
Abstract
Tamoxifen is a prodrug, and most of the therapeutic effect in treating breast cancer stems from its metabolite, endoxifen. Since cytochrome P450 (CYP) 2D6 is the most important enzyme in the production of endoxifen, drugs that inhibit CYP2D6 would be expected to reduce tamoxifen efficacy. In addition to drug–drug interactions (DDI) involving CYP2D6, there is growing evidence that enzyme inducers can substantially alter the disposition of endoxifen, reducing tamoxifen efficacy. Although the clinical evidence on the impact of CYP2D6 inhibitors on tamoxifen efficacy is mixed, there were serious flaws in many of the studies. Thus, there is a reasonable chance that CYP2D6 inhibitors do in fact inhibit tamoxifen efficacy. Tamoxifen has extraordinarily complex pharmacokinetics, with more than a dozen drug-metabolizing enzymes and transporters involved in its disposition. Enzyme inducers may increase the activity of several of these pathways, including phase II enzymes, ABC transporters, and various CYP enzymes other than CYP2D6. Based on current clinical evidence, one could argue that enzyme inducers are potentially more dangerous than CYP2D6 inhibitors in patients taking tamoxifen. Moreover, early evidence suggests that the combination of CYP2D6 inhibitors plus enzyme inducers may produce catastrophic inhibition of tamoxifen efficacy. One could argue that, given the available evidence, an agnostic “wait and see” position on tamoxifen DDI is ethically untenable, and that many women with breast cancer are currently being subjected to an unnecessary risk of cancer recurrence. Specific recommendations to reduce the risk of adverse tamoxifen DDI are offered for consideration.
Similar content being viewed by others

Tamoxifen drug–drug interactions (DDI) are controversial, but a careful evaluation of the clinical evidence suggests that CYP2D6 inhibitors and enzyme inducers may reduce tamoxifen efficacy. |
The consequences of falsely assuming tamoxifen DDI are important (type I error) are minor, but the consequences of falsely assuming they are not important (type II error) are potentially catastrophic. |
Given the ease with which many tamoxifen DDI can be avoided, reason dictates that we act to reduce the risk of tamoxifen DDI. |
Knowledge and wisdom, far from being one,
Have ofttimes no connection. Knowledge dwells
In heads replete with the thoughts of other men;
Wisdom in minds attentive to their own.
Knowledge, a rude unprofitable mass,
The mere materials with which wisdom builds,
Till smoothed and squared and fitted to its place
Does but encumber whom it seems to enrich.
William Cowper, The Task [1]
1 Background
No one knew about tamoxifen or cytochrome P450 (CYP) 2D6 when William Cowper (1731-1800) wrote his wonderful long poem, The Task, but for those of us who feel “encumbered” by the “rude unprofitable mass” of conflicting data regarding the effect of CYP2D6 activity on tamoxifen efficacy, Cowper’s words resonate across the centuries.
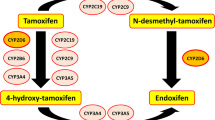
When it became clear over a decade ago that tamoxifen is a prodrug and is converted to the major active metabolite (endoxifen) primarily by the enzyme CYP2D6, papers began to appear describing reduced tamoxifen efficacy in breast cancer patients with reduced CYP2D6 activity due either to genotype or drugs that inhibit CYP2D6. But the plot thickened quickly as papers began to appear suggesting that reduced CYP2D6 activity did not increase the risk of recurrence and death due to breast cancer. We now have a substantial body of evidence on both sides of the issue.
Although much of the debate has centered on CYP2D6 activity and tamoxifen, it has become clear that the story involves much more than just CYP2D6. Tamoxifen efficacy may also be affected by ABC transporters (e.g., ABCB1, ABCC2), phase II metabolism such as glucuronidation, and possibly other CYP isoforms (e.g., CYP2B6, CYP2C9, CYP2C19, and CYP3A4) [2, 3]. Like CYP2D6, the activity of these enzymes and transporters can be affected by drugs and genotype, a fact that markedly increases the complexity of sorting out tamoxifen drug–drug interactions (DDI).
The tamoxifen controversy is fueled by the involvement of so many different disciplines: epidemiology, oncology, pathology, statistics, pharmacogenomics, pharmacology, and DDI. Although efforts to orchestrate this diverse group to achieve consensus have failed so far, untangling some of the errors and misconceptions about DDI is a necessary step if we are to succeed.
Moreover, despite the complexity and the many unanswered questions, I will argue in this commentary that although we do not have enough evidence to establish the importance of tamoxifen DDI with certainty, we clearly have enough data to act. Moreover, it appears that our failure to do so is putting patients on tamoxifen at unnecessary risk of therapeutic failure. In other words, the ambient presumption that we need not worry about CYP2D6 inhibitors or enzyme inducers in patients receiving tamoxifen has become ethically untenable.
2 Studies of CYP2D6 Genotype and Tamoxifen Efficacy
The interest in studying the possible effect of CYP2D6 inhibitors on tamoxifen efficacy came not only from the recognition that endoxifen was the active metabolite, but also from clinical studies suggesting that genetic difference in CYP2D6 activity may affect tamoxifen efficacy. For example, in 1325 patients receiving tamoxifen for breast cancer, those with the lowest CYP2D6 activity had the highest rate of cancer recurrence. The 609 patients who were CYP2D6 extensive metabolizers (EMs) had a cancer recurrence rate of 14.9%, the intermediate metabolizers (IMs) 20.9% and the poor metabolizers 29% [4]. The median follow-up was 6.3 years, but the authors did not collect data on the use of medications that affect CYP2D6 activity.
More recent studies have found similar relationship between CYP2D6 genotype and breast cancer recurrence. For example, in 95 breast cancer patients on tamoxifen, the odds ratio for recurrence and metastasis was 13-fold higher in CYP2D6 IMs compared to EMs [5].
Other studies have also found a relationship between CYP2D6 genotype and tamoxifen efficacy, but these results must be juxtaposed against several studies that failed to find such an association. Fortunately, since our focus is on tamoxifen drug interactions, there is no need to weigh in on the contentious CYP2D6 genotype debate. Moreover, there is general agreement on both sides of the debate that the CYP2D6 genotype studies published to date have one or more limitations, such as:
-
inadequate genotyping,
-
lack of adherence data,
-
inadequate duration of study,
-
retrospective, rather than prospective review,
-
lack of tamoxifen dose information,
-
failure to differentiate between premenopausal and postmenopausal patients,
-
methodological errors,
-
errors and omissions in dealing with concomitant drug therapy.
To study the effect of CYP2D6 genotype on tamoxifen efficacy one would want to adjust in some manner in the patients under study for the presence of concurrent therapy with all CYP2D6 inhibitors and other drugs that may affect tamoxifen efficacy. For some studies, the concurrent use of CYP2D6 inhibitors was not considered at all in the analysis [6]. Even when genotype studies tried to adjust their results for the use of CYP2D6 inhibitors, many of the studies used deeply flawed lists of inhibitors. There were two primary errors in these studies: (1) using lists of CYP2D6 inhibitors contaminated with non-CYP2D6 inhibitors (sometimes outnumbering the real CYP2D6 inhibitors), and (2) the failure to include many other CYP2D6 inhibitors available at the time the study was done. These problems will be discussed in detail below, so there is no need to cover them here.
It is not possible to precisely determine the degree to which using these flawed lists of CYP2D6 inhibitors affected the results of the genotype studies, but one can assume that it made it more difficult to ascertain whether CYP2D6 genotype affects tamoxifen efficacy.
3 CYP2D6 Inhibitors and Tamoxifen Efficacy
Let us briefly review the current status of the effect CYP2D6 inhibitors on tamoxifen pharmacokinetics and tamoxifen efficacy in breast cancer patients. First, there is general agreement that endoxifen is the primary active metabolite of tamoxifen, and that the formation of endoxifen is primarily via CYP2D6. Accordingly, the concurrent use of CYP2D6 inhibitors and tamoxifen would be expected to substantially reduce endoxifen concentrations, and there is clinical evidence to support this expectation [7,8,9]. For example in 80 patients with breast cancer started on tamoxifen 20 mg/day, the EMs who were also taking CYP2D6 inhibitors had endoxifen plasma concentrations that were 58% lower than EMs not on CYP2D6 inhibitors [7]. IMs on CYP2D6 inhibitors had endoxifen concentrations that were 38% lower than IMs not on CYP2D6 inhibitors.
In another study of 140 patients on tamoxifen for breast cancer, 46 patients were taking CYP2D6 inhibtors. In patients taking “weak” CYP2D6 inhibitors, endoxifen plasma concentrations were 24% lower than those not taking CYP2D6 inhibitors, but in those taking “potent” CYP2D6 inhibitors, endoxifen concentrations were 72% lower [8].
Moreover, switching patients from potent CYP2D6-inhibiting selective serotonin reuptake inhibitors (SSRIs; paroxetine or fluoxetine) to a different SSRI with little or no effect on CYP2D6 (escitalopram) results in substantial increases in endoxifen concentrations [10]. One would expect, therefore, that CYP2D6 inhibitors would inhibit the efficacy of tamoxifen in the treatment of breast cancer, but it turns out that some studies suggest such an association [11,12,13,14,15], while other studies do not [16,17,18,19].
In one study of 2430 women with breast cancer on tamoxifen and SSRIs, paroxetine (a potent CYP2D6 inhibitor) was associated with an increase in breast cancer mortality, and the greater the overlap of the two drugs, the higher was the breast cancer mortality [12]. Unexpectedly, another potent CYP2D6 inhibitor (fluoxetine) was not associated with increased breast cancer mortality. There are two ways to explain this anomaly. One explanation is that the study suggests that CYP2D6 activity is not important for tamoxifen efficacy. Another explanation is that there is some difference between paroxetine and fluoxetine that accounts for the anomaly. We know that paroxetine and fluoxetine differ regarding which CYP enzymes they inhibit, and their effect on transporters could vary as well. Given what we know from the 50-year history of DDI, the second explanation is as plausible as the first.
All of the studies of the effect of CYP2D6 inhibitors on tamoxifen efficacy—pro and con—have had one or more limitations, and many of these limitations were acknowledged by the authors. But in some studies, the limitations raise serious questions regarding the validity of the results. For example, as Juurlink [20] has pointed out, one recent study cited above [17] failed to find an association between the use of CYP2D6-inhibiting SSRIs and tamoxifen efficacy, but looked at total mortality instead of breast cancer deaths, and the study also had a median follow-up of only about 2 years. As with most studies of CYP2D6 activity and tamoxifen efficacy, the authors also did not differentiate between premenopausal and postmenopausal patients, although they did concede that CYP2D6 inhibitors may be more likely to affect tamoxifen efficacy in premenopausal patients. Although the study had a very large sample size, their attempt to adjust for other drugs taken by the patients that may affect CYP2D6 was seriously flawed. Of the 18 drugs on the list entitled “CYP2D6 enzyme inhibitors and inducers” less than half are likely to affect CYP2D6 activity. Moreover, as discussed below, CYP2D6 is resistant to enzyme induction, so the two enzyme inducers on the list probably have little or no effect on CYP2D6 activity. Presumably, they included enzyme inducers with the idea that they would offset any CYP2D6 inhibitors, but instead it is likely that the combination of CYP2D6 inhibitors with enzyme inducers would have an additive effect in reducing tamoxifen efficacy. We will discuss later the potential “double whammy” effect of patients on tamoxifen receiving CYP2D6 inhibitors and enzyme inducers simultaneously, a potentially disastrous combination.
One enzyme inducer, St. John’s wort, merits particular attention at this point. I am not aware of any study of CYP2D6 inhibitors on tamoxifen efficacy that corrected for use of St. John’s wort. Since this herbal product is used for some of the same reasons as the SSRIs paroxetine and fluoxetine, and could easily affect the results. Of course, St. John’s wort would not appear on computerized drug records and it would thus be difficult to determine which patients took it. This adds another confounding factor to the study of CYP2D6 inhibitors on tamoxifen efficacy.
Another recent study failing to find an effect of CYP2D6 inhibitors on tamoxifen efficacy had similar flaws [16]. Although the authors “adjusted for other CYP2D6 inhibitors”, the six drugs they listed do not make one optimistic that this part of the study was handled properly. For example, they included propranolol as a CYP2D6 inhibitor, presumably because it is a substrate for CYP2D6, but actually there is no evidence that it inhibits CYP2D6. They also included cimetidine, which is at best a modest CYP2D6 inhibitor, and they misspelled two of the six CYP2D6 inhibitor drugs as “amniodarone” and “chloroquinone”. This may sound like a minor point, but it suggests that the adjustments for CYP2D6 inhibitors may not have been optimal. Perhaps even more important, the median duration of tamoxifen administration was 2.7 years, which, as the authors acknowledged, may have been too short to find an association between CYP2D6 inhibitors and tamoxifen efficacy.
For some published reports of the effect of CYP2D6 inhibitors on tamoxifen efficacy, the lack of rigor in selecting CYP2D6 inhibitors was so egregious that it sheds serious doubts on the validity of the conclusions. For example, in two frequently cited tamoxifen studies that failed to find an effect of CYP2D6-inhibiting antidepressants on breast cancer recurrence, the researchers presented a drug list used to adjust the data for other medications that might affect CYP2D6 activity [18, 19]. Before even looking at the individual drugs in their list, it was clear from the title of the table “CYP2D6 inhibitors, substrates, and inducers” that trouble was afoot.
The authors repeatedly stated that CYP2D6 substrates can be considered CYP2D6 inhibitors. Actually, substrates for a given CYP isozyme are not necessarily inhibitors of that same enzyme. In fact, they often are not. This is a widely held fallacy, and not just in tamoxifen studies; one still sees this mistake regularly in the biomedical literature. One might call it the “Highway Fallacy” because people apparently assume that drug metabolism is like a highway, and when there are many cars on the highway, the traffic slows down. The theory appears to be that when two or more drugs use the same metabolic pathway, there must be inhibition of metabolism. Fortunately, and for a variety of reasons beyond the scope of this commentary, competition for metabolism between drugs using the same pathway is often clinically unimportant. The evidence is unequivocal: just because a drug is metabolized by a particular CYP isozyme in no way guarantees that it inhibits that enzyme. One despairs that we will ever see a stake driven into the heart of this fallacy—it has been with us for decades and shows no sign of dying.
The second problem with the table was that they included CYP2D6 “inducers”. CYP enzymes vary in the degree to which they can be induced, however, and it turns out that CYP2D6 is relatively resistant to enzyme induction [21]. A list of clinically important CYP2D6 inducers would be a tabula rasa. Hence, if one analyzes all 30 drugs on the list used in the two studies [18, 19], fewer than ten are actually known to be CYP2D6 inhibitors, about half are substrates for CYP2D6, but have not been shown to be inhibitors, and probably are not. There were also a couple of relatively modest CYP2D6 inhibitors, one drug (ranitidine) that does not inhibit CYP2D6 at all, one CYP3A4 inducer, and two unknown drugs. A charitable assessment, therefore, is that one-third of the drugs on the list could be considered legitimate CYP2D6 inhibitors. To make matters worse, there were also many CYP2D6 inhibitors that were not included in the list (see Table 1 for a current list of CYP2D6 inhibitors). Of course, some of the drugs in Table 1 were not on the market when these studies were done, but many were and should have been added to their list.
Note that Table 1 includes what are often considered both “strong” and “moderate” inhibitors of CYP2D6. Weak CYP2D6 inhibitors are not included. While it is true that some inhibitors of drug-metabolizing enzymes tend to be stronger than others, the extensive variability in magnitude of effect means that in one particular patient, a “moderate” inhibitor of a particular CYP enzyme may produce a greater interaction than a “strong” inhibitor in another patient. One of the striking and universal characteristics of DDI is the high intersubject variability in magnitude, regardless of whether the study is done in patients or healthy subjects, and regardless of whether the mechanism is pharmacokinetic, pharmacodynamic, or a mixture of the two. Making decisions about the clinical importance of a drug interaction based solely on the mean values from published studies is problematic. It is safest to assume, therefore, that any one of the drugs in Table 1 is capable of reducing tamoxifen efficacy in any given patient.
Finally, the study of potential DDI with tamoxifen and CYP2D6 inhibitors is made more difficult by the nature of the outcome of the interaction. As Juurlink has observed, it is not easy to assess drug interactions for agents like tamoxifen where the outcome is therapeutic failure, particularly when the failure may not be obvious for years [20]. For any given drug, there will always be patients who do not respond for a variety of reasons, so determining that a drug interaction was responsible for therapeutic failure is often difficult. The tamoxifen–CYP2D6 controversy is reminiscent of the decades-long debate on whether antibiotics are capable of reducing the efficacy of hormonal contraceptives, resulting in an increased risk of unintended pregnancy. Individual case reports (although there were many) were not very useful since contraceptive failure can occur with or without concurrent antibiotics. Epidemiologic studies of the interaction suffered from some of the same problems as the tamoxifen–CYP2D6 studies, where the lists of antibiotics were contaminated with antibiotics that would be expected to increase rather than decrease the levels of contraceptive hormones due to inhibition of CYP3A4. Yet authors of such flawed studies often declared that they had debunked the myth that antibiotics inhibit hormonal contraceptive efficacy. We still do not know with certainty if antibiotics impair oral contraceptive efficacy, and there is good reason to be skeptical. There is far more evidence supporting an interaction between tamoxifen and CYP2D6 inhibitors. In both cases, however, there are many potential causes of therapeutic failure; for tamoxifen, reduced CYP2D6 activity is only one such reason. Accordingly, we will now turn to other DDI that may affect tamoxifen efficacy.
4 Inducers of Enzymes/Transporters and Tamoxifen Efficacy
As if the lack of adequate handling of CYP2D6 inhibitors in many of the tamoxifen studies were not bad enough, as already mentioned, it turns out that other pathways for tamoxifen metabolism or transport are also likely to be important. These pathways were mostly ignored in the published studies, but the omission is understandable given that little was known at the time about how inhibition or induction of such alternate pathways would affect tamoxifen metabolism. Nonetheless, the failure to account for the effect of genotype or drug therapy on these other drug-metabolizing enzymes and transporters almost certainly complicated our ability to determine the effect of CYP2D6 activity (from genotype or drugs) on tamoxifen efficacy. It may turn out that CYP2D6 activity affects tamoxifen efficacy, but only in the presence of certain genotypes or drug therapy that affects the activity of transporters or other drug-metabolizing enzymes (including Phase II metabolism). This is a critical point, because it makes the conflicting data on CYP2D6 activity and tamoxifen efficacy more understandable, perhaps even inevitable.
Awareness of the possible impact of enzyme inducers on tamoxifen was advanced by a prospective study in which breast cancer patients on tamoxifen were given rifampin 600 mg/day for 15 days [23]. An interim analysis in four patients showed that endoxifen area under the concentration-time curve (AUC) decreased by about 70% after rifampin administration, leading the researchers to terminate the study prematurely to avoid harming patients. Tamoxifen AUC decreased by 84% following rifampin, which is consistent with an 86% decrease in tamoxifen AUC found previously when tamoxifen was given with rifampin to healthy subjects [24]. The previous study, however, did not measure endoxifen plasma concentrations.
A case report has also appeared showing a dramatic reduction in endoxifen plasma concentrations in a 39-year-old woman following rifampin therapy [25]. Her baseline endoxifen was in the therapeutic range (46 nM), but 2 weeks after completing 10 days of rifampin therapy (600 mg/day) endoxifen had declined to a subtherapeutic level of 15.75 nM. Ten weeks after the rifampin was stopped, her endoxifen concentrations had doubled, a positive dechallenge that supports the claim that rifampin was responsible. The case was well documented and the causal relationship between the rifampin therapy and the low endoxifen levels would be rated “Probable” by the Drug Interaction Probability Scale (DIPS) [26].
Tamoxifen, endoxifen, and tamoxifen metabolites are substrates for enzymes and transporters that are susceptible to induction, such as uridine diphosphate-glucuronosyltransferases (UGTs), a family of enzymes that catalyse the covalent addition of glucuronyltransferases, CYP3A4, CYP2C9/CYP2C19, CYP2B6, and efflux transporters such as P-glycoprotein (ABCB1) and multidrug resistance-associated protein 2 (ABCC2) [2, 27, 28]. Rifampin probably induces all of these enzymes and transporters [22, 29,30,31].
It seems likely, therefore, that the ability of rifampin (and other enzyme inducers, as described below) to reduce endoxifen AUC is via more than one pathway. Moreover, the effect of rifampin may have involved reduced tamoxifen bioavailability (due to induced ABCB1), or increased tamoxifen metabolism via induction of CYP2B6 or UGTs, thus shunting tamoxifen metabolism away from pathways that lead to the formation of endoxifen (see Fig. 1). There is some clinical evidence that patients on tamoxifen with genotypes that result in higher UGT2B15 activity have an increased risk of breast cancer recurrence and death [32]. Also, endoxifen appears to undergo glucuronide conjugation via UGT enzymes, so it is possible that rifampin can increase the phase II metabolism of endoxifen itself.

Potential effects of enzyme inducers on endoxifen. CYP cytochrome P450
As can be seen in Fig. 1, almost all of the pathways that are susceptible to enzyme induction would ultimately result in decreased plasma endoxifen concentrations. The problem could be compounded if the patient had reduced CYP2D6 activity due to genotype or therapy with CYP2D6 inhibitors, which would further depress endoxifen plasma concentrations (the “double whammy” effect mentioned earlier). Then, if, as the available evidence suggests, it turns out that increased activity of ABCB1 and/or ABCC2 increases the efflux of endoxifen out of the cancer cell as shown in Fig. 1, the reduction in tamoxifen efficacy by enzyme inducers could be devastating—a perfect storm of factors conspiring to negate tamoxifen’s anticancer effects.
What about enzyme inducers other than rifampin? Early evidence that enzyme inducers can reduce serum concentrations of tamoxifen and endoxifen appeared decades ago. In 1990, Lien et al. published a report of six postmenopausal patients in whom tamoxifen pharmacokinetics were studied before and after addition of aminoglutethimide for 6 weeks [33]. Like rifampin, aminoglutethimide substantially reduced tamoxifen AUC (by 73%), and the effect of aminoglutethimide on endoxifen AUC (93% decrease) was even greater than found for rifampin in the study cited above. When aminoglutethimide was added to tamoxifen, three of the six patients had endoxifen AUCs of zero (undetectable), and the authors observed that endoxifen “nearly disappeared in serum during aminoglutethimide treatment”. The study was small, but it is consistent with the rifampin data and the phenytoin data described below.
Aminoglutethimide is known to induce drug metabolism by various CYP enzymes and probably also induces glucuronidation [34, 35], but there is little clinically useful information regarding its effect on transporters. At this point, it is not possible to determine with certainty which enzymes or transporters are involved in the dramatic reductions in endoxifen AUC with aminoglutethimide. Note that aminoglutethimide is an aromatase inhibitor and has been used in the past to treat breast cancer. It is seldom used for any purpose today, so the importance of these findings is primarily by inference to other enzyme inducers.
Another enzyme inducer, phenytoin, may also markedly reduce endoxifen plasma concentrations. A 49-year-old woman who was an EM for CYP2D6 and on chronic phenytoin was started on tamoxifen 20 mg/day [36]. Two months later, her endoxifen level was 4.72 nmol/l, which was about sevenfold lower than the mean endoxifen for EMs in their clinic (33.0 nmol/l). The endoxifen levels in this patient were even lower than most CYP2D6 poor metabolizers in the clinic, and was about sixfold lower than eight matched controls. The AUC of the parent drug, tamoxifen, was only moderately decreased, suggesting that the dramatically reduced endoxifen concentrations were not due to lack of adherence. The case was well documented and the causal relationship between the phenytoin therapy and the low endoxifen levels would be rated “Probable” by the DIPS [26]. In a previous report of 23 male and female patients with glioma on high-dose tamoxifen, phenytoin was associated with a 60% reduction in tamoxifen AUC, but the decrease was not statistically significant [37]. The authors did not measure endoxifen concentrations, however, and some patients were receiving another enzyme inducer (dexamethasone) that could have confounded the results.
Taken together, the evidence on the effect of enzyme inducers (rifampin, aminoglutethimide, phenytoin) on tamoxifen pharmacokinetics suggests that these inducers may have a greater effect in reducing endoxifen concentrations than CYP2D6 inhibitors. The possibility that enzyme inducers can enhance transporter-induced efflux of endoxifen from breast cancer cells could compound the effect of reduced serum endoxifen concentrations (see Fig. 1). All in all, it may turn out that enzyme/transporter inducers are more dangerous than CYP2D6 inhibitors in patients taking tamoxifen. We will now turn to the transporters, which provide even more cause for concern about enzyme/transporter inducers.
4.1 P-glycoprotein (ABCB1)
Available evidence suggests that the transporter ABCB1 is involved in tamoxifen and endoxifen disposition [38]. There is growing evidence that altered ABCB1 activity may affect tamoxifen pharmacokinetics and possibly tamoxifen efficacy in breast cancer patients. In 71 premenopausal patients receiving tamoxifen for breast cancer, those with the ABCB1 polymorphism wild-type homozygous rs1045642 (CC) had worse outcomes than those with reduced ABCB1 activity due to a heterozygous (CT) or homozygous (TT) variant allele [14].
Another study of 95 breast cancer patients on tamoxifen found increased breast cancer recurrence and metastasis in patients with higher ABCB1 activity [39]. Using the homozygous wild type (CC) as a reference category with an odds ratio (OR) of 1.0, the OR of heterozygous patients (CT) was 0.58, and for homozygous mutant (TT) the OR was 0.21. Perhaps in those patients with higher ABCB1 activity, the efflux of endoxifen from the cancer cells was higher, thus reducing tamoxifen efficacy. It is also possible that those with higher ABCB1 activity have lower bioavailability of tamoxifen and/or increased tamoxifen elimination, although this may be less important than the effect on breast cancer cells.
There is also evidence of reduced tamoxifen efficacy with a combination of reduced CYP2D6 activity and higher ABCB1 activity. For example, in tamoxifen-treated breast cancer patients with decreased CYP2D6 activity (IMs) plus high ABCB1 activity, Teh et al. found the median time to develop recurrence and metastasis was only 12 months, compared to 48 months in patients with either intermediate CYP2D6 or higher ABCB1 activity alone [39]. The number of patients was, however, small and so more study is needed to determine if the reduced tamoxifen efficacy is as dramatic as noted in this report.
4.2 Multidrug Resistance-Associated Protein 2 (ABCC2)
Another transporter that may turn out to be important in the efficacy of tamoxifen is ABCC2. In a study of 282 patients receiving tamoxifen 20 mg/day for breast cancer, variant alleles of rs3740065 in ABCC2 that result in increased ABCC2 activity had a higher rate of cancer recurrence [40]. ABCC2 genotype GG (lowest ABCC2 activity) was used as a reference and was assigned an adjusted hazard ratio (HR) for cancer recurrence of 1.0. Patients with one ABCC2 “risk allele” (AG) and thus increased ABCC2 activity had an adjusted HR of 3.52, and those with two risk alleles (AA) resulting in the highest ABCC2 activity had an adjusted HR of 10.64. They also found that decreased CYP2D6 activity by itself increased the rate of cancer recurrence. When they designated EM patients as the reference with an adjusted HR of 1.0, heterozygous (IMs) had an adjusted HR of 4.44 and PMs an adjusted HR of 9.52.
The most astonishing (and alarming) results, however, were seen when they looked at patients with various combinations of reduced CYP2D6 activity and higher ABCC2 activity. They assigned the following points to the four “risk alleles” found in the patients: CYP2D6 IMs (1 point), CYP2D6 PMs (2 points), ABCC2-AG (1 point), ABCC2-AA (2 points). With the various combinations, they had patients with 0, 1, 2, 3 and 4 risk allele points. Designating the presence of 1 risk allele point as the reference with an adjusted HR of 1.0 for cancer recurrence, they found the following adjusted HRs: 2 risk allele points = 4.93; 3 risk allele points = 19.98; 4 risk allele points = 45.25. These results suggest that patients with both low CYP2D6 activity (PMs) and high ABCC2 activity may have a dramatic reduction in tamoxifen efficacy. More study of this potentially devastating effect is urgently needed, especially since the combination of reduced CYP2D6 activity and increased ABCC2 activity may be produced by concurrent therapy with CYP2D6 inhibitors and enzyme inducers.
Another study of 73 breast cancer patients also found reduced tamoxifen efficacy associated with a wild-type allele ABCC2 as compared to variant alleles with reduced ABCC2 activity [41]. Further, they found that the combination of higher ABCC2 activity (ABCC2 CC) with higher ABCB1 activity (ABCB1 CT + TT) was associated with reduced disease-free survival compared to any other combination of genotypes for ABCC2 and ABCB1.
Taken together, the evidence suggests that the activity of the transporters ABCB1 and ABCC2 may have a large influence on the efficacy of tamoxifen for breast cancer. Although it may not yet be practical to routinely genotype patients for ABCC1 or ABCC2, we could at least attempt to keep tamoxifen-treated patients from taking drugs that can potentially induce these transporters (see Table 2).
4.3 Enzyme Inducers of Concern
As already mentioned, there is specific and credible evidence that rifampin, aminoglutethimide and phenytoin interact with tamoxifen. A number of other drugs are also known inducers as shown in Table 2. For example, St John’s wort is a well-established inducer of CYP enzymes and ABC transporters [42,43,44]. In addition to a well-documented induction of ABCB1, there is some evidence that it may also induce ABCC2 [45, 46]. St John’s wort may also induce phase II metabolism, which is likely to reduce endoxifen concentrations. Since St John’s wort may be used to treat depression or hot flashes in place of SSRIs in patients on tamoxifen [47], this may be a critical issue. Is it possible that in studies of the effect of SSRIs on tamoxifen efficacy, patients who did not take SSRIs for their hot flashes or depression were more likely to take St John’s wort, thus leading to the false conclusion that SSRIs had no effect on tamoxifen? It does not appear that this possibility was considered in the studies of SSRIs effect on tamoxifen, and it could have reduced the likelihood of finding a real effect of CYP2D6 inhibition on tamoxifen efficacy.
Other inducers also increase the activity of many enzymes and transporters such as CYP enzymes (other than CYP2D6), UGTs, ABCB1 (P-glycoprotein) and ABCC2, so it is likely that many of the drugs in Table 2 will be able to reduce endoxifen plasma concentrations. Some of the drugs in Table 2 are better documented to be “broad-spectrum” inducers affecting multiple enzymes and transporters (e.g., carbamazepine, fosphenytoin, oxcarbazepine, phenobarbital, phenytoin, primidone rifabutin, rifampin, rifapentine, and St John’s wort). The extent to which the other inducers in Table 2 would affect tamoxifen and endoxifen pharmacokinetics is more speculative. Weak enzyme inducers are not included in Table 2.
Given that enzyme inducers have been shown to drastically reduce endoxifen serum concentrations and may also increase endoxifen efflux from breast cancer cells, as already mentioned, it may well turn out that enzyme inducers are more problematic than CYP2D6 inhibitors in patients on tamoxifen. It is possible that a catastrophic reduction in tamoxifen efficacy could occur in a patient on a potent CYP2D6 inhibitor and also taking a strong enzyme inducer at the same time. As mentioned previously, CYP2D6 is resistant to induction, so the inducer would not be able to counteract the effect of the CYP2D6 inhibitor. Pending additional study, one could argue that simultaneous administration of CYP2D6 inhibitors and enzyme inducers should be considered contraindicated in patients taking tamoxifen.
Note that endoxifen itself is being studied in patients with breast cancer, and the early results are promising [48]. Using endoxifen instead of the parent drug, tamoxifen, would avoid CYP2D6 interactions and also reduce the number of possible sites of interaction with enzyme inducers. There would still be the possibility, however, that inducers would increase the glucuronidation of endoxifen and increase the efflux of endoxifen from breast cancer cells; so, we need more study of these possibilities.
5 Other Cytochrome P450 Enzymes
In addition to CYP2D6, CYP3A4 and transporters, it is also possible that the activity of other CYP enzymes such as CYP2B6, CYP2C9, CYP2C19, and others may contribute to altered tamoxifen efficacy, although the evidence is limited. As with CYP2D6, the activity of these CYP enzymes can be altered by both polymorphisms and drug therapy, and, unlike CYP2D6 (which is resistant to enzyme induction) for CYP2C9 and CYP2C19, one must consider both enzyme inhibitors and enzyme inducers. But the point is that even if CYP2D6 activity ultimately proves to be important to tamoxifen efficacy (which may well turn out to be the case), it is likely that the CYP2D6 effect will be amplified or diminished by the activity of the various other drug-metabolizing enzymes and transporters that affect tamoxifen pharmacokinetics. Much more study is needed to establish the effect of the interplay of these various other enzymes and transporters on tamoxifen efficacy.
Many drugs inhibit CYP3A4, so if they affected tamoxifen efficacy it would be a serious concern. Almost all CYP3A4 inhibitors also inhibit ABCB1, so this increases the complexity of any potential interaction with tamoxifen. The concurrent use of tamoxifen with drugs that are combined CYP3A4/ABCB1 inhibitors, therefore, could theoretically either inhibit or enhance tamoxifen efficacy, because they might, (1) reduce endoxifen levels, since CYP3A4 is involved along with CYP2D6 in the formation of endoxifen, (2) reduce endoxifen efflux from breast cancer cells through inhibition of ABCB1 (and perhaps ABCC2), (3) counteract the detrimental effect of any CYP3A4/ABCB1 inducers the patient might be taking.
In one study of 80 patients with breast cancer, endoxifen concentrations did not appear to be affected by CYP3A4-inhibiting calcium channel blockers, although endoxifen plasma concentrations were 58% lower in the 24 patients who were taking CYP2D6 inhibitors [7]. More data are needed on the effect of CYP3A4 inhibitors on endoxifen, however, since only five patients took the CYP3A4 inhibitors, and calcium channel blockers generally produce only moderate inhibition of CYP3A4. Overall, however, there is not much evidence to suggest that CYP3A4 inhibitors affect tamoxifen efficacy.
6 Should We Take Action on Tamoxifen Drug Interactions?
I would argue that there is a disconnect between the available evidence on potentially serious tamoxifen DDI on the one hand, and the general degree of concern about them on the other. This is perhaps more of a problem in the USA than other countries, but considering just the drugs in Tables 1 and 2, we have well over 50 drugs that are probably capable of affecting endoxifen concentrations and tamoxifen efficacy in patients with breast cancer. We will now address what has been recommended in published studies and discuss whether we should listen to those who would dismiss the danger of tamoxifen DDI.
6.1 CYP2D6 Inhibitors
Based at least partly on the deeply flawed studies described above, a number of authors have suggested that there is no need to avoid the use of CYP2D6 inhibitors in patients on tamoxifen. Recommendations to ignore CYP2D6 inhibitors in patients on tamoxifen have come from several sources. One group of researchers claimed a “high level of evidence” that “there is no need to avoid CYP2D6 inhibitors in postmenopausal patients taking tamoxifen” [49]. Another group said in a callout under the title of their paper, “…available (studies) do not appear to support… a negative impact of CYP2D6 inhibitor use on breast cancer recurrence and mortality in women using tamoxifen” [50]. A study published in 2016 concluded, “In clinical practice, SSRI inhibition of CYP2D6 does not seem to reduce the effectiveness of tamoxifen” [17].
Another study from 2016 that found no effect of paroxetine on tamoxifen efficacy stated that the freedom from worry about CYP2D6-inhibiting SSRIs “will help improve the quality of life of breast cancer survivors given that thousands of survivors struggle with depression, sleep disturbance, and vasomotor symptoms while on tamoxifen treatment” [16]. That is a fine sentiment, but the argument ignores the fact that several other studies have found reduced tamoxifen efficacy with CYP2D6-inhibiting SSRIs. Given the potentially grave consequences of reduced tamoxifen efficacy, to assume that this study is correct and the others are wrong is, in my view, not reasonable or in the best interests of patients. Moreover, given that several SSRIs have little or no effect on CYP2D6 (citalopram, escitalopram, venlafaxine, desvenlafaxine), using potent CYP2D6 inhibitors when there are easily available alternatives is difficult to justify based on a rational assessment of the evidence.
Some authors, perhaps lacking imagination, conclude that although CYP2D6 inhibitors may interact with tamoxifen, in essence that it is “no big deal”. One group, while stating that the evidence is mixed, admitted that CYP2D6 inhibitors may “influence tamoxifen effectiveness” [51]. They then point out, however, that since citalopram and venlafaxine [non-interacting SSRIs] are “perhaps more widely accepted” in people on tamoxifen, “tamoxifen efficacy may not be greatly affected”. One could hardly take issue with the scientific accuracy of these statements. I do not think, however, it would be unfair to paraphrase their comments in this way: “It is possible that CYP2D6 inhibitors reduce tamoxifen efficacy, but since these days more women on tamoxifen receive citalopram and venlafaxine than potent CYP2D6 inhibitors, we aren’t killing as many women now as we used to”. I’m not trying to be harsh here, but I do not see any other way to interpret their comments. The “maybe the interactions occur but it is no big deal” approach to giving CYP2D6 inhibitors to women on tamoxifen is in essence saying that as long as reduced tamoxifen efficacy from drug interactions is not common, it can be ignored.
In another review of the effect of CYP2D6 genotype and CYP2D6 inhibitors on tamoxifen, the authors acknowledge that CYP2D6 inhibitors may be “most likely to affect recurrence risk in premenopausal women, where the full production of tamoxifen metabolites is necessary to compete with the abundant estrogen” [52]. They go on to state that the research findings “strongly suggest that the effect of CYP2D6 inhibition and tamoxifen effectiveness may be limited to premenopausal women”. They then state in their conclusion: “The evidence indicates that the effect of both drug-induced and/or gene-induced inhibition of CYP2D6 activity is probably null or small, or at most moderate in subjects carrying two reduced function alleles”. (italics added) So the authors clearly present the possibility that decreased CYP2D6 activity may reduce tamoxifen efficacy in some patients, but end with platitudes about the need for more research. Again, a fair paraphrase would be “The use of CYP2D6 inhibitors may be killing women on tamoxifen, but we don’t know how many, so we need to do additional research before we make any recommendations”. I would propose the following alternative wording: “There is evidence to suggest that CYP2D6 inhibitors may inhibit tamoxifen efficacy, so until the degree of risk is established, patients on tamoxifen should avoid all CYP2D6 inhibitors, whether SSRIs or other drugs”. Again, the intent is not to be polemical, but the stakes are so high that we need more than carefully couched scientific wording to help ensure that patients are not harmed by tamoxifen DDI.
6.2 Enzyme Inducers
As described above, enzyme/transporter inducers have been shown to markedly reduce endoxifen serum concentrations and may also increase endoxifen efflux from cancer cells. Although there is not as much data on enzyme inducers as for CYP2D6 inhibitors and the interactions are more theoretical, the available evidence suggests that enzyme/transporter inducers may produce catastrophic reductions in tamoxifen efficacy. The clinical evidence in patients with breast cancer, although limited, is consistent with the known effects of inducers on endoxifen levels as well as the biologically plausible effects on endoxifen efflux from breast cancer cells.
The question we are faced with, therefore, is whether we should wait for more definitive information before taking any decisive action on tamoxifen DDI. To help us answer this question, let us consider the consequences of making a type I error (assuming that tamoxifen DDI are clinically important but they are not) versus a type II error (assuming that tamoxifen DDI are not clinically important but they are). This decision not unlike the famous “Wager” of Blaise Pascal (1623–1662) in which he advises assuming that God exists because a type I error (falsely assuming God exists) is a minor inconvenience, while a type II error (falsely assuming God does not exist) meant (in the thinking of Pascal’s day) eternal damnation [53]. Few people today think Pascal’s Wager is good theology, but as a general model for making decisions when a type I error has only minor downsides, and a type II error can be catastrophic, it is exactly what we need.
Tamoxifen drug interactions also have a potentially catastrophic type II error, combined with a minimal downside of a type I error. For some reason, we seem obsessed with avoiding the type I error in which we believe that tamoxifen DDI are important, but it turns out in the long run that they are not. But as with Pascal’s example of believing in a non-existent God, a false belief in tamoxifen DDI means only that we have taken a few unnecessary precautions. This is not a big deal. What is a big deal is if we make a type II error, where we take no precautions to avoid DDI and in the long run they turn out to be real. Here, we have a type II error with a bite… some breast cancer patients will have an earlier recurrence and die as a result of our inaction.
Given the amount of evidence suggesting that DDI can reduce tamoxifen efficacy, even the most skeptical observer would have to admit that there is a non-trivial chance that the interactions are clinically important in some patients. This is an issue of probability. Even if one were to take a wildly conservative stance, and assume there is only a 30% chance that the tamoxifen DDI are real, Pascal would advise that it makes no logical sense to ignore the issue given the potentially tragic outcome (death). In his Wager, Pascal argued that nobody could know with certainty that God exists, or even know that God’s existence was probable. Even a low probability of God, however, gave the same result in his Wager, because a low probability multiplied by catastrophic consequences means that every effort should be made to avoid it. Thus, Pascal would urge us to act on tamoxifen DDI for the same reasons.
Accordingly, although many people apparently feel that we have insufficient evidence to act on tamoxifen DDI, I would argue that—given the potentially grave consequences of a type II error—the threshold for action was reached several years ago. In Fig. 2, bar A represents an issue where we have achieved scientific “certainty”—a situation where there is general agreement that we have enough scientific evidence to act on a particular scientific question (the word “certainty” is in quotes, of course, because every non-trivial scientific statement is in principle replaceable by something nearer to the truth). A drug interaction example of “certainty” (bar A) would be giving clarithromycin to a chronic renal failure patient on colchicine. The outcome is stochastic in that we do not know what will happen in any given patient, but we know mean colchicine concentrations substantially increase in groups given the combination. We also know that colchicine toxicity can be deadly, and that scores of fatalities have been reported from this drug interaction [54,55,56,57].

The threshold for action. Bar A is scientific “certainty” where there is general agreement on a scientific question. Bar B represents a lack of scientific certainty, but enough evidence so that action is necessary. Bar C represents a scientific issue where there is inadequate evidence for action
Unfortunately, if one reads the voluminous literature on tamoxifen DDI, it appears that many people are waiting for the issue to achieve the status of scientific “certainty” (Fig. 2, bar A). This sentiment is certainly understandable—we are reluctant to come to scientific conclusions with so many loose ends. Nevertheless, although I would agree that the tamoxifen drug interaction issue is not nearly at that level of certainty (bar A), I believe we have clearly reached the threshold for action (bar B). Admittedly, for many DDI the threshold for action is not easy to determine. The colchicine–clarithromycin interaction is an exception, and for most DDI we are dealing with evidence that is well below the level of scientific certainty. As Reinhold Niebuhr observed, however, most human endeavor consists in finding “proximate solutions for insoluble problems”. Given the grave consequences of the type II error for tamoxifen DDI, I do not think it is tenable to claim we are below the threshold for action (bar C in Fig. 2.)
To conclude the discussion let us consult with another brilliant mathematician who ventured into philosophy, William Kingdon Clifford (1845–1879). In his 1877 essay “Ethics of Belief”, Clifford describes the ethics of making decisions. He argues that if you have been diligent in your deliberations and have made a rational decision based on the best available evidence, your action is “right” no matter how things turn out. On the other hand, if you have not considered the best evidence, or have let your sloth or self-interest decide, you are “wrong” even if it turns out that you made the right choice by accident. Clifford observes, “When an action [or inaction] is once done, it is right or wrong for ever; no accidental failure of its good or evil fruits can possibly alter that”.
If we accept Clifford’s argument, it means that if it turns out in the long run that tamoxifen DDI with CYP2D6 inhibitors and enzyme/transporter inducers are not clinically important, we cannot look back and conclude that the naysayers were “right”. Failure to take measures to avoid potentially lethal DDI in patients on tamoxifen in the face of current evidence, I would argue, is “wrong forever” no matter how it turns out in the long run.
Before concluding, I must acknowledge the many researchers and clinicians who have spent so many hours studying the factors that may affect tamoxifen efficacy, on both sides of the controversial issues. Errors were probably inevitable, given that the topic of tamoxifen efficacy requires knowledge of so many different disciplines. Nevertheless, when one considers the empirical evidence on tamoxifen DDI as a whole, and applies a philosophical perspective to the data, it is hard to escape the conclusion that we can do much better in reducing the risk of tamoxifen therapeutic failure due to DDI. We must, of course, expect our scientific studies to be rigorous, and we need to avoid jumping to conclusions; in other words, we need to avoid “premature factulation” [58]. But for tamoxifen DDI, demanding an excessive degree of scientific certainty before acting is not in the best interests of patients. As John Locke famously said, “It is not possible to achieve certainty in our knowledge of the empirical world, but we can devise workable approximations and act on them”.
7 Conclusion
The papers suggesting that tamoxifen efficacy can be affected by reduced CYP2D6 activity notwithstanding, there is certainly contrary evidence suggesting that CYP2D6 activity is not important. Given the substantial limitations in all of the studies on both sides, it would be inappropriate for either side to claim certainty. For example, the fact that more than one study found a stepwise impairment of tamoxifen efficacy as CYP2D6 activity progressively decreased (due to drugs or genotype) cannot simply be dismissed out of hand, particularly given the significant limitations of the studies that did not find an effect of CYP2D6 inhibitors on tamoxifen efficacy. Also, it is possible that reduced CYP2D6 activity becomes a problem primarily in patients with one or more risk factors such as increased activity of ABCB1 and/or ABCC2 (from drugs or genotype), enzyme inducers, or premenopausal status.
With enzyme inducers and transporter activity the problem is not conflicting results, but rather the fact that we need more evidence for firm conclusions. Nonetheless, the evidence we do have on enzyme inducers and transporters is troubling. In the 50+ year history of DDI I cannot recall a single situation where compelling evidence coming from several different researchers involving a drug interacting with several different enzyme inducers has proven to be false.
As with DDI in general, assessing whether tamoxifen DDI are clinically important and worthy of concern can be considered from the standpoint of probability. In the chart below, I have summarized the various factors that potentially affect tamoxifen efficacy with an estimate of the probability. For the sake of discussion let us define a probability of 0–0.25 as “Unestablished”, 0.25–0.5 as “Possible”, 0.5–0.75 as “Probable” and 0.75–1.0 as “Established”. Given the controversial nature of the impact on tamoxifen efficacy from CYP2D6 inhibitors, it seems appropriate to put them in the “Possible” category.
Factors potentially affecting tamoxifen efficacy
Factor | Potential effect | Probability estimate | Comments |
|---|---|---|---|
CYP2D6 inhibitors | Reduced production of endoxifen | Possible | Conflicting results; significant limitations in all studies |
Genotype for reduced CYP2D6 function | Reduced production of endoxifen | Possible | Conflicting results; significant limitations in all studies |
Genotype for higher ABCB1 activity | Increased efflux of endoxifen from cancer cells | Possible | Two studies suggesting increased breast cancer recurrence with higher ABCB1 activity |
Genotype for higher ABCC2 activity | Increased efflux of endoxifen from cancer cells | Possible | Two studies suggesting increased breast cancer recurrence with higher ABCC2 activity |
Enzyme inducers | Decreased production and/or increased elimination of endoxifen | Probable | Dramatic reduction in endoxifen concentrations. Evidence from rifampin, aminoglutethimide, and phenytoin. |
Enzyme inducers | Increased activity of ABCB1 and ABCC2 with increased efflux of endoxifen from cancer cells | Unestablished | Theoretical, based on transport genotype studies and known inducer effects on transporters. |
Lower CYP2D activity combined with higher ABCB1 activity (both genotypes) | Reduced production of endoxifen AND increased efflux of endoxifen from cancer cells | Unestablished | Preliminary results suggest a dramatic reduction in the time of breast cancer recurrence and metastasis [39] |
Lower CYP2D activity combined with higher ABCC2 activity (both genotype) | Reduced production of endoxifen AND increased efflux of endoxifen from cancer cells | Unestablished | Preliminary results suggest a catastrophic increase in breast cancer recurrence (hazard ratio of over 45) with lowest CYP2D6 activity and highest ABCC2 activity [40] |
CYP2D6 inhibitors AND enzyme inducers | (1) Reduced production of endoxifen (2) Possible increased elimination of endoxifen (3) Increased efflux of endoxifen from cancer cells | Unestablished | Although the possibility of an additive effect is primarily theoretical, it seems likely that it would occur and is likely to be more problematic than either CYP2D6 inhibitors or enzyme inducers alone |
When deciding on a course of action on a given drug interaction, one must consider (1) the empirical evidence supporting the DDI, (2) the severity of the outcome, and (3) the ease with which the potentially interacting combinations can be avoided. Given the simplicity of treating a patient on tamoxifen with venlafaxine or citalopram instead of paroxetine, it is placing a very high value on one’s (fallible) opinions to give them paroxetine. Similarly, given the likelihood that enzyme inducers can reduce tamoxifen efficacy, not bothering to warn patients on tamoxifen to avoid St. John’s wort is inexcusable. It is not, therefore, that the tamoxifen DDI are an established fact; it is that we have reached the threshold for action.
We started this discussion with a distinction between knowledge and wisdom from William Cowper’s poem, The Task. For tamoxifen, “knowledge” tells us that the scientific evidence on tamoxifen DDI is not conclusive and there is still debate about the significance of these DDI to patients with breast cancer. On the other hand, “wisdom”—such as that acquired from Blaise Pascal and William K. Clifford—tells us that we have reached a threshold where failure to act represents a failure of judgment that may well lead to patient harm.
Anyone making such a call to action is obligated, of course, to state what action should be taken. I would like to submit the following recommendations for debate. It may be that these DDI are most important in premenopausal patients [52, 59], but pending more information I would propose that the recommendations apply to all patients on tamoxifen.
Recommendations
Avoid CYP2D6 Inhibitors. I argue that we have reached a point where agnosticism on avoiding CYP2D6 inhibitors in patients on tamoxifen is not an ethically justifiable position. In patients on tamoxifen, avoid adding or continuing CYP2D6 inhibitors (Table 1) unless there is no alternative therapy and the risk of avoiding the CYP2D6 inhibitor is greater than the increased risk of breast cancer recurrence. There should be very few situations where that is the case. When starting an antidepressant in a patient on tamoxifen, avoid CYP2D6 inhibitors and use agents with little or no effect on CYP2D6 such as venlafaxine, desvenlafaxine, escitalopram, or citalopram. If a patient on tamoxifen is already taking an antidepressant that is a moderate to strong CYP2D6 inhibitor, carefully switch her/him to another antidepressant or other therapy if at all possible. |
2. Avoid Enzyme/Transporter Inducers. In patients on tamoxifen, avoid adding or continuing enzyme/transporter inducers (Table 2) unless there is no alternative therapy and the risk of avoiding the inducer is greater than the increased risk of breast cancer recurrence. There should be very few situations where that is the case. The inducers that are well documented to be “broad-spectrum” inducers (those with an asterisk in Table 2) clearly should be avoided, because they are theoretically even more dangerous than CYP2D6 inhibitors in patients on tamoxifen. Whether the other drugs in Table 2 should be avoided is not as clear, and the issue should be debated before any firm recommendations are made. |
3. Advise Patients to Avoid Interacting Drugs Including OTCs and Herbal Products. Given that people often have more than one prescriber, every patient on tamoxifen should be given a list of CYP2D6 inhibitors and enzyme/transporter inducers (such as Tables 1, 2) to take with them. They should be advised to show the list to any other health professional who is prescribing a drug for them. Patients on tamoxifen should be specifically advised to avoid St John’s wort due to its ability to induce enzymes and transporters (there are currently no OTC drugs in the USA that are known to induce enzymes/transporters). They should also be advised to avoid herbals and OTCs that inhibit CYP2D6 such as berberine (goldenseal), diphenhydramine, and chlorpheniramine. (Note that berberine has been studied as an agent to suppress breast cancer cell proliferation, but it is too early to tell if this will have any clinical utility in patients with breast cancer) [60] |
Also, it may turn out that the use of dextromethorphan as a phenotyping probe will prove useful in selected patients, as shown in a study of 40 women on tamoxifen where the dextromethorphan AUC was significantly correlated with endoxifen AUC [61]. As the authors observe, however, additional validation and a more simplified methodology are needed before this can be recommended on a routine basis.
Most scientific debates eventually reach a denouement, but the interminable tamoxifen drama sometimes seems like the scientific equivalent of Wagner’s Ring Cycle. Our current failure to take decisive action to minimize the risk of tamoxifen DDI is, in this author’s view, a tragic abrogation of our duty to those whose lives depend on our judgment. Again, in William K. Clifford’s words: “When an action [or inaction] is once done, it is right or wrong forever; no accidental failure of its good or evil fruits can possibly alter that”. Patients receiving tamoxifen for breast cancer should not have to count on an “accidental failure of the evil fruits” of these DDI.
References
Cowper W. The task and other poems. Gloucestershire (UK): Dodo Press; 2007.
Kiyotani K, Mushiroda T, Nakamura Y, Zembutsu H. Pharmacogenomics of tamoxifen: roles of drug metabolizing enzymes and transporters. Drug Metab Pharmacokinet. 2012;27:122–31.
Powers JL, Buys SS, Fletcher D, Melis R, Johnson-Davis KL, Lyon E, et al. Multigene and drug interaction approach for tamoxifen metabolite patterns reveals possible involvement of CYP2C9, CYP2C19, and ABCB1. J Clin Pharmacol. 2016;56:1570–81.
Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, et al. Assocaition between CYP2D6 polymorphisms and outcomes among women with early state breast cancer treated with tamoxifen. JAMA. 2009;302:1429–36.
Teh LK, Mohamed NI, Salleh MZ, Rohaizak M, Shahrun NS, et al. The risk of recurrence in breast cancer patients treated with tamoxifen: polymorphisms of CYP2D6 and ABCB1. AAPS J. 2012;14:52–9.
Hertz DL, McLeod HL, Irvin WJ Jr. Tamoxifen and CYP2D6: a contradiction of data. Oncologist. 2012;17:620–30.
Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee K-H, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97:30–9.
Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80:61–74.
Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. 2003;95:1758–64.
Binkhorst L, Bannink M, de Bruijn P, Ruit J, Droogendijk H, van Alphen RJ, et al. Augmentation of endoxifen exposure in tamoxifen-treated women following SSRI switch. Clin Pharmacokinet. 2016;55:249–55.
Goetz MP, Knox SK, Suman VJ, Rae JM, Safgren SL, Ames MM, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat. 2007;101:113–21.
Kelley CM, Juurlink DN, Gomes T, Duong-Hua M, Pritchard KI, Austin PC, et al. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: a population based cohort study. BMJ. 2010;340:c693. https://doi.org/10.1136/bmj.c693.
Chubak J, Bowles EJA, Yu O, Buist DSM, Fujii M, Boudreau DM. Breast cancer recurrence in relation to antidepressant use. Cancer Causes Control. 2016;27:125–36.
Argalacsova S, Slanar O, Vitek P, Tesarova P, Bakhouche H, Drazdakova M, et al. Contribution of ABCB1 and CYP2D6 genotypes to the outcome of tamoxifen adjuvant treatment in premenopausal women with breast cancer. Physiol Res. 2015;64(Suppl 4):S539–47.
Newman WG, Hadfield KD, Latif A, Roberts SA, Shenton A, McHague C, et al. Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clin Cancer Res. 2008;14:5913–8.
Haque R, Shi J, Schottinger JE, Ahmed SA, Cheetham TC, Chung J, et al. Tamoxifen and antidepressant drug interaction among a cohort of 16,887 breast cancer survivors. J Natl Cancer Inst. 2016;108. https://doi.org/10.1093/jnci/djv337.
Donneyong MM, Bykov, K, Bosco-Levy P, Dong Y-H, Levin R, et al. Risk of mortality with concomitant use of tamoxifen and selective serotonin reuptake inhibitors: multi-database cohort study. BMJ. 2016;354:i5014. https://doi.org/10.1136/bmj.i5014.
Lash TL, Cronin-Fenton D, Ahern TP, Rosenberg CL, Lunetta KL, Silliman RA, et al. Breast cancer recurrence risk related to concurrent use of SSRI antidepressants and tamoxifen. Acta Oncol. 2010;49:305–12.
Lash TL, Cronin-Fenton D, Ahern TP, Rosenberg CL, Lunetta KL, Silliman RA, et al. CYP2D6 inhibition and breast cancer recurrence in a population-based study in Denmark. J Natl Cancer Inst. 2011;103:489–500.
Juurlink D. Revisiting the drug interaction between tamoxifen and SSRI antidepressants. BMJ. 2016;354:i5309. https://doi.org/10.1136/bmj.i5309.
He Z-X, Chen X-W, Zhou Z-W, Zhou S-F. Impact of physiological, pathological and environmental factors on the expression and activity of human cytochrome P450 2D6 and implications in precision medicine. Drug Metab Rev. 2015;47:470–519.
Hansten PD, Horn JR. The top 100 drug interactions. A guide to patient management. Freeland (WA): H&H Publications; 2017.
Binkhorst L, van Gelder T, Loos WJ, de Jongh FE, Hamberg P, Ghobadi Moghaddam-Helmantel IM, et al. Effects of CYP induction by rifampicin on tamoxifen exposure. Clin Pharmacol Ther. 2012;92:62–7.
Kivisto KT, Villikka K, Nyman L, Anttila M, Neuvonen PJ. Tamoxifen and toremifene concentrations in plasma are greatly decreased by rifampin. Clin Pharmacol Ther. 1998;64:648–54.
Henderson SL, Teft WA, Kim RB. Profound reduction in tamoxifen active metabolite endoxifen in a breast cancer patient treated with rifampin prior to initiation of an anti-TNFα biologic for ulcerative colitis: a case report. BMC Cancer 2016;304. https://doi.org/10.1186/s12885-016-2342-x.
Horn JR, Hansten PD, Chan L-N. Proposal for a new tool to evaluate drug interaction cases. Ann Pharmacotherapy. 2007;41:674–80.
ter Heine R, Binkhorst L, de Graan AJM, de Bruijn P, Beijnen JH, Mathijssen RHJ, et al. Population pharmacokinetic modeling to assess the impact of CYP2D6 and CYP3A metabolic phenotypes on the pharmacokinetics of tamoxifen and endoxifen. Br J Clin Pharmacol. 2014;78:572–86.
Binkhorst L, Mathijssen RHJ, Jager A, van Gelder T. Individualization of tamoxifen therapy: much more than just CYP2D6 genotyping. Cancer Treat Rev. 2015;41:289–99.
Williamson B, Dooley KE, Zhang Y, Back DJ, Owen A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob Agents Chemother. 2013;57:6366–9.
Benson EA, Eadon MT, Desta Z, Liu Y, Lin H, Burgess KS, et al. Rifampin regulation of drug transporters gene expression and the association of MicroRNAs in human hepatocytes. Front Pharmacol. 2016;7:111. https://doi.org/10.3389/fphar.2016.00111.
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–53.
Nowell SA, Ahn J, Rae JM, Scheys JO, Trovato A, Sweeney C, et al. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Res Treat. 2005;91:249–58.
Lien EA, Anker G, Lonning PE, Solheim E, Ueland PM. Decreased serum concentrations of tamoxifen and its metabolites induced by aminoglutethimide. Cancer Res. 1990;50:5851–7.
Lonning PE, Kvinnsland S, Bakke OM. Effect of aminoglutethimide onantipyrine, theophylline, and digitoxin disposition in breast cancer. Clin Pharmacol Ther. 1984;36:796–802.
Lonning PE. Aminoglutethimide enzyme induction: pharmacological and endocrinological implications. Cancer Chemother Pharmacol. 1990;26:241–4.
Gryn SE, Teft WA, Kim RB. Profound reduction in the tamoxifen active metabolite endoxifen in a patient on phenytoin for epilepsy compared with a CYP2D6 genotype matched cohort. Pharmacogenet Genomics. 2014;24:367–9.
Ducharme J, Fried K, Shenouda G, Leyland-Jones B, Wainer IW. Tamoxifen metabolic patterns with a glioma patient population treated with high-dose tamoxifen. Br J Clin Pharmacol. 1997;43:189–93.
Teft WA, Mansell SE, Kim RB. Endoxifen, the active metabolite of tamoxifen, is a substrate of the efflux transporter P-glycoprotein (multidrug resistance 1). Drug Metab Dispos. 2011;39:558–62.
Teh LK, Mohamed NI, Salleh MZ, Rohaizak M, Shahrun NS, Saladina JJ, et al. The risk of recurrence in breast cancer patients treated with tamoxifen: polymorphisms of CYP2D6 and ABCB1. AAPS J. 2012;14:52–9.
Kiyotani K, Mushiroda T, Imamura CK, Hosono N, Tsunoda T, Kubo M, et al. Significant effect of polymorphisms in CYP2D6 and ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients. J Clin Oncol. 2010;28:1287–93.
Sensorn I, Sukasem C, Sirachainan E, Chamnanphon M, Pasomsub E, Trachu N, et al. ABCB1 and ABCC2 and the risk of distant metastasis in Thai breast cancer patients treated with tamoxifen. Onco Targets Ther. 2016;9:2121–9.
Collado-Borrell R, Escudero-Vilaplana V, Romero-Jimenez R, Iglesias-Peinado I, Herranz-Alonso A, Sanjurjo-Saez M. Oral antineoplastic agent interaction with medicinal plants and food: an issue to take into account. J Cancer Res Clin Oncol. 2016;142:2319–30.
Marchetti S, Mazzanti R, Beijnen JH, Schellens JH. Concise review: clinical relevance of drug drug and herb drug interactions medicated by the ABC transporter ABCB1 (MDR1, P-glycoprotein). Oncologist. 2007;12:927–41.
Soleymani S, Bahramsoltani R, Rahimi R, Abdollahi. Clinical risks of St John’s wort (Hypericum perforatum) co-administration. Expert Opin Drug Metab Toxicol. 2017;13:1047–62.
Shibayama Y, Ikeda R, Motoya T, Yamada K. St. John’s wort (Hypericum perforatum) induces overexpression of multidrug resistance protein 2 (MRP2) in rats: a 30-day ingestion study. Food Chem Toxicol. 2004;42:995–1002.
Shibayama Y, Kawachi A, Onimaru S, Tokunaga J, Ikeda R, Nishida K, et al. Effect of pre-treatment with St. John’s wort on nephrotoxicity of cisplatin in rats. Life Sci. 2007;81:103–8.
L’Esperance S, Frenette S, Dionne A, Dionne J-Y. Pharmacological and non-hormonal treatment of hot flashes in breast cancer survivors: CEPO review and recommendations. Support Care Cancer. 2013;21:1461–74.
Goetz MP, Suman VJ, Reid JM, Northfelt DW, Mahr MA, Ralya AT, et al. First-in-human phase I study of the tamoxifen metabolite Z-endoxifen in women with endocrine-refractory metastatic breast cancer. J Clin Oncol. 2017;35:3391–400.
Rae JM, Drury S, Hayes DF, Stearns V, Thibert JN, Haynes BP, et al. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst. 2012;104:452–60.
Cronin-Fenton D, Lash TL, Sorensen HT. Selective serotonin reuptake inhibitors and adjuvant tamoxifen therapy: risk of breast cancer recurrence and mortality. Future Oncol. 2010;6:877–80.
Ahern TP, Hertz DL, Damkier P, Ejlertsen B, Hamilton-Dutoit SJ, et al. Cytochrome P-450 2D6 (CYP2D6) genotype and breast cancer recurrence in tamoxifen-treated patients: evaluating the importance of loss of heterozygosity. Am J Epidemiol. 2017;185:75–85.
Cronin-Fenton DP, Damkier P, Lash TL. Metabolism and transport of tamoxifen in relation to its effectiveness: new perspectives on an ongoing controversy. Future Oncol. 2014;10:107–22.
Pensées Pascal B, Writings Other. Honor Levi, translator. Oxford: Oxford University Press; 1995.
Hung IFN, Wu AK, Cheng VC, Tang BS, To KW, Yeung CK, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis. 2005;41:291–300.
Rollot F, Pajot O, Chauvelot-Moachon L, Nazal EM, Kelaidi C, Blanche P. Acute colchicine intoxication during clarithromycin administration. Ann Pharmacother. 2004;38:2074–7.
Dogukan A, Oymak FS, Taskapan H, Guven M, Tokgoz B, Utas C. Acute fatal colchicine intoxication in a patient on continuous ambulatory peritoneal dialysis (CAPD). Possible role of clarithromycin administration. Clin Nephrol. 2001;55:181–2.
FDA, Docket No. FDA-2010-P-0614.
Hansten PD. Premature factulation: the ignorance of certainty and the ghost of montaigne. Port Ludlow: Philoponus Press; 2009.
Saladores P, Murdter T, Eccles D, Chowbay B, Zgheib NK, Winter S, et al. Tamoxifen metabolism predicts drug concentrations and outcome in premenopausal patients with early breast cancer. Pharmacogenomics J. 2015;15:84–94.
Du J, Sun Y, Lu YY, Lau E, Zhao M, Zhou QM, et al. Berberine and evodiamine act synergistically against human breast cancer MCF-7 cells by inducing cell cycle arrest and apoptosis. Anticancer Res. 2017;37:6141–51.
de Graan A-JM, Teunissen SF, de Vos FYFL, Loos WJ, van Schaik RHN, de Jongh FE, et al. Dextromethorphan as a phenotyping test to predict endoxifen exposure in patients on tamoxifen treatment. J Clin Oncol. 2011;29:3240–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No source of funding was used to prepare this article.
Conflict of interest
The author declares that he has no competing interests.
Author’s information
PDH has worked in the field of drug–drug interactions for 50 years. His books on drug interactions have sold more than 1 million copies since 1971, and have been translated into Dutch, French, German, Japanese, Korean, Portuguese, and Spanish. He is currently Professor Emeritus at the University of Washington in Seattle and has held positions in the Division of Clinical Pharmacology at Stanford University School of Medicine (working with Professor Stanley N. Cohen) and at Washington State University. He has taught philosophy of science at the University of Washington for over a decade and is currently in the process of setting up a similar class at the University of California San Francisco. He is also currently working as a consultant on grants at the University of Arizona which focus on patient-specific clinical decision support for drug interactions.
Author details
Professor Emeritus, School of Pharmacy, University of Washington, Seattle, Washington, USA.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hansten, P.D. The Underrated Risks of Tamoxifen Drug Interactions. Eur J Drug Metab Pharmacokinet 43, 495–508 (2018). https://doi.org/10.1007/s13318-018-0475-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-018-0475-9