
Abstract
Biologic therapies targeting B-cells are emerging as an effective strategy to treat a variety of immune-mediated diseases. One of the most studied B-cell-targeted therapies is rituximab, an anti-CD20 monoclonal antibody that exemplifies B-cell depletion therapy and has served as the prototype for other anti-CD20 monoclonal antibodies and the development of biosimilars. While there are multiple studies on the use of rituximab in dermatology, a comprehensive review of rituximab therapy in autoimmune skin conditions is lacking. In this literature review, we summarize indications, treatment efficacy, and safety of rituximab among common autoimmune diseases of the skin: pemphigus vulgaris, cutaneous lupus erythematous, dermatomyositis, systemic sclerosis, thyroid dermopathy, autoimmune pemphigoid diseases, and cutaneous vasculitis diseases. Existing data on rituximab support the approach of rituximab, biosimilars, and newer B-cell-targeting therapies in immune-mediated cutaneous diseases. Overall, rituximab, which targets CD20, provides an effective alternative or concomitant option to traditional immunosuppressants in the management of various autoimmune diseases of the skin. Further studies are necessary to expand the understanding and possible utility of B-cell-targeted therapies among autoimmune skin diseases.
Similar content being viewed by others


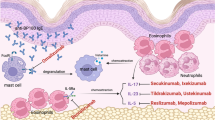
B-cell-targeted therapy is an emerging effective treatment for autoimmune skin diseases. |
Rituximab, a prototype anti-CD20 monoclonal antibody, has shown favorable results in pemphigus vulgaris, autoimmune pemphigoid diseases, cutaneous lupus erythematosus, dermatomyositis, systemic sclerosis, thyroid dermatopathy of Graves’ disease, and cutaneous vasculitic diseases. |
Rituximab is generally safe and well tolerated and can effectively augment or replace conventional therapies for autoimmune skin diseases. |
1 Introduction
Autoimmune diseases affecting the skin can cause significant morbidity, and the effects can be profoundly agonizing, debilitating, and disfiguring. Effective treatment has historically been challenging due to diseases becoming refractory to conventional therapies. The pathogenesis of many severe cutaneous autoimmune diseases, such as blistering diseases, lupus erythematous, dermatomyositis (DM), and rheumatoid arthritis (RA), are multifactorial. These disorders have dysfunctions of both the innate and adaptive immune system, manifested by the production of autoantibodies. However, the etiologic basis of clinical symptoms among common autoimmune skin diseases remains poorly defined. Recent success with rituximab, an anti-CD20 monoclonal antibody, provides evidence that B cells contribute significantly in the pathogenesis of several autoimmune skin disorders. The marked clinical response and successful remission seen in many patients after treatment with rituximab is often associated with complete or almost complete B-cell depletion (Table 1) [1]. While there have been favorable responses to rituximab among many autoimmune skin diseases, the role of rituximab remains controversial among others. Nevertheless, the success from rituximab has allowed for the use and development of highly specific therapy to B cells and provided new treatment options for patients with refractory autoimmune skin disease. In this literature review, we summarize indications, treatment efficacy, and the safety profile of rituximab as the prototype for anti-CD20 monoclonal antibody treatment in autoimmune skin diseases.
2 Rituximab: B-Cell-Targeted Therapy
B-cells are an essential component of the adaptive immune system, and are continuously generated from the bone marrow, eliminated for autoreactivity, and matured into the circulatory and lymphatic system to populate secondary lymphoid organs. Exposure of naïve B cells to antigens initiates B-cell activation, resulting in the formation of antibody-producing, plasma, and memory B cells [1]. However, loss of self-tolerance during normal B-cell development may lead to immune-mediated diseases through the formation of autoreactive antibodies or cytokines. B-cell dysregulation may also lead to dysfunctional antigen-presenting cells or uncontrolled clonal B-cell proliferation [2].
Controlling unwanted functions of autoreactive immunity has been a goal of many traditional immunosuppressive therapies, including corticosteroids and cytotoxic drugs; however, these drugs are often associated with significant adverse effects towards non-target organs. Over the past decade, monoclonal antibody technology has allowed for the development of therapeutic antibodies with high specificity with reduced adverse effects compared with traditional immunosuppressive drugs. The most studied B-cell-targeted therapy in autoimmune diseases is rituximab (Rituxan; Genentech, San Francisco, CA, USA). Rituximab is a chimeric murine/human monoclonal antibody that targets CD20 transmembrane protein and induces depletion of B cells. Rituximab has been approved by the US FDA for non-Hodgkin’s lymphoma, leukemia, RA unresponsive to tumor necrosis alpha antagonists, granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and moderate to severe pemphigus vulgaris (PV) [3]. Numerous off-label reports have described the success of rituximab in treating immune diseases in dermatology, rheumatology, solid organ transplantation, nephrology, neuromuscular disorders, and endocrinology [4]. The two most widely used infusion protocols for rituximab in autoimmune diseases are the lymphoma protocol (four weekly 375 mg/m2 infusions) or the RA protocol (two 1000 mg infusions separated by 2 weeks) [5]. The advantage of the lymphoma protocol over the RA protocol is the flexibility to adjust dose using body surface area (BSA), tailoring to patients of different sizes. Furthermore, additional doses of rituximab administered as maintenance therapy to treat disease relapse increase options to control disease.
Existing research on rituximab has provided a foundation for the understanding of other B-cell-targeted therapies. Rituximab biosimilar drugs have been developed, including rituximab-abbs (Truxima), rituximab-pvvr (Ruxience), and rituximab-arrx (Riabni) [6,7,8]. Other types of B-cell-directed monoclonal antibodies include obexelimab and epratuzumab, which target CD19 and CD22, respectively. Since the development of rituximab, newer anti-CD20 biologics have emerged, such as ofatumumab and veltuzumab, which are type II humanized anti-CD20 monoclonal antibodies [9]. Other emerging B-cell-targeted biologics include belimumab, which is currently the only approved biologic agent for systemic lupus erythematosus (SLE). Belimumab is a human immunoglobulin (Ig) G1 monoclonal antibody that inhibits B-cell-activating factor (BAFF), a cytokine that promotes the survival of B cells, including autoreactive B cells) [10]. However, this review will focus on data from rituximab.
3 Autoimmune Diseases of the Skin
3.1 Pemphigus Vulgaris
PV is a rare, potentially lethal, autoimmune bullous disease characterized by the development of pruritic, flaccid blisters and painful erosions of skin and mucous membranes. PV is mediated by the production of IgG autoantibodies targeting desmogleins (Dsg) 1 and 3 of epidermal keratinocytes. According to the Dsg compensation theory, pathogenic autoantibodies to Dsg1 cause cutaneous disease, while anti-Dsg3 antibodies are responsible for mucosal dominant disease; however, other factors including non-Dsg pathways were also suggested to be involved [11, 12].
Standard first-line therapy for PV includes systemic corticosteroid monotherapy or in combination with rituximab or conventional immunosuppressants, such as azathioprine and mycophenolate mofetil [13]. Downregulation of both autoreactive B and T cells is thought to mediate response to rituximab in PV. Recent studies have demonstrated the involvement of BAFF in the pathogenesis of PV and the response to rituximab. A study of 50 patients with PV compared with 56 healthy controls revealed that the BAFF level was significantly higher at baseline in PV patients than controls (p = 0.0005), which is likely explained by overactivation of B cells. After treatment with rituximab, there was a significant increase in the BAFF level at months 3 (p = 0.033) and 6 (p = 0.0134). The post-rituximab increase in BAFF concentration may be reflected in a decrease in BAFF receptor due to B-cell depletion [14]. A study investigating gene expression and prognostic biomarkers for PV and rituximab reported a significant decrease in expressions of IL22, IL9, EBI3, TNFSF13B, FCGR3A, CTLA4, and PDCD1 in PV patients (n = 48) compared with controls (n = 32) [p < 0.05]. The study also demonstrated that PDCD1, EBI3, IL21, and IL22 were significantly overexpressed 3 months post-rituximab (p < 0.05) [15].
Rituximab became the first FDA biologic agent approved for moderate to severe PV. FDA approval was based on a 2017 prospective, multicenter, open-label, randomized controlled trial of rituximab (two injections, 1 g on weeks 0 and 2, with maintenance 0.5 g rituximab infusions at months 12 and 18) in combination with low-dose prednisone versus prednisone alone in patients with moderate and severe PV (Ritux 3) [16]. Complete remission, defined as re-epithelization of the lesions and absence of new lesions without the use of corticosteroids for over 2 months, was achieved in 89.5% of patients compared with only 27.8% receiving high-dose prednisone. For the rituximab group, the number needed to treat was 1.82 patients (95% confidence interval [CI] 1.39–2.60) and the relapse rate (24%) was lower than the steroid-only group (45%). The cumulative dose of prednisone in the rituximab group (6143.1 mg) was significantly lower than the prednisone-alone group (17,973.6 mg) [p < 0.0001]. In addition, the rituximab group had greater improvements in the Dermatology Life Quality Index and Skindex scores compared with the prednisone-alone group (p = 0.0411 and p = 0.0137, respectively) [16].
Published in 2021, the PEMPHIX trial (NCT02383589) was a randomized, controlled trial that compared rituximab (n = 62; 1000 mg on days 1, 15, 168, and 182) and mycophenolate mofetil (n = 63; 2 g/day) in patients with moderate-to-severe PV (both groups also received glucocorticoid in a 1:1 ratio). At week 52, sustained complete remission, defined as the healing of lesions with no new active lesions for at least 16 weeks without glucocorticoid use, was observed more frequently in the rituximab group (25/62) than the mycophenolate mofetil group (6/63) [p < 0.001]. The rituximab group reported six disease flares, while the mycophenolate mofetil group had 44 flares (adjusted rate ratio 0.12; p < 0.001). The mean change in the Dermatology Life Quality Index score was also significantly greater in the rituximab group (p = 0.001). In addition, the mean cumulative glucocorticoid dose was significantly lower in the rituximab group compared with the mycophenolate mofetil group (3545 mg and 5140 mg, respectively; p < 0.001) [17].
The efficacy of rituximab in PV has also been demonstrated in a number of case reports and retrospective studies since 2002 [18]. The complete remission rate ranged from 47 to 89.5% and the relapse rate ranged from 18 to 52% [19,20,21,22,23,24,25]. A systematic review of 114 publications and 1085 PV patients summarized general lessons from the literature: rituximab monotherapy is effective and well tolerated in refractory PV; the majority of adult and juvenile patients responded well; and relapse after 6–10 months can be treated with additional rituximab infusions [19]. A randomized control trial (n = 22) and open series study (n = 15) demonstrated that low-dose (500 mg) rituximab protocols lead to adequate response [22, 26]. However, a greater decrease in severity scores was associated with high-dose rituximab (1 g every 2 weeks; n = 11) compared with the low-dose protocol (500 mg every 2 weeks; n = 11) [p = 0.049] [26]. For treatment-resistant PV, intralesional rituximab was also shown to be beneficial, with no significant difference in effect compared with intralesional triamcinolone, in a randomized clinical trial of 21 patients (p > 0.05) [27]. A cohort study of 112 patients demonstrated that patients who received the lymphoma dosing (n = 75) were 2.70-fold more likely to achieve complete remission off-treatment compared with patients with RA dosing (n = 37) [p = 0.04]. The study also indicated that young age and a BMI over 35 were negative prognostic factors for achieving remission after rituximab [28]. In addition, relapse in 8/11 patients after 6 months suggests early maintenance infusions (at month 6) may be more beneficial than at 12 months [16]. Further studies are needed to optimize infusion protocols.
Newer biologics for B-cell depletion may provide even higher efficacy and convenience in treating PV [29]. Successful treatment of ofatumumab, a type II humanized anti-CD20 monoclonal antibody, was reported in a few patients with refractory PV who could not tolerate rituximab [30, 31]. However, the phase III clinical trial of ofatumumab in patients with PV (NCT01920477) was terminated for non-safety reasons [32]. Veltuzumab (a second-generation humanized anti-CD20 antibody) can be administered subcutaneously, potentially providing greater treatment convenience than parenteral rituximab, which requires pharmacy preparation and delivery at infusion sites. A case of refractory PV treated with veltuzumab (two subcutaneous doses of 320 mg [188 mg/m2] 2 weeks apart) resulted in complete response, with relapse 2 years after treatment [33]. A case of PV was treated with four cycles of belimumab and led to markedly decreased Pemphigus Disease Area Index score and autoantibody level [34]. Further studies of belimumab independently and in combination with rituximab are necessary to understand the role of BAFF and B cells in the pathogenesis and treatment of PV.
Overall, B-cell-targeted therapy exemplified by rituximab has revolutionized PV treatment with the reduction of glucocorticoid use and its associated adverse effects (Table 2). Future developments in the biologic drugs for B-cell-targeted therapy and optimization of rituximab protocols may further improve PV treatment success.
3.2 Autoimmune Pemphigoid Diseases
The group of autoimmune pemphigoid diseases (APDs) includes bullous pemphigoid (BP), mucous membrane pemphigoid (MMP), linear IgA and IgA/IgG bullous dermatosis (LABD and LAGBD, respectively), pemphigoid gestationis (PG), and a group of sublamina densa blistering diseases featuring epidermolysis bullosa acquisita (EBA), bullous SLE, and anti-p200 pemphigoid. Presence of pathogenic autoantibodies and the level of such antibodies identify clinical presentation and disease activity for the pemphigoid diseases [35, 36]. Disruption of the basement membrane zone (BMZ) components by autoantibodies causes subepidermal separation, leading to formation of tense blisters and vesicles [37]. Therapy for APD includes topical and systemic steroids as well as steroid-sparing therapy such as intravenous immunoglobulin (IVIG), azathioprine, mycophenolate mofetil, cyclophosphamide, and methotrexate. Patients with IgA antibody-dominant diseases (LABD, LAGBD) and MMP benefit from dapsone therapy [38]. While rituximab is considered a first-line option for PV, rituximab is not considered standard therapy for BP and other APDs.
3.2.1 Bullous Pemphigoid
BP is characterized by autoantibodies targeting the BP180 antigen (type XVII collagen) and less commonly BP230. Typically, IgG1 and IgG4 anti-BP180 autoantibodies are predominant and correlate with disease severity or duration, however IgG2 and IgG3 autoantibodies may also be pathogenic [39].
Randomized controlled trials of rituximab for BP are lacking, however several retrospective studies and case series have been published. Results of a recently published retrospective cohort study of 84 patients with BP suggested that rituximab as an adjuvant therapy within 12 weeks of initiating systemic corticosteroids was associated with a more rapid and frequent complete remission rate. Median time to complete remission was 215 days (95% CI 176.9–253.1) for patients receiving both rituximab and steroids versus 529 days (95% CI 338.6–719.4) for those receiving steroids alone [40]. In another retrospective case-control study of 32 patients with moderate to severe BP, first-line rituximab (500 mg weekly for 4 weeks) plus prednisolone (0.5 mg/kg/day) therapy (n = 13) was compared with prednisolone alone (n = 19). Complete remission rate for rituximab/prednisolone patients (92%) was significantly greater than the prednisolone-alone group (61%) [p = 0.02]. In the rituximab group, 61% of these patients remained in remission off therapy for over 2 years, and 30% of patients experienced mild disease recurrence [41]. Polansky et al. demonstrated similar results of rituximab in a retrospective study of 20 patients with recalcitrant or severe BP treated with rituximab (RA protocol: n = 19; lymphoma protocol: n = 1). The decrease in serum anti-BP180 antibody levels was associated with clinical response. The study achieved a 75% remission rate, reasonable adverse effect profile, and steroid-sparing effect of rituximab [42]. A retrospective study of 12 patients with recalcitrant BP demonstrated that the combination of rituximab (lymphoma protocol for 8 weeks, then monthly for 4 months) with IVIG resulted in clinical clearance in all 12 patients after an average of 4.6 months. Two of 12 patients relapsed after 1 year; however, response was observed after retreatment with another cycle of rituximab. All patients remained in remission without adverse events for 6 years [43].
In another retrospective study of BP patients, 48 rituximab-treated patients reported a remission rate of 79% and relapse rate of 29%, with a median time of 5.6 months to relapse [44]. A retrospective review of eight patients with recalcitrant BP treated with rituximab reported a disease control rate of 83.3%, partial remission rate of 62.5%, and a complete remission rate of 12.5%, with a relapse rate of 71.4% [45]. Individual case reports have demonstrated rituximab efficacy in treating recalcitrant BP, including one patient with both BP and psoriasis [35, 46,47,48]. Rituximab depletion of B-cell and IgG autoantibodies have been linked to clinical response, while relapses were associated with an inadequate total B-cell depletion [35, 39, 41, 42]. Overall, despite a lack of randomized controlled trials, the existing data demonstrate that rituximab is a valuable steroid-sparing therapy option for moderate to severe BP (Table 3).
3.2.2 Mucous Membrane Pemphigoid
MMP is a rare disease characterized by autoantibodies most commonly targeting the C-terminus of BP180, and less often BP230, laminin 332, or the β4 subunit of α6β4 integrin. The consequential damage to BMZ at various mucosal surfaces results in severe erosions, bullae, and tissue scarring in severe cases. Conjunctival and laryngeal involvement may result in blindness and airway constriction. Ocular MMP is sometimes referred to as ocular cicatricial pemphigoid (OCP) [49].
Multiple literature reviews and retrospective case-control studies demonstrated that rituximab (both RA and lymphoma protocols) in MMP patients results in a 71–100% disease control rate [50,51,52,53]. Repeated rituximab cycles were reported to increase the response rate [52, 53]. No correlation was established between the onset of clinical relapse and recovery of peripheral blood B cells [52]. The largest retrospective study (n = 49) demonstrated a significant difference in disease control rate between rituximab (n = 24/24) and conventional treatment (n = 10/25) for MMP (p < 0.01). Time to disease control was also shorter for the rituximab group (10.17 months) compared with the control group (37.7 months) [p = 0.02]. Notably, there was no significant difference between the rituximab and conventional treatment groups in the number of patients off prednisone after disease control has been established (n = 16/24 and n = 12/25, respectively) [p = 0.15] [50]. Rituximab treatment had a lower rate of adverse events compared with systemic immunosuppression alone; therapy complications were associated with disease severity and long histories of immunosuppression prior to rituximab [50, 52].
The available data from retrospective studies show that rituximab administered early in the disease may prevent scarring and blindness in MMP patients (Table 4) [53, 54]. The high recurrence rate for MMP (up to 50%) may require continuation of the immunosuppressive therapy or additional cycles of rituximab [50,51,52,53]. The positive response with rituximab suggests a need for well-designed trials to confirm the safety and efficacy of rituximab in MMP.
3.2.3 Linear Immunoglobulin (Ig) A Bullous Dermatosis and Linear IgA/IgG Bullous Dermatosis
Both LABD and LAGBD are blistering diseases caused by deposition of autoantibodies targeting integral components of BMZ, similar to BP. BP, LAGBD, and LABD are believed to be on a spectrum, and for LAGBD, the clinical presentation and pathological findings are determined by predominance of either IgG or IgA deposited along the BMZ [55]. In LABD, the IgA autoantibodies are formed against the 97 kDa or 120 kDa fractions of BP180, and sometimes collagen VII (COL7) [36]. In LAGBD, IgG and IgA antibodies target BP180, laminin-332, and BP230. For LABD/LAGBD diseases, dapsone and topical corticosteroids are the first-line therapy [56, 57].
The available literature on rituximab in LABD/LAGBD is limited. Several published cases of patients with refractory LABD to dapsone and corticosteroids reported complete clearance and remission of their disease after rituximab [58, 59]. LAGBD therapy with rituximab (lymphoma protocol) resulted in complete skin clearance initially, which relapsed after 9 months; fortunately, an additional cycle of rituximab restored remission [60].
A retrospective review of rituximab therapy in 28 patients with various pemphigoid diseases included a single case of LABD that did not reach disease control after a more than 5-year follow-up. The study concluded that IgA-dominant pemphigoid disease may have a lower disease control rate with rituximab compared with IgG-dominant diseases [45]. Overall, rituximab may benefit patients with LABD and LAGBD disease refractory to first-line therapy; however, randomized trials are needed to better understand the utility of rituximab in IgA-dominant pemphigoid conditions.
3.2.4 Epidermolysis Bullosa Acquisita
EBA is caused by deposition of IgG and C3 (IgA or IgM are less common) autoantibodies to COL7 along the BMZ [36, 61]. Clinical presentations include pruritus, tense blisters, and skin fragility, and may resemble other autoimmune bullous dermatoses. Mucocutaneous involvement has been frequently reported [61, 62]. A meta-analysis of existing EBA therapies summarized 1159 cases of EBA published between 1971 and 2016, including 16 cases treated with rituximab. The study failed to find statistical significance between complete remission of EBA and the use of conventional therapies (corticosteroids and various corticosteroid-sparing medications). However, there was a significant association between EBA complete remission and the use of IVIG (p = 0.0047) and rituximab (p = 0.0114), making them likely candidates for combination EBA therapy [61].
In a case series of three patients with EBA refractory to standard therapy, several cycles of rituximab (both RA and lymphoma protocols were utilized) resulted in two patients achieving complete disease control and one patient with partial disease control, allowing for dramatically decreased prednisone dose [63]. In a retrospective study, four patients with resistant EBA were treated with rituximab, IVIg and colchicine (lymphoma protocol) and all demonstrated a decrease in mean skin involvement scores compared with pretreatment baseline [64]. Additionally, multiple single case reports demonstrated that rituximab is an effective treatment for recalcitrant EBA, leading to a complete clinical remission [62, 65, 66].
3.2.5 Pemphigoid Gestationis
PG is a rare disease caused by the loss of immune tolerance and cross-reactivity to placental BP180. The formation of C3 and IgG autoantibodies against BP180 in the maternal hemidesmosomes causes blistering disease in the late pregnancy or early postpartum periods. While most PG cases resolve spontaneously, PG flare or persistent disease may need treatment with corticosteroids and other immunosuppressive agents [67].
Case reports described rituximab for PG refractory to conventional immunosuppression (corticosteroids, azathioprine, and dapsone) and recurrent PG with fetal loss in a previous pregnancy [68, 69]. Rituximab provided a complete clearance of refractory PG followed by a mild relapse and prevented the recurrent PG, allowing the patient to have normal gestation with a healthy full-term baby. In both cases, a decrease in serum anti-BP180 antibody levels was observed. Rituximab was well tolerated, without adverse effects [68, 69].
Rituximab is a category C in pregnancy due to limited data on its safety. The transfer of IgG across the placenta poses the highest risk for fetuses after the first trimester of pregnancy and may result in fetal B-cell depletion, lymphopenia, and thrombocytopenia [70, 71]. Rituximab was detected in breast milk in animal studies; however, the data for or against its use during breast feeding in humans are insufficient [72]. Overall, PG tends to resolve spontaneously, and rituximab therapy has a very limited range of application to prevent a recurrence of PG for refractory PG.
3.3 Cutaneous Vasculitic Diseases
Anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (AAV), cryoglobulinemia-associated vasculitis (CV), IgA vasculitis (IgAV), and other vasculitides are all inflammatory conditions secondary to inflammation of blood vessels with subsequent ischemia and presenting with cutaneous and systemic manifestations [73]. Management of these vasculitides may be challenging due to adverse effects or contraindications to systemic immunosuppression used as first-line treatment. A significant portion of patients would relapse with decreasing immunosuppressive treatments or present with a disease refractory to the treatment [74]. Rituximab has become a valuable option in managing these vasculitides.
3.3.1 Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-Associated Vasculitis
AAV includes GPA, eosinophilic granulomatous with polyangiitis (EGPA), and MPA. All of these are characterized by the involvement of small- to medium-sized vessels and the presence of IgG anti-neutrophil circulating antibodies directed against components of both primary granules of neutrophils and monocyte lysosomes. Common cutaneous manifestations include palpable purpura, erythematous macules, subcutaneous nodules, and ulceration [75]. The Birmingham Vasculitis Activity Score (BVAS) is often used to quantify AAV severity based on assessment of nine organ systems to capture a broad spectrum of clinical disease manifestations [76]. The standard treatment approach is based on systemic immunosuppression with corticosteroids and/or cyclophosphamide, or azathioprine. In addition, rituximab is FDA-approved and a first-line option for induction and maintenance therapy for GPA and MPA (Table 5) [74].
The RAVE study is a randomized controlled trial that compared glucocorticoids plus either rituximab (lymphoma protocol) or cyclophosphamide for remission induction in 197 patients with severe AAV (GPA = 148, MPA = 48; other = 1). A higher percentage of rituximab patients achieved remission by 6 months (64% vs. 53%, p < 0.001) and rituximab was shown to be non-inferior to cyclophosphamide. Rituximab demonstrated higher efficiency than cyclophosphamide for inducing remission of the relapsing disease (67% vs. 42%, p = 0.01). There was no significant difference in the rate of disease flares between the cyclophosphamide and rituximab groups. By 6 months, 47% of rituximab patients became ANCA-negative; however, the loss of ANCA reactivity was not associated with remission induction [77].
The RITUXVAS study is an open-label, randomized trial that compared glucocorticoids plus either rituximab (lymphoma protocol) and intravenous cyclophosphamide (RTTX group) or intravenous cyclophosphamide followed by azathioprine (control group) for remission induction in 44 patients [104]. No significant difference was found in sustained remission rates (BVAS of 0 for 6 months) between the rituximab group (76%) and the control group (82%). Follow-up in the RITUXVAS study at 24 months found no significant difference between the rates of death, end-stage renal disease, and relapse between the rituximab and control groups. All relapses were associated with the return of B cells [78].
Another randomized clinical trial revealed that rituximab combined with reduced-dose prednisolone (0.5 mg/kg/day; n = 70) was not inferior to rituximab combined with high-dose glucocorticoids (1 mg/kg/day; n = 70) in patients with ANCA-associated vasculitis (p = 0.003 for non-inferiority) [79].
The MAINRITSAN study demonstrated the effectiveness of rituximab as maintenance therapy for ANCA-associated vasculitis. A total of 115 patients with ANCA-associated vasculitis were randomized to receive rituximab (n = 58) or azathioprine (n = 57). With a follow-up duration of 28 months, the rituximab group exhibited significantly fewer major relapses (5%) compared with the azathioprine group (29%) [p = 0.002] [80]. A follow-up study, the MAINRITSAN2, explored differing rituximab regimens for maintenance therapy: tailored infusions (n = 81, initial 500 mg infusion and re-infusion only when CD19+B lymphocytes or ANCA reappeared) versus fixed number of infusions (n = 81, 500 mg on days 0 and 14, then at 6, 12, and 18 months). The tailored arm received less infusions (total 248, median three per patient) compared with the fixed arm (total 381, median five per patient). There was no significant difference in relapse rate between the groups (tailored arm, 17.3% vs. fixed arm, 9.9%; p = 0.22). The MAINRITSAN2 study results indicated that patients receiving individually tailored regimens benefit from fewer rituximab infusions, without an increased rate of relapse [81].
3.3.2 Cryoglobulinemia-Associated Vasculitis
CV is a disease mediated by immune complex deposition, mostly in small vessels, causing purpura, arthralgia, and weakness, and sometimes involving the kidneys and the peripheral nervous system [82]. The disease is classified as type I (deposition of monoclonal IgG or IgM), type II (IgG and IgM-RF of monoclonal origin), or type III (IgG and IgM-RF of polyclonal origin). Types II and III are also called mixed cryoglobulinemia (MC). Treatment of MC includes treatment of the underlying disease when appropriate (e.g., hepatitis C virus [HCV], hematologic malignancy), systemic immunosuppression, and plasmapheresis [83]. Rituximab depletion of B cells producing monoclonal or polyclonal cryoglobulins was recommended in cases of severe vasculitis, skin ulcers, neuropathy, and nephropathy [84].
Several studies have analyzed rituximab therapy for CV, both as monotherapy and in combination with antiviral drugs to target HCV. Complete response rate has been reported in 50–62% of cases with clinical improvement observed in the majority of cases [82, 85, 86]. Rituximab infusion is associated with reduced levels of cryoglobulins, rheumatoid factor, and IgM. Rituximab monotherapy for active MC has demonstrated a clinical improvement rate of 74% for skin purpuric lesions and 87% for non-healing vasculitic leg ulcers [86].
A multicenter, phase III, randomized controlled trial of 57 patients with either HCV‐related or ‐unrelated type II CV demonstrated higher survival rates at 12 months in rituximab-treated (RA protocol; 64.3%) patients compared with patients receiving conventional immunosuppression (3.5%) [p < 0.0001]. In addition, all rituximab-treated patients with skin ulcers at baseline experienced a complete response (n = 5/5). BVAS scores were not significantly different between groups (p = 0.076), but the BVAS score was significantly lower compared with baseline for the rituximab group starting at 2 months (p < 0.001) [87]. In a randomized clinical trial of rituximab (lymphoma protocol) for HCV-associated CV in patients who failed to achieve remission with antiviral therapy, the rituximab groups achieved an 83% remission rate compared with 8% in the control group treated with the best available immunosuppressive therapy (p < 0.001) [88]. BVAS scores were significantly lower in the rituximab group (p < 0.02) [87, 88]. Multiple trials demonstrated that low-dose rituximab (two infusions administered at 250 mg/m2) for relapsing MC is an efficient, well tolerated, and cost-effective option, with most patients demonstrating clinical improvement [89, 90]. Rituximab combined with Peg-interferon-alpha2b/ribavirin was shown to be effective in treating severe refractory HCV-related MC vasculitis [85].
Overall, rituximab has demonstrated efficacy and safety in treating MC both related and unrelated to HCV (Table 6). Rituximab was shown to improve both dermatological and systemic disease manifestations [89]. Rituximab efficacy was low in cases with plasmocytic proliferation [82, 91]. However the success from direct-acting antivirals for HCV and HCV-associated MC requires re-evaluation of the role of rituximab in HCV-associated CV [89, 92].
3.3.3 IgA Vasculitis
IgAV, also referred to as Henoch–Schönlein purpura, is a small-vessel leukocytoclastic vasculitis most frequently affecting pediatric patients following an infection. Major disease manifestations are palpable purpura of the lower extremities, arthralgia, abdominal pain associated with melena, and neurological and renal involvement. The disease typically has a transient course, with most patients recovering spontaneously; however, occasional refractory cases may require intravenous corticosteroids and plasmapheresis [93].
The effectiveness of rituximab to treat adult-onset IgAV has been demonstrated in prospective studies. In a prospective study of 22 patients with adult-onset IgAV treated with rituximab, a remission rate of 90.9% and relapse rate of 35% was observed. Patients had significant reductions in 24-h proteinuria (p < 0.0001), C-reactive protein (CRP) levels (p = 0.0005), and Birmingham Vasculitis Activity Score (p < 0.0001) [94]. Another study demonstrated complete response in 10/12 rituximab-treated patients and no response in 1/12 rituximab-treated patients with adult-onset IgAV after 6 months [95].
A case series of eight pediatric patients with chronic steroid-dependent Henoch–Schonlein purpura reported remission in seven patients. The number of patients requiring hospitalization decreased from 7 to 2 after rituximab treatment. In addition, the median oral corticosteroid burden decreased from 0.345 mg/kg/day to 0 mg/kg/day at 6 months (p = 0.078), 1 year (p = 0.0625), and 2 years (p = 0.03) [96].
A systematic review of rituximab therapy for IgAV identified 35 cases treated with rituximab following either the RA or lymphoma protocol. Most patients (93.4%) improved after initial rituximab; the recurrence rate was 37.1%. Sustained remission was achieved by 74.3% of patients [97]. Overall, rituximab is an effective and well-tolerated option for refractory IgAV, especially if conventional immunosuppression therapy is contraindicated. However, more studies are necessary in both the adult- and pediatric-onset populations.
3.4 Dermatomyositis
DM is an autoimmune disease characterized by inflammation of the skin and muscles. The etiology of DM is unknown and is thought to be multifactorial. It is thought that injury in DM is due to antibody- and complement-mediated capillary damage [98]. Recent studies hinted a role for B cells in DM. One study reported that DM patients have more naïve B cells and fewer memory B cells compared with healthy controls [99]. Other studies have suggested that regulatory B-cell (Breg) deficiency contributes to the pathogenesis of DM since clinical improvement and remission of DM has been associated with a return or increase in Bregs [100, 101].
Common systemic therapies for the cutaneous manifestations of DM include hydroxychloroquine, methotrexate, mycophenolate mofetil, azathioprine, and IVIG [102]. The literature has reported mixed responses to rituximab treatment among DM patients. The Rituximab in Myositis (RIM) Trial was a major randomized control trial that evaluated the safety and efficacy of rituximab in refractory adult and juvenile DM (JDM) and adult polymyositis patients (n = 76, n = 48, n = 76, respectively) over a study period of 44 weeks. Patients were randomized into early (week 0 and 1) and late (week 8 and 9) rituximab treatment arms. Rituximab was administered as 575 mg/m2 per infusion for children with a BSA ≤1.5 m2 and 750 mg/m2 (up to 1 g) per infusion for adults and children with BSA >1.5 m2 [21]. The study found no significant difference between the early and late treatment arms for its primary outcome: time to achieve the International Myositis Assessment and Clinical Studies Group preliminary definition of improvement (DOI) [p = 0.74; median time to DOI: 20.2 and 20.0 weeks for the early and late arms, respectively]. Despite not meeting the primary endpoint, 83% of patients met the DOI [103]. The authors of the RIM trial published an additional study further outlining the improvement in cutaneous findings of their study population (adult DM, n = 72; JDM, n = 48). The trial utilized the Myositis Disease Activity Assessment Tool (MDAAT) and Myositis Damage Index (MDI) to assess cutaneous disease activity and cutaneous damage, respectively [104, 105]. Rituximab demonstrated significant improvement in cutaneous visual analog scale disease activity from baseline in both adult DM and JDM (p = 0.0002 and p < 0.0001, respectively). A significant decrease in frequency of the following symptoms was seen in adult DM patients: erythroderma, erythematous rashes without secondary changes, heliotrope rash, Gottron sign and papules, periungual erythema, diffuse alopecia, and mechanics hands (p = 0.002, p < 0.001, p < 0.001, p < 0.001, p < 0.001, p = 0.028, and p = 0.008, respectively). Among adult DM patients, there were no significant improvements in cutaneous ulceration, panniculitis, erythematous rash with ulceration or necrosis, focal alopecia, calcinosis, cutaneous scarring or atrophy, poikiloderma, or lipodystrophy. Similar findings were seen in the pediatric DM cohort, except for additional significant improvements in cutaneous ulcerations (p = 0.02) and focal (not diffuse) alopecia (p = 0.028). The cutaneous disease activity score improved in 67% of adult DM patients and 75% of JDM patients, and worsened in 12% of adult DM patients and 11% of JDM patients. The frequency of any DM rash decreased by 13% (89–76% decrease) for adult DM patients (p = 0.047) and 18% (100–82% decrease) for JDM patients (p = 0.002) at week 36 [106, 107].
One open-label study of eight adult DM patients with rituximab treatment showed no significant change in skin scores from baseline (RA protocol) [108]. In contrast, another open-label study of seven refractory adult DM patients demonstrated major clinical improvement in strength and cutaneous DM as early as 12 weeks after initial rituximab infusion (lymphoma protocol). All patients with baseline rash (n = 5) showed improvement and patients with alopecia had hair regrowth (n = 2). However, disease relapse was observed in four of six patients by weeks 24–36, which was associated with the return of B cells [109].
Since small vessel vasculopathy is thought to play a role in DM, decreased nailfold capillary density has been studied as a potential measure of DM disease activity. A retrospective study suggested the ability of rituximab (n = 10) to reverse nailfold capillary changes in adult DM patients compared with other immunosuppressive therapies (n = 25; prednisone, methotrexate, mycophenolate mofetil, and IVIG). Of those patients treated with rituximab, 80% had normal-appearing nailfold capillaries at 6 months, and 100% at 2 years. In contrast, patients receiving other immunosuppressants had no improvement in nailfold capillaries at 6 months or 2 years [110, 111].
Despite existing controversial findings of rituximab therapy in DM, recent studies have provided evidence that rituximab may provide benefit to refractory DM patients (Table 7).
3.5 Systemic Sclerosis (Scleroderma)
Systemic sclerosis (SSc), also known as scleroderma, is a rare connective tissue disease involving endothelial and vascular damage of the skin and internal organs with progressive fibrosis. The pathogenesis of SSc is complex and the precise mechanisms of the disease are not fully understood, but likely arise from a combination of autoimmunity, vascular defects, and fibroblast dysfunction. The clinical heterogeneity among patients with SSc further suggest that additional variables may vary among each patient [112]. Previous studies have suggested the involvement of B-cell dysfunction in the pathogenesis of fibrosis in SSc. Patients with SSC have been shown to have hyperactive memory B cells that overproduce profibrotic cytokines. These profibrotic B cells infiltrate the skin and lungs of patients with SSc, leading to the characteristic thick, hard skin and diminished lung function seen in SSc [113].
Based on the extent of skin involvement, SSc is classified as either diffuse cutaneous SSc or limited cutaneous SSc. In addition to progressive skin fibrosis, other cutaneous symptoms of SSc include pruritus, edema, capillary changes at the nail beds, digital ulceration, calcinosis, and telangiectasia [112]. The modified Rodnan skin score (mRSS) is typically used in clinical trials as a measure of skin fibrosis and to assess trial outcomes [114]. Treatment of diffuse skin sclerosis includes methotrexate or mycophenolate mofetil; unfortunately, the efficacy of these drugs have been modest. Another commonly used drug in SSc is cyclophosphamide, but it is typically preserved for refractory disease or for patients with interstitial lung disease (ILD) [115, 116].
Although limited, the data on rituximab use in SSc have demonstrated the potential of B-cell-targeting therapy in SSc. Improvements in both cutaneous and pulmonary symptoms have been demonstrated in studies of SSc patients treated with rituximab. A multicenter case-control study by the European Scleroderma Trial and Research Group (EUSTAR) of 63 SSc patients demonstrated improvement in skin fibrosis and prevented worsening lung fibrosis in rituximab-treated patients compared with matched controls. The mRSS decreased significantly from baseline in the 46 rituximab-treated patients after a mean follow-up of 7 months (p = 0.0002) [24]. The improvement in mRSS after rituximab was most pronounced in patients with severe, diffuse cutaneous SSc (n = 25, p = 0.0001). The mRSS was also improved significantly in the rituximab group compared with matched controls in patients with severe, diffuse cutaneous SSc (n = 25 each, p = 0.03) [117]. In addition, there was a measurable difference in functional vital capacity (FVC) change between rituximab and matched control groups among SSc patients with ILD (n = 9 each, p = 0.02) [118]. More recently, the EUSTAR database was utilized in a prospective cohort study of 254 rituximab-treated SSc patients. Compared with matched controls, rituximab-treated patients showed greater skin fibrosis improvement (p = 0.002), and those with a baseline mRSS ≥10 (n = 131) had significantly higher improvement in mRSS scores (p < 0.0001) [119].
Rituximab treatment may be considered an alternative or combination treatment to cyclophosphamide in refractory SSc or SSc with ILD. An open-label clinical trial of 60 diffuse SSc patients with positive anti-Scl70 antibody randomly assigned patients to receive intravenous cyclophosphamide (n = 30) or rituximab (RA protocol) with concurrent prednisolone (n = 30). Significant improvement in the percentage-predicted FVC, the study’s primary outcome, was only seen in the rituximab group (p = 0.002) compared with the cyclophosphamide group (p = 0.496). Furthermore, greater improvement in mean mRSS was achieved in the rituximab group (−9.67) compared with the cyclophosphamide group (−5.5) after 6 months (p ≤ 0.001) [115].
A small randomized controlled study of 14 patients with SSc achieved a similar mRSS score decrease of 38% and noted a significant reduction in collagen deposition in the papillary dermis but not the reticular dermis [120]. Individual case reports showed that rituximab can improve SSc-related cutaneous calcinosis [121, 122]. In a case series of eight patients with cutaneous calcinosis, four patients had a clinical response [122].
However, several small, open-label trials and retrospective studies of rituximab in SSc patients lacked significant changes in mRSS scores [123]. An open-label trial of 15 patients with diffuse SSc showed no significant difference in mRSS from baseline to 6 months (p = 0.82) or 12 months (p = 0.83) [124]. A small retrospective study of six patients with SSc showed stabilization or improvement of skin involvement, but the change in mRSS between baseline and 12-month follow-up was minimal [125]. In a retrospective study, a rituximab biosimilar (CT-P10, Truxima) demonstrated a significant improvement in mean mRSS scores in SSc patients, in both patients naïve to rituximab (n = 17, p < 0.024) and those previously treated with rituximab (n = 16, p < 0.031) [126].
In summary, studies have demonstrated beneficial effects of rituximab on both skin and lung function in patients with SSc (Table 8). However, larger-scale clinical trials of rituximab with longer evaluations are necessary to better assess its long-term clinical efficacy in patients with varying cutaneous features of SSc.
3.6 Cutaneous Lupus Erythematosus
Cutaneous lupus erythematosus (CLE) is an autoimmune disease that can present independently or in association with SLE. The main CLE subsets are acute CLE (ACLE), subacute CLE (SCLE), and chronic CLE (CCLE). The most common subset is CCLE, in which the majority of CCLE (up to 80%) is discoid lupus erythematosus (DLE) [127]. The disease mechanism of SLE is complex and multifactorial, including both genetic and environmental factors, such as ultraviolet radiation exposure and smoking. SLE is characterized by autoantibodies to intracellular antigens that lead to formation of immune complexes, causing damage to various organs [1]. The pathophysiology of CLE is thought to include defects of both innate and adaptive immune cells [128]. Anti-SSA/Ro and anti-LA are known to be associated with CLE; however, the pathogenic role of autoantibodies in CLE remains unclear [5, 129].
The current management of CLE includes strict sun protection, topical corticosteroids, antimalarials, and corticosteroid-sparing immunosuppressing therapies. Most recommended therapies for CLE are derived from SLE, however many CLE patients are recalcitrant to the current treatment options. Clinical trials for SLE treatments often exclude CLE patients who do not meet the criteria for SLE; thus, CLE patients often miss out on opportunities for emerging lupus treatments [130]. There are currently no FDA-approved treatments specifically for CLE. One of the two FDA-approved medications for SLE targets B cells, i.e. belimumab, a monoclonal antibody against BAFF [10]. Although belimumab has shown success in SLE, the efficacy for skin disease is unclear. The original trial lacked skin-specific outcomes measures and evaluations by a dermatologist; thus, the results of the trial cannot be extended to CLE patients. Studies have suggested belimumab may lead to improved skin manifestations; however, the CLE-specific evidence is limited and additional trials are required in CLE patients. Recent studies have demonstrated success of belimumab in decreasing anti-dsDNA antibody levels in SLE patients after B-cell depletion by rituximab, as well as coadministration of belimumab and rituximab [131, 132]. Further studies are indicated to understand the effect of combining belimumab and rituximab on cutaneous manifestations of lupus erythematous.
B-cell-targeted therapy using rituximab for SLE has shown mixed results. Multiple authors have demonstrated rituximab as an effective treatment for SLE [133, 134]. However, two large, randomized, placebo-controlled clinical trials (EXPLORER, LUNAR) failed to achieve their primary endpoints of overall cutaneous response at 6 and 12 months and renal response at week 52, respectively) [135, 136]. A systematic review identified several potential predictive and prognostic factors of rituximab outcomes in SLE, including clinical phenotype and severity, anti-ENA, anti-Ro antibodies, post-rituximab B-cell depletion and earlier B-cell repopulation; however, validation of these factors is lacking [137].
The benefits of rituximab on cutaneous specific manifestations of lupus erythematous remains controversial. A number of case studies have demonstrated rituximab efficacy in the treatment of bullous SLE and refractory SCLE [138,139,140,141,142,143]. Previous prospective and retrospective studies of rituximab treatment in CLE patients have shown variable results: 23–76% of patients demonstrated at least a partial response and 29–48% of patients demonstrated a complete response. Relapses were observed in 39–46% of patients [144,145,146,147]. In addition, the efficacy of rituximab in CLE has been shown to vary among subgroups. In a retrospective study of 17 patients, two of three SLE patients with non-specific lesions (66.6%), two of three ACLE patients (66.6%), two of three SCLE patients (66.6%), and three of eight CCLE patients (37.5%) resulted in cutaneous response to rituximab [146]. A prospective study of 26 patients revealed that the mucocutaneous response to rituximab at 6 months was best in ACLE patients (6/14, 42.9%), compared with 0% response in CCLE patients (0/8) [145]. In contrast, a retrospective study of 50 rituximab-treated CLE patients showed no statistically significant difference in response among CLE subtypes [147]. Notably, post-rituximab flares of CCLE in these studies were associated with lack of B-cell repletion [145, 147].
Overall, rituximab has shown promising but variable results in the treatment of CLE (Table 9). The decreased response in and lack of B-cell repletion in flares of CCLE suggest that innate and T-cell-dependent autoimmunity may potentially account for non-response to rituximab in CCLE patients [11,12,13]. The mixed results of existing rituximab studies in CLE and its subtypes indicate that additional trials are necessary to better understand the clinical utility of B-cell-targeted therapy in CLE.
3.7 Thyroid Dermopathy of Graves’ Disease
Thyroid dermopathy (TD), or pretibial myxedema, is a rare manifestation of Graves’ disease (GD) that typically develops within the first 2 years after hyperthyroidism diagnosis. It affects 1–4% of GD patients; the majority of these patients also develop Graves’ orbitopathy (GO) [148]. It presents as a localized, waxy skin thickening, usually in the pretibial area, but may occur anywhere on the skin, including extensor areas, back, and head and neck areas [149].
Production of anti-thyroid stimulating hormone (TSH) receptor autoantibodies binding TSH receptors by B cells is associated with GD [148]. The exact pathogenesis of the TD is unclear, however it has been demonstrated that normal dermal fibroblasts express TSH receptor protein and may be stimulated by a circulating factor. Additionally, fibroblasts may be stimulated by inflammatory cytokines such as tumor necrosis factor-alpha and gamma interferon secreted by T-helper (Th) 1 cells specific to TSH receptor antigen [148, 149]. Mild TD often resolves over time without treatment, however severe dermopathy may be refractory to treatment [150]. The initial therapy relies on topical or intralesional corticosteroids and normalization of thyroid function.
Recently, B-cell-targeted therapy with rituximab has been found to be helpful in severely affected patients with thyroid orbitopathy; however, no large-scale trials of rituximab in patients with TD have yet been reported [151]. A case series reported data from five patients with TD treated with rituximab. Objective improvement was observed in one of five patients, and stabilization of the disease was noted in three of five patients. The authors noted that a limited duration of rituximab benefits suggested the need for repeated infusions [152]. Several case reports demonstrated improvement in patients with treatment resistant TD and GO after one cycle of rituximab [153, 154]. A patient with severe TD that progressed to elephantiasic dermopathy was treated with a combination of plasmapheresis and rituximab (a total of 29 weekly rituximab doses over 3.5 years). The patient had improvement in the subcutaneous tissue thickness and resolution of the macrodactyly, which coincided with a decrease in the levels of anti-TSH autoantibodies, supporting a hypothesis for the role of pathogenic autoantibodies in the TD [150].
Although there are preliminary data, additional well-designed trials to confirm the safety and efficacy of rituximab in TD is needed; however, the available data suggest that rituximab may provide a well-tolerated option in patients with severe TD. Notably, data on the use of rituximab for GO show that better response is achieved early in the course of the extrathyroidal GD [155, 156].
3.8 Lichen Planus
Lichen planus is a chronic, recurrent inflammatory condition that affects the skin, oral mucosa, genital mucosa, scalp, and nails. The erosive variant is characterized with painful ulcerations and scarring of the mucosa and skin. The pathogenesis of LP remains unclear, but is likely T-cell-mediated, with CD8+T cells directed against basal keratinocytes [157]. However multiple case reports have described rituximab use for LP. Improvement of LP due to rituximab suggests B cells are also involved in the pathogenesis of LP.
A report of a patient with generalized mucocutaneous LP with esophageal involvement showed rapid resolution to rituximab (lymphoma protocol). The patient had dramatic improvement at months 3 and 6. Endoscopy also demonstrated complete remission of esophageal involvement at month 3 [158]. Three additional case reports of four patients with refractory oral and vulvovaginal erosive LP reported successful treatment with rituximab [159,160,161]. One case report of lichen planopilaris in a patient with juvenile chronic arthritis described rapid and complete resolution with rituximab treatment [162]; however, a retrospective study of five patients with refractory erosive LP reported failure or transient minimal improvement with rituximab. Three patients had no response to rituximab, and one patient had minimal reduction of pain and number of erosive lesions. Another patient had mild improvement of genital and skin involvement without oral improvement, and relapses treated with repeated courses of rituximab did not lead to clinical improvement [163]. Incidentally, there have been reported cases of anti-CD20 therapies causing lichenoid reactions [164,165,166]. Larger-scale studies are therefore required to understand the role of rituximab in the treatment and development of lichenoid conditions.
4 Safety of Rituximab
Overall, rituximab is well tolerated and serious adverse reactions are rare. Among the indications discussed in this review, rituximab has demonstrated a favorable safety profile compared with conventional therapies. There were less frequent or comparable frequencies of adverse effects in rituximab groups compared with controls among studies of the discussed indications. In the randomized trial for PV by Werth et al., the total number of adverse events was lower for rituximab (85%) versus mycophenolate mofetil (88%); however, the number of serious adverse events was greater in the rituximab group (22%) compared with the mycophenolate mofetil group (15%) [17].
The most common adverse reaction to rituximab was infusion reaction during the first treatment, which may be prevented or minimized with concomitant, corticosteroid, acetaminophen or diphenhydramine premedication [167]. Other reported adverse effects included neutropenia, hypogammaglobulinemia, hypertension, rash, gastrointestinal upset, cardiac disease, cough, and upper respiratory tract infections. Severe adverse effects included mucocutaneous reactions (including lichenoid dermatitis and Stevens–Johnson syndrome) and serious infections [71, 168, 169]. When treating lymphoma patients, tumor lysis syndrome can be seen. Rituximab is associated with hepatitis B virus (HBV) reactivation, and screening for subclinical HBV prior to initiating rituximab is essential [170]. Progressive multifocal leukoencephalopathy (PML) brain infection caused by reactivation of the JC virus, as well as neurologic examination, are important to monitor for developing symptoms, which necessitate cessation [171]. Rare development of various mucocutaneous and skin conditions, including psoriasis, oral lichenoid reaction, scar sarcoidosis, and cutaneous vasculitis have also been reported after initiating treatment with rituximab [164, 172,173,174,175]. Case reports of rituximab-treated PV have reported reticulate pigmentation over the face and paradoxical worsening of pemphigus presenting as figurate bullous eruption [176, 177].
There are significant concerns on the use of rituximab during the coronavirus disease 2019 (COVID-19) pandemic. Early data suggested poorer outcomes in rituximab-treated patients who were hospitalized due to COVID-19 [178]. In multiple case reports, patients receiving rituximab for rheumatological diseases experienced severe forms of COVID-19 [179,180,181,182,183]. However, these associations may be skewed due to the pre-existing risk factors that rituximab-treated patients generally have, such as higher rates of interstitial lung disease and other known factors associated with poorer outcomes of COVID-19. A single-center retrospective study of patients with COVID-19 and receiving rituximab for any indication (n = 49) reported that the duration between the last rituximab infusion and COVID-19 diagnosis did not significantly affect rates of hospitalization, admission to intensive care units (ICUs), or death. In the analysis, patients received their last rituximab dose <3 months (57.1%), 3–6 months (26.5%), or >6 months (16.3%) prior to their COVID-19 diagnosis. There was no significant difference in median time from the last rituximab infusion to COVID-19 diagnosis between those who developed COVID-19 antibodies (51.7%) and those who did not (48.3%) [p = 0.323]. The study also found that in comparison with patients receiving rituximab as cancer therapy, patients who were treated with rituximab for non-malignant indications had higher rates of ICU stays for COVID-19 (9.5% and 35.7%, respectively; p = 0.035). Interestingly, of the 14 patients with negative COVID-19 antibody titers, 11 patients survived COVID-19 [184]. This may suggest that antibody development is not necessary for recovery from COVID-19.
Data on the safety of vaccinations in rituximab-treated patients are limited. Rituximab is known to be associated with an impaired humoral response to the PPSV-23 and influenza vaccines [185, 186]. In addition, live vaccinations are not recommended during rituximab treatment. While there are no standard guidelines on COVID-19 vaccinations in rituximab-treated patients, it is generally recommended to vaccinate before initiating rituximab or after at least 6 months post-rituximab infusion. If the need for vaccination is urgent, consider delaying rituximab if there is a low risk of disease flare [187]. A study of 126 patients with lymphoma treated with anti-CD20 agents reported only 55% of patients developed an antibody response to the COVID-19 vaccination. If rituximab was initiated after a vaccinated individual mounted an antibody response, they tended to maintain their antibody titers. For those who were vaccinated after initiating rituximab, time since the last dose of anti-CD20 was a significant independent predictor of antibody response to the vaccine. Antibody response was detected in 0/31 patients who last received anti-CD20 within 6 months prior to vaccination [188]. There is evidence that rituximab is associated with an impaired but inducible response to the COVID-19 vaccine. In a study of 74 rituximab-treated patients, only 39% developed antibodies against COVID-19 after two vaccinations with BioNTech/Pfizer BNT162b2 or Moderna mRNA-1273. Only 1/36 patients without detectable CD19+ peripheral B cells developed antibodies against COVID-19. Antibody levels correlated with the amount of circulating B cells in patients (p < 0.001); however, some patients with <1% of B cells mounted detectable antibody responses to the vaccine. A total of 58% of patients had detectable COVID-19-specific T cells, which was independent of humoral response [189].
5 Conclusion
Targeting B cells with high specificity using anti-CD20 monoclonal antibodies, best shown by robust data from rituximab, has demonstrated the efficacy of therapy with ability to deplete pathogenic B cells in the treatment of autoimmune disease. Our review highlights the use of anti-CD20 for the following autoimmune diseases affecting the skin: CLE, DM, SSc, TD, PV, APD, and cutaneous vasculitic diseases. Rituximab is currently only FDA-approved for non-Hodgkin’s lymphoma, leukemia, RA, GPA, MPA, and PV [3]. The off-label use of rituximab in cutaneous autoimmune diseases has shown favorable results, in which rituximab can effectively augment or replace conventional therapies with undesirable adverse effects or in refractory disease. Rituximab is generally safe and well tolerated, with the most common adverse reaction being infusion-related reactions. While rituximab is associated with occasional severe-to-fatal adverse reactions, these events are extremely rare. Further trials are required to develop guidelines for rituximab and other anti-CD20 biosimilars in dermatological autoimmune diseases. With promising results in the literature, the use of anti-CD20 monoclonal antibodies in autoimmune diseases involving the skin will likely expand in the future.
References
Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. 2021;20(3):179–99.
Bonasia CG, Abdulahad WH, Rutgers A, Heeringa P, Bos NA. B cell activation and escape of tolerance checkpoints: recent insights from studying autoreactive B cells. Cells. 2021;10:1190.
Genentech. RITUXAN (rituximab) [package insert]. 2019. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/103705s5467lbl.pdf
Sarsour K, Beckley-Kartey S, Melega S, Odueyungbo A, Kirchner P, Khalife N, et al. Rituximab utilization for approved and off-label nononcology indications and patients’ experiences with the patient alert card. Pharmacol Res Perspect. 2020;8: e00555.
Gürcan HM, Keskin DB, Stern JNH, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9:10–25. Available at: https://www.sciencedirect.com/science/article/pii/S1567576908003196
Cobb P, Niederwieser D, Cohen S, Hamm C, Burmester G, Seo N, et al. A review of the totality of evidence in the development of ABP 798, a rituximab biosimilar. Immunotherapy. 2022;14:727–40.
Deeks ED. CT-P10 (TruximaTM): a rituximab biosimilar. BioDrugs. 2017;31:275–8.
Sharman JP, Liberati AM, Ishizawa K, Khan T, Robbins J, Alcasid A, et al. A randomized, double-blind, efficacy and safety study of PF-05280586 (a rituximab biosimilar) compared with rituximab reference product (MabThera®) in subjects with previously untreated cd20-positive, low-tumor-burden follicular lymphoma (LTB-FL). BioDrugs. 2020;34:171–81.
Bag-Ozbek A, Hui-Yuen JS. Emerging B-cell therapies in systemic lupus erythematosus. Ther Clin Risk Manag. 2021;17:39–54.
Furie R, Petri M, Zamani O. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2022;63:3918–30.
Frampton JE. Rituximab: a review in pemphigus vulgaris. Am J Clin Dermatol. 2020;21(1):149–56.
Ahmed AR, Carrozzo M, Caux F. Monopathogenic vs multipathogenic explanations of pemphigus pathophysiology. Exp Dermatol. 2016;25(11):839–46.
Melchionda V, Harman KE. Pemphigus vulgaris and pemphigus foliaceus: an overview of the clinical presentation, investigations and management. Clin Exp Dermatol. 2019;44:740–6.
Daneshvar E, Tavakolpour S, Mahmoudi H, Daneshpazhooh M, Teimourpour A, Aslani S, et al. Elevated serum level of B-cell activating factor (BAFF) after rituximab therapy in pemphigus vulgaris patients suggests a possible therapeutic efficacy of B-cell depletion therapies combined with anti-BAFF agents. Int J Dermatol. 2022. https://doi.org/10.1111/ijd.16363.
Tavakolpour S, Mahmoudi H, Karami F, Elikaei Behjati S, Balighi K, Abbasi M, et al. Investigating expression pattern of eight immune-related genes in pemphigus patients compared with the healthy controls and after rituximab therapy: potential roles of CTLA4 and FCGR3A genes expression in outcomes of rituximab therapy. Dermatol Ther. 2020;33: e14380.
Joly P, Maho-Vaillant M, Prost-Squarcioni C. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031–40.
Werth VP, Joly P, Mimouni D, Maverakis E, Caux F, Lehane P, et al. Rituximab versus mycophenolate mofetil in patients with pemphigus vulgaris. N Engl J Med. 2021;384:2295–305.
Salopek TG, Logsetty S, Tredget EE. Anti-CD20 chimeric monoclonal antibody (rituximab) for the treatment of recalcitrant, life-threatening pemphigus vulgaris with implications in the pathogenesis of the disorder. J Am Acad Dermatol. 2022;47(5):785–8.
Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: a systematic review. Int Immunopharmacol. 2018;54:131–8.
Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–9.
Joly P, Mouquet H, Roujeau J-C. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–52.
Horváth B, Huizinga J, Pas HH, Mulder AB, Jonkman MF. Low-dose rituximab is effective in pemphigus. Br J Dermatol. 2012;166:405–12.
Balighi K, Daneshpazhooh M, Khezri S, Mahdavi-nia M, Hajiseyed-javadi M, Chams-Davatchi C. Adjuvant rituximab in the treatment of pemphigus vulgaris: a phase II clinical trial. Int J Dermatol. 2013;52:862–7.
Loi C, Magnano M, Ravaioli GM, Sacchelli L, Patrizi A, Bardazzi F. Rituximab therapy in pemphigus: a long-term follow-up. Dermatol Ther. 2019;32(1): e12763.
Wang H-H, Liu C-W, Li Y-C, Huang Y-C. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95:928–32.
Kanwar AJ, Vinay K, Sawatkar GU. Clinical and immunological outcomes of high- and low-dose rituximab treatments in patients with pemphigus: a randomized, comparative, observer-blinded study. Br J Dermatol. 2014;170:1341–9.
Iraji F, Danesh F, Faghihi G. Comparison between the efficacy of intralesional rituximab versus intralesional triamcinolone in the treatment refractory Pemphigus Vulgaris lesions: a randomized clinical trial. Int Immunopharmacol. 2019;73:94–7.
Kushner CJ, Wang S, Tovanabutra N, Tsai DE, Werth VP, Payne AS. Factors associated with complete remission after rituximab therapy for pemphigus. JAMA Dermatol. 2019;155:1404–9.
Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris: rituximab and beyond. J Am Acad Dermatol. 2016;74:746–53.
Klufas DM, Amerson E, Twu O, Clark L, Shinkai K. Refractory pemphigus vulgaris successfully treated with ofatumumab. JAAD Case Rep. 2020;6:734–6.
Rapp M, Pentland A, Richardson C. Successful treatment of pemphigus vulgaris with ofatumumab. J Drugs Dermatol. 2018;17(12):1338–9.
Novartis Pharmaceuticals. Efficacy and Safety of Ofatumumab in Treatment of Pemphigus Vulgaris. 2019. Available at http://clinicaltrials.gov
Ellebrecht CT, Choi EJ, Allman DM. Subcutaneous veltuzumab, a humanized anti-CD20 antibody, in the treatment of refractory pemphigus vulgaris. JAMA Dermatol. 2014;150:1331–5.
Ruan J, Zhang X, Zheng M. Belimumab for the treatment of pemphigus. Dermatol Ther. 2022;35(8): e15621.
Hall RP, Streilein RD, Hannah DL. Association of serum B-cell activating factor level and proportion of memory and transitional B cells with clinical response after rituximab treatment of bullous pemphigoid patients. J Invest Dermatol. 2013;133:2786–8.
Thomas RM, Colon A, Motaparthi K. Rituximab in autoimmune pemphigoid diseases: indications, optimized regimens, and practice gaps. Clin Dermatol. 2020;38:384–96.
Kneisel A, Hertl M. Autoimmune bullous skin diseases. Part 1: Clinical manifestations. J Dtsch Dermatol Ges. 2011;9(10):844–57.
Kneisel A, Hertl M. Autoimmune bullous skin diseases. Part 2: diagnosis and therapy. JDDG J der Dtsch Dermatologischen Gesellschaft. 2011;9:927–47.
Ronaghy A, Streilein RD, Hall RP. Rituximab decreases without preference all subclasses of IgG anti-BP180 autoantibodies in refractory bullous pemphigoid (BP). J Dermatol Sci. 2014;74(1):93–4.
Tsai Y-J, Cho Y-T, Chu C-Y. Clinical effectiveness and safety of initial combination therapy with corticosteroids and rituximab in bullous pemphigoid: a retrospective cohort study. Am J Clin Dermatol. 2022;23:571–85.
Cho YT, Chu CY, Wang LF. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. 2015;173:302–4.
Polansky M, Eisenstadt R, DeGrazia T, Zhao X, Liu Y, Feldman R. Rituximab therapy in patients with bullous pemphigoid: A retrospective study of 20 patients. J Am Acad Dermatol. 2019;81:179–86.
Ahmed AR, Shetty S, Kaveri S, Spigelman ZS. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-8.e3.
Yoo DS, Lee JH, Kim S-C, Kim JH. Mortality and clinical response of patients with bullous pemphigoid treated with rituximab. Br J Dermatol. 2021;185:210–2.
Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horváth B. Effectiveness and Safety of Rituximab in Recalcitrant Pemphigoid Diseases. Front Immunol. 2018;9:248–248.
Silva N, Costa A, Salvador F, Serradeiro E. Bullous pemphigoid successfully treated with rituximab. Acta Med Port. 2022;30:243–6.
Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2022;65:552–8.
Wang T-S, Tsai T-F. Remission of bullous pemphigoid after rituximab treatment in a psoriasis patient on regular low-dose methotrexate. Acta Derm Venereol. 2022;94:108–9.
Xu H-H, Werth VP, Parisi E, Sollecito TP. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611–30.
Maley A, Warren M, Haberman I, Swerlick R, Kharod-Dholakia B, Feldman R. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP). J Am Acad Dermatol. 2013;74:835–40.
Shetty S, Ahmed AR. Critical analysis of the use of rituximab in mucous membrane pemphigoid: a review of the literature. J Am Acad Dermatol. 2015;68:499–506.
Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol. 147:843–849.
Dastmalchi DA, Moslemkhani S, Bayat M, et al. The efficacy of rituximab in patients with mucous membrane pemphigoid. J Dermatolog Treat. 2022;33(2):1084–90.
Foster CS, Chang PY, Ahmed AR. Combination of rituximab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid: a preliminary report. Ophthalmology. 2010;117:861–9.
Li Z, Jing K, Li S, Feng S. Antigen recognition in the pathogenesis of immunoglobulin A-related autoimmune bullous diseases. Postep Dermatol Alergol. 2018;35:338–43.
Matsumoto T, Nakamura S, Ishii N. Erythrodermic linear IgA/IgG bullous dermatosis. Eur J Dermatol. 2019;29:220–1.
Sakaguchi M, Bito T, Oda Y. Three cases of linear IgA/IgG bullous dermatosis showing IgA and IgG reactivity with multiple antigens, particularly laminin-332. JAMA Dermatol. 2013;149:1308–13.
İslamoĝlu ZGK, Akyürek FT. A case of recalcitrant linear IgA bullous dermatosis: successfully treated with rituximab. Dermatol Ther. 2019;32(3): e12911.
Pinard C, Hebert V, Lecuyer M, Sacre L, Joly P. Linear IgA bullous dermatosis treated with rituximab. JAAD Case Rep. 2019;5:124–6.
Nedosekin D. Immunologic overlap in a case of linear IgG/IgA bullous dermatosis responsive to rituximab: a case report and discussion. JAAD case reports. 2021;9:57–60.
Iwata H, Vorobyev A, Koga H. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13(1):153.
Yang A, Kim M, Craig P, Murrell DF. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Acta Dermatovenerol Croat. 2018;26(4):325–8.
Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31(6): e12726.
Oktem A, Akay BN, Boyvat A. Long-term results of rituximab-intravenous immunoglobulin combination therapy in patients with epidermolysis bullosa acquisita resistant to conventional therapy. J Dermatolog Treat. 2016;28:50–4.
McKinley SK, Huang JT, Tan J, Kroshinsky D, Gellis S. A case of recalcitrant epidermolysis bullosa acquisita responsive to rituximab therapy. Pediatr Dermatol. 2014;31:241–4.
Saha M, Cutler T, Bhogal B, Black MM, Groves RW. Refractory epidermolysis bullosa acquisita: successful treatment with rituximab. Clin Exp Dermatol. 2009;34(8):e979–80.
Lipozenčić J, Ljubojevic S, Bukvić-Mokos Z. Pemphigoid gestationis. Clin Dermatol. 2012;30:51–5.
Cianchini G, Masini C, Lupi F, Corona R, Pità O, Puddu P. Severe persistent pemphigoid gestationis: long-term remission with rituximab. Br J Dermatol. 2007;157:388–9.
Tourte M, Brunet-Possenti F, Mignot S, Gavard L, Descamps V. Pemphigoid gestationis: a successful preventive treatment by rituximab. J Eur Acad Dermatol Venereol. 2017;31(4):e206–7.
Chakravarty EF, Murray ER, Kelman A, Farmer P. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499–506.
Kasi PM, Tawbi HA, Oddis CV, Kulkarni HS. Clinical review: serious adverse events associated with the use of rituximab—a critical care perspective. Crit Care. 2012;16(4):231.
Bragnes Y, Boshuizen R, de Vries A, Lexberg Å, Østensen M. Low level of Rituximab in human breast milk in a patient treated during lactation. Rheumatology. 2017;56:1047–8. https://doi.org/10.1093/rheumatology/kex039.
Silva-Fernández L, Loza E, Martínez-Taboada VM. Biological therapy for systemic vasculitis: a systematic review. Semin Arthritis Rheum. 2014;43:542–57.
Taha R, El-Haddad H, Almuallim A, Alshaiki F, Obaid E, Almoallim H. Systematic review of the role of rituximab in treatment of antineutrophil cytoplasmic autoantibody-associated vasculitis, hepatitis C virus-related cryoglobulinemic vasculitis, Henoch-Schönlein purpura, ankylosing spondylitis, and Raynaud’s phenomenon. Open Access Rheumatol. 2017;9:201–14.
Yates M, Watts R. ANCA-associated vasculitis. Clin Med. 2017;17:60–4.
Mukhtyar C, Lee R, Brown D, Carruthers D, Dasgupta B, Dubey S, et al. Modification and validation of the Birmingham vasculitis activity score (version 3). Ann Rheum Dis. 2009;68:1827–32.
Stone JH, Merkel PA, Spiera R. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–32.
Jones RB, Tervaert JWC, Hauser T. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–20.
Furuta S, Nakagomi D, Kobayashi Y, Hiraguri M, Sugiyama T, Amano K, et al. Effect of reduced-dose vs high-dose glucocorticoids added to rituximab on remission induction in ANCA-associated vasculitis: a randomized clinical trial. JAMA. 2021;325:2178–87.
Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaître O, Cohen P, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371:1771–80.
Charles P, Terrier B, Perrodeau É, Cohen P, Faguer S, Huart A, et al. Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis. 2018;77:1143–9.
Wink F, Houtman PM, ThA J. Rituximab in cryoglobulinaemic vasculitis, evidence for its effectivity: a case report and review of literature. Clin Rheumatol. 2010;30:293–300.
Silva F, Pinto C, Barbosa A, Borges T, Dias C, Almeida J. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105: 102313.
Pietrogrande M, Vita S, Zignego AL. Recommendations for the management of mixed cryoglobulinemia syndrome in hepatitis C virus-infected patients. Autoimmun Rev. 2011;10:444–54.
Saadoun D, Resche-Rigon M, Sene D, Perard L, Karras A, Cacoub P. Rituximab combined with Peg-interferon-ribavirin in refractory hepatitis C virus-associated cryoglobulinaemia vasculitis. Ann Rheum Dis. 2008;67(10):1431–6.
Ferri C, Cacoub P, Mazzaro C. Treatment with rituximab in patients with mixed cryoglobulinemia syndrome: Results of multicenter cohort study and review of the literature. Autoimmun Rev. 2010;11:48–55.
Vita S, Quartuccio L, Isola M. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheum. 2015;64:843–53.
Sneller MC, Hu Z, Langford CA. A randomized controlled trial of rituximab following failure of antiviral therapy for hepatitis C virus-associated cryoglobulinemic vasculitis. Arthritis Rheum. 2013;64:835–42.
Colantuono S, Mitrevski M, Yang B. Efficacy and safety of long-term treatment with low-dose rituximab for relapsing mixed cryoglobulinemia vasculitis. Clin Rheumatol. 2021;36:617–23.
Visentini M, Ludovisi S, Petrarca A. A phase II, single-arm multicenter study of low-dose rituximab for refractory mixed cryoglobulinemia secondary to hepatitis C virus infection. Autoimmun Rev. 2016;10:714–9.
Harel S, Mohr M, Jahn I. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168(5):671–8.
Gragnani L, Visentini M, Fognani E. Prospective study of guideline-tailored therapy with direct-acting antivirals for hepatitis C virus-associated mixed cryoglobulinemia. Hepatology. 2016;64:1473–82.
Ozen S, Marks SD, Brogan P. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatol (Oxford). 2019;58:1607–16.
Maritati F, Fenoglio R, Pillebout E, Emmi G, Urban ML, Rocco R, et al. Brief report: rituximab for the treatment of adult-onset iga vasculitis (Henoch-Schönlein). Arthritis Rheumatol (Hoboken, NJ). 2018;70:109–14.
Fenoglio R, Sciascia S, Naretto C, De Simone E, Del Vecchio G, Ferro M, et al. Rituximab in severe immunoglobulin-A vasculitis (Henoch-Schönlein) with aggressive nephritis. Clin Exp Rheumatol. 2020;38(Suppl 1):195–200.
Crayne CB, Eloseily E, Mannion ML, Azerf SP, Weiser P, Beukelman T, et al. Rituximab treatment for chronic steroid-dependent Henoch-Schonlein purpura: 8 cases and a review of the literature. Pediatr Rheumatol Online J. 2018;16:71.
Hernández-Rodríguez J, Carbonell C, Mirón-Canelo J-A, Diez-Ruiz S, Marcos M, Chamorro AJ. Rituximab treatment for IgA vasculitis: a systematic review. Autoimmun Rev. 2020;19(4): 102490.
DeWane ME, Waldman R, Lu J. Dermatomyositis: clinical features and pathogenesis. J Am Acad Dermatol. 2020;82:267–81.
Sasaki H, Takamura A, Kawahata K, Takashima T, Imai K, Morio T, et al. Peripheral blood lymphocyte subset repertoires are biased and reflect clinical features in patients with dermatomyositis. Scand J Rheumatol. 2019;48:225–9.
Wang W-M, Guo L, Jin H-Z. Role of B cells in immune-mediated dermatoses. Mol Immunol. 2020;126:95–100.
Li W, Tian X, Lu X, Peng Q, Shu X, Yang H, et al. Significant decrease in peripheral regulatory B cells is an immunopathogenic feature of dermatomyositis. Sci Rep. 2016;6:1–11.
Waldman R, DeWane ME, Lu J. Dermatomyositis: diagnosis and treatment. J Am Acad Dermatol. 2020;82:283–96.
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–24.
Sultan SM, Allen E, Oddis CV, Kiely P, Cooper RG, Lundberg IE, et al. Reliability and validity of the myositis disease activity assessment tool. Arthritis Rheum. 2008;58:3593–9.
Sultan SM, Allen E, Cooper RG, Agarwal S, Kiely P, Oddis C V, et al. Interrater reliability and aspects of validity of the myositis damage index. Ann Rheum Dis. 2011;70:1272–1276. Available at: http://ard.bmj.com/content/70/7/1272.abstract
Aggarwal R, Bandos A, Reed AM. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2011;66:740–9.
Aggarwal R, Loganathan P, Koontz D, Qi Z, Reed AM, Oddis CV. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatol (Oxford). 2015;56:247–54.
Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol. 2007;143:763–7.
Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2022;52:601–7.
Schmeling H, Stephens S, Goia C, Manlhiot C, Schneider R, Luthra S, et al. Nailfold capillary density is importantly associated over time with muscle and skin disease activity in juvenile dermatomyositis. Rheumatology. 2011;50:885–93. https://doi.org/10.1093/rheumatology/keq407.
Argobi Y, Smith GP. Tracking changes in nailfold capillaries during dermatomyositis treatment. J Am Acad Dermatol. 2019;81(1):275–6.
Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Front Immunol. 2015;6:272.
Yoshizaki A. Pathogenic roles of B lymphocytes in systemic sclerosis. Immunol Lett. 2018;195:76–82.
Khanna D, Furst DE, Clements PJ. Standardization of the modified Rodnan skin score for use in clinical trials of systemic sclerosis. J Scleroderma Relat Disord. 2017;2:11–8.
Sircar G, Goswami RP, Sircar D, Ghosh A, Ghosh P. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: open label, randomized, controlled trial. Rheumatology. 2018;57:2106–13.
Thoreau B, Chaigne B, Renaud A, Mouthon L. Treatment of systemic sclerosis. Presse Med. 2021;50: 104088.
Jordan S, Distler JHW, Maurer B. Effects and safety of rituximab in systemic sclerosis: an analysis from the European scleroderma trial and research (EUSTAR) group. Ann Rheum Dis. 2015;74(6):1188–94.
McQueen FM, Solanki K. Rituximab in diffuse cutaneous systemic sclerosis: should we be using it today? Rheumatology. 2015;54:757–67.
Elhai M, Boubaya M, Distler O, Smith V, Matucci-Cerinic M, Alegre Sancho JJ, et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis. 2019;78:979–87.
Daoussis D, Liossis S-NC, Tsamandas AC. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology. 2010;49:271–80.
de Paula DR, Klem FB, Lorencetti PG, et al. Rituximab-induced regression of CREST-related calcinosis. Clin Rheumatol. 2013;32:281–3.
Narváez J, Pirola JP, LLuch J, Juarez P, Nolla JM, Valenzuela A. Effectiveness and safety of rituximab for the treatment of refractory systemic sclerosis associated calcinosis: A case series and systematic review of the literature. Autoimmun Rev. 2019;18:262–9.
Boonstra M, Meijs J, Dorjée AL, Marsan NA, Schouffoer A, Ninaber MK, et al. Rituximab in early systemic sclerosis. RMD Open. 2017;3:e000384. Available at: http://rmdopen.bmj.com/content/3/2/e000384.abstract
Lafyatis R, Kissin E, York M, Farina G, Viger K, Fritzler MJ, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009;60:578.
YarkanTuğsal H, Zengin B, Kenar G, Önen F, Birlik M. Rituximab on lung, skin, and joint involvement in patients with systemic sclerosis: a case series study and review of the literature. Int J Rheum Dis. 2022;25:755–68. https://doi.org/10.1111/1756-185X.14333.
Campochiaro C, Luca G, Lazzaroni MG. Safety and efficacy of rituximab biosimilar (CT-P10) in systemic sclerosis: an Italian multicentre study. Rheumatology (Oxford). 2020;59(12):3731–6.
Ribero S, Sciascia S, Borradori L, Lipsker D. The cutaneous spectrum of lupus erythematosus. Clin Rev Allergy Immunol. 2017;53:291–305.
Achtman JC, Werth VP. Pathophysiology of cutaneous lupus erythematosus. Arthritis Res Ther. 2015;17:182. https://doi.org/10.1186/s13075-015-0706-2.
Biazar C, Sigges J, Patsinakidis N, Ruland V, Amler S, Bonsmann G, et al. Cutaneous lupus erythematosus: first multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun Rev. 2013;12:444–54.
Chen KL, Krain RL, Werth VP. Advancing understanding, diagnosis, and therapies for cutaneous lupus erythematosus within the broader context of systemic lupus erythematosus. F1000Research. 2019;8:2.
Teng YKO, Bruce IN, Diamond B, Furie RA, van Vollenhoven RF, Gordon D, et al. Phase III, multicentre, randomised, double-blind, placebo-controlled, 104-week study of subcutaneous belimumab administered in combination with rituximab in adults with systemic lupus erythematosus (SLE): BLISS-BELIEVE study protocol. BMJ Open. 2019;9: e025687.
Jones A, Muller P, Dore CJ, Ikeji F, Caverly E, Chowdhury K, et al. Belimumab after B cell depletion therapy in patients with systemic lupus erythematosus (BEAT Lupus) protocol: a prospective multicentre, double-blind, randomised, placebo-controlled, 52-week phase II clinical trial. BMJ Open. 2019;9: e032569.
Lu TY, Ng KP, Cambridge G, Leandro MJ, Edwards JCW, Ehrenstein M, et al. A retrospective seven-year analysis of the use of B cell depletion therapy in systemic lupus erythematosus at university college london hospital: the first fifty patients. Arthritis Care Res. 2009;61:482–7.
Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: a systematic review of off-label use in 188 cases. Lupus. 2009;18:767–76.
Merrill JT, Neuwelt CM, Wallace DJ. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–33.
Rovin BH, Furie R, Latinis K. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis assessment with Rituximab study. Arthritis Rheum. 2012;64:1215–26.
Pirone C, Mendoza-Pinto C, Windt DA, Parker B, Sullivan O, M B, et al. Predictive and prognostic factors influencing outcomes of rituximab therapy in systemic lupus erythematosus (SLE): a systematic review. Semin Arthritis Rheum. 2017;47:384–96.
Alsanafi S, Kovarik C, Mermelstein AL, Werth VP. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142–4.
Lowe CD, Brahe CA, Green B, Lam TK, Meyerle JH. Bullous systemic lupus erythematosus successfully treated with rituximab. Cutis. 2019;103(6):e5-E7.
Penha MÁ, S LR, Miot HA. Rituximab in the treatment of extensive and refractory subacute cutaneous lupus erythematosus. An Bras Dermatol. 2018;93:467–9.
Dodds M, Hobday P, Schultz B, Maguiness S. Successful treatment of severe subacute cutaneous lupus erythematosus with rituximab in an adolescent. Pediatr Dermatol. 2108;35(3):189–190.
Kieu V, O’Brien T, Yap L-M. Refractory subacute cutaneous lupus erythematosus successfully treated with rituximab. Australas J Dermatol. 2010;50:202–6.
Uthman I, Taher A, Abbas O, Menassa J, Ghosn S. Successful treatment of refractory skin manifestations of systemic lupus erythematosus with rituximab: report of a case. Dermatology. 2019;216:257–9.
Terrier B, Amoura Z, Ravaud P. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum. 2013;62:2458–66.
Vital EM, Wittmann M, Edward S. Brief report: responses to rituximab suggest B cell-independent inflammation in cutaneous systemic lupus erythematosus. Arthritis Rheumatol. 2022;67:1586–91.
Hofmann SC, Leandro MJ, Morris SD, Isenberg DA. Effects of rituximab-based B-cell depletion therapy on skin manifestations of lupus erythematosus—report of 17 cases and review of the literature. Lupus. 2013;22:932–9.
da Quelhas CR, Aguirre-Alastuey ME, Isenberg DA, et al. Assessment of response to B-cell depletion using rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154:1432–40.
Antonelli A, Fallahi P, Elia G. Graves’ disease: Clinical manifestations, immune pathogenesis (cytokines and chemokines) and therapy. Best Pr Res Clin Endocrinol Metab. 2020;34(1): 101388.
Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of graves disease (pretibial myxedema. Review of 150 cases. Medicine. 1994;73(1):1–7.
Heyes C, Nolan R, Leahy M, Gebauer K. Treatment-resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53(1):e1-4.
Genere N, Stan MN. Current and emerging treatment strategies for graves. Orbitopathy Drugs. 2019;79:109–24.
Kotwal A, Turcu AF, Sonawane V. Clinical experience with rituximab and intravenous immunoglobulin for pretibial myxedema: a case series. Thyroid. 2019;29:692–9.
Mitchell AL, Gan EH, Morris M. The effect of B cell depletion therapy on anti-TSH receptor antibodies and clinical outcome in glucocorticoid-refractory Graves’ orbitopathy. Clin Endocrinol (Oxf). 2013;79(3):437–42.
Ferreira-Hermosillo A, Casados-V R, Paúl-Gaytán P, Mendoza-Zubieta V. Utility of rituximab treatment for exophthalmos, myxedema, and osteoarthropathy syndrome resistant to corticosteroids due to Graves’ disease: a case report. J Med Case Rep. 2021;12:38–38.
Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS. Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J Clin Endocrinol Metab. 2022;100:432–41.
Salvi M, Vannucchi G, Currò N. Efficacy of B-cell targeted therapy with rituximab in patients with active moderate to severe Graves’ orbitopathy: a randomized controlled study. J Clin Endocrinol Metab. 2015;100:422–31.
Lehman JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682–94. https://doi.org/10.1111/j.1365-4632.2009.04062.x.
Parmentier L, Bron B-A, Prins C, Samson J, Masouyé I, Borradori L. Mucocutaneous lichen planus with esophageal involvement: successful treatment with an anti-CD20 monoclonal antibody. Arch Dermatol. 2008;144:1427–30. https://doi.org/10.1001/archderm.144.11.1427.
Lagerstedt M, Kotaniemi-Talonen L, Antonen J, Vaalasti A. Erosive vulvo–vaginal lichen planus treated with rituximab: a case report. Int J Gynecol Obstet. 2022;156:172–3. https://doi.org/10.1002/ijgo.13814.
Heelan K, McAleer MA, Roche L, McCreary C, Murphy M. Intractable erosive lichen planus treated successfully with rituximab. Br J Dermatol. 2015;172:538–40. https://doi.org/10.1111/bjd.13537.
Goñi Esarte S, Arín Letamendía A, Vila Costas JJ, Jiménez Pérez FJ, Ruiz-Clavijo García D, Carrascosa Gil J, et al. Rituximab as rescue therapy in refractory esophageal lichen planus [in Spanish]. Gastroenterol Hepatol. 2013;36(4):264–7.
Erras S, Mouna Z, Akhdari N, Belaabidia B, Essaadouni L. Rapid and complete resolution of lichen planopilaris in juvenile chronic arthritis treated with rituximab. Eur J Dermatol. 2011;21(1):116–7.
Tétu P, Monfort J-B, Barbaud A, Francès C, Chasset F. Failure of rituximab in refractory erosive lichen planus. Br J Dermatol. 2018;179:980–1. https://doi.org/10.1111/bjd.16704.
Giudice A, Liborio F, Averta F, Barone S, Fortunato L. Oral lichenoid reaction: an uncommon side effect of rituximab. Case Rep Dent. 2019;2019:3154856.
Bakkour W, Coulson IH. GA101 (a novel anti-CD20 monoclonal antibody)-induced lichenoid eruption. Dermatol Ther (Heidelb). 2012;2:3.
Kuten-Shorrer M, Hochberg EP, Woo S-B. Lichenoid mucosal reaction to rituximab. Oncologist. 2014;19:e12–3.
Paul F, Cartron G. Infusion-related reactions to rituximab: frequency, mechanisms and predictors. Expert Rev Clin Immunol. 2019;15:383–9.
Makatsori M, Kiani-Alikhan S, Manson AL. Hypogammaglobulinaemia after rituximab treatment-incidence and outcomes. QJM. 2014;107:821–8.
Dunleavy K, Tay K, Wilson WH. Rituximab-associated neutropenia. Semin Hematol. 2010;47:180–6.
Tsutsumi Y, Yamamoto Y, Ito S. Hepatitis B virus reactivation with a rituximab-containing regimen. World J Hepatol. 2015;7:2344–51.
Berger JR, Malik V, Lacey S, et al. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event [published correction appears in J Neurovirol. 2018;24(3):332]. J Neurovirol. 2018;24(3):323–31.
Dass S, Vital EM, Emery P. Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum. 2007;56:2715–8.
Kim MJ, Kim HO, Kim HY, Park YM. Rituximab-induced vasculitis: a case report and review of the medical published work. J Dermatol. 2009;36:284–7.
Karamanakos A, Vergou T, Panopoulos S, Tektonidou MG, Stratigos AJ. Psoriasis as an adverse reaction to biologic agents beyond anti-TNF-α therapy. Eur J Dermatol. 2021;31:307–17.
Vesely NC, Thomas RM, Rudnick E, Longo MI. Scar sarcoidosis following rituximab therapy. Dermatol Ther. 2020;33: e13693.
Sanke S, Mendiratta V, Jassi R, Yadav J, Chander R. Rituximab induced reticulate pigmentation over face in pemphigus vulgaris. Dermatol Ther. 2020;33:e13752. Available at: https://onlinelibrary-wiley-com.libproxy.uams.edu/doi/full/https://doi.org/10.1111/dth.13752
Mahmoudi H, Nili A, Salehi Farid A, Tavakolpour S, Daneshpazhooh M. Paradoxical worsening of pemphigus after rituximab presenting as figurate bullous eruption. Br J Dermatol. 2021;185:e2. Available at: https://onlinelibrary-wiley-com.libproxy.uams.edu/doi/full/https://doi.org/10.1111/bjd.20054
Nuño L, Navarro MN, Bonilla G, Franco-Gómez K, Aguado P, Peiteado D, et al. Clinical course, severity and mortality in a cohort of patients with COVID-19 with rheumatic diseases. Ann Rheum Dis. 2020;79:1659–61.
Tepasse P, Hafezi W, Lutz M, Kühn J, Wilms C, Wiewrodt R, et al. Persisting SARS-CoV-2 viraemia after rituximab therapy: two cases with fatal outcome and a review of the literature. Br J Haematol. 2020;190:185–8.
Guilpain P, Le Bihan C, Foulongne V, Taourel P, Pansu N, Maria ATJ, et al. Rituximab for granulomatosis with polyangiitis in the pandemic of covid-19: lessons from a case with severe pneumonia. Ann Rheum Dis. 2021;80: e10.
Schulze-Koops H, Krueger K, Vallbracht I, Hasseli R, Skapenko A. Increased risk for severe COVID-19 in patients with inflammatory rheumatic diseases treated with rituximab. Ann Rheum Dis. 2021;80:e67–e67.
Avouac J, Airó P, Carlier N, Matucci-Cerinic M, Allanore Y. Severe COVID-19-associated pneumonia in 3 patients with systemic sclerosis treated with rituximab. Ann Rheum Dis. 2021;80:37.
Rodriguez-Pla A, Vikram HR, Khalid V, Wesselius LJ. COVID-19 pneumonia in a patient with granulomatosis with polyangiitis on rituximab: case-based review. Rheumatol Int. 2022;41:1509–14.
Levavi H, Lancman G, Gabrilove J. Impact of rituximab on COVID-19 outcomes. Ann Hematol. 2021;100:2805–12.
Winthrop KL, Silverfield J, Racewicz A, Neal J, Lee EB, Hrycaj P, et al. The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis. 2016;75:687–95.
França ILA, Ribeiro ACM, Aikawa NE, Saad CGS, Moraes JCB, Goldstein-Schainberg C, et al. TNF blockers show distinct patterns of immune response to the pandemic influenza A H1N1 vaccine in inflammatory arthritis patients. Rheumatology. 2012;51:2091–8.
Arnold J, Winthrop K, Emery P. COVID-19 vaccination and antirheumatic therapy. Rheumatology (Oxford). 2021;60:3496–502.
Shree T, Shankar V, Lohmeyer JJK, Czerwinski DK, Schroers-Martin JG, Rodriguez GM, et al. CD20-targeted therapy ablates de novo antibody response to vaccination but spares preestablished immunity. Blood Cancer Discov. 2022;3:95–102.
Mrak D, Tobudic S, Koblischke M, Graninger M, Radner H, Sieghart D, et al. SARS-CoV-2 vaccination in rituximab-treated patients: B cells promote humoral immune responses in the presence of T-cell-mediated immunity. Ann Rheum Dis. 2021;80(10):1345–50.
Agarwal A, Hall R, Banez L, Cardones A. Comparison of rituximab and conventional adjuvant therapy for pemphigus vulgaris: a retrospective analysis. PLoS ONE. 2018;13:2.
Tovanabutra N, Payne AS. Clinical outcome and safety of rituximab therapy for pemphigoid diseases. J Am Acad Dermatol. 2020;82:1237–9. Available at: https://www.sciencedirect.com/science/article/pii/S0190962219331135
Rübsam A, Stefaniak R, Worm M, Pleyer U. Rituximab preserves vision in ocular mucous membrane pemphigoid. Expert Opin Biol Ther. 2015;15:927–33.
Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, Caux F, Pascal F, Doan S, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol. 2011;147:843–9.
Heelan K, Walsh S, Shear NH. Treatment of mucous membrane pemphigoid with rituximab. J Am Acad Dermatol. 2013;69:310–1.
Kuye IO, Smith GP. The use of rituximab in the management of refractory dermatomyositis. J Drugs Dermatol. 2017;16:162–6.
Bosello SL, De Luca G, Rucco M, Berardi G, Falcione M, Danza FM, et al. Long-term efficacy of B cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin Arthritis Rheum. 2015;44:428–36.
Smith V, Van Praet JT, Vandooren B, Van der Cruyssen B, Naeyaert J-M, Decuman S, et al. Rituximab in diffuse cutaneous systemic sclerosis: an open-label clinical and histopathological study. Ann Rheum Dis. 2010;69:193–7.
Md Yusof MY, Shaw D, El-Sherbiny YM, Dunn E, Rawstron AC, Emery P, et al. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann Rheum Dis. 2017;76:1829–36.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author contributions
All authors contributed to the study design. The literature search was performed by DN and SL. The manuscript was drafted by DN and SL, and critically revised by HKW, DN, and SL.
Funding
No funding was received for this study.
Conflicts of interest
Henry Wong, Dmitry Nedosekin, and Sophia Ly have no conflicts of interest to declare.
Ethics approval
Not applicable.
Consent to participate/publish
Not applicable.
Availability of data and material
No datasets were generated and/or analyzed during the current study
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Ly, S., Nedosekin, D. & Wong, H.K. Review of an Anti-CD20 Monoclonal Antibody for the Treatment of Autoimmune Diseases of the Skin. Am J Clin Dermatol 24, 247–273 (2023). https://doi.org/10.1007/s40257-022-00751-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-022-00751-7