
Abstract
Palladium nanoparticles supported on carbon Vulcan XC72 (Pd/C) and biocarbon (Pd/BC) synthesized by sodium borohydride process were used as catalysts for ethanol electro-oxidation in alkaline media. The biocarbon (BC) from coconut shell with mesoporous and high surface area (792 m2 g−1) was obtained by carbonization at 900 °C and the hydrothermal treatment in a microwave oven. The D-band and G-band intensity ratio (ID/IG) from Raman analysis showed high disorder of the biocarbon, while X-ray photoelectron spectroscopy (XPS) suggests higher percentage of oxygen groups on the surface of biocarbon than of Vulcan XC72. From X-ray diffraction (XRD), it was observed peaks in 2θ degree related to the face centered cubic (fcc) structure of palladium and the mean crystallite sizes calculated based on the diffraction peak of Pd (220) were 5.6 nm for Pd/C and 5.3 nm for Pd/BC. Using Transmission Electron Microscope (TEM), it was observed particles well dispersed on both carbons support materials. The electrocatalytic activity of the materials was investigated by cyclic voltammetry (CV) and chronoamperometry (CA) experiments. The peak current density (on CV experiments) from ethanol electro-oxidation on Pd/BC was 50% higher than on Pd/C, while the current density measured at 15 min of CA experiments was 80% higher on Pd/BC than on Pd/C. The higher catalytic activity of Pd/BC might be related to the large surface area of the biocarbon (792 m2 g−1) vs (239 m2 g−1) of Vulcan carbon, the defects of the biocarbon structure and higher amount of oxygen on the surface than Carbon Vulcan XC 72.
Similar content being viewed by others

Introduction
The search for new energy sources based on the concept of clean and renewable energy has been intensified in the recently years [1,2,3]. Fuel cells are pointed out as a promising technology for clean energy generation and its concept is based on the conversion of chemical energy into electricity [4,5,6]. Liquid fuels cells (LFC) are considerably more convenient in terms of easy handling than gaseous hydrogen [7], and low temperature fuel cells based on ion exchange membrane can be fed with different fuels, such as methanol, ethanol, formic acid, etc., [8,9,10]. Ethanol has been recognized as a promising fuel, since it can be produced directly from the fermentation of biomass; thus, it is a renewable fuel that does not promote alterations on the natural balance of carbon dioxide in the atmosphere [11,12,13].
It has been reported that the ethanol electro-oxidation kinetic in alkaline media is enhanced compared to the acid media [14, 15]. However, catalysts are required to promote ethanol electro-oxidation. Palladium is pointed out as the metal with the highest catalytic activity for ethanol electro-oxidation in alkaline media [16,17,18] and catalysts are usually synthesized as nanoparticles in order to increase the surface area which increases the catalytic rate compared to the bulk materials [19, 20].
Support materials for catalysts nanoparticles are very important in the ethanol electro-oxidation process. An appropriate support must attend some requirements, such as low cost, large surface area, high electrical conductivity and stability in the catalysis process [21,22,23]. Carbon attend satisfactorily all these requirements [22, 23]. Carbon with different structures and morphology have been investigated in the literature as support material for nanoparticles, such as carbon nanotubes [24,25,26], carbon nanofibers [27] graphene [28] and carbon black [22, 23].
Considering support materials, carbon from biomass has been considered as a suitable option [1, 29,30,31]. Among different sources, biomass coconut shell seems to be a good choice, because it is abundant, cheap, environmentally safe, commercially available and sustainable; moreover, it is suitable for the preparation of porous carbons due to its excellent natural structure [32, 33]. Additionally, it presents high surface area ranging from 800 to 1500 m2 g−1 [33, 34], which is much higher than that reported for carbon Vulcan XC-72 (239 m2 g−1) [35] (commercial support usually used). The mesoporous structure is highly desirable for the faster diffusion of the electrolyte and larger molecules (such as ethanol) into the internal porous surface of carbons. Consequently, this structure provides numerous accessible active sites and facilitates efficient mass transport in the catalyst layers [36, 37].
Thus, the coconut shell is a very interesting option to be used as support for catalysts nanoparticles for ethanol electro-oxidation.
In the present work, palladium nanoparticles were supported on high surface area porous biocarbon from coconut shell and on carbon Vulcan XC-72. The materials were used as catalysts for ethanol electro-oxidation in alkaline media. As far as we know, this is the first report related to the ethanol electro-oxidation in alkaline media on palladium nanoarticles supported on porous biocarbon from coconut shell.
Experimental
The biocarbon from coconut shell was obtained by carbonization at temperature of 900 °C for 40 min at heating rate of 10 °C/min and in a second step the hydrothermal treatment in a microwave oven at 75 °C for 20 min in 0.3 mol L−1 HNO3. Carbon Vulcan XC72 Cabot was previous treated at in a tubular oven at 800 °C under argon atmosphere as reported in our recently publications [38, 39].
Palladium nanoparticles were synthesized by the sodium borohydride reduction process [22], using Pd(NO3)2.2H2O (Sigma-Aldrich). In the synthesis, carbon Vulcan XC-72 or biocarbon from coconut shell was first dispersed in isopropanol/water solution (50/50, v/v). The mixture was homogenized under stirring and then the metal precursor was added to obtain 20 wt% of metal loading, and placed in an ultrasonic bath for 5 min. Then, 10 mL of 0.15 M NaBH4 in 0.1 mol L−1 KOH was added in one portion under stirring at room temperature. The resulting colloidal solution was stirred for 15 min more before filtering and washing the solids with water and then dried at 70 °C for 2 h.
A Rigaku diffractometer model Miniflex II using Cu Kα radiation source (0.15406 nm) was used to characterize the synthesized materials by X-ray diffraction (XRD). The X-ray diffraction patterns were recorded with a step size of 0.05° and a scan time of 2 s per step from 2θ = 20° to 90°. Raman measurements of carbon supports were performed on LAMULT (Xplora) da Horiba spectrometer with a laser wavelength of 532 nm.
XPS experiments were performed in a K-alpha surface analysis (Thermo Scientific) equipment with an Al-Kα X-ray source (1486.6 eV) and a flood gun. The investigated area was an ellipse of approximately 300 μm in diameter and three different areas of each sample were examined. Peaks were fitted using the Avantage software (Thermo Scientific) using a Gaussian–Lorentzian product function and integrated at their full widths at half maximum (FWHM) for quantification. The binding energies (BE) of the spectra were corrected with that of adventitious carbon C 1 s (C–C, C–H) at 284.8 eV.
A JEOL transmission electron microscope (TEM-FEG) JEM-2100F operated at 200 kV was used to obtain information about the distribution and sizes of the nanoparticles. A JEOL JSM6010 LA scanning electron microscope (SEM) was used to obtain information of the carbon morphology and Brunauer–Emmett–Teller (BET) analysis was performed in a Quantachrome, ChemBET 3000 to obtain the surface area of the biocarbon (792 m2 g−1).
Electrochemical measurements were done with a bipotentiostat/galvanostat μStat 400 DropSens at room temperature and in a three-compartment electrochemical cell. A platinum foil was used as counter electrode and the Hg/HgO as reference electrode. A Glassy carbon (GC) with the geometric area of 0.031 cm2 was used as working electrodes to support the synthesized materials. Alumina (1 μm) was employed to polish the GC support before each experiment. In all experimental procedures, Ultrapure water obtained from a Milli-Q system (Millipore®) was used.
The working electrodes were constructed by dispersing 3 mg of the electrocatalyst powder in 900 μL of water, 100 of μL isopropyl alcohol and 20 μL of 5% Nafion®. Then, the mixture was dispersed in an ultrasonic bath for 30 min. Shortly thereafter, aliquots of 5 μL of the dispersion fluid were deposited onto the GC surface (The catalyst loading on the working electrode was 0.47 mg cm−2) and dried for 20 min at 60 °C. 1 mol L−1 KOH solution was used in all the electrochemical measurements.
Cyclic voltammograms (CV) in ethanol-free solutions were carried out at the potential range of − 0.85 V to 0.1 V vs Hg/HgO at a scan rate of 20 mV s−1. The electrocatalysts were cycled for ten consecutive cycles resulting in the reproducible shape of the CVs. The CVs in 1 mol L−1 KOH + 1 mol L−1 ethanol were carried out at a scan rate of 20 mV s−1 from − 0.85 to 0.1 V. The electrocatalysts were cycled for three consecutive cycles and the third cycle is shown. Chronoamperometric experiments were carried out at − 0.35 V for 15 min.
Results and discussion
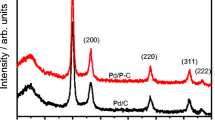
Figure 1 shows the XRD patterns of the electrocatalysts Pd/C and Pd/BC. In all XRD patterns, a broad peak at about 25º 2θ due to the (022) reflection of the hexagonal structure of carbon can be seen [40, 41]. Furthermore, it was seen peaks related to the palladium face centered cubic (fcc) structure at around 2θ = 39°, 46°, 67° and 81°, corresponding to (111), (200), (220) and (311) planes, respectively [42, 43]. The mean crystallite size estimated using Scherrer equation and (220) peak [4, 44] was 5.6 nm for Pd/C and 5.3 nm for Pd/BC, which is in agreement with the palladium nanoparticles synthesized using sodium borohydrate process [45, 46].

X-ray diffraction patterns for Pd/C and Pd/BC electrocatalysts
The carbon supports were also characterized by Raman spectroscopy (Fig. 2). The ratio of intensities of D-band (~ 1335 cm−1) and G-band (~ 1590 cm−1) was used to measure the carbon disorder [30, 47]. The G-band reveals the presence of graphitic in carbon materials and the D-band provides information about the structure defects and disorder in the carbon structures [30]. The ID/IG of the biocarbon was 1.25, and of carbon Vulcan XC 72 was 0.71. Higher ID/IG values are associated with the higher disorder in the biocarbon support material [30, 48].

Raman spectra of carbon Vulcan XC 72 and biocarbon
X-ray photoelectron spectroscopy analysis was performed to obtain chemical information of the surface of the materials. Figure 3a shows the spectra of Pd/BC and Pd/C electrocatalysts. Pd 3d region exhibits a doublet at binding energies of ~ 335.7 assigned to Pd 3d5\2 and 340.9 assigned to Pd 3d3\2 with a spin-orbit splitting of about ~ 5.2 eV, in agreement with the literature [49,50,51] and the tails on the left side of each peak suggest the presence of oxide palladium with metallic palladium on both samples. The components located at ~ 335.3, ~ 336.2 and ~ 337.3–336.6 eV and attributed to metallic Pd, Pd(II) and Pd(IV) phases, respectively [52, 53].

XPS results of the Pd 3d region (a), C 1 s (b) and O 1 s (c)
It was not possible to observe any considerable shift of the binding energies in the spectra of Pd nanoparticles supported on both support materials. In the Fig. 3b, it is possible to see the C 1 s deconvoluted spectrum. The dominant peak at about 284.4 eV is assigned to graphitic carbon phase, whereas the peak at around 286 eV is related to hydrocarbons (C–H) from defects in the graphitic structure [54,55,56]. The peak at around 286.7 eV is related to the epoxy carbon, and at ~ 288 eV associated with the carbonyl carbon C=O [57]. It is important to point out that the graphitic carbon phase in biocarbon was ~ 77%, while in Vulcan carbon was ~ 73%. The O 1 s peak (Fig. 3c) of the biocarbon consists of ~ 3.50% of the total species, while on carbon Vulcan ~ 2.1%. Thus, the biocarbon has higher amount of oxygen on the surface than Carbon Vulcan XC 72.
In Fig. 4a, b, the SEM micrographs of the biocarbon are shown. As can be seen, the biocarbon consists of high degree of porosity. The presence of mesoporous is characteristic of coconut shell [32, 33, 58]. Figure 4c shows the SEM micrograph of palladium supported on the biocarbon; it is possible to observe the presence of palladium on the surface and into the porous of the biocarbon. In the dark field micrograph from STEM (Fig. 4d), it is possible to observe nanoparticles from 5 to 8 nm supported on the biocarbon, and in Fig. 4e (TEM micrograph) it is possible to see the palladium nanoparticle of 6 nm. From SEM micrograph (Fig. 4f), it is possible to see that carbon Vulcan XC 72 consists of clusters of spheres [59]. Thus, the morphology is completely different from the biocarbon from coconut shell. Figure 4g shows the TEM micrograph of palladium nanoparticles supported on carbon Vulcan XC 72 from 3 to 8 nm; as suggested by Scherrer equation the palladium particle sizes supported on both materials are very close and lower than 8 nm.

SEM micrographs of biocarbon (a) and (b), Pd/BC (c), STEM micrograph of Pd/BC (d), TEM micrograph of Pd/BC (e), SEM micrographs of carbon Vulcan XC 72 (f) and TEM micrograph of Pd/C (g)
In Fig. 5, the results from cyclic voltammetry experiments in 1 mol L−1 KOH in the potential range of − 0.85 to 0.1 V vs Hg/HgO are shown. The shape of the CVs of palladium catalysts supported on both materials is similar and in agreement with the results from the literature [52, 60, 61]. The region associated with palladium oxide formation in the forward scan from − 0.20 to 0.1 V, and the palladium oxide reduction at around − 0.2 V in the backward scan, was observed [52, 62]. Additionally, the peak at ~ − 0.4 V in the forward scan due to OH adsorption on palladium can be seen [46, 52]. Thus, as expected the carbon support from the different sources has not promoted modifications of the CV shape of palladium catalysts.

Voltammograms of Pd/C and Pd/BC in 1 mol L−1 KOH at 20 mVs−1
In Fig. 6, the results from CVs experiments in 1 mol L−1 KOH + 1 mol L−1 ethanol are shown. The Pd/BC shows higher catalytic activity towards ethanol electro-oxidation than Pd/C. Using the Pd/BC, the onset potential from ethanol electro-oxidation was 60 mV lower than on Pd/C, and the peak current density from ethanol electro-oxidation was about 50% higher than on Pd/C. The higher catalytic activity might be related to the large surface area of the biocarbon (792 m2 g−1) vs (239 m2 g−1) of Vulcan carbon [35], which could increase the density of the catalyst active sites accessible to reactants [63], and also due to the mesoporous structures, since it is reported that the presence of porous facilitate the diffusion of the electrolyte in the material [33] and consequently the ethanol. This enhancement in the diffusion may increase the mass transport process of ethanol [63] and, therefore, the ethanol electro-oxidation rate. The influence of the carbon structure in the catalyst activity for different reaction can be seen in the literature [27, 29, 30, 64], which is associated with the surface area, defects in the structure and the functional groups on the carbon support. As shown in the XPS results, the biocarbon has higher amount of oxygen species on the surface than carbon Vulcan XC 72. The peak current density from ethanol electro-oxidation on Pd/BC was about twice higher than that one obtained for electro-oxidation on Pd/CNT reported in the literature [65].

Voltammograms of Pd/C and Pd/BC in 1 moL L−1 KOH + 1 moL L−1 ethanol at 20 mVs−1
Figure 7 displays the results from chronoamperometry experiments at − 0.35 V during 15 min in the presence of 1 mol L−1 ethanol + 1 mol L−1 KOH. In the CA experiments, the current value decreases faster in the first minutes due to the instability of the nanoparticles and the poisoning of the surface sites [4]. As in the CV experiments, using Pd/BC was obtained better result than on Pd/C electrocatalyst. The current density from ethanol electro-oxidation at the end of the CA experiments on Pd/BC was ~ 80% higher than on Pd/C. Thus, it is obvious the improvement in the catalytic activity using biocarbon as support for the palladium nanoparticles.

Chronoamperometric results at − 0.35 V of Pd/C and Pd/BC in 1 moL L−1 KOH + 1 moL L−1 ethanol
As can be seen, in both CV and CA experiments, biocarbon improves the catalytic activity of palladium towards ethanol electro-oxidation. Thus, this suitable material could be used in the future works as support for bimetallic or multimetallic Pd-based materials catalysts for ethanol electro-oxidation.
Conclusions
The result of this work showed that biocarbon from coconut shell is a suitable support for palladium nanoparticles towards ethanol electro-oxidation reaction in alkaline media. Mesoporous of biocarbon was observed in the micrograph from SEM and according to TEM micrographs the palladium nanoparticles supported on both carbon support (carbon Vulcan XC 72 and biocarbon) are smaller than 8 nm with very narrow size. The ID/IG from Raman analysis showed higher disorder of the biocarbon (1.25) than that of carbon Vulcan (0.71) and according to the BET experiments the biocarbon has surface area (792 m2 g−1) ~ 3.4 higher than carbon Vulcan XC-72 (236 m2 g−1).
In CV experiments, it was seen that the onset potential of ethanol electro-oxidation was 60 mV lower on Pd/BC than on Pd/C and the peak current density from ethanol electro-oxidation on Pd/BC was 50% higher than on Pd/C. In the CA analysis, the current density measured at the end of the experiment was 80% higher on Pd/BC than on Pd/C. The enhancement in the catalytic activity might be related to the higher surface area, higher disorder of biocarbon, and also higher percentage of oxygen groups on the surface (as suggested by XPS analysis) and the mesoporous structures that facilitate the diffusion of the ethanol into the support material.
References
Wang, G., Peng, H., Qiao, X., Du, L., Li, X., Shu, T., Liao, S.: Biomass-derived porous heteroatom-doped carbon spheres as a high-performance catalyst for the oxygen reduction reaction. Int J Hydrog Energy 41(32), 14101–14110 (2016). https://doi.org/10.1016/j.ijhydene.2016.06.023
Chen, C.-Y., Lai, W.-H., Yan, W.-M., Chen, C.-C., Hsu, S.-W.: Effects of nitrogen and carbon monoxide concentrations on performance of proton exchange membrane fuel cells with Pt–Ru anodic catalyst. J Power Sources 243, 138–146 (2013). https://doi.org/10.1016/j.jpowsour.2013.06.003
Crisafulli, R., Antoniassi, R.M., Oliveira Neto, A., Spinacé, E.V.: Acid-treated PtSn/C and PtSnCu/C electrocatalysts for ethanol electro-oxidation. Int J Hydrog Energy 39(11), 5671–5677 (2014). https://doi.org/10.1016/j.ijhydene.2014.01.111
Silva, J.C.M., Anea, B., De Souza, R.F.B., Assumpcao, M.H.M.T., Calegaro, M.L., Neto, A.O., Santos, M.C.: Ethanol oxidation reaction on IrPtSn/C electrocatalysts with low Pt content. J Braz Chem Soc 24(10), 1553–1560 (2013). https://doi.org/10.5935/0103-5053.20130196
Maffei, N., Pelletier, L., McFarlan, A.: A high performance direct ammonia fuel cell using a mixed ionic and electronic conducting anode. J Power Sources 175(1), 221–225 (2008). https://doi.org/10.1016/j.jpowsour.2007.09.040
Bonesi, A.R., Moreno, M.S., Triaca, W.E., Luna, A.M.C.: Modified catalytic materials for ethanol oxidation. Int J Hydrog Energy 35(11), 5999–6004 (2010). https://doi.org/10.1016/j.ijhydene.2009.12.093
Zignani, S.C., Baglio, V., Linares, J.J., Monforte, G., Gonzalez, E.R., Aricò, A.S.: Performance and selectivity of PtxSn/C electro-catalysts for ethanol oxidation prepared by reduction with different formic acid concentrations. Electrochim Acta 70, 255–265 (2012). https://doi.org/10.1016/j.electacta.2012.03.055
Rizo, R., Sebastián, D., Lázaro, M.J., Pastor, E.: On the design of Pt-Sn efficient catalyst for carbon monoxide and ethanol oxidation in acid and alkaline media. Appl Catal B 200, 246–254 (2017). https://doi.org/10.1016/j.apcatb.2016.07.011
Yu, E.H., Scott, K.: Development of direct methanol alkaline fuel cells using anion exchange membranes. J Power Sources 137(2), 248–256 (2004). https://doi.org/10.1016/j.jpowsour.2004.06.004
Matsuoka, K., Iriyama, Y., Abe, T., Matsuoka, M., Ogumi, Z.: Alkaline direct alcohol fuel cells using an anion exchange membrane. J Power Sources 150, 27–31 (2005). https://doi.org/10.1016/j.jpowsour.2005.02.020
da Silva, S.G., Assumpção, M.H.M.T., de Souza, R.F.B., Buzzo, G.S., Spinacé, E.V., Neto, A.O., Silva, J.C.M.: Electrochemical and fuel cell evaluation of PtIr/C electrocatalysts for ethanol electrooxidation in alkaline medium. Electrocatalysis 5(4), 438–444 (2014). https://doi.org/10.1007/s12678-014-0213-2
Switzer, E.E., Olson, T.S., Datye, A.K., Atanassov, P., Hibbs, M.R., Cornelius, C.J.: Templated Pt-Sn electrocatalysts for ethanol, methanol and CO oxidation in alkaline media. Electrochim Acta 54(3), 989–995 (2009)
Gao, H., Liao, S., Liang, Z., Liang, H., Luo, F.: Anodic oxidation of ethanol on core-shell structured Ru@PtPd/C catalyst in alkaline media. J Power Sources 196(15), 6138–6143 (2011). https://doi.org/10.1016/j.jpowsour.2011.03.031
Santasalo-Aarnio, A., Tuomi, S., Jalkanen, K., Kontturi, K., Kallio, T.: The correlation of electrochemical and fuel cell results for alcohol oxidation in acidic and alkaline media. Electrochim Acta 87, 730–738 (2013). https://doi.org/10.1016/j.electacta.2012.09.100
Cui, G., Song, S., Shen, P.K., Kowal, A., Bianchini, C.: First-principles considerations on catalytic activity of Pd toward ethanol oxidation. J Phys Chem C 113(35), 15639–15642 (2009). https://doi.org/10.1021/jp900924s
Ma, L., Chu, D., Chen, R.: Comparison of ethanol electro-oxidation on Pt/C and Pd/C catalysts in alkaline media. Int J Hydrog Energy 37(15), 11185–11194 (2012). https://doi.org/10.1016/j.ijhydene.2012.04.132
Zhang, F., Zhou, D., Zhou, M.: Ethanol electrooxidation on Pd/C nanoparticles in alkaline media. J Energy Chem 25(1), 71–76 (2016). https://doi.org/10.1016/j.jechem.2015.10.013
Nguyen, S.T., Ling Tan, D.S., Lee, J.-M., Chan, S.H., Wang, J.Y., Wang, X.: Tb promoted Pd/C catalysts for the electrooxidation of ethanol in alkaline media. Int J Hydrog Energy 36(16), 9645–9652 (2011). https://doi.org/10.1016/j.ijhydene.2011.05.049
Han, S.-B., Song, Y.-J., Lee, J.-M., Kim, J.-Y., Park, K.-W.: Platinum nanocube catalysts for methanol and ethanol electrooxidation. Electrochem Commun 10(7), 1044–1047 (2008). https://doi.org/10.1016/j.elecom.2008.04.034
Maillard, F., Savinova, E.R., Simonov, P.A., Zaikovskii, V.I., Stimming, U.: Infrared spectroscopic study of CO adsorption and electro-oxidation on carbon-supported Pt nanoparticles: interparticle versus intraparticle heterogeneity. J Phys Chem B 108(46), 17893–17904 (2004). https://doi.org/10.1021/jp0479163
Nguyen, S.T., Yang, Y., Wang, X.: Ethanol electro-oxidation activity of Nb-doped-TiO2 supported PdAg catalysts in alkaline media. Appl Catal B 113–114, 261–270 (2012). https://doi.org/10.1016/j.apcatb.2011.11.046
Silva, J.C.M., Piasentin, R.M., Spinacé, E.V., Neto, A.O., Baranova, E.A.: The effect of antimony-tin and indium-tin oxide supports on the catalytic activity of Pt nanoparticles for ammonia electro-oxidation. Mater Chem Phys 180, 97–103 (2016). https://doi.org/10.1016/j.matchemphys.2016.05.047
Antolini, E.: Nitrogen-doped carbons by sustainable N- and C-containing natural resources as nonprecious catalysts and catalyst supports for low temperature fuel cells. Renew Sustain Energy Rev 58, 34–51 (2016). https://doi.org/10.1016/j.rser.2015.12.330
Hiltrop, D., Masa, J., Maljusch, A., Xia, W., Schuhmann, W., Muhler, M.: Pd deposited on functionalized carbon nanotubes for the electrooxidation of ethanol in alkaline media. Electrochem Commun 63, 30–33 (2016). https://doi.org/10.1016/j.elecom.2015.11.010
Habibi, B., Mohammadyari, S.: Facile synthesis of Pd nanoparticles on nano carbon supports and their application as an electrocatalyst for oxidation of ethanol in alkaline media: the effect of support. Int J Hydrog Energy 40(34), 10833–10846 (2015). https://doi.org/10.1016/j.ijhydene.2015.07.021
Karousis, N., Tagmatarchis, N., Tasis, D.: Current progress on the chemical modification of carbon nanotubes. Chem Rev 110(9), 5366–5397 (2010). https://doi.org/10.1021/cr100018g
Maiyalagan, T., Scott, K.: Performance of carbon nanofiber supported Pd–Ni catalysts for electro-oxidation of ethanol in alkaline medium. J Power Sources 195(16), 5246–5251 (2010). https://doi.org/10.1016/j.jpowsour.2010.03.022
Ghosh, S., Remita, H., Kar, P., Choudhury, S., Sardar, S., Beaunier, P., Roy, P.S., Bhattacharya, S.K., Pal, S.K.: Facile synthesis of Pd nanostructures in hexagonal mesophases as a promising electrocatalyst for ethanol oxidation. J Mater Chem A 3(18), 9517–9527 (2015). https://doi.org/10.1039/C5TA00923E
Zhang, M., Jin, X., Wang, L., Sun, M., Tang, Y., Chen, Y., Sun, Y., Yang, X., Wan, P.: Improving biomass-derived carbon by activation with nitrogen and cobalt for supercapacitors and oxygen reduction reaction. Appl Surf Sci 411, 251–260 (2017). https://doi.org/10.1016/j.apsusc.2017.03.097
Zheng, X., Cao, X., Wu, J., Tian, J., Jin, C., Yang, R.: Yolk-shell N/P/B ternary-doped biocarbon derived from yeast cells for enhanced oxygen reduction reaction. Carbon 107, 907–916 (2016). https://doi.org/10.1016/j.carbon.2016.06.102
Zhao, X., Zhu, J., Liang, L., Li, C., Liu, C., Liao, J., Xing, W.: Biomass-derived N-doped carbon and its application in electrocatalysis. Appl Catal B 154–155, 177–182 (2014). https://doi.org/10.1016/j.apcatb.2014.02.027
Sun, L., Tian, C., Li, M., Meng, X., Wang, L., Wang, R., Yin, J., Fu, H.: From coconut shell to porous graphene-like nanosheets for high-power supercapacitors. J Mater Chem A 1(21), 6462–6470 (2013). https://doi.org/10.1039/C3TA10897J
Mi, J., Wang, X.-R., Fan, R.-J., Qu, W.-H., Li, W.-C.: Coconut-shell-based porous carbons with a tunable micro/mesopore ratio for high-performance supercapacitors. Energy Fuels 26(8), 5321–5329 (2012). https://doi.org/10.1021/ef3009234
Guo, S., Peng, J., Li, W., Yang, K., Zhang, L., Zhang, S., Xia, H.: Effects of CO2 activation on porous structures of coconut shell-based activated carbons. Appl Surf Sci 255(20), 8443–8449 (2009). https://doi.org/10.1016/j.apsusc.2009.05.150
Thanh Ho, V.T., Pillai, K.C., Chou, H.-L., Pan, C.-J., Rick, J., Su, W.-N., Hwang, B.-J., Lee, J.-F., Sheu, H.-S., Chuang, W.-T.: Robust non-carbon Ti0.7Ru0.3O2 support with co-catalytic functionality for Pt: enhances catalytic activity and durability for fuel cells. Energy Environ Sci 4(10), 4194–4200 (2011). https://doi.org/10.1039/c1ee01522b
Kaur, P., Singh, S., Verma, G.: Facile synthesis of mesoporous carbon material from treated kitchen waste for energy applications. Mater Renew Sustain Energy 7(2), 9 (2018). https://doi.org/10.1007/s40243-018-0116-x
Lebedeva, M.V., Yeletsky, P.M., Ayupov, A.B., Kuznetsov, A.N., Yakovlev, V.A., Parmon, V.N.: Micro–mesoporous carbons from rice husk as active materials for supercapacitors. Mater Renew Sustain Energy 4(4), 20 (2015). https://doi.org/10.1007/s40243-015-0061-x
Ribeiro, V.A., de Freitas, I.C., Neto, A.O., Spinacé, E.V., Silva, J.C.M.: Platinum nanoparticles supported on nitrogen-doped carbon for ammonia electro-oxidation. Mater Chem Phys 200, 354–360 (2017). https://doi.org/10.1016/j.matchemphys.2017.07.088
Silva, J.C.M., de Freitas, I.C., Neto, A.O., Spinacé, E.V., Ribeiro, V.A.: Palladium nanoparticles supported on phosphorus-doped carbon for ethanol electro-oxidation in alkaline media. Ionics 24(4), 1111–1119 (2018). https://doi.org/10.1007/s11581-017-2257-9
Assumpção, M.H.M.T., da Silva, S.G., de Souza, R.F.B., Buzzo, G.S., Spinacé, E.V., Neto, A.O., Silva, J.C.M.: Direct ammonia fuel cell performance using PtIr/C as anode electrocatalysts. Int J Hydrog Energy 39(10), 5148–5152 (2014). https://doi.org/10.1016/j.ijhydene.2014.01.053
Silva, J.C.M., da Silva, S.G., De Souza, R.F.B., Buzzo, G.S., Spinacé, E.V., Neto, A.O., Assumpção, M.H.M.T.: PtAu/C electrocatalysts as anodes for direct ammonia fuel cell. Appl Catal A 490, 133–138 (2015). https://doi.org/10.1016/j.apcata.2014.11.015
Li, J., Tian, Q., Jiang, S., Zhang, Y., Wu, Y.: Electrocatalytic performances of phosphorus doped carbon supported Pd towards formic acid oxidation. Electrochim Acta 213, 21–30 (2016). https://doi.org/10.1016/j.electacta.2016.06.041
Neto, A.O., da Silva, S.G., Buzzo, G.S., de Souza, R.F.B., Assumpção, M.H.M.T., Spinacé, E.V., Silva, J.C.M.: Ethanol electrooxidation on PdIr/C electrocatalysts in alkaline media: electrochemical and fuel cell studies. Ionics 21(2), 487–495 (2015). https://doi.org/10.1007/s11581-014-1201-5
Lomocso, T.L., Baranova, E.A.: Electrochemical oxidation of ammonia on carbon-supported bi-metallic PtM (M = Ir, Pd, SnOx) nanoparticles. Electrochim Acta 56(24), 8551–8558 (2011). https://doi.org/10.1016/j.electacta.2011.07.041
Neto, A.O., da Silva, S.G., Buzzo, G.S., de Souza, R.F.B., Assumpção, M.H.M.T., Spinacé, E.V., Silva, J.C.M.: Ethanol electrooxidation on PdIr/C electrocatalysts in alkaline media: electrochemical and fuel cell studies. Ionics 21(2), 487–495 (2015). https://doi.org/10.1007/s11581-014-1201-5
Assumpção, M.H.M.T., da Silva, S.G., De Souza, R.F.B., Buzzo, G.S., Spinacé, E.V., Santos, M.C., Neto, A.O., Silva, J.C.M.: Investigation of PdIr/C electrocatalysts as anode on the performance of direct ammonia fuel cell. J Power Sources 268, 129–136 (2014). https://doi.org/10.1016/j.jpowsour.2014.06.025
Santos Pereira, V., da Silva, J.C.M., Oliveira Neto, A., Spinacé, E.V.: PtRu nanoparticles supported on phosphorous-doped carbon as electrocatalysts for methanol electro-oxidation. Electrocatalysis 8(3), 245–251 (2017). https://doi.org/10.1007/s12678-017-0360-3
Anstey, A., Vivekanandhan, S., Rodriguez-Uribe, A., Misra, M., Mohanty, A.K.: Oxidative acid treatment and characterization of new biocarbon from sustainable Miscanthus biomass. Sci Total Environ 550((Supplement C)), 241–247 (2016). https://doi.org/10.1016/j.scitotenv.2016.01.015
Lamb, R.N., Ngamsom, B., Trimm, D.L., Gong, B., Silveston, P.L., Praserthdam, P.: Surface characterisation of Pd–Ag/Al2O3 catalysts for acetylene hydrogenation using an improved XPS procedure. Appl Catal A 268(1–2), 43–50 (2004). https://doi.org/10.1016/j.apcata.2004.03.041
Batista, J., Pintar, A., Mandrino, D., Jenko, M., Martin, V.: XPS and TPR examinations of γ-alumina-supported Pd-Cu catalysts. Appl Catal A 206(1), 113–124 (2001). https://doi.org/10.1016/S0926-860X(00)00589-5
Hasik, M., Bernasik, A., Drelinkiewicz, A., Kowalski, K., Wenda, E., Camra, J.: XPS studies of nitrogen-containing conjugated polymers–palladium systems. Surf Sci 507–510, 916–921 (2002). https://doi.org/10.1016/S0039-6028(02)01372-9
Geraldes, A.N., da Silva, D.F., Pino, E.S., da Silva, J.C.M., de Souza, R.F.B., Hammer, P., Spinacé, E.V., Neto, A.O., Linardi, M., dos Santos, M.C.: Ethanol electro-oxidation in an alkaline medium using Pd/C, Au/C and PdAu/C electrocatalysts prepared by electron beam irradiation. Electrochim Acta 111, 455–465 (2013). https://doi.org/10.1016/j.electacta.2013.08.021
Yu, W., Hou, H., Xin, Z., Niu, S., Xie, Y., Ji, X., Shao, L.: Nanosizing Pd on 3D porous carbon frameworks as effective catalysts for selective phenylacetylene hydrogenation. RSC Adv 7(25), 15309–15314 (2017). https://doi.org/10.1039/c7ra00123a
Zhang, L., Li, F.: Helical nanocoiled and microcoiled carbon fibers as effective catalyst supports for electrooxidation of methanol. Electrochim Acta 55(22), 6695–6702 (2010). https://doi.org/10.1016/j.electacta.2010.06.002
Assumpção, M.H.M.T., Moraes, A., De Souza, R.F.B., Reis, R.M., Rocha, R.S., Gaubeur, I., Calegaro, M.L., Hammer, P., Lanza, M.R.V., Santos, M.C.: Degradation of dipyrone via advanced oxidation processes using a cerium nanostructured electrocatalyst material. Appl Catal A 462–463, 256–261 (2013). https://doi.org/10.1016/j.apcata.2013.04.008
dos Reis, F.V.E., Antonin, V.S., Hammer, P., Santos, M.C., Camargo, P.H.C.: Carbon-supported TiO2–Au hybrids as catalysts for the electrogeneration of hydrogen peroxide: investigating the effect of TiO2 shape. J Catal 326, 100–106 (2015). https://doi.org/10.1016/j.jcat.2015.04.007
Wu, K., Zhang, Q., Sun, D., Zhu, X., Chen, Y., Lu, T., Tang, Y.: Graphene-supported Pd–Pt alloy nanoflowers: in situ growth and their enhanced electrocatalysis towards methanol oxidation. Int J Hydrog Energy 40(20), 6530–6537 (2015). https://doi.org/10.1016/j.ijhydene.2015.03.115
Jurewicz, K., Babeł, K.: Efficient capacitor materials from active carbons based on coconut shell/melamine precursors. Energy Fuels 24(6), 3429–3435 (2010). https://doi.org/10.1021/ef901554j
Ma, Y., Wang, H., Ji, S., Goh, J., Feng, H., Wang, R.: Highly active Vulcan carbon composite for oxygen reduction reaction in alkaline medium. Electrochim Acta 133((Supplement C)), 391–398 (2014). https://doi.org/10.1016/j.electacta.2014.04.080
Peng, C., Hu, Y., Liu, M., Zheng, Y.: Hollow raspberry-like PdAg alloy nanospheres: high electrocatalytic activity for ethanol oxidation in alkaline media. J Power Sources 278, 69–75 (2015). https://doi.org/10.1016/j.jpowsour.2014.12.056
Monyoncho, E.A., Ntais, S., Soares, F., Woo, T.K., Baranova, E.A.: Synergetic effect of palladium–ruthenium nanostructures for ethanol electrooxidation in alkaline media. J Power Sources 287, 139–149 (2015). https://doi.org/10.1016/j.jpowsour.2015.03.186
Modibedi, R.M., Masombuka, T., Mathe, M.K.: Carbon supported Pd–Sn and Pd–Ru–Sn nanocatalysts for ethanol electro-oxidation in alkaline medium. Int J Hydrog Energy 36(8), 4664–4672 (2011). https://doi.org/10.1016/j.ijhydene.2011.01.028
Cheon JY, Kim T, Choi Y, Jeong HY, Kim MG, Sa YJ, Kim J, Lee Z, Yang T-H, Kwon K, Terasaki O, Park G-G, Adzic RR, Joo SH (2013) Ordered mesoporous porphyrinic carbons with very high electrocatalytic activity for the oxygen reduction reaction. 3:2715. https://doi.org/10.1038/srep02715, https://www.nature.com/articles/srep02715#supplementary-information
Parreira, L.S., Silva, J.C.M., Simões, F.R., Cordeiro, M.A.L., Sato, R.H., Leite, E.R., dos Santos, M.C.: PtSn Electrocatalyst supported on MWCNT-COOH: investigating the ethanol oxidation reaction. ChemElectroChem 4(8), 1950–1958 (2017). https://doi.org/10.1002/celc.201700326
Qi, J., Benipal, N., Liang, C., Li, W.: PdAg/CNT catalyzed alcohol oxidation reaction for high-performance anion exchange membrane direct alcohol fuel cell (alcohol = methanol, ethanol, ethylene glycol and glycerol). Appl Catal B 199, 494–503 (2016). https://doi.org/10.1016/j.apcatb.2016.06.055
Acknowledgements
The authors wish to thank FAPESP (Proc. no 2014/09087-4 and 2017/15469-5) and CAPES for the financial support. The use of TEM facilities (JEOL JEM-2100F) of LNNano-CNPEM is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ferreira, J.C., Cavallari, R.V., Bergamaschi, V.S. et al. Palladium nanoparticles supported on mesoporous biocarbon from coconut shell for ethanol electro-oxidation in alkaline media. Mater Renew Sustain Energy 7, 23 (2018). https://doi.org/10.1007/s40243-018-0130-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-018-0130-z