d). The rationally designed Cu-Al electrocatalysts show a high C2H4 FE of 80% at a current density of 400 mA/cm2. Thus, Sargent’s group [123] demonstrated the essential role of high-throughput screening based on the adsorption energy of key intermediates in the development of Cu-based bimetallic catalysts, and they accelerated the targeted design of catalysts with high C2+ selectivity. Weitzner et al. [119] calculated the *CO dimerization energies on different Cu-based alloys, and they found that Cu-Al alloys favor the dimerization reaction of *CO. In general, alloying is a promising approach to tuning the adsorption energy of intermediates by manipulating the electronic structure of Cu catalysts. Thus, alloying is extremely helpful in facilitating the efficient generation of C2+ products.
Chemical State Effects
The adjustment of the valence state of Cu can modify the electronic structure of Cu-based catalysts, thus improving the catalytic performance. Some studies suggest the presence of oxidized Cu plays a crucial role in converting CO2 to C2+ products via ex situ characterizations [95, 124]. However, Cu is highly susceptible to reoxidation at the open circuit potential [71], and the real valence state of Cu under operating conditions remains controversial, limiting the mechanistic understanding of Cu-based catalysts. To clarify whether the oxidized Cu exists at an extremely negative potential during the CO2RR, isotopic labeling was used in the preparation process of Cu oxides, and only < 1% 18O remanence in the 18OD-Cu was discovered after a 5 h CO2RR electrolysis (Fig. 9a) [125]. Lei et al. [126] investigated the distribution of various Cu species on electrodes by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and electron energy loss spectroscopy (EELS) and found that oxidized Cu was completely reduced to metallic Cu during CO2RR, regardless of the initial state (Fig. 9b). Further, based on DFT modeling, Mandal et al. [127] suggested that the reduction of Cu2O is kinetically and energetically more favorable than that of CO2RR, implying that oxidized Cu should be reduced to metallic Cu before forming CO2RR products.

Reproduced with permission from Ref. [125]. Copyright 2018 Wiley–VCH. b High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and electron energy loss spectroscopy (EELS) mapping of the cross section of HQ-Cu after 1 h of CO2RR. Reproduced with permission from Ref. [126]. Copyright 2020 American Chemical Society. c Extended X-ray absorption fine structure spectra of different catalysts under operando conditions. d Summary of the activity of plasma-treated Cu electrodes. Reproduced with permission from Ref. [129]. Copyright 2016 Springer Nature. e The DFT calculation of CO dimerization and CO hydrogenation toward CHO over mixed Cu0/Cu+ catalyst. Reproduced with permission from Ref. [132]. Copyright 2017 National Academy of Sciences
Improvement of C2+ selectivity by chemical state effects. a 18O percentage of OD-Cu catalysts at different CO2 reduction catalytic times.
Conversely, using in situ characterization techniques, some researchers found that Cu+ and residual subsurface oxygen can exist even at negative potentials [128, 129]. Cuenya and coworkers [129] provided experimental evidence for the survival of Cu+ species during CO2RR via operando X-ray absorption spectroscopy (Fig. 9c). They revealed that Cu+ significantly inhibits CH4 production and thus increases the FE of C2H4 (Fig. 9d). Yang et al. [130] synthesized Cu nanocavity catalysts to confine carbonaceous intermediates, which in turn cover the catalyst surface to protect the Cu+ species, and they achieved a C2+ FE of 75.2%. In addition, the coexistence of Cu0 and Cu+ on the surface affects the CO2RR selectivity by changing the CO adsorption configuration. The top-adsorbed CO (COatop) intermediates are mainly observed on Cu+ sites, while the bridge-adsorbed CO (CObridge) intermediates are mainly observed on Cu0 sites [131]. The adjacent-adsorbed CObridge and COatop are negatively and positively charged, respectively, because of which they are beneficial for CO dimerization (Fig. 9e) [132]. Despite these promising results, the catalytic role of oxidized Cu still remains a debated issue, and further advances in operando techniques are required to resolve this issue.
Electric Field Effects
The stabilizing effect of electric fields on intermediates (e.g., *CO or *COCO), as demonstrated by theoretical calculations, makes it a promising method to manipulate the reaction pathways by regulating the applied electric field [47]. Sargent’s group [133] investigated the effect of an intensified electric field on localized CO2 enrichment. They used a metallic nanotip electrode to generate a locally high electric field at low overpotentials. In their work, the cations were concentrated around the sharp tip, leading to an increased local concentration of CO2 near the active surface for CO2RR. The electric field-induced effect on the concentration of reagents around the tips was also analyzed using kinetic simulations, indicating that the electric field-induced aggregation effect can provide additional CO2 for improving the CO2RR performance [134, 135]. Recently, Liu’s group [136, 137] also reported that the high electric field created locally by high-curvature Cu nanoneedle (Cu NN) arrays can optimize *CO adsorption and reduce C−C coupling energy barriers (Fig. 10a). Besides, encapsulating the Cu NNs in polytetrafluoroethylene (PTFE) conformal coatings was proposed as a typical strategy to enhance the local electric and thermal fields simultaneously (Fig. 10b) [138, 139]. DFT calculations indicated that the enhanced electric field reduces the Gibbs free energy of the C−C coupling, while increasing the thermal field intensity boosts the reaction rate of C−C coupling; thus, a CO2-to-C2 FE of over 86% can be achieved (Fig. 10c). To summarize, a locally enhanced electric field effectively enhances the adsorption strength of CO2 and intermediates and reduces the C−C coupling energy barriers, thus enabling the highly selective production of C2+ at the sharp tip catalytic hotspots.

Reproduced with permission from Ref. [137]. Copyright 2022 American Chemical Society. b The electric field distribution on a pristine Cu NN (left) and on a Cu NN with 99% PTFE coverage (Cu-PTFE NN) (right). c Product distribution and corresponding FEs. Reproduced with permission from Ref. [138]. Copyright 2022 American Chemical Society
Improvement of C2+ selectivity by electric field effects. a Schematic diagram of C2 formation process on the single tip of Cu nanoneedle (NN) arrays.
Substrate Effects
Although catalyst substrates are primarily used to load catalysts and provide electron-conductive channels, their structures also affect CO2RR performance, resulting in the so-called “substrate effects.” Specifically, the substrates can not only affect the reconstruction of Cu nanoparticles, but also have electronic interactions with active Cu sites [140]. Because of the outstanding chemical and electrochemical stability of carbon materials, they are the most widely used substrates. Since N has strong CO2 adsorption capability, N-doped substrates can increase the local CO2 concentration and enrich the key intermediates of C−C coupling [141]. For example, N-doped nanodiamonds (N-ND) were used as substrates for sputtered Cu nanoparticles, and an FE of 63% was realized for C2 oxygenates at − 0.5 V vs. RHE (Fig. 11a) [142]. DFT calculations indicate that the increased *CO binding at the interface between the Cu nanoparticles and an N-doped nanodiamond substrate suppresses the *CO desorption and reduces the barriers to *CO dimerization, thus promoting C2 production (Fig. 11b, c). The similar synergy between N-doped carbon nanospikes and loaded Cu nanoparticles was demonstrated for the highly selective conversion of CO2 to C2H5OH [143].

Reproduced with permission from Ref. [142]. Copyright 2020 Springer Nature. d Results of 40 h stability test at − 300 mA/cm2, where FE is shown as a function of time for unsupported Cu nanocubes (U-NC) and for C-supported nanocubes (S-NC). Reproduced with permission from Ref. [145]. Copyright 2020 Wiley–VCH
Substrate effect for improving C2+ selectivity. a Schematic illustration of the preparation of N-doped nanodiamond (N-ND)/Cu composite materials. b Free energy diagram for CO coupling. c FE values for carbon-containing products by N-ND/Cu electrodes.
The substrate also affects the reconstruction process of Cu during CO2RR, thereby altering product selectivity. Cu cubes on carbon paper were observed to undergo drastic morphological and compositional changes during the CO2RR with selective inhibition of the C2+ products relative to CH4 [144]. This is caused by the physical separation of the active sites on the carbon substrates with high specific surface areas. However, as the electrolysis duration increases, the C-supported Cu nanocubes also gradually agglomerate, leading to the transition of CO2RR products from C1 to C2 again (Fig. 11d) [145]. Hence, a reasonable selection of substrates and the dispersion of active sites are crucial to be able to make full use of the substrate effects.
Confinement Effects
Spatial confinement effects can change the mass transfer process and optimize the local distribution of intermediates [146]. Constructing porous Cu electrodes is a typical method of altering the mass transfer based on the specific confinement effect of pores. Increasing the pore depth results in locally higher pH and lower CO2 concentration inside the pore, and this effect can be intensified by increasing the cathodic potential [147, 148]. Changes in the local environment caused by mass transfer further affect the product distribution of the CO2RR. For example, C1 products strongly depend on the local concentration of CO2, while C2 products tend to be formed in a locally high pH environment [63]. Besides, confinement effects can also restrict the diffusion of intermediates via the formation of closed spaces. Cu nanocavities can limit the efflux of C2 intermediates, thereby increasing the coverage and retention time of the intermediates required for C3 production. Finite element simulations revealed that the cavity-opening angles play critical roles in the C3/C2 selectivity ratio (Fig. 12a). An opening of ~ 60° is the most favorable for C3 production, where the enrichment of C2 intermediates is not limited by the reduced CO availability (Fig. 12b, c) [149]. Hollow porous Cu nanospheres, hollow multishelled Cu, and multishelled CuO microboxes were also reported to show similar confinement effects. The high C2+ selectivity is attributed to the C−C coupling facilitated by the high coverage of carbonaceous intermediates in the cavity [150,151,152].

Reproduced with permission from Ref. [149]. Copyright 2018 Springer Nature. d Schematic representation and compositional characterization of the Ag core/porous Cu shell particle. Reproduced with permission from Ref. [153]. Copyright 2021 Wiley–VCH. e The C2+/C1 product selectivity on the Ag@Cu catalysts with different average pore diameters at 400 mA/cm2. Reproduced with permission from Ref. [154]. Copyright 2022 American Chemical Society
Improvement of C2+ selectivity by confinement effect. a CO (left), C2 (middle), and C3 (right) concentrations (color scale, in millimoles) and flux distributions (arrows) on the cavity confinement structure. b Representative scanning electron microscopy images of different catalysts. c C3/C2 product selectivity on different catalysts obtained from experiments and from finite element simulations.
Confinement effects can also be exploited to design tandem catalysts to reutilize the products of CO-selective catalysts. For example, nanoparticles with Ag cores and porous Cu shells can simultaneously use the CO overflow from the Ag cores and confinement effects of the porous Cu channels (Fig. 12d) [153]. In this system, CO2 is selectively reduced to CO on the Ag core at the bottom of the porous Cu channel, while a prolonged diffusion time provides more opportunities for the porous Cu shell to reduce the substrate-enriched CO to C2+ products. The C2+/C1 product selectivity can be further increased by optimizing the diameter of the channels (Fig. 12e) [154]. Therefore, the confinement effects that can alter the electrolyte environment and locally enriched intermediates are a highly effective strategy to improve the utilization of carbonaceous intermediates for C2+ products.
Local Microenvironment Effects
Regulation of the local microenvironment near the catalyst surface is another promising strategy to facilitate the electroreduction of CO2 to C2+ products. However, many influencing factors have complex effects on the local microenvironment [155]. In this section, the effects of electrolyte composition and concentration on the local microenvironment properties, such as local pH and electrostatic interactions, are mainly discussed.
Although KHCO3 solution is widely used in CO2RR experiments to maintain neutral pH, a pH gradient exists near the electrode surface because of the OH− formed during the reduction reaction [156, 157]. The local pH near the electrode plays an important role in the *CO/*H coverage, which determines whether *CO subsequently undergoes dimerization or hydrogenation. At lower pH values, *CO prefers to form CH4 in the presence of abundant *H. At higher pH, where *H is scarce and *CO coverage is high, ethylene formation dominates over ethane formation [158]. As the rate of OH− production depends on the applied potential, it is necessary to probe the local pH near the electrode under operational conditions. Lu et al. [159] designed a flow Raman electrochemical cell to measure the local pH near the gas diffusion electrode during CO2RR (Fig. 13a). By analyzing the CO32− and HCO3− concentrations in different electrolyte regions near the electrode via in situ microarea Raman spectroscopy, the corresponding pH values can be deduced from the equilibrium between HCO3− and CO32−. A much higher local pH (11.9) than that of the electrolyte bulk was observed near the cathode surface (Fig. 13b). This accurate estimation of the local pH facilitated the further investigation of the pH effect during the CO2RR. In addition, anions with buffering capacity can also alter the local pH, thus affecting the pH-sensitive reactions at the catalyst surface [160]. Buffering anions increase the selectivity of H2 and CH4 in the following order of decreasing pKa: HCO3− (10.33) > H3BO3 (9.23) > HPO42− (7.21) [161]. This finding suggests that buffer anions with pKa lower than water can provide hydrogen directly to the electrode surface. Notably, special adsorption additives can also modulate the local proton feeding microenvironment. Our group [39] has explored boosting proton feeding toward Cu surfaces using EDTMPA additives. The adsorbed EDTMPA serves as a proton-delivering medium that accelerates the dissociation of water, providing abundant *H to assist *CO protonation toward *CHO. Typically, one H atom is transferred from the adsorbed methanephosphonic acid (*MPA, a fragment of EDTMPA) to Cu (110), and the *MPA that loses one H (*MPA − H) subsequently captures one H atom from the adjacent H2O molecule to become an *MPA again (Fig. 13c).

Reproduced with permission from Ref. [159]. Copyright 2020 American Chemical Society. c Kinetic energy diagrams of an H atom transferred from *MPA to Cu (110) and an H atom compensated from H2O to *MPA − H. Reproduced with permission from Ref. [39]. Copyright 2022 Springer Nature. d Partial current densities of ethylene at different potentials as a function of the electrolyte metal cation on Cu (100). Reproduced with permission from Ref. [162]. Copyright 2017 American Chemical Society. e Cyclic voltammetric curves of Cu electrodes in Ar and CO2 atmosphere after CO2RR in 1 mmol/L H2SO4 with or without Cs+. f Schematic representation of the interaction of the cation with the negatively charged CO2− intermediate. Reproduced with permission from Ref. [40]. Copyright 2021 Springer Nature
Modulation of intermediate adsorption by local microenvironment. a Fitted concentrations of HCO3−, CO32−, CO2 (aq), and OH− and b pH profile in 1 mol/L KHCO3 with respect to the distance from the GDE surface.
In addition to local pH, the electrostatic interactions between cations and negatively charged CO2 intermediates in the local microenvironment also have a considerable impact on CO2RR. Bell’s group [162] measured the CO2RR performance of Cu catalysts in bicarbonate electrolytes with different alkali metal cations. The activity of ethylene formation on Cu follows the trend of atomic radius increasing in order: Li+ < Na+ < K+ < Rb+ < Cs+ (Fig. 13d). Theoretical calculations suggest that the presence of solvated cations in the outer Helmholtz plane stabilizes the negatively charged reaction intermediates such as *CO2− and *OCCO–. The difference in activity when using various cations is attributed to the broader coverage of cations as the cation size increases. Recently, Koper’s group [40] found that CO2RR can only proceed on Cu when metal cations are present in the electrolyte (Fig. 13e). DFT simulations confirmed that the partially desolated metal cations stabilized the CO2− intermediates by short-range electrostatic interactions, thus allowing its reduction (Fig. 13f). These works highlight the necessity of cations and water in the electrochemical activation of CO2.
In situ Characterization Techniques
To deeply understand the CO2RR mechanisms, various in situ characterization techniques were developed to capture real-time information under actual operating conditions. To date, in situ infrared (IR) spectroscopy, in situ Raman spectroscopy, in situ mass spectrometry (MS), and in situ isotope tracer technology have been widely used to characterize the reaction intermediates to propose possible reaction pathways.
In situ IR spectroscopy was extensively used to reveal the CO2RR mechanisms by detecting the chemisorbed species on the electrode surface [163]. Then, the adsorbed species with unique vibrational modes such as *CO, adsorbed CO32−, and *CO dimers can be analyzed in real time during the CO2RR. Hori and coworkers [164] first identified *CO at a Cu electrode during CO2RR by performing in situ IR spectroscopy. Heyes et al. [56] detected the vibration bands of *CO and *H on Cu at 2060 and 2090 cm−1, respectively, by performing surface-enhanced infrared absorption spectroscopy (SEIRA). Further, peak deconvolution revealed that *H can partially replace *CO, while *CO cannot replace *H (Fig. 14a). Moradzaman et al. [165] assigned the band at ~ 1610 cm−1 to the CO2-dimer radical anion, which subsequently decomposed into CO32− and *CO. The adsorbed carbonate was distinguished from the dissolved carbonate because of its strongly potential-dependent peak position in the range of 1510–1570 cm−1. Koper’s group [166] found that the vibrational bands at 1191 and 1584 cm−1 corresponding to the C–O–H and C=O stretching modes of the hydrogenated dimer (*COCOH), respectively, suggesting that the stable adsorption of *COCOH is responsible for the high C2H4 selectivity on Cu (100).

Reproduced with permission from Ref. [56]. Copyright 2016 American Chemical Society. b Comparison of the steady-state Raman spectra during reduction at various potentials. Reproduced with permission from Ref. [101]. Copyright 2021 Wiley–VCH. c Hypothetical scenario for the reduction in a mixture of 13CO and 12CO2. Reproduced with permission from Ref. [172]. Copyright 2019 Springer Nature. d, e MS signal of the relative abundance of the liquid phase products generated on Cu. The solid lines represent the relative abundances of the liquid phase products in a traditional H-cell when analyzing the bulk electrolyte. Reproduced with permission from Ref. [61]. Copyright 2018 American Chemical Society
In situ intermediate characterization techniques. a Time-resolved surface-enhanced infrared absorption spectroscopy (SEIRA)-integrated peak areas for *CO and *H bands after peak deconvolution, reported as a percentage of the largest *H peak. CO introduced on the H-saturated Cu surface (left). *H accumulation on a CO-saturated Cu surface (right).
In situ Raman spectroscopy can effectively complement IR spectroscopy because of its relatively low response to water, which is usually used to detect varying rotational or vibrational states of different molecules on the catalyst surface [167]. The high-speed data acquisition in the case of Raman spectroscopy helps distinguish the real-time reaction intermediates. *CO is the most common CO2RR reaction intermediate observed by in situ Raman spectroscopy, and a low-intensity carbonate signal can also be detected [168, 169]. Zhan et al. [170] found that the ratio of the intensity of the Cu–CO stretching band to that of the CO rotational band follows a volcano-shaped trend, which depends on the potential-dependent surface coverage of CO. At high CO surface coverage, the mixed adsorption conformation of CO at the top and bridge sites kinetically and thermodynamically favors C–C coupling, thereby effectively improving the C2+ selectivity. An et al. [101] used subsecond in situ time-resolved surface-enhanced Raman spectroscopy to reveal the dynamics of CO intermediates during the CO2RR on Cu. A highly dynamic *CO with characteristic vibration below 2060 cm−1 is associated with C–C coupling and C2H4 production, while the isolated and stationary *CO with a distinct vibration at 2092 cm−1 favors the generation of gaseous CO (Fig. 14b).
Quantitative isotope measurements are indispensable for understanding the CO2RR mechanisms at the molecular and atomic levels. Because of the different kinetic radii of different isotopes, the kinetic isotope effect (KIE) is commonly used to investigate the effect of isotopic substitution on the reaction rate to verify the potential RDS. In addition, the isotopic labeling method is usually used to reveal the origin of the intermediates or final products or both by detecting the location of the isotopically labeled atoms, thus providing suggestions of potential reaction pathways [171]. For the precise quantification of different isotopic atoms, nuclear magnetic resonance and MS are usually applied in the isotopic analysis. Ma et al. [111] reported that fluorine-modified Cu promotes the activation of water supported by a drastically reduced KIE of H/D (H/D is defined as the ratio of the rate of ethylene formation in H2O and D2O), and a KIE value close to 1 indicates that dissociation of H2O is no longer involved in the RDS. Ager and coworkers [172] used 12CO2/13CO co-feed experiments to identify three product-specific active sites on OD-Cu electrocatalysts; these three sites lead to the production of ethylene, ethanol or acetate, and n-propanol (Fig. 14c). Chang et al. [62] elucidated the formation mechanism of the C3 products by combining isotopic labeling with in situ spectroscopy. Since only 36% of n-propanol is derived from the cross-coupling of CO with acetaldehyde, the adsorbed methylcarbonyl intermediate is proposed to be the bifurcation point of the reaction pathway for the C2 and C3 products.
In situ MS is typically used to achieve the real-time detection and semiquantification of gaseous and evaporable liquid products based on mass-to-charge ratios. By improving the time resolution of in situ MS, the formation of intermediates and products under transient operation can be detected, and the potential correlations between them can be established [173, 174]. Koper and coworkers [54] proposed two possible reaction mechanisms for ethylene formation using online electrochemical MS. In one pathway, C2H4 shares an intermediate with CH4 on the Cu (111) and Cu (100) surfaces, while in the other pathway, the selective reduction of CO to C2H4 occurs on Cu (100) at a relatively low overpotential, presumably via the formation of a CO dimer. Clark et al. [61] found that the ratio of aldehydes to alcohols detected in the region close to the Cu electrode was much higher than that in the bulk electrolyte. With increasing overpotential, the relative abundance of ethanol rises along with a decrease in propionaldehyde, suggesting that acetaldehyde is a bifurcated intermediate for ethanol and propionaldehyde formation, while n-propanol is formed via propionaldehyde (Fig. 14d, e).
Summary and Perspectives
This review focuses on strategies to improve the selectivity of CO2RR to C2+ products through manipulation of the intermediates on the surface of the Cu-based catalysts. A systematic discussion of C2+ formation mechanisms revealed that the binding energies and competitive adsorption of intermediates on the catalyst surface are crucial for C2+ selectivity as these factors can affect the C–C coupling and hydrodeoxygenation processes. Subsequently, we provided deep insights into the manipulating strategies and factors influencing intermediate adsorption, covering topics such as surface structural effects, introduction of additional elements, chemical state effects, electric field effects, substrate effects, confinement effects, and local microenvironment effects. Table 1 summarizes the electrochemical CO2-to-C2+ performance of Cu-based electrocatalysts according to the intermediate manipulation strategies. Through an optimal combination of these strategies, C2+ production can be further improved in practical applications. For example, the simultaneous utilization of doping and confinement strategies improves the selectivity of C2+ products without any catalytic activity degradation [107]. Specifically, quasigraphitic C shells can stabilize the crystal size of Cu nanoparticles based on the confinement effects, while doping with p-block elements can drastically improve the C2+ selectivity and cathodic power conversion efficiency via the modulation of the binding properties of the Cu catalysts.
As the CO2RR involves complex reaction steps and numerous intermediates, the use of advanced in situ characterization techniques is important for elucidating the relationship among structures, intermediates, and performance. In particular, the operando characterization of intermediates is the key to accurately revealing the reaction pathways and RDS of the CO2RR, and thus, such studies can provide useful guidance for optimizing catalysts. Therefore, we comprehensively discussed various in situ characterization techniques to identify the intermediates and their adsorption states under operating conditions. These techniques included IR spectroscopy, Raman spectroscopy, MS, and quantitative isotope measurements. In addition, theoretical calculations and simulations that closely match the actual operating conditions are also irreplaceable critical tools for understanding the catalytic mechanisms in detail. By considering the adsorption intermediate as a bridge, the correlation between the catalyst structure and catalytic performance was established to promote the practical application of CO2RR.
Although the activity and selectivity of CO2 reduction to C2+ products on Cu-based catalysts have greatly improved in recent years, their in-depth mechanisms are still unclear, and the industrial application of the CO2RR is still challenging. On the one hand, most reaction mechanisms for the conversion of CO2 to C2+ products are presumed based on the final product distribution. Such an inference from the effect to the cause makes it difficult to objectively and accurately understand the actual reaction process. On the other hand, for meeting the industrial requirements for the CO2RR, the focus is on greatly reducing the cost of the catalytic systems rather than further improving catalyst selectivity, activity, and stability. Note that cofeeding of CO2 with other feedstocks provides a promising pathway for the generation of high-value-added products, and this pathway should be actively explored. To this end, efforts should be focused on the following aspects in future research:
-
1)
Clarification of the CO2RR mechanisms. A combination of in situ experimental evidence and theoretical calculations can provide insights into the actual reaction pathways of the CO2RR, thus directing the rational design of catalysts. In particular, experimental observations can provide modeling guidance for theoretical calculations, in turn enabling the high-throughput screening of highly active catalysts with intermediates with suitable binding energies [176]. To enable the precise customization of active sites on Cu-based catalysts, there is an urgent need to develop high temporal and spatial resolution characterization techniques to accurately detect the intermediates [177, 178].
-
2)
Rational design of electrolyzers. Recently, it was established that high-efficiency gas diffusion electrodes and membrane electrode assembly are more promising electrolytic devices for the industrial application of the CO2RR [179]. The optimization of electrolyzers can not only reduce the working voltage, but also significantly increase the current density (by up to 1.6 A/cm2), which is close to the technoeconomic requirements for the industrial application of CO2RR [111, 180]. However, the problems of flooding and salt precipitation that occur at gas diffusion electrodes over long-term electrolysis should be addressed to avoid a rapid decrease in selectivity and current density. Moreover, the formation and crossover of carbonates in alkaline electrolytes also affects the single-pass conversion of CO2 and contaminates the O2 stream released from anodes, thereby increasing the cost of feedstock and separation. Impressively, the solid-electrolyte reactor, which decouples the ion conduction and product collection, successfully recovers the substantial carbon loss that occurs during CO2RR electrolysis and avoids the requirement for cost-intensive downstream product-separation processes [181,182,183]. Besides, acid electrolytes [184] and pulsed electrolysis [185] were also used to avoid carbonate accumulation, offering promising ways to realize long-term electrolysis.
-
3)
Cofeeding of CO2 and other reactants. Currently, high-purity CO2, which requires considerable energy to capture and purify, is the main feedstock used in experimental research. However, because of its high cost, high-purity CO2 is impractical for industrial applications of CO2RRs. In this situation, cofeeding CO2 with other chemicals can decrease the cost of CO2 purification and/or create high-value products, in turn improving the technical economy of the catalytic systems. For example, flue gas is a potential source of CO2 and can be adopted to reduce the purification cost; however, more deliberate catalyst design and pressurization are required to overcome the competition reaction caused by the oxidizing gases [186, 187]. Besides, the formation of C–N compounds by the coelectrolysis of CO2 and nitrogen-containing reactants also holds great promise [188,189,190]. For example, the coactivation of N2 and CO2 enables the electrosynthesis of desirable urea [191].
In conclusion, the CO2RR provides a promising solution for converting CO2 emissions into valuable feedstocks. Encouragingly, much progress has been achieved toward the electroreduction of CO2 to C2+ products using Cu-based catalysts during the last decades. However, many problems still persist for industrial applications. Together with a mechanistic exploration supported by advanced in situ characterization techniques and theoretical simulations, the optimization of electrolyzers and CO2 feeding modes is expected to further advance the industrial applications of the CO2RR.
References
Ling YF, Ma QL, Yu YF et al (2021) Optimization strategies for selective CO2 electroreduction to fuels. Trans Tianjin Univ 27(3):180–200
Li L, Li XD, Sun YF et al (2022) Rational design of electrocatalytic carbon dioxide reduction for a zero-carbon network. Chem Soc Rev 51(4):1234–1252
Mallapaty S (2020) How China could be carbon neutral by mid-century. Nature 586(7830):482–483
Hepburn C, Adlen E, Beddington J et al (2019) The technological and economic prospects for CO2 utilization and removal. Nature 575(7781):87–97
De Luna P, Hahn C, Higgins D et al (2019) What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364(6438): eaav3506
Endrődi B, Kecsenovity E, Samu A et al (2020) High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell. Energy Environ Sci 13(11):4098–4105
Ren SX, Joulié D, Salvatore D et al (2019) Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 365(6451):367–369
Jiao JQ, Lin R, Liu SJ et al (2019) Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat Chem 11(3):222–228
Zhao Y, Liu XL, Chen DC et al (2021) Atomic-level-designed copper atoms on hierarchically porous gold architectures for high-efficiency electrochemical CO2 reduction. Sci China Mater 64(8):1900–1909
Li JC, Kuang Y, Meng YT et al (2020) Electroreduction of CO2 to formate on a copper-based electrocatalyst at high pressures with high energy conversion efficiency. J Am Chem Soc 142(16):7276–7282
Fan L, Xia C, Zhu P et al (2020) Electrochemical CO2 reduction to high-concentration pure formic acid solutions in an all-solid-state reactor. Nat Commun 11:3633
Weng Z, Wu YS, Wang MY et al (2018) Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat Commun 9:415
Zhu HL, Huang JR, Zhang XW et al (2021) Highly efficient electroconversion of CO2 into CH4 by a metal-organic framework with trigonal pyramidal Cu(I)N3 active sites. ACS Catal 11(18):11786–11792
Zhang L, Li XX, Lang ZL et al (2021) Enhanced cuprophilic interactions in crystalline catalysts facilitate the highly selective electroreduction of CO2 to CH4. J Am Chem Soc 143(10):3808–3816
García de Arquer FP, Dinh CT, Ozden A et al (2020) CO2 electrolysis to multicarbon products at activities greater than 1 A/cm2. Science 367(6478):661–666
Dinh CT, Burdyny T, Kibria MG et al (2018) CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360(6390):783–787
Zhang BX, Zhang JL, Hua ML et al (2020) Highly electrocatalytic ethylene production from CO2 on nanodefective Cu nanosheets. J Am Chem Soc 142(31):13606–13613
Li YC, Wang ZY, Yuan TG et al (2019) Binding site diversity promotes CO2 electroreduction to ethanol. J Am Chem Soc 141(21):8584–8591
Luo MC, Wang ZY, Li YC et al (2019) Hydroxide promotes carbon dioxide electroreduction to ethanol on copper via tuning of adsorbed hydrogen. Nat Commun 10:5814
Wang X, Wang ZY, García de Arquer FP et al (2020) Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat Energy 5(6):478–486
Peng C, Luo G, Zhang JB et al (2021) Double sulfur vacancies by lithium tuning enhance CO2 electroreduction to n-propanol. Nat Commun 12:1580
Tang MT, Peng HJ, Stenlid JH et al (2021) Exploring trends on coupling mechanisms toward C3 product formation in CO(2)R. J Phys Chem C 125(48):26437–26447
Jiang K, Sandberg RB, Akey AJ et al (2018) Metal ion cycling of Cu foil for selective C-C coupling in electrochemical CO2 reduction. Nat Catal 1(2):111–119
Bushuyev OS, de Luna P, Dinh CT et al (2018) What should we make with CO2 and how can we make it? Joule 2(5):825–832
Fan L, Xia C, Yang FQ et al (2020) Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2+ products. Sci Adv 6(8): eaay3111
Bagger A, Ju W, Varela AS et al (2017) Electrochemical CO2 reduction: a classification problem. ChemPhysChem 18(22):3266–3273
Kuhl KP, Cave ER, Abram DN et al (2012) New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ Sci 5(5):7050–7059
Ross MB, de Luna P, Li YF et al (2019) Designing materials for electrochemical carbon dioxide recycling. Nat Catal 2(8):648–658
Ma WC, He XY, Wang W et al (2021) Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts. Chem Soc Rev 50(23):12897–12914
Birdja YY, Pérez-Gallent E, Figueiredo MC et al (2019) Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat Energy 4(9):732–745
Zhang YJ, Sethuraman V, Michalsky R et al (2014) Competition between CO2 reduction and H2 evolution on transition-metal electrocatalysts. ACS Catal 4(10):3742–3748
Kortlever R, Shen J, Schouten KJP et al (2015) Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J Phys Chem Lett 6(20):4073–4082
Zhi X, Vasileff A, Zheng Y et al (2021) Role of oxygen-bound reaction intermediates in selective electrochemical CO2 reduction. Energy Environ Sci 14(7):3912–3930
Mariano RG, Kang M, Wahab OJ et al (2021) Microstructural origin of locally enhanced CO2 electroreduction activity on gold. Nat Mater 20(7):1000–1006
Verdaguer-Casadevall A, Li CW, Johansson TP et al (2015) Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts. J Am Chem Soc 137(31):9808–9811
Mariano RG, McKelvey K, White HS et al (2017) Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 358(6367):1187–1192
Zhou YS, Che FL, Liu M et al (2018) Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat Chem 10(9):974–980
Xiang KS, Liu YC, Li CF et al (2021) Microenvironmental feeding and stabilization of C2H4 intermediates by iodide-doped copper nanowire arrays to boost C2H6 formation. Energy Fuels 35(19):15987–15994
Han ZS, Han DL, Chen Z et al (2022) Steering surface reconstruction of copper with electrolyte additives for CO2 electroreduction. Nat Commun 13:3158
Monteiro MCO, Dattila F, Hagedoorn B et al (2021) Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat Catal 4(8):654–662
Hori Y, Wakebe H, Tsukamoto T et al (1994) Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim Acta 39(11–12):1833–1839
Hori Y, Takahashi I, Koga O et al (2003) Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J Mol Catal A Chem 199(1–2):39–47
Shi C, Hansen HA, Lausche AC et al (2014) Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys Chem Chem Phys 16(10):4720–4727
Nitopi S, Bertheussen E, Scott SB et al (2019) Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem Rev 119(12):7610–7672
Wang L, Nitopi SA, Bertheussen E et al (2018) Electrochemical carbon monoxide reduction on polycrystalline copper: effects of potential, pressure, and pH on selectivity toward multicarbon and oxygenated products. ACS Catal 8(8):7445–7454
Zhong DZ, Zhao ZJ, Zhao Q et al (2021) Coupling of Cu(100) and (110) facets promotes carbon dioxide conversion to hydrocarbons and alcohols. Angew Chem Int Ed Engl 60(9):4879–4885
Sandberg RB, Montoya JH, Chan KR et al (2016) CO-CO coupling on Cu facets:coverage, strain and field effects. Surf Sci 654:56–62
Jeong HM, Kwon Y, Won JH et al (2020) Atomic-scale spacing between copper facets for the electrochemical reduction of carbon dioxide. Adv Energy Mater 10(10):1903423
Luo WJ, Nie XW, Janik MJ et al (2016) Facet dependence of CO2 reduction paths on Cu electrodes. ACS Catal 6(1):219–229
de Luna P, Quintero-Bermudez R, Dinh CT et al (2018) Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat Catal 1(2):103–110
Wang X, Ou PF, Ozden A et al (2022) Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag–Ru–Cu catalyst. Nat Energy 7(2):170–176
Li J, Chang K, Zhang HC et al (2019) Effectively increased efficiency for electroreduction of carbon monoxide using supported polycrystalline copper powder electrocatalysts. ACS Catal 9(6):4709–4718
Santatiwongchai J, Faungnawakij K, Hirunsit P (2021) Comprehensive mechanism of CO2 electroreduction toward ethylene and ethanol: the solvent effect from explicit water-Cu(100) interface models. ACS Catal 11(15):9688–9701
Schouten KJP, Qin ZS, Pérez Gallent E et al (2012) Two pathways for the formation of ethylene in CO reduction on single-crystal copper electrodes. J Am Chem Soc 134(24):9864–9867
Chang XX, Li J, Xiong HC et al (2022) C-C coupling is unlikely to be the rate-determining step in the formation of C2+ products in the copper-catalyzed electrochemical reduction of CO. Angew Chem Int Ed Engl 61(2):e202111167
Heyes J, Dunwell M, Xu BJ (2016) CO2 reduction on Cu at low overpotentials with surface-enhanced in situ spectroscopy. J Phys Chem C 120(31):17334–17341
Cheng T, Xiao H, Goddard WA (2017) Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc Natl Acad Sci USA 114(8):1795–1800
Hoang TT, Verma S, Ma SC et al (2018) Nanoporous copper-silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol. J Am Chem Soc 140(17):5791–5797
Wang X, Wang ZY, Zhuang TT et al (2019) Efficient upgrading of CO to C3 fuel using asymmetric C−C coupling active sites. Nat Commun 10:5186
Hori Y, Takahashi R, Yoshinami Y et al (1997) Electrochemical reduction of CO at a copper electrode. J Phys Chem B 101(36):7075–7081
Clark EL, Bell AT (2018) Direct observation of the local reaction environment during the electrochemical reduction of CO2. J Am Chem Soc 140(22):7012–7020
Chang XX, Malkani A, Yang X et al (2020) Mechanistic insights into electroreductive C−C coupling between CO and acetaldehyde into multicarbon products. J Am Chem Soc 142(6):2975–2983
Veenstra FLP, Ackerl N, Martín AJ et al (2020) Laser-microstructured copper reveals selectivity patterns in the electrocatalytic reduction of CO2. Chem 6(7):1707–1722
de Gregorio GL, Burdyny T, Loiudice A et al (2020) Facet-dependent selectivity of Cu catalysts in electrochemical CO2 reduction at commercially viable current densities. ACS Catal 10(9):4854–4862
Zhang G, Zhao ZJ, Cheng DF et al (2021) Efficient CO2 electroreduction on facet-selective copper films with high conversion rate. Nat Commun 12:5745
Chang CC, Ku MS (2021) Role of high-index facet Cu(711) surface in controlling the C2 selectivity for CO2 reduction reaction—a DFT study. J Phys Chem C 125(20):10919–10925
Hahn C, Hatsukade T, Kim YG et al (2017) Engineering Cu surfaces for the electrocatalytic conversion of CO2: controlling selectivity toward oxygenates and hydrocarbons. Proc Natl Acad Sci USA 114(23):5918–5923
Chen Y, Fan ZX, Wang J et al (2020) Ethylene selectivity in electrocatalytic CO2 reduction on Cu nanomaterials: a crystal phase-dependent study. J Am Chem Soc 142(29):12760–12766. https://doi.org/10.1021/jacs.0c04981
Liu JZ, Guo L (2021) In situ self-reconstruction inducing amorphous species:a key to electrocatalysis. Matter 4(9):2850–2873
Weng Z, Zhang X, Wu YS et al (2017) Self-cleaning catalyst electrodes for stabilized CO2 reduction to hydrocarbons. Angew Chem Int Ed Engl 56(42):13135–13139
Lee SH, Lin JC, Farmand M et al (2021) Oxidation state and surface reconstruction of Cu under CO2 reduction conditions from in situ X-ray characterization. J Am Chem Soc 143(2):588–592
Zhu CY, Zhao SW, Shi GS et al (2022) Structure-function correlation and dynamic restructuring of Cu for highly efficient electrochemical CO2 conversion. Chemsuschem 15(7):e202200068
Chen JY, Wang L (2022) Effects of the catalyst dynamic changes and influence of the reaction environment on the performance of electrochemical CO2 reduction. Adv Mater 34(25):e2103900
Raaijman SJ, Arulmozhi N, Koper MTM (2021) Morphological stability of copper surfaces under reducing conditions. ACS Appl Mater Interfaces 13(41):48730–48744
Arán-Ais RM, Rizo R, Grosse P et al (2020) Imaging electrochemically synthesized Cu2O cubes and their morphological evolution under conditions relevant to CO2 electroreduction. Nat Commun 11:3489
Simon GH, Kley CS, Roldan Cuenya B (2021) Potential-dependent morphology of copper catalysts during CO2 electroreduction revealed by in situ atomic force microscopy. Angew Chem Int Ed Engl 60(5):2561–2568
Kim YG, Baricuatro JH, Javier A et al (2014) The evolution of the polycrystalline copper surface, first to Cu(111) and then to Cu(100), at a fixed CO2RR potential: a study by operando EC-STM. Langmuir 30(50):15053–15056
Kim YG, Baricuatro JH, Soriaga MP (2018) Surface reconstruction of polycrystalline Cu electrodes in aqueous KHCO3 electrolyte at potentials in the early stages of CO2 reduction. Electrocatalysis 9(4):526–530
Wang HX, Liang Z, Tang M et al (2019) Self-selective catalyst synthesis for CO2 reduction. Joule 3(8):1927–1936
Wang YH, Wang ZY, Dinh CT et al (2020) Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis. Nat Catal 3(2):98–106
Kim JY, Park W, Choi C et al (2021) High facets on nanowrinkled Cu via chemical vapor deposition graphene growth for efficient CO2 reduction into ethanol. ACS Catal 11(9):5658–5665
Vollmer S, Witte G, Wöll C (2001) Determination of site specific adsorption energies of CO on copper. Catal Lett 77(1–3):97–101
Li J, Che FL, Pang YJ et al (2018) Copper adparticle enabled selective electrosynthesis of n-propanol. Nat Commun 9:4614
Piqué O, Low QH, Handoko AD et al (2021) Selectivity map for the late stages of CO and CO2 reduction to C2 species on copper electrodes. Angew Chem Int Ed Engl 60(19):10784–10790
Kim T, Palmore GTR (2020) A scalable method for preparing Cu electrocatalysts that convert CO2 into C2+ products. Nat Commun 11:3622
Jiang K, Huang YF, Zeng GS et al (2020) Effects of surface roughness on the electrochemical reduction of CO2 over Cu. ACS Energy Lett 5(4):1206–1214
Loiudice A, Lobaccaro P, Kamali EA et al (2016) Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew Chem Int Ed Engl 55(19):5789–5792
Handoko AD, Ong CW, Huang Y et al (2016) Mechanistic insights into the selective electroreduction of carbon dioxide to ethylene on Cu2O-derived copper catalysts. J Phys Chem C 120(36):20058–20067
Reske R, Mistry H, Behafarid F et al (2014) Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J Am Chem Soc 136(19):6978–6986
Rong WF, Zou HY, Zang WJ et al (2021) Size-dependent activity and selectivity of atomic-level copper nanoclusters during CO/CO2 electroreduction. Angew Chem Int Ed Engl 60(1):466–472
Li CW, Kanan MW (2012) CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J Am Chem Soc 134(17):7231–7234
Li CW, Ciston J, Kanan MW (2014) Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 508(7497):504–507
Feng XF, Jiang KL, Fan SS et al (2016) A direct grain-boundary-activity correlation for CO electroreduction on Cu nanoparticles. ACS Cent Sci 2(3):169–174
Chen CJ, Yan XP, Wu YH et al (2021) The in situ study of surface species and structures of oxide-derived copper catalysts for electrochemical CO2 reduction. Chem Sci 12(16):5938–5943
Chen CJ, Sun XF, Yan XP et al (2020) A strategy to control the grain boundary density and Cu+/Cu0 ratio of Cu-based catalysts for efficient electroreduction of CO2 to C2 products. Green Chem 22(5):1572–1576
Yang C, Shen HC, Guan AX et al (2020) Fast cooling induced grain-boundary-rich copper oxide for electrocatalytic carbon dioxide reduction to ethanol. J Colloid Interface Sci 570:375–381
Chen ZQ, Wang T, Liu B et al (2020) Grain-boundary-rich copper for efficient solar-driven electrochemical CO2 reduction to ethylene and ethanol. J Am Chem Soc 142(15):6878–6883
Sun H, Chen L, Xiong LK et al (2021) Promoting ethylene production over a wide potential window on Cu crystallites induced and stabilized via current shock and charge delocalization. Nat Commun 12:6823
Cheng DF, Zhao ZJ, Zhang G et al (2021) The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat Commun 12:395
Mi YY, Shen SB, Peng XY et al (2019) Selective electroreduction of CO2 to C2 products over Cu3N-derived Cu nanowires. ChemElectroChem 6(9):2393–2397
An HY, Wu LF, Mandemaker LDB et al (2021) Sub-second time-resolved surface-enhanced Raman spectroscopy reveals dynamic CO intermediates during electrochemical CO2 reduction on copper. Angew Chem Int Ed Engl 60(30):16576–16584
Kim Y, Park S, Shin SJ et al (2020) Time-resolved observation of C−C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ Sci 13(11):4301–4311
Pang YJ, Li J, Wang ZY et al (2019) Efficient electrocatalytic conversion of carbon monoxide to propanol using fragmented copper. Nat Catal 2(3):251–258
Zhuang TT, Liang ZQ, Seifitokaldani A et al (2018) Steering post-C–C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat Catal 1(6):421–428
Wang SS, Ding PC, Li Z et al (2021) Subsurface-carbon-induced local charge of copper for an on-surface displacement reaction. Angew Chem Int Ed 60(43):23123–23127
Li MH, Ma YY, Chen J et al (2021) Residual chlorine induced cationic active species on a porous copper electrocatalyst for highly stable electrochemical CO2 reduction to C2+. Angew Chem Int Ed Engl 60(20):11487–11493
Kim JY, Hong D, Lee JC et al (2021) Quasi-graphitic carbon shell-induced Cu confinement promotes electrocatalytic CO2 reduction toward C2+ products. Nat Commun 12:3765
Zhi X, Jiao Y, Zheng Y et al (2021) Directing the selectivity of CO2 electroreduction to target C2 products via non-metal doping on Cu surfaces. J Mater Chem A 9(10):6345–6351
Song YF, Junqueira JRC, Sikdar N et al (2021) B-Cu-Zn gas diffusion electrodes for CO2 electroreduction to C2+ products at high current densities. Angew Chem Int Ed Engl 60(16):9135–9141
Chen CJ, Sun XF, Lu L et al (2018) Efficient electroreduction of CO2 to C2 products over B-doped oxide-derived copper. Green Chem 20(20):4579–4583
Ma WC, Xie SJ, Liu TT et al (2020) Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C-C coupling over fluorine-modified copper. Nat Catal 3(6):478–487
Chen RZ, Cheng L, Liu JZ et al (2022) Toward high-performance CO2-to-C2 electroreduction via linker tuning on MOF-derived catalysts. Small 18(18):2200720
Clark EL, Hahn C, Jaramillo TF et al (2017) Electrochemical CO2 reduction over compressively strained CuAg surface alloys with enhanced multi-carbon oxygenate selectivity. J Am Chem Soc 139(44):15848–15857
Xiao H, Cheng T, Goddard WA (2017) Atomistic mechanisms underlying selectivities in C1 and C2 products from electrochemical reduction of CO on Cu(111). J Am Chem Soc 139(1):130–136
Lum Y, Cheng T, Goddard WA et al (2018) Electrochemical CO reduction builds solvent water into oxygenate products. J Am Chem Soc 140(30):9337–9340
Li J, Xu AN, Li FW et al (2020) Enhanced multi-carbon alcohol electroproduction from CO via modulated hydrogen adsorption. Nat Commun 11:3685
He JF, Johnson NJJ, Huang AX et al (2018) Electrocatalytic alloys for CO2 reduction. Chemsuschem 11(1):48–57
Hitt JL, Li YC, Tao SS et al (2021) A high throughput optical method for studying compositional effects in electrocatalysts for CO2 reduction. Nat Commun 12:1114
Weitzner SE, Akhade SA, Kashi AR et al (2021) Evaluating the stability and activity of dilute Cu-based alloys for electrochemical CO2 reduction. J Chem Phys 155(11):114702
Xu YZ, Li CL, Xiao YQ et al (2022) Tuning the selectivity of liquid products of CO2RR by Cu-Ag alloying. ACS Appl Mater Interfaces 14(9):11567–11574
Hu F, Yang L, Jiang YW et al (2021) Ultrastable Cu catalyst for CO2 electroreduction to multicarbon liquid fuels by tuning C−C coupling with CuTi subsurface. Angew Chem Int Ed 60(50):26122–26127
Li YL, Tian ZQ, Chen L (2021) Theoretical understanding of the interface effect in promoting electrochemical CO2 reduction on Cu-Pd alloys. J Phys Chem C 125(39):21381–21389
Zhong M, Tran K, Min YM et al (2020) Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581(7807):178–183
Kim C, Cho KM, Park K et al (2021) Cu/Cu2O interconnected porous aerogel catalyst for highly productive electrosynthesis of ethanol from CO2. Adv Funct Mater 31(32):2102142
Lum Y, Ager JW (2018) Stability of residual oxides in oxide-derived copper catalysts for electrochemical CO2 reduction investigated with 18O labeling. Angew Chem Int Ed 57(2):551–554
Lei Q, Zhu H, Song KP et al (2020) Investigating the origin of enhanced C2+ selectivity in oxide-/hydroxide-derived copper electrodes during CO2 electroreduction. J Am Chem Soc 142(9):4213–4222
Mandal L, Yang KR, Motapothula MR et al (2018) Investigating the role of copper oxide in electrochemical CO2 reduction in real time. ACS Appl Mater Interfaces 10(10):8574–8584
Cavalca F, Ferragut R, Aghion S et al (2017) Nature and distribution of stable subsurface oxygen in copper electrodes during electrochemical CO2 reduction. J Phys Chem C 121(45):25003–25009
Mistry H, Varela AS, Bonifacio CS et al (2016) Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat Commun 7:12123
Yang PP, Zhang XL, Gao FY et al (2020) Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J Am Chem Soc 142(13):6400–6408
Chou TC, Chang CC, Yu HL et al (2020) Controlling the oxidation state of the Cu electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J Am Chem Soc 142(6):2857–2867
Xiao H, Goddard WA, Cheng T et al (2017) Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc Natl Acad Sci USA 114(26):6685–6688
Liu M, Pang YJ, Zhang B et al (2016) Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 537(7620):382–386
Jiang HJ, Hou ZH, Luo Y (2017) Unraveling the mechanism for the sharp-tip enhanced electrocatalytic carbon dioxide reduction: the kinetics decide. Angew Chem Int Ed 56(49):15617–15621
Huo H, Wang J, Fan QK et al (2021) Cu-MOFs derived porous Cu nanoribbons with strengthened electric field for selective CO2 electroreduction to C2+ fuels. Adv Energy Mater 11(42):2102447
Li H, Zhou HM, Zhou YJ et al (2022) Electric-field promoted C−C coupling over Cu nanoneedles for CO2 electroreduction to C2 products. Chin J Catal 43(2):519–525
Zhou YJ, Liang YQ, Fu JW et al (2022) Vertical Cu nanoneedle arrays enhance the local electric field promoting C2 hydrocarbons in the CO2 electroreduction. Nano Lett 22(5):1963–1970
Yang BP, Liu K, Li H et al (2022) Accelerating CO2 electroreduction to multicarbon products via synergistic electric-thermal field on copper nanoneedles. J Am Chem Soc 144(7):3039–3049
An PD, Wei L, Li H et al (2020) Enhancing CO2 reduction by suppressing hydrogen evolution with polytetrafluoroethylene protected copper nanoneedles. J Mater Chem A 8(31):15936–15941
Suominen M, Kallio T (2021) What we currently know about carbon-supported metal and metal oxide nanomaterials in electrochemical CO2 reduction. ChemElectroChem 8(13):2397–2406
Zhang JG, Guo YT, Shang B et al (2021) Unveiling the synergistic effect between graphitic carbon nitride and Cu2O toward CO2 electroreduction to C2H4. Chemsuschem 14(3):929–937
Wang HX, Tzeng YK, Ji YF et al (2020) Synergistic enhancement of electrocatalytic CO2 reduction to C2 oxygenates at nitrogen-doped nanodiamonds/Cu interface. Nat Nanotechnol 15(2):131–137
Song Y, Peng R, Hensley DK et al (2016) High-selectivity electrochemical conversion of CO2 to ethanol using a copper nanoparticle/N-doped graphene electrode. ChemistrySelect 1(19):6055–6061
Grosse P, Gao DF, Scholten F et al (2018) Dynamic changes in the structure, chemical state and catalytic selectivity of Cu nanocubes during CO2 electroreduction: size and support effects. Angew Chem Int Ed 57(21):6192–6197
Möller T, Scholten F, Thanh TN et al (2020) Electrocatalytic CO2 reduction on CuOx nanocubes: tracking the evolution of chemical state, geometric structure, and catalytic selectivity using operando spectroscopy. Angew Chem Int Ed 59(41):17974–17983
Jaugstetter M, Blanc N, Kratz M et al (2022) Electrochemistry under confinement. Chem Soc Rev 51(7):2491–2543
Pang YJ, Burdyny T, Dinh CT et al (2017) Joint tuning of nanostructured Cu-oxide morphology and local electrolyte programs high-rate CO2 reduction to C2H4. Green Chem 19(17):4023–4030
Yang KD, Ko WR, Lee JH et al (2017) Morphology-directed selective production of ethylene or ethane from CO2 on a Cu mesopore electrode. Angew Chem Int Ed Engl 56(3):796–800
Zhuang TT, Pang YJ, Liang ZQ et al (2018) Copper nanocavities confine intermediates for efficient electrosynthesis of C3 alcohol fuels from carbon monoxide. Nat Catal 1(12):946–951
Liu B, Cai C, Yang BP et al (2021) Intermediate enrichment effect of porous Cu catalyst for CO2 electroreduction to C2 fuels. Electrochim Acta 388:138552
Liu CX, Zhang ML, Li JW et al (2022) Nanoconfinement engineering over hollow multi-shell structured copper towards efficient electrocatalytical C−C coupling. Angew Chem Int Ed Engl 61(3):e202113498
Tan DX, Zhang JL, Yao L et al (2020) Multi-shelled CuO microboxes for carbon dioxide reduction to ethylene. Nano Res 13(3):768–774
Junqueira JRC, O’Mara PB, Wilde P et al (2021) Combining nanoconfinement in Ag core/porous Cu shell nanoparticles with gas diffusion electrodes for improved electrocatalytic carbon dioxide reduction. ChemElectroChem 8(24):4848–4853
Zhong YZ, Kong XD, Song ZM et al (2022) Adjusting local CO confinement in porous-shell Ag@Cu catalysts for enhancing C−C coupling toward CO2 eletroreduction. Nano Lett 22(6):2554–2560
Bui JC, Kim C, King AJ et al (2022) Engineering catalyst-electrolyte microenvironments to optimize the activity and selectivity for the electrochemical reduction of CO2 on Cu and Ag. Acc Chem Res 55(4):484–494
Gupta N, Gattrell M, MacDougall B (2006) Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J Appl Electrochem 36(2):161–172
Henckel DA, Counihan MJ, Holmes HE et al (2021) Potential dependence of the local pH in a CO2 reduction electrolyzer. ACS Catal 11(1):255–263
Moradzaman M, Mul G (2021) Optimizing CO coverage on rough copper electrodes: effect of the partial pressure of CO and electrolyte anions (pH) on selectivity toward ethylene. J Phys Chem C 125(12):6546–6554
Lu X, Zhu CQ, Wu ZS et al (2020) In situ observation of the pH gradient near the gas diffusion electrode of CO2 reduction in alkaline electrolyte. J Am Chem Soc 142(36):15438–15444
Hori Y, Murata A, Takahashi R (1989) Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J Chem Soc Faraday Trans 85(8):2309–2326
Resasco J, Lum Y, Clark E et al (2018) Effects of anion identity and concentration on electrochemical reduction of CO2. ChemElectroChem 5(7):1064–1072
Resasco J, Chen LD, Clark E et al (2017) Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J Am Chem Soc 139(32):11277–11287
Liu HJ, Qi ZM, Song L (2021) In situ electrocatalytic infrared spectroscopy for dynamic reactions. J Phys Chem C 125(44):24289–24300
Hori Y, Koga O, Yamazaki H et al (1995) Infrared spectroscopy of adsorbed CO and intermediate species in electrochemical reduction of CO2 to hydrocarbons on a Cu electrode. Electrochim Acta 40(16):2617–2622
Moradzaman M, Mul G (2020) Infrared analysis of interfacial phenomena during electrochemical reduction of CO2 over polycrystalline copper electrodes. ACS Catal 10(15):8049–8057. https://doi.org/10.1021/acscatal.0c02130
Pérez-Gallent E, Figueiredo MC, Calle-Vallejo F et al (2017) Spectroscopic observation of a hydrogenated CO dimer intermediate during CO reduction on Cu(100) electrodes. Angew Chem Int Ed 56(13):3621–3624
Kurouski D, Dazzi A, Zenobi R et al (2020) Infrared and Raman chemical imaging and spectroscopy at the nanoscale. Chem Soc Rev 49(11):3315–3347
Jiang S, Klingan K, Pasquini C et al (2018) New aspects of operando Raman spectroscopy applied to electrochemical CO2 reduction on Cu foams. J Chem Phys 150(4):041718
Gao J, Zhang H, Guo XY et al (2019) Selective C−C coupling in carbon dioxide electroreduction via efficient spillover of intermediates as supported by operando Raman spectroscopy. J Am Chem Soc 141(47):18704–18714
Zhan C, Dattila F, Rettenmaier C et al (2021) Revealing the CO coverage-driven C-C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via operando Raman spectroscopy. ACS Catal 11(13):7694–7701
Lin YH, Deng CY, Wu L et al (2020) Quantitative isotope measurements in heterogeneous photocatalysis and electrocatalysis. Energy Environ Sci 13(9):2602–2617
Lum Y, Ager JW (2019) Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction. Nat Catal 2(1):86–93
Khanipour P, Löffler M, Reichert AM et al (2019) Electrochemical real-time mass spectrometry (EC-RTMS): monitoring electrochemical reaction products in real time. Angew Chem Int Ed Engl 58(22):7273–7277
Ye K, Zhang GR, Ma XY et al (2022) Resolving local reaction environment toward an optimized CO2-to-CO conversion performance. Energy Environ Sci 15(2):749–759
Pan Y, Li HD, Xiong J et al (2022) Protecting the state of Cu clusters and nanoconfinement engineering over hollow mesoporous carbon spheres for electrocatalytical C-C coupling. Appl Catal B Environ 306:121111
Zhang N, Yang BP, Liu K et al (2021) Machine learning in screening high performance electrocatalysts for CO2 reduction. Small Methods 5(11):e2100987
Zhu CY, Zhang ZB, Zhong LX et al (2021) Product-specific active site motifs of Cu for electrochemical CO2 reduction. Chem 7(2):406–420
Guo SX, Bentley CL, Kang M et al (2022) Advanced spatiotemporal voltammetric techniques for kinetic analysis and active site determination in the electrochemical reduction of CO2. Acc Chem Res 55(3):241–251
Wakerley D, Lamaison S, Wicks J et al (2022) Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers. Nat Energy 7(2):130–143
Kibria MG, Edwards JP, Gabardo CM et al (2019) Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv Mater 31(31):1807166
Xia C, Zhu P, Jiang Q et al (2019) Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat Energy 4(9):776–785
Zhu P, Wang HT (2021) High-purity and high-concentration liquid fuels through CO2 electroreduction. Nat Catal 4(11):943–951
Kim JY, Zhu P, Chen FY et al (2022) Recovering carbon losses in CO2 electrolysis using a solid electrolyte reactor. Nat Catal 5(4):288–299
Huang JE, Li FW, Ozden A et al (2021) CO2 electrolysis to multicarbon products in strong acid. Science 372(6546):1074–1078
Xu Y, Edwards JP, Liu SJ et al (2021) Self-cleaning CO2 reduction systems: unsteady electrochemical forcing enables stability. ACS Energy Lett 6(2):809–815
Cheng YY, Hou J, Kang P (2021) Integrated capture and electroreduction of flue gas CO2 to formate using amine functionalized SnOx nanoparticles. ACS Energy Lett 6(9):3352–3358
Xu Y, Edwards JP, Zhong JJ et al (2020) Oxygen-tolerant electroproduction of C2 products from simulated flue gas. Energy Environ Sci 13(2):554–561
Li JN, Kornienko N (2022) Electrochemically driven C-N bond formation from CO2 and ammonia at the triple-phase boundary. Chem Sci 13(14):3957–3964
Tao ZX, Rooney CL, Liang YY et al (2021) Accessing organonitrogen compounds via C-N coupling in electrocatalytic CO2 reduction. J Am Chem Soc 143(47):19630–19642
Rooney CL, Wu YS, Tao ZX et al (2021) Electrochemical reductive N-methylation with CO2 enabled by a molecular catalyst. J Am Chem Soc 143(47):19983–19991
Yuan ML, Chen JW, Xu Y et al (2021) Highly selective electroreduction of N2 and CO2 to urea over artificial frustrated Lewis pairs. Energy Environ Sci 14(12):6605–6615
Acknowledgements
We gratefully appreciate the support of the National Natural Science Foundation of China (Nos. 51972223, 51932005 and 22109116), the Natural Science Foundation of Tianjin (No. 20JCYBJC01550), the Fundamental Research Funds for the Central Universities, and the Haihe Laboratory of Sustainable Chemical Transformations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, G., Han, D., Han, Z. et al. Rational Manipulation of Intermediates on Copper for CO2 Electroreduction Toward Multicarbon Products. Trans. Tianjin Univ. 28, 265–291 (2022). https://doi.org/10.1007/s12209-022-00330-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-022-00330-1