
Abstract
Introduction
Detecting upper gastrointestinal (GI) cancers in primary care is challenging, as cancer symptoms are common, often non-specific, and most patients presenting with these symptoms will not have cancer. Substantial investment has been made to develop biomarkers for cancer detection, but few have reached routine clinical practice. We aimed to identify novel biomarkers for upper GI cancers which have been sufficiently validated to be ready for evaluation in low-prevalence populations.
Methods
We systematically searched MEDLINE, Embase, Emcare, and Web of Science for studies published in English from January 2000 to October 2019 (PROSPERO registration CRD42020165005). Reference lists of included studies were assessed. Studies had to report on second measures of diagnostic performance (beyond discovery phase) for biomarkers (single or in panels) used to detect pancreatic, oesophageal, gastric, and biliary tract cancers. We included all designs and excluded studies with less than 50 cases/controls. Data were extracted on types of biomarkers, populations and outcomes. Heterogeneity prevented pooling of outcomes.
Results
We identified 149 eligible studies, involving 22,264 cancer cases and 49,474 controls. A total of 431 biomarkers were identified (183 microRNAs and other RNAs, 79 autoantibodies and other immunological markers, 119 other proteins, 36 metabolic markers, 6 circulating tumour DNA and 8 other). Over half (n = 231) were reported in pancreatic cancer studies. Only 35 biomarkers had been investigated in at least two studies, with reported outcomes for that individual marker for the same tumour type. Apolipoproteins (apoAII-AT and apoAII-ATQ), and pepsinogens (PGI and PGII) were the most promising biomarkers for pancreatic and gastric cancer, respectively.
Conclusion
Most novel biomarkers for the early detection of upper GI cancers are still at an early stage of matureness. Further evidence is needed on biomarker performance in low-prevalence populations, in addition to implementation and health economic studies, before extensive adoption into clinical practice can be recommended.
Similar content being viewed by others

We aimed to identify novel biomarkers which had been validated and showed sufficient promise to warrant further evaluation in low-prevalence populations. |
We identified 431 unique biomarkers; only 35 of which had been investigated in at least two studies, with outcomes for that individual marker for the same tumour type - four of these were identified as the most promising for future studies. |
This review highlights the need for more biomarker studies that consider primary care/community settings as their intended populations. |
Findings also indicate we still need better reporting to facilitate knowledge translation; we also need more consistency in the use of biomarkers. |
Research collaborations are vital to reduce duplicate efforts and ensure appropriate samples sizes when studying low-prevalence populations. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13214843.
Introduction
Gastrointestinal (GI) cancers represented more than 25% (4.8 million) of cancer cases and over a third (3.4 million) of cancer-related deaths worldwide in 2018 [1]. Upper GI cancers contribute an important proportion of these, with over 2.1 million new cases of cancers of the stomach, oesophagus, pancreas and biliary tract diagnosed worldwide in 2018 [1, 2]. Prognosis is often poor as upper GI cancers are generally not detected until the disease is advanced and less amenable to curative treatment [1].
Primary care plays a key role in the early detection of upper GI cancers, as more than 90% of patients present with symptoms [3,4,5], and screening tests for asymptomatic populations are not yet widely established. Early detection of upper GI cancers is challenging, as initial symptoms such as indigestion, abdominal discomfort or fatigue are common, often intermittent, and most patients presenting with them will not have cancer [6, 7].
There is growing demand to improve early cancer detection through better diagnostic and triage approaches, particularly for use in primary care or other community settings where cancer prevalence is low [5]. New diagnostic approaches, applied either among asymptomatic at-risk populations or to triage patients presenting with cancer symptoms, could be transformational. Electronic health records and large population-based surveys have been used to develop cancer risk prediction models to identify those requiring investigation for cancer [8]; diagnostic pathways have also been implemented in different countries in an effort to improve timely cancer diagnosis [5]. Innovative strategies applying artificial intelligence techniques to imaging and other medical data are also promising [5, 9]. For cancers with non-specific symptom signatures, like most upper GI cancers, we also need better biomarkers to support diagnostic assessment [10]. Biomarkers such as carcinoembryonic antigen (CEA) and CA19-9 are used in clinical practice predominantly for surveillance following treatment of upper GI cancers [9, 11]. Substantial investment has been made into developing new biomarkers for early cancer detection; most such biomarker research has been conducted in laboratory and specialist clinical settings [12, 13], where cancer prevalence is higher compared to community settings [14, 15].
The distinction between care settings is important, as the diagnostic performance characteristics of a test are strongly determined by the prevalence and severity of the target disease and of other diseases within the study population [14]. In populations in which the prevalence of the target disease is low (e.g. primary care), positive predictive values are lower than in high-prevalence populations seen in specialist cancer centres. Tests evaluated in high-prevalence populations tend to have lower sensitivity and higher specificity when used in low-prevalence populations [15, 16]. This is known as the spectrum effect or spectrum bias [14, 15] and has crucial implications for translating results from one care setting to another. To gain an accurate understanding of how a test will perform within a low incidence setting, it must ultimately be evaluated within that setting.
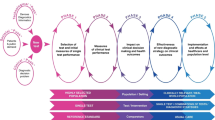
In recognition of this, the CanTest Framework has been developed, proposing a 5-phase translational pathway for diagnostic tests, from new test development to health system implementation in low-prevalence populations [15]. The framework highlights the importance of evaluating not only clinical performance but also the feasibility and acceptability of implementation, patient safety and quality of care, and cost-effectiveness in the chosen clinical setting. Understanding and addressing these issues is vital, as test performance alone, even if evaluated in the target populations, does not guarantee clinical utility nor improved patient outcomes [12].
This review set out to systematically identify novel biomarkers for the early detection of upper GI cancers which have been validated and show sufficient promise to warrant further evaluation in low-prevalence populations.
Methods
Search Strategy and Inclusion/Exclusion Criteria
This systematic review followed the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [17], and the protocol was registered in PROSPERO (CRD42020165005). We searched MEDLINE, Embase, Emcare and Web of Science from 1 January 2000 to 31 October 2019 for primary studies published in English. The search strategy (Online supplementary file 1) was developed with the assistance of a medical librarian and refined until it identified all relevant core publications known by the senior authors. Reference lists of included studies were also screened. Articles that were not available online were ordered via the British Library.
Studies were included if they reported on at least one measure of diagnostic performance: sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), false positive, false negative or area under the curve (AUC) for biomarkers used to detect oesophageal, gastric, pancreatic or biliary tract cancers. We included adult populations (mean/median age ≥ 18); we accepted individuals aged < 18 if these were outliers in large samples. The search strategy also included terms for lower GI (colorectal and anal) cancers for the purposes of a parallel review of novel biomarkers for the early detection of lower GI cancers, to be reported separately. Non-specified GI cancers, neuroendocrine cancers and studies only reporting on familial populations at risk of hereditary cancers were excluded.
Novel biomarkers were considered both individually and as part of a combination/panel test. Studies reporting only the performance of a single, established biomarker (i.e. CEA and CA19-9 for pancreatic cancer) were not eligible for inclusion [9]. We included studies reporting on performance for established biomarkers if these were in combination with additional novel biomarkers.
We aimed to identify studies situated within Phase 2 (measures of diagnostic accuracy in high-prevalence settings) and Phase 3 of the CanTest framework (measures of diagnostic accuracy or clinical utility, acceptability and feasibility in intended low-prevalence settings) (Fig. 1) [15]. We included studies if they reported more than preliminary measures of performance calculated in a discovery phase; this required additional measures of diagnostic performance in an independent cohort. If no references to previous studies evaluating performance were available and the study provided only one set of measures, the study was excluded. Panels with previously investigated biomarkers were included even if the biomarkers had not been investigated as part of a panel. As larger sample sizes are required beyond the biomarker discovery phase [13, 18], studies had to include at least 50 cancer cases and at least one group of 50 non-cancer controls with similar clinical characteristics (healthy, or with non-malignant or pre-malignant conditions). Similar criterion has been adopted by previous reviews that informed our study [13, 19].

Reproduced with permission from [15]
The CanTest Framework
We only included biomarkers which are feasible to use in a community setting, i.e. blood (serum and plasma), urine, faecal, salivary or breath samples. Observational studies (cross-sectional or longitudinal, prospective or retrospective) and trials were eligible for inclusion. We included all recruitment settings, as we expected that very few studies would have been carried out in community settings.
We used the online tool Covidence [20] to facilitate title and abstract screening and study selection. Two reviewers (any two of NC, PED, CS, KMM, DB or RB) independently screened titles and abstracts. Then, two reviewers (any two of the above) independently evaluated full-text articles for inclusion. Titles and abstracts of reference lists of included studies were reviewed by one author (NC); full-text articles selected at this stage were independently assessed by two reviewers (any two of NC, PED, RB or DB). Disagreements were resolved by consensus; when this could not be reached a senior, third reviewer (FMW or JE) was consulted.
Data Extraction and Analysis
Data extraction was piloted to ensure consistency and was carried out by one of seven reviewers (NC, PED, RB, DB, JMG, JO and SS). We extracted information on: study characteristics (publication year, country of population of interest, recruitment setting, study aims and design); populations (numbers included, age, sex, tumour staging for cases and health status for controls); biomarkers (type of sample, biomarker name, biomarker category); and summary measures of diagnostic performance (sensitivity, specificity, PPV, NPV, false positives, false negatives and AUC, with 95% confidence intervals when available, for all comparisons). When studies reported on different phases of biomarker development, we only extracted data from the eligible phases (i.e. biomarkers and measures beyond the discovery phase). When studies had more than one eligible phase, we extracted data from all phases. Extracted data were collated and checked for consistency and inaccuracies (NC).
Biomarkers were categorised according to a modified version of Uttley et al.’s classification [19], which included: microRNAs and other RNAs, autoantibodies and other immunological markers, other proteins (that did not fit into other categories), metabolic markers, circulating tumour DNA, and other biomarkers. Controls were classified as: normal/healthy, having non-malignant, or pre-malignant conditions. Biomarkers and control populations were coded by one author (NC) and checked by other authors (PED, KMM and MM; and PED, FMW and JE, respectively). Controls described as being healthy were coded as such unless studies described underlying conditions. Patients with cancer were ineligible as controls. Full details of the classification of controls are available (online supplementary table S1). Microsoft Excel 2015 and SPSS v.23 (IBM) were used for data extraction and data analysis.
Quality Assessment and Risk of Bias
Risk of bias [21] was not assessed as described in the original protocol, following independent piloting. Appraisal was hindered by the use of diverse methods across studies and incomplete reporting, resulting in a large number of “unclear” assessments. Instead, a list of issues identified in the studies was prepared (Online supplementary file 2). As spectrum bias is a key issue when translating results from high- to low-prevalence populations, all included studies were classified as either single-gate or two-gate designs. In single-gate designs, cases and controls are recruited through a single route of entry and with the same inclusion criteria (e.g. all cases and controls presented with symptoms). In two-gate designs, participants are recruited through different routes and different inclusion criteria exist for cases and controls. In this situation, controls can be either normal/healthy or with an alternative diagnosis, which can produce symptoms and signs similar to patients with cancer [16]. One author (NC) classified all studies and another (PED) checked the classification. A full description of this classification and how it approaches some of the issues covered by the critical appraisal tool is available (Online supplementary file 3).
Data Synthesis
Included studies were heterogeneous and rarely evaluated the same biomarkers in the same way, often using different cut-off points, populations and/or biomarker combinations in panels. Therefore, we were unable to undertake meta-analysis. Instead, we used narrative synthesis to summarise data across studies [22]. First, we developed an overview of the available evidence, describing key characteristics of included studies, their populations and biomarkers, and outcome measures. Then, we looked for similarities that would allow for subgroup analyses, namely the same biomarker, for the same tumour type, with similar designs, outcome measures and populations.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Results
Database searches identified 16,597 records; 9172 were retained after removing duplicates. During title and abstract screening, 8179 ineligible records were excluded. The full texts of the remaining 993 records were assessed for eligibility; 731 were excluded (Fig. 2). A total of 262 studies from database searches met inclusion criteria; 25 additional studies were identified in reference lists. Of these, 149 included studies referred to upper GI cancers and were included in our narrative synthesis.

Study selection
Characteristics of Included Studies
Key characteristics of included studies are described in Table 1 and 2. Most studies recruited participants from a single country (n = 142). China was the most common country (n = 77), followed by Japan and South Korea (n = 15 each), the USA (n = 12) and Germany (n = 9). The most common recruitment settings were hospital or other secondary care institutions (n = 125), biobanks, reference sets, databases or archived samples (n = 20), general population cohorts or cohorts from population screening programmes (n = 11) and cohorts from previous trials or observational studies (n = 9). Several studies recruited from more than one setting. Gastric cancer was the most commonly investigated tumour type (n = 69), followed by pancreatic (n = 54), oesophageal (n = 24) and biliary tract cancers (n = 3). Four studies investigated more than one type of upper GI cancer (Table 1).
Characteristics of Cases and Controls
Overall, the included studies reported on 22,264 cancer cases (10,589 gastric, 7964 pancreatic, 3258 oesophageal and 290 biliary tract cancers, and 163 oesophago-gastric cancers, not distinguishing between oesophageal and gastric cancer). The minimum age for cases was 16 while the oldest patient was aged 93. Most cases were male (68%) across all tumour types. Over 50% of cancers had been diagnosed at stages III and IV (median 55.5%, interquartile range 47.0–68.1%; data available for 106 included studies). The included studies reported on 49,474 controls (38,955 normal/healthy, 9042 with non-malignant conditions, 1106 with pre-malignant conditions, and 371 with either normal or non-malignant conditions). Pancreatitis and gastritis were the most commonly reported non-malignant conditions (online supplementary Figure S1). Over half of the studies (n = 83) investigated more than one type of control population. Normal healthy controls were the majority across all tumour types, except for biliary tract cancers. The minimum age for controls was 16 while the maximum age was 94. Overall, most controls were male (74%); this was the case for all tumour types except for biliary tract cancers.
Types of Biomarkers
Biomarkers were most commonly sampled from blood (145 studies; 107 investigated serum, 33 plasma and 5 both); two studies analysed urine [28, 36], one breath [169] and another saliva [47]. Most studies (n = 128) investigated more than one biomarker. A total of 431 biomarkers were identified (online supplementary table S2). These were most often microRNA and other RNAs (n = 183), other proteins (n = 119) and autoantibodies and other immunological markers (n = 79). Less than a third of studies (n = 44) included biomarkers from different categories. This was often due to use of established biomarkers (proteins CA19-9 and CEA) in combination with novel biomarkers. Studies of pancreatic cancer reported on over half of identified biomarkers (n = 231) (Fig. 3). Only about a fifth (n = 90) of all identified biomarkers were reported in more than one study; 72 of these were reported in more than one study for the same tumour type (Table 3).

Types of biomarkers, overall and by tumour type. aFive proteins; bthese refer to volatile organic compounds and platelets; autoab autoantibodies, ctDNA circulating tumour DNA, miRNA microRNA
Measures of Diagnostic Performance
The most commonly reported measures of diagnostic performance were sensitivity (n = 136), specificity (n = 129) and AUC (n = 123). PPV and NPV were each reported by 40 studies, while false positives and false negatives were least often reported (11 studies each). Outcome data on individual biomarkers were available in most studies (n = 121); the remaining 28 studies only reported on performance for a combination/panel. Over half of the included studies (n = 83) reported on measures of performance for biomarkers both individually and in combinations. Outcome data were not available for all control populations; only 95 studies provided outcome data for cancers versus normal controls, 54 provided outcome data for cancers versus non-malignant controls, and 10 provided measures for cancers versus pre-malignant conditions (online supplementary table S3).
Individual measures of diagnostic performance were available for 35 biomarkers mentioned more than once, for the same tumour type (online supplementary table S4). We were not able to synthesise outcomes further due to heterogeneity in biomarker combinations, in control populations and subgroup analyses, and variations in reported cut-off points and diagnostic accuracy data (see online supplementary table S5 for a textual description of outcomes).
Only four novel biomarkers were reported on studies adopting a single-gate design (Table 4). Apolipoproteins AII-AT and AII-ATQ had poor sensitivity (range 4–25%) but good AUCs (range 52–94.6%) reported for pancreatic cancer in three studies (same first author for all) [104,105,106]. Their diagnostic accuracy increased when combined with CA19-9 (sensitivity range 7–95.4%, specificity range 96–98%, AUC range 56–78%). Pepsinogen I (PGI) and PGI/PGII ratio had a wide range of sensitivity and specificity (ranges 27–77.9% and 20.2–92%, respectively) and good AUC (range 70–76%) reported for gastric cancer across four studies [29, 40, 41, 76]. When evaluated with other novel biomarkers (including miR-1290, MIC-1, ULBP2 and CA125), one established biomarker, CA19-9, also showed some promise (sensitivity range 23.1–88%, specificity range 71.6–96.6%, AUC 92–98%) for pancreatic cancer [121, 132, 138]. There were also two studies reporting panels rather than individual biomarkers using a single-gate, reversed-flow design (Table 4) [89, 119].
Discussion
Our systematic review identified 149 studies reporting on 431 different biomarkers for gastric, pancreatic, oesophageal and biliary tract cancers. Only a fifth of biomarkers were reported by more than one study, and from these only four novel biomarkers, apoAII-AT and apoAII-ATQ (pancreatic cancer) and pepsinogen I and II (gastric cancer), plus one established biomarker (CA19-9 combined with other novel biomarkers), were reported with individual measures of diagnostic performance, adopting a recommended single-gate design. Heterogeneity in methods, populations, biomarkers, outcomes and comparisons precluded meta-analysis. Applying novel biomarkers for the early detection of upper GI cancers is therefore at an early stage of matureness: few have been extensively evaluated and evaluations have almost exclusively focussed on high-prevalence populations. Further evaluation of the most promising biomarkers in low-prevalence populations is needed before extensive adoption into routine clinical practice can be recommended.
While other reviews have investigated biomarkers used for early cancer detection [19, 172], few have considered the evidence in the context of future application of tests in low-prevalence populations, the likely target for clinical application [12, 13]. To our knowledge, this is the first review to do so for upper GI cancers. The four novel and one established biomarkers we highlight in this review were evaluated in a mix of high- and low-prevalence populations, including hospital patients, general population cohorts, screening populations (both high and average cancer risk), and patients presenting with symptoms. We did not identify any studies reporting outcomes relevant to feasibility, acceptability, benefits and harms, nor health economics as initially planned in the review protocol (i.e. phase 3 studies and beyond in the CanTest framework). The best performing biomarkers for pancreatic cancer, with an AUC between 56% and 94%, were ApoAII-ATQ/AT alone, CA19-9 plus miR-1290, MIC-1 and ULPB2, and Mellby et al.’s [119] 29-panel signature. These may be ready for trials and other phase 3 studies, single or in combination, in low-prevalence populations. We did not identify any novel biomarkers with similar AUCs for gastric, biliary tract or oesophageal cancers.
A previous review investigating the role of pepsinogens in early detection of gastric cancers reported that they had only moderate capacity to detect gastric cancer [173]. Another review on early pancreatic cancer detection highlighted that no single biomarker has yet translated to clinical use and suggested the use of ‘robust panels of biomarkers’ [9]. This review confirms that more research is required before we have sufficient evidence about biomarkers for upper GI cancers to warrant their adoption into clinical practice.
We identified several important methodological limitations within the biomarker studies to date. These include large numbers of biomarkers analysed in parallel during discovery studies, increasing risk of falsely positive results; limited sample sizes; evaluation of “extreme” cases; limited external, independent validation; and selective reporting for validation (several alternatives analyses and combinations, use of several cut-off points and over-optimistic interpretation of the data) [12]. Together with use of two-gate rather than recommended single-gate designs, these could all lead to over-inflated measures of performance. Population characteristics were often provided as supplementary data, with little discussion of potential selection bias and other sources of uncertainty. We also excluded relevant studies when we could not obtain sufficient information on an individual tumour type; this was the case for the CancerSeek tool [174]. Adoption of reporting guidelines [175] and development of early cancer detection collaborations [15, 18] could be useful strategies to address these issues.
This review offers a comprehensive overview of the available evidence. It benefitted from having a multidisciplinary team of experts, a broad search strategy, independent screening, and classifications checked by senior team members. Since meta-analysis was not feasible nor appropriate, we had to use text and tables to synthesise the evidence. We did not include studies investigating biomarkers as part of risk prediction models or risk assessment tools. These studies have strong potential to be used in the community and should be investigated in a separate systematic review. Recent reviews indicate that only including studies in English has minimal impact on review conclusions [176, 177]. We believe this is also the case for this review, particularly due to the overall lack of evidence on biomarkers ready to be evaluated in low-prevalence settings. Although we did not formally appraise risk of bias, we identified several quality and methodological issues, indicating that challenges already highlighted in the literature persisted over time [12]. Finally, due to the large amount of evidence on biomarker development and evaluation, we believe the field could benefit from a “living systematic review”; this refers to high quality, up-to-date online summaries of evidence which can be constantly updated as new research becomes available [178].
The studies we identified focused on measures of diagnostic performance, which is reasonable given the phase of development for most of them. The CanTest Framework [15] can help guide studies aiming to build much needed evidence on later phases of biomarker development, focussing on impact on clinical decision-making, patient, health system and economic outcomes.
Conclusion
There is a large body of evidence on biomarkers being developed for the detection of upper GI cancers, but relatively few have yet to demonstrate their validity or clinical utility in settings where cancer prevalence is low. Early detection of colorectal cancer already benefits from biomarkers that can be used across different populations. This is the case for the faecal immunochemical test (FIT), which is recommended for use in primary care in Spain, Australia and the United Kingdom, in addition to being effective at mass population screening programmes, using different cut-off points [179, 180]. It took several decades from FIT development to generate evidence for its cost-effectiveness as a screening test for colorectal cancer. Its role in the assessment of patients in primary care with lower GI symptoms is still being evaluated. Biomarkers for upper GI cancer remain in their infancy but there are a few which show promise and require further evaluations. Ultimately, they may be able to contribute to improving outcomes for upper GI cancers through earlier detection.
References
Arnold M, Abnet CC, Neale RE, et al. Global burden of 5 major types of gastrointestinal cancer. Gastroenterology. 2020;159(1):335–49.
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol. 2014;11(1):38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol. 2015;16(12):1231–72.
Rubin G, Walter F, Emery J, et al. Reimagining the diagnostic pathway for gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2018;15(3):181–8.
Hamilton W. The CAPER studies: five case-control studies aimed at identifying and quantifying the risk of cancer in symptomatic primary care patients. Br J Cancer. 2009;101(Suppl 2):S80–6.
Rasmussen S, Haastrup PF, Balasubramaniam K, et al. Predictive values of upper gastrointestinal cancer alarm symptoms in the general population: a nationwide cohort study. BMC Cancer. 2018;18(1):440.
Hippisley-Cox J, Coupland C. Development and validation of risk prediction algorithms to estimate future risk of common cancers in men and women: prospective cohort study. BMJ Open. 2015;5(3):e007825.
Pereira SP, Oldfield L, Ney A, et al. Early detection of pancreatic cancer. Lancet Gastroenterol Hepatol. 2020;5(7):698–710.
Lyratzopoulos G, Wardle J, Rubin G. Rethinking diagnostic delay in cancer: how difficult is the diagnosis? BMJ Br Med J. 2014;349:g7400.
Hall C, Clarke L, Pal A, et al. A review of the role of carcinoembryonic antigen in clinical practice. Ann Coloproctol. 2019;35(6):294–305.
Ioannidis JPA, Bossuyt PMM. Waste, leaks, and failures in the biomarker pipeline. Clin Chem. 2017;63(5):963–72.
Worm Ørntoft MB. Review of blood-based colorectal cancer screening: how far are circulating cell-free DNA methylation markers from clinical implementation? Clin Colorectal Cancer. 2018;17(2):e415–33.
Usher-Smith JA, Sharp SJ, Griffin SJ. The spectrum effect in tests for risk prediction, screening, and diagnosis. BMJ. 2016;353:i3139.
Walter FM, Thompson MJ, Wellwood I, et al. Evaluating diagnostic strategies for early detection of cancer: the CanTest framework. BMC Cancer. 2019;19(1):586.
Rutjes AW, Reitsma JB, Vandenbroucke JP, et al. Case-control and two-gate designs in diagnostic accuracy studies. Clin Chem. 2005;51(8):1335–41.
Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097.
Pepe MS, Etzioni R, Feng Z, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93(14):1054–61.
Uttley L, Whiteman BL, Woods HB, et al. Building the evidence base of blood-based biomarkers for early detection of cancer: a rapid systematic mapping review. EBioMedicine. 2016;10:164–73.
Covidence systematic review software VHI, Melbourne, Australia. Available at www.covidence.org (Accessed 31 July 2020).
Whiting PF, Rutjes AW, Westwood ME, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med. 2011;155(8):529–36.
Popay J, Roberts H, Sowden A, et al. Guidance on the conduct of narrative synthesis in systematic reviews. a product from the ESRC methods programme: ESRC Methods Programme; 2006.
Cai H, Yuan Y, Hao YF, et al. Plasma microRNAs serve as novel potential biomarkers for early detection of gastric cancer. Med Oncol. 2013;30(1):452.
Chen T, Sun L, He C, et al. Serum OPN expression for identification of gastric cancer and atrophic gastritis and its influencing factors. PLoS ONE. 2014;9(12):e114005.
Chen C, Chen Q, Zhao Q, et al. Value of combined detection of serum CEA, CA72-4, CA19-9, CA15-3 and CA12-5 in the diagnosis of gastric cancer. Ann Clin Lab Sci. 2017;47(3):260–3.
Chung HW, Kim JJ, Choi JI, et al. A disintegrin and metalloproteinase 8 as a potential blood biomarker for early diagnosis of gastric cancer. Yonsei Med J. 2019;60(8):713–9.
Ding J, Che C, Liang YY, et al. Serum long non-coding RNA signatures serve as novel noninvasive biomarkers for diagnosis and prognosis of gastric cancer. Int J Clin Exp Pathol. 2017;10(5):5227–39.
Dong X, Wang G, Zhang G, et al. The endothelial lipase protein is promising urinary biomarker for diagnosis of gastric cancer. Diagn Pathol. 2013;8:45.
Gantuya B, Oyuntsetseg K, Bolor D, et al. Evaluation of serum markers for gastric cancer and its precursor diseases among high incidence and mortality rate of gastric cancer area. Gastric Cancer. 2019;22(1):104–12.
Gwak HK, Lee JH, Park SG. Preliminary evaluation of clinical utility of CYFRA 21–1, CA 72–4, NSE, CA19-9 and CEA in stomach cancer. Asian Pac J Cancer Prev. 2014;15(12):4933–8.
He CZ, Zhang KH, Li Q, et al. Combined use of AFP, CEA, CA125 and CAl9-9 improves the sensitivity for the diagnosis of gastric cancer. BMC Gastroenterol. 2013;13:87.
Hoshino I, Nagata M, Takiguchi N, et al. Panel of autoantibodies against multiple tumor-associated antigens for detecting gastric cancer. Cancer Sci. 2017;108(3):308–15.
Huang Z, Zhang X, Lu H, et al. Serum trefoil factor 3 is a promising non-invasive biomarker for gastric cancer screening: a monocentric cohort study in China. BMC Gastroenterol. 2014;14(1):74.
Huang S, Wang J, Li J, et al. Serum microRNA expression profile as a diagnostic panel for gastric cancer. Jpn J Clin Oncol. 2016;46(9):811–8.
Huang Z, Zhu D, Wu L, et al. Six serum-based miRNAs as potential diagnostic biomarkers for gastric cancer. Cancer Epidemiol Biomarkers Prev. 2017;26(2):188–96.
Iwasaki H, Shimura T, Yamada T, et al. A novel urinary microRNA biomarker panel for detecting gastric cancer. J Gastroenterol. 2019;25:25.
Ji B, Huang Y, Gu T, et al. Potential diagnostic and prognostic value of plasma long noncoding RNA LINC00086 and miR-214 expression in gastric cancer. Cancer Biomark. 2019;24(2):249–55.
Juan Cai W, Yin L, Kang Q, et al. The serum pepsinogen test as a predictor of kazakh gastric cancer. Sci Rep. 2017;7:43536.
Kaise M, Miwa J, Tashiro J, et al. The combination of serum trefoil factor 3 and pepsinogen testing is a valid non-endoscopic biomarker for predicting the presence of gastric cancer: a new marker for gastric cancer risk. J Gastroenterol. 2011;46(6):736–45.
Kang JM, Kim N, Yoo JY, et al. The role of serum pepsinogen and gastrin test for the detection of gastric cancer in Korea. Helicobacter. 2008;13(2):146–56.
Kikuchi R, Abe Y, Iijima K, et al. Low serum levels of pepsinogen and gastrin 17 are predictive of extensive gastric atrophy with high-risk of early gastric cancer. Tohoku J Exp Med. 2011;223(1):35–44.
Kim M, Kim HJ, Choi BY, et al. Identification of potential serum biomarkers for gastric cancer by a novel computational method, multiple normal tissues corrected differential analysis. Clin Chim Acta. 2012;413(3):428–33.
Kurilovich S, Belkovets A, Reshetnikov O, et al. Stomach-specific biomarkers (GastroPanel) can predict the development of gastric cancer in a caucasian population: a longitudinal nested case–control study in Siberia. Anticancer Res. 2016;36(1):247–53.
Li BS, Zhao YL, Guo G, et al. Plasma microRNAs, miR-223, miR-21 and miR-218, as novel potential biomarkers for gastric cancer detection. PLoS ONE. 2012;7(7):e41629.
Li Q, Shao Y, Zhang X, et al. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumour Biol. 2015;36(3):2007–12.
Li FX, Guo YN, Liu JT, et al. The significance of elevated plasma expression of microRNA 106b similar to 25 clusters in gastric cancer. PLoS ONE. 2017;12(5):12.
Li F, Yoshizawa JM, Kim KM, et al. Discovery and validation of salivary extracellular RNA biomarkers for noninvasive detection of gastric cancer. Clin Chem. 2018;64(10):1513–21.
Li J, Xu L, Run ZC, et al. Multiple cytokine profiling in serum for early detection of gastric cancer. World J Gastroenterol. 2018;24(21):2269–78.
Lim JB, Kim DK, Chung HW. Clinical significance of serum thymus and activation-regulated chemokine in gastric cancer: potential as a serum biomarker. Cancer Sci. 2014;105(10):1327–33.
Lim JB, Chung HW. Serum ENA78/CXCL5, SDF-1/CXCL12, and their combinations as potential biomarkers for prediction of the presence and distant metastasis of primary gastric cancer. Cytokine. 2015;73(1):16–22.
Lin LY, Yang L, Zeng Q, et al. Tumor-originated exosomal lncUEGC1 as a circulating biomarker for early-stage gastric cancer. Mol Cancer. 2018;17(1):84.
Liu R, Zhang C, Hu Z, et al. A five-microRNA signature identified from genome-wide serum microRNA expression profiling serves as a fingerprint for gastric cancer diagnosis. Eur J Cancer. 2011;47(5):784–91.
Liu WL, Liu D, Cheng K, et al. Evaluating the diagnostic and prognostic value of circulating cathepsin S in gastric cancer. Oncotarget. 2016;7(19):28124–38.
Liu J, Wang J, Song Y, et al. A panel consisting of three novel circulating lncRNAs, is it a predictive tool for gastric cancer? J Cell Mol Med. 2018;22(7):3605–13.
Meistere I, Werner S, Zayakin P, et al. The prevalence of cancer-associated autoantibodies in patients with gastric cancer and progressive grades of premalignant lesions. Cancer Epidemiol Biomarkers Prev. 2017;26(10):1564.
Mroczko B, Groblewska M, Lukaszewicz-Zajac M, et al. Pre-treatment serum and plasma levels of matrix metalloproteinase 9 (MMP-9) and tissue inhibitor of matrix metalloproteinases 1 (TIMP-1) in gastric cancer patients. Clin Chem Lab Med. 2009;47(9):1133–9.
Ning S, Wei W, Li J, et al. Clinical significance and diagnostic capacity of serum TK1, CEA, CA 19–9 and CA 72–4 levels in gastric and colorectal cancer patients. J Cancer. 2018;9(3):494–501.
Oue N, Sentani K, Noguchi T, et al. Serum olfactomedin 4 (GWI 12, hGC4) in combination with Reg IV is a highly sensitive biomarker for gastric cancer patients. Int J Cancer. 2009;125(10):2383–92.
Pan YQ, Ruan YY, Peng JB, et al. Diagnostic significance of soluble human leukocyte antigen-G for gastric cancer. Hum Immunol. 2016;77(4):317–24.
Park KU, Lee HE, Nam SK, et al. The quantification of HER2 and MYC gene fragments in cell-free plasma as putative biomarkers for gastric cancer diagnosis. Clin Chem Lab Med. 2014;52(7):1033–40.
Parvaee P, Sarmadian H, Khansarinejad B, et al. Plasma level of microRNAs, MiR-107, MiR-194 and MiR-210 as potential biomarkers for diagnosis intestinal-type gastric cancer in human. Asian Pac J Cancer Prev. 2019;20(5):1421–6.
Qin J, Wang S, Shi J, et al. Using recursive partitioning approach to select tumor-associated antigens in immunodiagnosis of gastric adenocarcinoma. Cancer Sci. 2019;110(6):1829–41.
Qiu X, Zhang J, Shi W, et al. Circulating microRNA-26a in plasma and its potential diagnostic value in gastric cancer. PLoS ONE. 2016;11(3):e0151345.
Song MY, Pan KF, Su HJ, et al. Identification of serum microRNAs as novel non-invasive biomarkers for early detection of gastric cancer. PLoS ONE. 2012;7(3):e33608.
Su Y, Shen J, Qian H, et al. Diagnosis of gastric cancer using decision tree classification of mass spectral data. Cancer Sci. 2007;98(1):37–43.
Sun L, Tu H, Chen T, et al. Three-dimensional combined biomarkers assay could improve diagnostic accuracy for gastric cancer. Sci Rep. 2017;7(1):11621.
Tsalikidis C, Papachristou F, Pitiakoudis M, et al. Soluble E-cadherin as a diagnostic and prognostic marker in gastric carcinoma. Folia Med (Plovdiv). 2013;55(3–4):26–32.
Wang CS, Wu TL, Tsao KC, et al. Serum TIMP-1 in gastric cancer patients: a potential prognostic biomarker. Ann Clin Lab Sci. 2006;36(1):23–30.
Wang J, Zhang H, Zhou X, et al. Five serum-based miRNAs were identified as potential diagnostic biomarkers in gastric cardia adenocarcinoma. Cancer Biomark. 2018;23(2):193–203.
Wang S, Qin J, Ye H, et al. Using a panel of multiple tumor-associated antigens to enhance autoantibody detection for immunodiagnosis of gastric cancer. Oncoimmunology. 2018;7(8):e1452582.
Wang M, Wang J, Jiang H. Diagnostic value of apolipoprotein C-I, transthyretin and apolipoprotein C-III in gastric cancer. Oncol Lett. 2019;17(3):3227–32.
Werner S, Chen H, Butt J, et al. Evaluation of the diagnostic value of 64 simultaneously measured autoantibodies for early detection of gastric cancer. Sci Rep. 2016;6:25467.
Wu J, Li G, Yao Y, et al. MicroRNA-421 is a new potential diagnosis biomarker with higher sensitivity and specificity than carcinoembryonic antigen and cancer antigen 125 in gastric cancer. Biomarkers. 2015;20(1):58–63.
Wu X, Wang Q, Zhu Z, et al. Serum cell free DNA by branched DNA in patients with gastric cancer. Int J Clin Exp Med. 2016;9(6):11175–83.
Wu Y, Jiang M, Qin Y, et al. Single and combined use of neutrophil-lymphocyte ratio, platelet-lymphocyte ratio and carcinoembryonic antigen in diagnosing gastric cancer. Clin Chim Acta. 2018;481:20–4.
Yanaoka K, Oka M, Mukoubayashi C, et al. Cancer high-risk subjects identified by serum pepsinogen tests: outcomes after 10-year follow-up in asymptomatic middle-aged males. Cancer Epidemiol Biomarkers Prev. 2008;17(4):838–45.
Yang JJ, Yang JH, Kim J, et al. Soluble c-Met protein as a susceptible biomarker for gastric cancer risk: a nested case-control study within the Korean multicenter cancer cohort. Int J Cancer. 2013;132(9):2148–56.
Yang Z, Sun Y, Liu R, et al. Plasma long noncoding RNAs PANDAR, FOXD2-AS1, and SMARCC2 as potential novel diagnostic biomarkers for gastric cancer. Cancer Manag Res. 2019;11:6175–84.
Yoon JH, Park YG, Nam SW, et al. The diagnostic value of serum gastrokine 1 (GKN1) protein in gastric cancer. Cancer Med. 2019;8(12):5507–14.
Yun ZY, Li N, Zhang X, et al. Mean platelet volume, platelet distribution width and carcinoembryonic antigen to discriminate gastric cancer from gastric ulcer. Oncotarget. 2017;8(37):62600–5.
Zayakin P, Ancans G, Silina K, et al. Tumor-associated autoantibody signature for the early detection of gastric cancer. Int J Cancer. 2013;132(1):137–47.
Zhang Y, Qiu L, Wang Y, et al. Unsaturated free fatty acids: a potential biomarker panel for early detection of gastric cancer. Biomarkers. 2014a;19(8):667–73.
Zhang KC, Xi HQ, Cui JX, et al. Hemolysis-free plasma miR-214 as novel biomarker of gastric cancer and is correlated with distant metastasis. Am J Cancer Res. 2015;5(2):821–9.
Zhang K, Shi H, Xi H, et al. Genome-wide lncRNA microarray profiling identifies novel circulating lncRNAs for detection of gastric cancer. Theranostics. 2017;7(1):213–27.
Zhou X, Ji G, Chen H, et al. Clinical role of circulating miR-223 as a novel biomarker in early diagnosis of cancer patients. Int J Clin Exp Med. 2015;8(9):16890–8.
Zhou X, Zhu W, Li H, et al. Diagnostic value of a plasma microRNA signature in gastric cancer: a microRNA expression analysis. Sci Rep. 2015;5:11251.
Zhou X, Yin C, Dang Y, et al. Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci Rep. 2015;5:11516.
Akita H, Ritchie SA, Takemasa I, et al. Serum metabolite profiling for the detection of pancreatic cancer: results of a large independent validation study. Pancreas. 2016;45(10):1418–23.
Balasenthil S, Huang Y, Liu S, et al. A plasma biomarker panel to identify surgically resectable early-stage pancreatic cancer. J Natl Cancer Inst. 2017;109(8):01.
Brand RE, Nolen BM, Zeh HJ, et al. Serum biomarker panels for the detection of pancreatic cancer. Clin Cancer Res. 2011;17(4):805–16.
Cao Z, Liu C, Xu J, et al. Plasma microRNA panels to diagnose pancreatic cancer: results from a multicenter study. Oncotarget. 2016;7(27):41575–83.
Capello M, Bantis LE, Scelo G, et al. Sequential validation of blood-based protein biomarker candidates for early-stage pancreatic cancer. J Natl Cancer Inst. 2017;109(4):01.
Chung HW, Lim JB. Clinical significance of serum levels of immune-associated molecules, uric acid and soluble MHC class I chain-related molecules A and B, as diagnostic tumor markers for pancreatic ductal adenocarcinoma. Cancer Sci. 2011;102(9):1673–9.
Chung HW, Jang S, Lim JB. Clinical implications and diagnostic usefulness of correlation between soluble major histocompatibility complex class I chain-related molecule a and protumorigenic cytokines in pancreatic ductal adenocarcinoma. Cancer. 2013;119(1):233–44.
Deng T, Yuan Y, Zhang C, et al. Identification of circulating MiR-25 as a potential biomarker for pancreatic cancer diagnosis. Cell Physiol Biochem. 2016;39(5):1716–22.
Duraker N, Hot S, Polat Y, et al. CEA, CA 19–9, and CA 125 in the differential diagnosis of benign and malignant pancreatic diseases with or without jaundice. J Surg Oncol. 2007;95(2):142–7.
Firpo MA, Gay DZ, Granger SR, et al. Improved diagnosis of pancreatic adenocarcinoma using haptoglobin and serum amyloid A in a panel screen. World J Surg. 2009;33(4):716–22.
Fukutake N, Ueno M, Hiraoka N, et al. A novel multivariate index for pancreatic cancer detection based on the plasma free amino acid profile. PLoS ONE. 2015;10(7):e0132223.
Gao L, He SB, Li DC. Effects of miR-16 plus CA19-9 detections on pancreatic cancer diagnostic performance. Clin Lab. 2014;60(1):73–7.
David VG, David EM, Zhiliang Y, et al. New MUC1 serum immunoassay differentiates pancreatic cancer from pancreatitis. J Clin Oncol. 2006;24(2):252–8.
Gold DV, Gaedcke J, Ghadimi BM, et al. PAM4 enzyme immunoassay alone and in combination with CA 19–9 for the detection of pancreatic adenocarcinoma. Cancer. 2013;119(3):522–8.
Groblewska M, Mroczko B, Wereszczyńska-Siemiatkowska U, et al. Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients. Clin Chem Lab Med. 2007;45(1):30–4.
Guo X, Lv XH, Fang C, et al. Dysbindin as a novel biomarker for pancreatic ductal adenocarcinoma identified by proteomic profiling. Int J Cancer. 2016;139(8):1821–9.
Honda K, Okusaka T, Felix K, et al. Altered plasma apolipoprotein modifications in patients with pancreatic cancer: protein characterization and multi-institutional validation. PLoS ONE. 2012;7(10):e46908.
Honda K, Kobayashi M, Okusaka T, et al. Plasma biomarker for detection of early stage pancreatic cancer and risk factors for pancreatic malignancy using antibodies for apolipoprotein-AII isoforms. Sci Rep. 2015;5:15921.
Honda K, Katzke VA, Husing A, et al. CA19-9 and apolipoprotein-A2 isoforms as detection markers for pancreatic cancer: a prospective evaluation. Int J Cancer. 2019;144(8):1877–87.
Jiang JT, Wu CP, Deng HF, et al. Serum level of TSGF, CA242 and CA19-9 in pancreatic cancer. World J Gastroenterol. 2004;10(11):1675–7.
Kaur S, Smith LM, Patel A, et al. A combination of MUC5AC and CA19-9 improves the diagnosis of pancreatic cancer: a multicenter study. Am J Gastroenterol. 2017;112(1):172–83.
Kim J, Bamlet WR, Oberg AL, et al. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci Transl Med. 2017;9(398):12.
Kuwatani M, Kawakami H, Kubota Y, et al. Verification of the effectiveness of fucosylated haptoglobin as a pancreatic cancer marker in clinical diagnosis. Pancreatology. 2019;19(4):569–77.
Le Calvez-Kelm F, Foll M, Wozniak MB, et al. KRAS mutations in blood circulating cell-free DNA: a pancreatic cancer case-control. Oncotarget. 2016;7(48):78827–40.
Lee JH. The feasibility of serum multiple tumor markers test between patients with primary pancreatic cancer and those with benign pancreatic cystic disease. Clin Lab. 2019;65(10):01.
Liao WC, Wu MS, Wang HP, et al. Serum heat shock protein 27 is increased in chronic pancreatitis and pancreatic carcinoma. Pancreas. 2009;38(4):422–6.
Liu J, Gao J, Du Y, et al. Combination of plasma microRNAs with serum CA19-9 for early detection of pancreatic cancer. Int J Cancer. 2012;131(3):683–91.
Liu R, Chen X, Du Y, et al. Serum microRNA expression profile as a biomarker in the diagnosis and prognosis of pancreatic cancer. Clin Chem. 2012;58(3):610–8.
Liu F, Du F, Chen X. Multiple tumor marker protein chip detection system in diagnosis of pancreatic cancer. World J Surg Oncol. 2014;12:333.
Matsubara J, Honda K, Ono M, et al. Reduced plasma level of CXC chemokine ligand 7 in patients with pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2011;20(1):160–71.
Mayerle J, Kalthoff H, Reszka R, et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut. 2018;67(1):128–37.
Mellby LD, Nyberg AP, Johansen JS, et al. Serum biomarker signature-based liquid biopsy for diagnosis of early-stage pancreatic cancer. J Clin Oncol. 2018;36(28):2887–94.
Mizuno S, Isayama H, Nakai Y, et al. Diagnostic yield of the plasma free amino acid index for pancreatic cancer in patients with diabetes mellitus. Pancreatology. 2019;19(5):695–8.
O’Brien DP, Sandanayake NS, Jenkinson C, et al. Serum CA19-9 is significantly upregulated up to 2 years before diagnosis with pancreatic cancer: implications for early disease detection. Clin Cancer Res. 2015;21(3):622–31.
Park HD, Kang ES, Kim JW, et al. Serum CA19-9, cathepsin D, and matrix metalloproteinase-7 as a diagnostic panel for pancreatic ductal adenocarcinoma. Proteomics. 2012;12(23–24):3590–7.
Park J, Lee E, Park KJ, et al. Large-scale clinical validation of biomarkers for pancreatic cancer using a mass spectrometry-based proteomics approach. Oncotarget. 2017;8(26):42761–71.
Peng HY, Chang MC, Hu CM, et al. Thrombospondin-2 is a highly specific diagnostic marker and is associated with prognosis in pancreatic cancer. Ann Surg Oncol. 2019;26(3):807–14.
Poruk KE, Firpo MA, Scaife CL, et al. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas. 2013;42(2):193–7.
Ritchie SA, Chitou B, Zheng Q, et al. Pancreatic cancer serum biomarker PC-594: diagnostic performance and comparison to CA19-9. World J Gastroenterol. 2015;21(21):6604–12.
Rychlíková J, Vecka M, Jáchymová M, et al. Osteopontin as a discriminating marker for pancreatic cancer and chronic pancreatitis. Cancer Biomark. 2016;17(1):55–65.
Sakai Y, Honda M, Matsui S, et al. Development of novel diagnostic system for pancreatic cancer, including early stages, measuring mRNA of whole blood cells. Cancer Sci. 2019;110(4):1364–88.
Song J, Sokoll LJ, Pasay JJ, et al. Identification of serum biomarker panels for the early detection of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2019;28(1):174–82.
Tachezy M, Zander H, Marx AH, et al. ALCAM (CD166) expression and serum levels in pancreatic cancer. PLoS ONE. 2012;7(6):e39018.
Talar-Wojnarowska R, Gasiorowska A, Olakowski M, et al. Clinical value of serum neopterin, tissue polypeptide-specific antigen and CA19-9 levels in differential diagnosis between pancreatic cancer and chronic pancreatitis. Pancreatology. 2010;10(6):689–94.
Tavano F, Gioffreda D, Valvano MR, et al. Droplet digital PCR quantification of miR-1290 as a circulating biomarker for pancreatic cancer. Sci Rep. 2018;8(1):16389.
Ward DG, Wei W, Buckels J, et al. Detection of pancreatic adenocarcinoma using circulating fragments of fibrinogen. Eur J Gastroenterol Hepatol. 2010;22(11):1358–63.
Xu J, Cao Z, Liu W, et al. Plasma miRNAs effectively distinguish patients with pancreatic cancer from controls: a multicenter study. Ann Surg. 2016;263(6):1173–9.
Zhang P, Zou M, Wen X, et al. Development of serum parameters panels for the early detection of pancreatic cancer. Int J Cancer. 2014;134(11):2646–55.
Zhang Y, Qiu L, Wang Y, et al. High-throughput and high-sensitivity quantitative analysis of serum unsaturated fatty acids by chip-based nanoelectrospray ionization-Fourier transform ion cyclotron resonance mass spectrometry: early stage diagnostic biomarkers of pancreatic cancer. Analyst. 2014b;139(7):1697–706.
Zhong A, Qin R, Qin W, et al. Diagnostic significance of serum IgG galactosylation in CA19-9-negative pancreatic carcinoma patients. Front Oncol. 2019;9:114.
Zhou YF, Xu LX, Huang LY, et al. Combined detection of serum UL16-binding protein 2 and macrophage inhibitory cytokine-1 improves early diagnosis and prognostic prediction of pancreatic cancer. Oncol Lett. 2014;8(5):2096–102.
Zhou CY, Dong YP, Sun X, et al. High levels of serum glypican-1 indicate poor prognosis in pancreatic ductal adenocarcinoma. Cancer Med. 2018;7(11):5525–33.
Zhou X, Lu Z, Wang T, et al. Plasma miRNAs in diagnosis and prognosis of pancreatic cancer: a miRNA expression analysis. Gene. 2018;673:181–93.
Bagaria B, Sood S, Sharma R, et al. Diagnostic precision of carcinoembryonic antigen level in esophageal carcinoma. Biomed Res (Aligarh). 2013a;24(3):353–8.
Bai Y, Lin H, Fang Z, et al. Plasma microRNA-19a as a potential biomarker for esophageal squamous cell carcinoma diagnosis and prognosis. Biomark Med. 2017;11(5):431–41.
Bagaria B, Bagaria A, Singh M, et al. Diagnostic sensitivity of serum carcinoembryonic antigen, carbohydrate antigen 19–9, alpha-fetoprotein, and beta-human chorionic gonadotropin in esophageal carcinoma (receiver operating characteristic curve analysis). Clin Cancer Investig J. 2015;4(3):312–7.
Brockmann JG, St Nottberg H, Glodny B, et al. Analysis of serum CYFRA 21–1 concentrations in patients with esophageal cancer. Anticancer Res. 2000;20(6D):4899–904.
Huang Z, Zhang L, Zhu D, et al. A novel serum microRNA signature to screen esophageal squamous cell carcinoma. Cancer Med. 2017;6(1):109–19.
Jia K, Li W, Wang F, et al. Novel circulating peptide biomarkers for esophageal squamous cell carcinoma revealed by a magnetic bead-based MALDI-TOFMS assay. Oncotarget. 2016;7(17):23569–80.
Liao Y, Xing S, Xu B, et al. Evaluation of the circulating level of fibroblast activation protein alpha for diagnosis of esophageal squamous cell carcinoma. Oncotarget. 2017;8(18):30050–62.
Lukaszewicz-Zajac M, Mroczko B, Kozlowski M, et al. Stem cell factor in the serum of patients with esophageal cancer in relation to its histological types. Arch Med Sci. 2017;13(6):1357–64.
Lv H, He Z, Wang H, et al. Differential expression of miR-21 and miR-75 in esophageal carcinoma patients and its clinical implication. Am J Transl Res. 2016;8(7):3288–98.
Pan J, Zheng QZ, Li Y, et al. Discovery and validation of a serologic autoantibody panel for early diagnosis of esophageal squamous cell carcinoma. Cancer Epidemiol Biomark Prev. 2019;28(9):1454–60.
Peng YH, Xu YW, Guo H, et al. Combined detection of serum Dickkopf-1 and its autoantibodies to diagnose esophageal squamous cell carcinoma. Cancer Med. 2016;5(7):1388–96.
Sudo K, Kato K, Matsuzaki J, et al. Development and validation of an esophageal squamous cell carcinoma detection model by large-scale MicroRNA profiling. JAMA Netw Open. 2019;2(5):e194573.
Wang C, Guan S, Liu F, et al. Prognostic and diagnostic potential of miR-146a in oesophageal squamous cell carcinoma. Br J Cancer. 2016;114(3):290–7.
Xing S, Zheng X, Wei LQ, et al. Development and validation of a serum biomarker panel for the detection of esophageal squamous cell Carcinoma through RNA transcriptome sequencing. J Cancer. 2017;8(12):2346–55.
Xu YW, Peng YH, Chen B, et al. Autoantibodies as potential biomarkers for the early detection of esophageal squamous cell carcinoma. Am J Gastroenterol. 2014;109(1):36–45.
Xu YW, Chen H, Guo HP, et al. Combined detection of serum autoantibodies as diagnostic biomarkers in esophagogastric junction adenocarcinoma. Gastric Cancer. 2019;22(3):546–57.
Yan L, Dong X, Gao J, et al. A novel rapid quantitative method reveals stathmin-1 as a promising marker for esophageal squamous cell carcinoma. Cancer Med. 2018;7(5):1802–13.
Zhang T, Wang Q, Zhao D, et al. The oncogenetic role of microRNA-31 as a potential biomarker in oesophageal squamous cell carcinoma. Clin Sci. 2011;121(10):437–47.
Zhang B, Zhang Z, Zhang X, et al. Serological antibodies against LY6K as a diagnostic biomarker in esophageal squamous cell carcinoma. Biomarkers. 2012;17(4):372–8.
Zhang T, Zhao D, Wang Q, et al. MicroRNA-1322 regulates ECRG2 allele specifically and acts as a potential biomarker in patients with esophageal squamous cell carcinoma. Mol Carcinog. 2013;52(8):581–90.
Zhang HF, Qin JJ, Ren PF, et al. A panel of autoantibodies against multiple tumor-associated antigens in the immunodiagnosis of esophageal squamous cell cancer. Cancer Immunol Immunother. 2016;65(10):1233–42.
Zhang JB, Cao M, Chen J, et al. Serum anti-TOPO48 autoantibody as a biomarker for early diagnosis and prognosis in patients with esophageal squamous cell carcinoma. Clin Res Hepatol Gastroenterol. 2018;42(3):276–84.
Zheng X, Xing S, Liu XM, et al. Establishment of using serum YKL-40 and SCCA in combination for the diagnosis of patients with esophageal squamous cell carcinoma. BMC Cancer. 2014;14:490.
Zhou SL, Yue WB, Fan ZM, et al. Autoantibody detection to tumor-associated antigens of P53, IMP1, P16, cyclin B1, P62, C-myc, Survivn, and Koc for the screening of high-risk subjects and early detection of esophageal squamous cell carcinoma. Dis Esophagus. 2014;27(8):790–7.
Deng YW, Zhong RH, Xie XY, et al. Serum CEA, CA125, CA19-9, and CA724 levels for the diagnosis and staging of cholangiocarcinoma. Biomed Res (Aligarh). 2017;28(3):1413–8.
Leelawat K, Narong S, Wannaprasert J, et al. Prospective study of MMP7 serum levels in the diagnosis of cholangiocarcinoma. World J Gastroenterol. 2010;16(37):4697–703.
Wang YF, Feng FL, Zhao XH, et al. Combined detection tumor markers for diagnosis and prognosis of gallbladder cancer. World J Gastroenterol. 2014;20(14):4085–92.
Bagaria B, Sood S, Sharma R, et al. Comparative study of CEA and CA19-9 in esophageal, gastric and colon cancers individually and in combination (ROC curve analysis). Cancer Biol Med. 2013b;10(3):148–57.
Markar SR, Wiggins T, Antonowicz S, et al. Assessment of a noninvasive exhaled breath test for the diagnosis of oesophagogastric cancer. JAMA Oncol. 2018;4(7):970–6.
Ren S, Zhang Z, Xu C, et al. Distribution of IgG galactosylation as a promising biomarker for cancer screening in multiple cancer types. Cell Res. 2016;26(8):963–6.
Schneider J, Bitterlich N, Schulze G. Improved sensitivity in the diagnosis of gastro-intestinal tumors by fuzzy logic-based tumor marker profiles including the tumor M2-PK. Anticancer Res. 2005;25(3A):1507–15.
Schultz NA, Dehlendorff C, Jensen BV, et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA. 2014;311(4):392–404.
Huang Y-K, Yu J-C, Kang W-M, et al. Significance of serum pepsinogens as a biomarker for gastric cancer and atrophic gastritis screening: a systematic review and meta-analysis. PLoS ONE. 2015;10(11):e0142080.
Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926.
Bossuyt PM, Reitsma JB, Bruns DE, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. BMJ. 2015;351:h5527.
Jüni P, Holenstein F, Sterne J, et al. Direction and impact of language bias in meta-analyses of controlled trials: empirical study. Int J Epidemiol. 2002;31(1):115–23.
Nussbaumer-Streit B, Klerings I, Dobrescu AI, et al. Excluding non-english publications from evidence-syntheses did not change conclusions: a meta-epidemiological study. J Clin Epidemiol. 2020;118:42–54.
Elliott JH, Turner T, Clavisi O, et al. Living systematic reviews: an emerging opportunity to narrow the evidence-practice gap. PLoS Med. 2014;11(2):e1001603.
Westwood M, Lang S, Armstrong N, et al. Faecal immunochemical tests (FIT) can help to rule out colorectal cancer in patients presenting in primary care with lower abdominal symptoms: a systematic review conducted to inform new NICE DG30 diagnostic guidance. BMC Med. 2017;15(1):189.
van Melle M, Yep Manzano SI, Wilson H, et al. Faecal immunochemical test to triage patients with abdominal symptoms for suspected colorectal cancer in primary care: review of international use and guidelines. Fam Pract. 2020 (epub 8 May 2020).
Acknowledgements
Funding
This study and the journal’s rapid service fee were supported by the CanTest Collaborative (funded by Cancer Research UK C8640/A23385) of which Fiona M. Walter is Director, Jon Emery is an Associate Director, Mike Messenger is co-investigator, and Natalia Calanzani and Garth Funston are researchers. The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. Paige Druce, Kristi Milley and Jon Emery are supported by the Cancer Australia Primary Care Collaborative Cancer Clinical Trials Group (PC4). Mike Messenger is funded by the NIHR Leeds In Vitro Diagnostic Co-operative (UK). No Open Access Fee was received by the journal for the publication of this article.
Other Assistance
We thank Veronica Phillips, Assistant Librarian, University of Cambridge Medical Library, and Jim Berryman, Liaison Librarian, Brownless Biomedical Library University of Melbourne for expert input when developing the search strategy.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Mike Messenger is a paid consultant for Cepheid Inc in the field of bladder cancer diagnostics. Garth Funston is on the Editorial Board of Advances in Therapy but has nothing further to disclose. Natalia Calanzani, Paige E. Druce, Claudia Snudden, Kristi M. Milley, Rachel Boscott, Dawnya Behiyat, Smiji Saji, Javiera Martinez-Gutierrez, Jasmeen Oberoi, Jon Emery and Fiona M Walter have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Calanzani, N., Druce, P.E., Snudden, C. et al. Identifying Novel Biomarkers Ready for Evaluation in Low-Prevalence Populations for the Early Detection of Upper Gastrointestinal Cancers: A Systematic Review. Adv Ther 38, 793–834 (2021). https://doi.org/10.1007/s12325-020-01571-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01571-z