
Abstract
Background
The efficacy, safety and immunogenicity risk of switching between an originator biologic and a biosimilar or from one biosimilar to another are of potential concern.
Objectives
The aim was to conduct a systematic literature review of the outcomes of switching between biologics and their biosimilars and identify any evidence gaps.
Methods
A systematic literature search was conducted in PubMed, EMBASE and Cochrane Library from inception to June 2017. Relevant societal meetings were also checked. Peer-reviewed studies reporting efficacy and/or safety data on switching between originator and biosimilar products or from one biosimilar to another were selected. Studies with fewer than 20 switched patients were excluded. Data were extracted on interventions, study population, reason for treatment switching, efficacy outcomes, safety and anti-drug antibodies.
Results
The systematic literature search identified 63 primary publications covering 57 switching studies. The reason for switching was reported as non-medical in 50 studies (23 clinical, 27 observational). Seven studies (all observational) did not report whether the reasons for switching were medical or non-medical. In 38 of the 57 studies, fewer than 100 patients were switched. Follow-up after switching went beyond 1 year in eight of the 57 studies. Of the 57 studies, 33 included statistical analysis of disease activity or patient outcomes; the majority of these studies found no statistically significant differences between groups for main efficacy parameters (based on P < 0.05 or predefined acceptance ranges), although some studies observed changes for some parameters. Most studies reported similar safety profiles between groups.
Conclusions
There are important evidence gaps around the safety of switching between biologics and their biosimilars. Sufficiently powered and appropriately statistically analysed clinical trials and pharmacovigilance studies, with long-term follow-ups and multiple switches, are needed to support decision-making around biosimilar switching.
Similar content being viewed by others
We believe that sufficiently powered and appropriately statistically analysed trials and pharmacovigilance studies, with long-term follow-ups and multiple switching sequences, are still needed to support decision-making around biosimilar interchangeability. |
In the interim, switching should remain a clinical decision made by the treating physician and the patient based on available evidence and individual patient circumstances. |
1 Introduction
Biological medicines (‘biologics’) are derived from living organisms, often by using recombinant DNA technology. Biologics differ from traditional, small molecule medicines, which are generally structurally simple, chemically synthesised and easily characterised analytically. Post-translational modifications of biologics are particularly vulnerable to manufacturing process conditions, and maintaining batch-to-batch consistency is essential [1]. Small process changes (‘evolution’) or unintentional deviations (‘drift’) can lead to changes in the biological end product and, ultimately, to product divergence, a concept that has particular relevance in the setting of biosimilars [2].
Quality assessment of biologics in terms of efficacy and safety requires physicochemical and functional assays, pharmacokinetic and pharmacodynamic evaluations, toxicity testing in animals and clinical pharmacology [3,4,5]. Unwanted immune responses are a concern, both in terms of adverse events (e.g. anaphylaxis, infusion reactions) and neutralisation of either the therapeutic biologic or the endogenous counterparts by neutralising antibodies, and have terminated development of some biologics [6].
Biosimilars have established similarity to the biologic reference product in terms of safety and efficacy. There are stringent regulatory requirements for demonstration of biosimilarity—including demonstration of comparable physicochemical characteristics, biological activity, efficacy and safety/immunogenicity—and these are laid down in the respective US Food and Drug Administration (FDA) and European Union (EU) guidance documents, and the approval process of biosimilars in these highly regulated markets is rigorous [4, 5, 7,8,9,10]. However, unlike generics of small molecule medicines (which can be fully characterised structurally, thus allowing for the same drug substance to be produced), biosimilars of biologic reference products can differ in terms of the overall structure of their drug substance and are similar to, but not identical to, the originator product. This is because biologics are large and structurally complex, meaning that, unlike the characterisation of generics, current analytical methodology may not be able to detect or characterise all relevant structural and functional differences between biologics, a distinction that is also noted in the FDA guidance on demonstrating biosimilarity and the Australian Therapeutic Goods Administration (TGA) biologic nomenclature consultation [3, 11]. Data on the efficacy and safety of initiating therapy with a biosimilar versus with an originator biologic, generated as part of regulatory approval and post-approval phases, have been widely reviewed [12,13,14] and are not the focus of the current article.
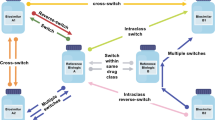
‘Interchangeability’ is the medical practice of changing one medicine for another that is expected to achieve the same clinical effect in a given clinical setting and in any patient, on the initiative, or with the agreement, of the prescriber [15, 16]. ‘Switching’ refers to the treating physician exchanging one medicine with another medicine with the same therapeutic intent in a given patient [17]. Substitution is the practice of dispensing one medicine instead of another equivalent and interchangeable medicine at the pharmacy level without consulting the prescriber [15]. Implied in this is that a biosimilar has been shown to produce the same result as the originator in any patient and that there are no signals suggesting loss of efficacy or increased toxicity if multiple switches are made in the cases in which the medicine must be administered on many occasions.
Definitions of biosimilarity and interchangeability used in the USA, the EU and Australia are summarised in Supplementary Table 1 (see the electronic supplementary material) [4, 5, 7,8,9,10]. For a reference product and a biosimilar to be considered interchangeable, the FDA requires sponsors to demonstrate that the risk in terms of side effects or diminished efficacy of switching is not greater than the risk of using the reference product without such alternating or switching [18]. Recently published FDA draft guidance on the specific data required to achieve a designation of interchangeability states that for products that are administered more than once, marketing applications are expected to include data from switching studies in appropriate conditions of use [19]. In the EU, substitution policies are within the remit of each member state [4]. However, officials from Finnish, Dutch, German and Norwegian national regulatory agencies concluded in a recent ‘current opinion’ article, published in BioDrugs, that “biosimilars licensed in the EU are interchangeable” [17]. In Australia, the TGA has responsibility for marketing authorisation (focusing on safety, efficacy and quality), with the Pharmaceutical Benefit Advisory Committee (PBAC) advising the government about whether a drug should be subsidised (focusing on cost-benefit) [20]. The PBAC has proposed that biosimilar substitution at the pharmacy level is acceptable when there is “absence of data to suggest significant differences in clinical effectiveness or safety compared with the originator product” that argue against such action [8]. The interchangeability of an originator biologic and a biosimilar or between biosimilars is considered by the PBAC on a case-by-case basis [8].
Switching between an originator biologic and a biosimilar medicine or between biosimilars can be categorised as either medical or non-medical. Medical switching is initiated by the prescriber because of medical concerns such as adverse events, or convenience of dosing or administration. For example, the incidence of injection site reactions in phase 3 clinical trials was lower with biosimilar than originator tumour necrosis factor (TNF) inhibitors, a difference that was explained by a lack of l-arginine in the formulation and of latex in the needle shield of the biosimilar products [21]. Non-medical switching occurs because of non-medical concerns, including treatment cost or availability. Recommendations from professional societies around switching are summarised in Supplementary Table 2 [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. Importantly, across the USA, Canada, Europe and Australia, professional societies state that the decision to switch has to be taken by the treating physician. In the USA, for example, the American Academy of Dermatology (AAD) and the American College of Rheumatology (ACR) support switching only if it is deemed suitable by the prescribing provider [22, 26], and the Australian Rheumatology Association (ARA) position is that the decision to prescribe any medication should rest with the prescriber, in consultation with an informed patient, and substitution should not occur without the knowledge and consent of the patient [35]. The Australian Diabetes Society and Australian Diabetes Educators Association (in a collective statement with Diabetes Australia), the Gastroenterological Society of Australia and the Australian Inflammatory Bowel Disease Association strongly oppose recommendations of biosimilars as interchangeable on the grounds of patient safety [23, 33]. This is in stark contrast with the Pharmaceutical Society of Australia and the Pharmacy Guild of Australia, who support substitution by the pharmacist if the patient agrees and if the prescribing physician has not specifically indicated that no substitution should occur [38, 39].
Ongoing areas of scientific debate and potential concern around biosimilars are (1) extrapolation of approval of biosimilars across multiple clinical indications, (2) presence of impurities or chemical alterations not detected with standard comparability testing [40,41,42], and (3) whether the efficacy, safety and immunogenicity risk of therapy are affected when patients are switched between originator and biosimilar products or from one biosimilar to another, in particular when multiple switching occurs. This article will focus on the third point, namely the key principles around biosimilarity and interchangeability, and analyse the available evidence around switching between originator biologics and biosimilars or between biosimilars based on a systematic review of the published literature, identifying any significant evidence gaps.
2 Methods
2.1 Systematic Literature Searches
A systematic literature review was conducted in PubMed, EMBASE and Cochrane Library to 10 June 2017 to identify studies reporting efficacy and/or safety data on switching between originator biologics and biosimilars or between biosimilars, using the following search string: ((biosimilars OR biosimilar OR biosimilarity OR “subsequent entry biologic” OR “subsequent entry biologics” OR “similar biotherapeutic product” OR “similar biotherapeutic products” OR “similar biological medicinal product” OR “similar biological medicinal products” OR “follow-on biologic” OR “follow-on biologics”) AND (equivalent OR equivalence OR comparability OR substitute OR substitution OR substitutability OR switch OR switched OR switching OR interchange OR interchanged OR interchangeable OR interchangeability)). Records in PubMed were retrieved using the title and abstract (‘tiab’) search filter. Records in EMBASE were retrieved using the ‘ex/mj’ syntax to capture synonyms and apply major focus for all terms.
The following meetings from the main societies representing key therapeutic areas for biosimilars were checked for relevant abstracts published in 2015 or 2016: American Association of Clinical Endocrinologists (AACE); AAD; ACR; American Diabetes Association; American Gastroenterology Association; American Society of Clinical Oncology (ASCO); American Society of Nephrology; European Crohn’s and Colitis Organisation; European Renal Association/European Dialysis and Transplant Association European Society for Medical Oncology; and European League against Rheumatism. In addition, ASCO was also checked for relevant abstracts published in 2017.
The reference lists of identified primary articles and literature reviews were hand searched for any additional, potentially relevant articles. To supplement the systematic searches, an exploratory literature search was undertaken to support the overview section around the principles of biologics, biosimilarity and interchangeability.
2.2 Study Eligibility and Data Extraction
Studies were excluded if they presented data on switching only between different classes of originator biologics and biosimilars or between different classes of biosimilars. Pharmacokinetic/pharmacodynamic cross-over studies in healthy volunteers were included if more than one dose of study drug was administered and if data were reported separately for the post-washout/post-switch study period. Studies comprising fewer than 20 switch patients were excluded. No exclusion criteria were applied regarding study type (clinical or observational), retrospective or prospective analysis or publication date.
Data were extracted on treatment, indication, study design, comparators, patient numbers and demographics (age, sex), reason for treatment switching, efficacy, safety and anti-drug antibodies (ADAs).
All data were tabulated separately by study type (clinical or observational) and reason for switching (medical or non-medical). No meta-analyses were performed given the heterogeneity of study designs, interventions, populations and methods of analysis.
2.3 Role of Funding Source
The funding source had no role in the conduct of the present study.
3 Results
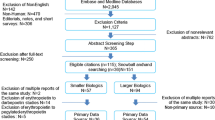
The systematic literature search identified 63 primary publications, covering 57 switching studies that reported efficacy and/or safety data [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105]. A flow chart of the systematic searches is shown in Figure 1. Forty-one of the identified studies were reported as full articles; the remaining 16 studies were covered only in abstract form. In terms of design, 23 were clinical studies [22 randomised controlled trials (RCTs), including 11 open-label extensions of RCTs; and one non-RCT cross-over study] and 34 were observational (24 prospective, 10 retrospective).

Flow chart of systematic literature searches. RCT randomised controlled trial
Patient demographics and other study characteristics are summarised in Supplementary Table 3 [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105]. The number of included patients per study ranged from 20 to 802, except for a retrospective chart review with N = 3018 [72] and an insurance database study with N = 6177 [76]. The average age, where reported, ranged from younger than 10 years to 71 years, and the proportion of men, where reported, ranged from 0% to 87%.
The reason for switching was reported as non-medical in 50 studies, of which 23 were clinical studies (Table 1) and 27 were observational studies (Table 2). The remaining seven studies did not report whether the reasons for switching were medical or non-medical; all were observational (Table 3). None of the studies reported having only medical reasons for switching.
3.1 Non-medical Switching: Clinical Efficacy and Safety Data
Of the 23 clinical, non-medical switching studies (Table 1) [53, 55, 56, 60, 61, 69, 70, 73,74,75, 81, 82, 84,85,86, 91,92,93,94,95,96,97,98,99, 101,102,103,104], 22 reported data on main efficacy parameters in switched versus non-switched or pre-switch versus post-switch groups. The originator biologics/biosimilars assessed were infliximab (six studies [53, 55, 56, 61, 69, 70, 73]), erythropoietin-stimulating agents (ESAs) (four studies [74, 75, 81, 82]), filgrastim [93,94,95] (three studies), insulin [84,85,86], adalimumab [96,97,98], rituximab [60, 99] and etanercept [101,102,103] (two studies each), and genotropin [83, 92] and follicle stimulating hormone [104] (one study each).
Of these, 15 included statistical analyses of disease activity or patient outcomes in switched versus non-switched groups, pre-switch versus post-switch groups or observed versus predicted outcomes [53, 60, 61, 73, 74, 81, 82, 84,85,86, 91,92,93,94,95, 99], with five studies specifying a formal equivalence or non-inferiority margin [53, 74, 81, 82, 95]. There were no statistically significant differences between groups (based on P < 0.05 or predefined acceptance ranges), with the exception of a significant weight gain that was observed in patients with type 1 diabetes mellitus switched to biosimilar compared with patients remaining on originator insulin (+ 1.0 kg vs + 0.2 kg, respectively; P < 0.05) in the open-label extension of the ELEMENT 1 RCT, although the range of weight changes was wide in both groups [84]. The NOR-SWITCH infliximab trial was conducted across six indications and used a non-inferiority margin of 15% (adapted from rheumatoid arthritis studies), with results suggesting no loss of efficacy and no unexpected treatment-emergent adverse events (TEAEs) [53]. Eight studies reported similar outcomes between groups, but failed to provide a formal statistical analysis of between-group comparisons [55, 56, 69, 70, 75, 98, 102,103,104].
Most studies reported similar safety profiles between groups. An exception was the PLANETAS extension study of infliximab in patients with ankylosing spondylitis, in which TEAEs were considerably more frequent in the switched group [71% (60 patients out of 84) vs 49% (44 patients out of 90) in the group remaining on originator biologic] and included TEAEs that were considered related to the study drug [39% (33 patients out of 84) vs 22% (20 patients out of 90)] [61].
3.2 Non-medical Switching: Real-world Efficacy and Safety Data
All of the 27 observational, non-medical switching studies reported data on main efficacy parameters after switching compared with baseline/before switching (Table 2) [43,44,45,46,47,48,49,50,51,52, 54, 58, 62, 63, 65,66,67,68, 71, 72, 77, 79, 83, 87,88,89,90, 105]. The originator biologics/biosimilars assessed were infliximab (20 studies [43,44,45,46,47,48,49,50,51,52, 54, 58, 62, 63, 65,66,67,68, 71, 72]), ESAs (two studies [77, 79]), insulin (two studies [83, 87]) and growth hormone (three studies [88,89,90]).
Of 16 studies that reported statistical analyses, 13 found no statistically significant differences for main efficacy parameters [43, 46,47,48,49, 58, 62, 67, 68, 71, 77, 79, 87], although six of these observed changes for some parameters (in favour of the pre-switch and post-switch treatments in five studies and one study, respectively). There was a need for additional medication to control increased disease activity after switching from originator to biosimilar infliximab in four of 83 patients with inflammatory bowel disease (IBD) [68] and in 13 of 28 patients undergoing chemotherapy and switching epoetins [77]; a significant increase in median C-reactive protein values (from 1.95 to 4.0 mg/L; P < 0.05) and median pain scores (from 28.8 to 38.1 mm on a scale of 0–100; P < 0.05) in patients with IBD switching from originator to biosimilar infliximab [43]; a significant increase in disease activity (mean Bath Ankylosing Spondylitis Activity Index, from 3.8 to 4.3; P < 0.05) in patients with spondyloarthritis after switching to biosimilar infliximab [71]; a small, significant increase in total daily insulin dose (from 0.62 to 0.65 U/kg/day; P < 0.05) in patients with type 1 or type 2 diabetes mellitus switched to a biosimilar insulin [87]; but a significant decrease (i.e. improvement) in duration of morning stiffness [median duration, from 7.2 to 5.8 (no units provided); P = 0.02] in patients with arthritis switching from originator to biosimilar infliximab [47]. In the three remaining studies that included statistical analyses, IBD disease activity significantly improved in paediatric patients switched from originator to biosimilar infliximab (P < 0.05; actual values not reported) [66], but significantly worsened in adults who underwent a similar switch (median IBD-Control-8 score, from 11 to 14; P < 0.05) [63], and primary failure was significantly lower in patients with IBD switched from originator to biosimilar infliximab than in patients switched from other biologics or who were treatment naïve (0 vs 11 and 10%, respectively; P < 0.05) [50].
Eight studies reported that switching generally had no noticeable effect, but did not provide a formal statistical analysis of between-group comparisons or based this conclusion on modelling [44, 45, 52, 65, 83, 88,89,90]. Two studies reported on switching from infliximab originator to biosimilar. In one, relapse was reported in seven of 23 patients with rheumatic diseases within about 2 months after switching [51], and in the other, three of 36 patients with IBD discontinued within 1 year of switching because of loss of efficacy [54].
In terms of safety data after switching, eight studies reported no concerns or similar safety profiles before and after switching [43, 44, 47, 50, 62, 63, 79, 87, 89], six studies reported no general safety data [45, 51, 72, 77, 83, 90], and 12 studies reported adverse events such as injection site pain, acute hypersensitivity reactions, rash and infusion reactions after switching (although most did not provide comparative data from before switching) [46, 48, 49, 52, 54, 58, 65,66,67,68, 71, 88].
3.3 Switching, Reasons Not Reported: Efficacy and Safety Data
All seven observational studies that did not include reasons for switching assessed efficacy parameters after switching compared with baseline/before switching (Table 3) [57, 59, 64, 76, 78, 80, 100]. Three involved infliximab [57, 59, 64], three ESAs [76, 78, 80] and one rituximab [100].
Two studies included statistical analyses, both of which found no significant differences for main efficacy parameters [57, 78]. In one of these studies, on ESAs in patients on haemodialysis, switching to a biosimilar resulted in higher mean drug dose requirements (from 8765 to 10,886 IU/week; P < 0.05) to maintain haemoglobin levels [78]. Of the five studies not reporting a formal statistical analysis of between-group comparisons, four reported that switching had no noticeable effect [64, 76, 80, 100] and one reported disease worsening in five of 46 evaluable patients with IBD (11%) during 30 weeks of follow-up after switching from infliximab originator to biosimilar [59].
In terms of safety data after switching, one study reported similar patterns to before switching [57], one (in patients with IBD) reported three cases of infusion reaction and one of pneumonia in the switch group, which comprised 60 patients (vs none in the treatment-naive group, which comprised 113 patients) [59], one reported adverse events (cutaneous leishmaniasis) only when these were a reason for treatment discontinuation [64], and four studies did not report safety data [76, 78, 80, 100].
3.4 Switching: Immunogenicity Data
The relative frequency of ADAs in biologic originator and biosimilars was reported in 27 studies, of which 18 were clinical studies [53, 55, 56, 60, 61, 69, 70, 73, 81, 82, 84,85,86, 91, 92, 95,96,97,98,99, 101,102,103,104] and nine were observational [46,47,48, 52, 57, 63, 67, 68, 71, 105]. In 26 studies (17 clinical: follow-up 12–52 weeks [53, 55, 56, 60, 61, 69, 70, 73, 81, 82, 84, 85, 91, 92, 95,96,97,98,99, 101,102,103]; nine observational: follow-up 6–13 months [46,47,48, 52, 57, 63, 67, 68, 71, 105]), there were either no cases of ADAs, or the incidence of ADAs was similar in both groups.
A significantly higher proportion of patients with ADAs in the switched group than the non-switched group (19% vs 8% overall; P = 0.01) was reported in a clinical study of insulin in type 2 diabetes mellitus (ELEMENT 2) during 24 weeks of follow-up [84,85,86].
Comparative data regarding neutralising ADAs were reported in six studies (all clinical); none of which observed any clinically relevant [60, 96, 97, 104] or significant [61, 73, 96] differences between groups. No studies reported any treatment failure related to the development of ADAs.
4 Discussion
4.1 Clinical Considerations
This systematic literature review reveals important evidence gaps around the safety of switching between originator biologics and biosimilars or between biosimilars. There is a paucity of data from large studies of sufficient statistical power for efficacy, safety and immunogenicity risk considerations. There is little information on immunogenicity outside of clinical studies, with only nine of 34 included observational studies (26%) reporting such data. There is a general lack of robustness of evidence and no long-term data, with only eight of the included studies having a post-switch follow-up period beyond 1 year. This is important because patients may be exposed to treatment long-term and, although safety issues such as ADAs occur most commonly early on during treatment with biologics [106], they have been reported to arise only after the first year of treatment in some patients [107, 108]: a meta-analysis of 68 studies on incidence of ADAs to TNF inhibitors found that these first developed as early as 2 weeks, but also as late as 3 years, after treatment initiation [108]. There is large heterogeneity in the design of available studies including the number of switches and permutations of switch scenarios, use of formal equivalence or non-inferiority margins, and assessment of safety and immunogenicity risk, making it difficult to draw conclusions from the data.
Demonstration of interchangeability is a complex process that requires clinical studies in addition to those conducted to establish biosimilarity [109]. In the absence of specifically designed cross-over or multiple switch studies, demonstration of biosimilarity is not adequate to establish interchangeability [109]. The study duration needs to be clinically informed and will not be the same in every setting. Long-term studies are needed if treatment is chronic. A particular challenge when designing interchangeability studies is how to define the non-inferiority or equivalence margin, a topic that is covered by the recent FDA draft guidance [19]. In most studies in the current review, analyses around interchangeability were fairly limited, for example, taking a lack of a statistically significant difference as a lack of need for concern even when confidence intervals were wide [60]. Only five studies specified a formal equivalence or non-inferiority margin [53, 74, 81, 82, 95]. Formal, standardised definitions around margins are needed.
To demonstrate interchangeability, a study needs to show that even multiple switches between products deemed biosimilar do not affect patient safety or treatment effectiveness. The FDA draft guidance on interchangeability states that the switching arm should incorporate at least two separate exposure periods to each of the two products (i.e. at least three switches, with each switch crossing over to the alternate product) [19]. The EGALITY (etanercept) and NOR-SWITCH (infliximab) trials focused specifically on the impact of switching between these TNF-α originator biologics and their corresponding biosimilars, but NOR-SWITCH was not designed to assess the effects of multiple switches [53, 103]. In the EGALITY study, patients with chronic plaque psoriasis either continued with their original therapy or underwent three treatment switches at 6-weekly intervals [103]. TEAEs were shown to be similar between groups, ADAs were low titre and transient, and no neutralising antibodies were detected. Although the study follow-up period was short and numbers small (200 switched patients), the study is an important addition to the evidence.
In NOR-SWITCH, 240 patients were switched across six diseases, with numbers per disease indication ranging from 16 patients for psoriatic arthritis to 77 patients for Crohn’s disease. Disease worsening, the primary endpoint in NOR-SWITCH, was defined differently across the six diseases, with the data then pooled across indications for analysis. The study showed similar results for switched and non-switched groups for disease worsening and safety although, as the authors of the study commented, it was not powered to demonstrate non-inferiority within each diagnostic group, and thus the pooled analysis used does not allow conclusions about individual diseases [53, 110].
Both EGALITY and NOR-SWITCH were relatively small, short-duration studies, and larger studies with longer-term follow-up are still needed, in particular for patients with rheumatic diseases and IBD who are exposed to treatments over many years. In general, once interchangeability is deemed to be established, the relevant information will need to be passed on to healthcare providers to allow them to make individualised treatment decisions for their patients.
Differences exist regarding the regulatory approval pathways for biosimilars in different parts of the world. The regulatory framework around biosimilars is robust in the highly regulated markets of Europe, the USA, Canada, Australia and Japan, with South Korea also having positioned itself as a high-quality global biosimilars leader, while in emerging markets in south-east Asia, Latin America and Africa the regulatory pathways for biosimilars are still evolving [111]. These differences need to be taken into account when interpreting results from switching studies. Almost all of the studies identified for inclusion in the current review originated from highly regulated markets, with the exception of seven studies that included emerging markets [75, 83, 87, 93, 94, 99, 100], of which six reported similar outcomes for switched and non-switched groups and one, from South Africa, reported a requirement for increased insulin dose post-switch [87].
While not the focus of the current review, changing from one originator biologic to another is relatively common in clinical practice, most likely because of inefficacy or intolerability issues with the initial treatment [112, 113]. It is important to note, however, that this scenario is very different from switching between an originator biologic and its biosimilar or between different biosimilars of the same biologic: for example, two originator biologics targeting a given molecule may bind different epitopes, whereas a biologic and its biosimilar or two biosimilars of the same biologic should bind the same epitope. This difference probably explains why ADAs to infliximab demonstrate identical reactivity towards its biosimilar, whereas ADAs to adalimumab do not cross-react with infliximab or its biosimilar [114].
4.2 Glycosylation Patterns
Originator biologics and their biosimilars are often glycoproteins, which may be inherently immunogenic, whereas small molecules and their generics are only sometimes immunogenic, usually due to haptenating endogenous proteins [115]. There are no well-designed investigations into the clinical implications of glycosylation differences that may occur in production of biosimilars [41, 116]. The terminal sugars of glycans attached to immunoglobulin heavy chain have been shown to be critical for safety and/or efficacy of therapeutic monoclonal antibodies. For example, removal of fucose residues from these glycans can result in up to a 100-fold increase in affinity of antibodies to natural killer cells, which mediate antibody-dependent cellular cytotoxicity [117, 118]. In addition, a high degree of galactosylation has been shown to promote activation of complement in vitro [119]. Different patterns of glycosylation have been shown to influence the pharmacodynamic and pharmacokinetic behaviour of biologics, sometimes resulting in a reduction of the serum half-life of therapeutic antibodies [120]. Such changes have been used purposely as part of the product development process, such as when two extra glycosylation sites were engineered into recombinant human erythropoietin (EPO), resulting in a new, stand-alone product—darbepoetin alfa, with a threefold longer half-life—that could be administered less frequently [121]. A recent study has also demonstrated that the biological activity of EPO glycoforms varies depending on the glycosylation site occupancy pattern [122].
At present, no clinical studies have shown adverse effects of immunogenicity associated with therapeutics with high levels of non-glycosylated heavy chains; however, Fc glycan structural elements may be involved in adverse immune reactions. A high prevalence of hypersensitivity reactions to cetuximab, a monoclonal antibody approved for use in colorectal cancer and squamous-cell carcinoma of the head and neck, has been reported [123]. NeuGc, a non-human sialic acid that is found on some therapeutic proteins, can reduce efficacy due to rapid clearance of the biotherapeutic, and has been reported to cause hypersensitivity. In summary, production processes can change the glycosylation profiles of product biosimilars [41], which may potentially affect safety and efficacy. To date, the clinical implications of these changes are not well understood.
4.3 Immunogenicity Risk
If glycoproteins are sufficiently different from endogenous analogues, then patients may not exhibit immunological tolerance to them, thus rendering such glycoproteins immunogenic. In addition, variations can occur as a result of insertion of xenogeneic amino acid sequences when biologics are made in xenogeneic cells, which can enhance their immunogenicity [124, 125]. Even when fully humanised and glycosylated, therapeutic monoclonal antibodies can be immunogenic because they form immune complexes with target proteins, undergo phagocytosis and potentially activate inflammation [126]. Even within the human population, genes encoding the constant domains of immunoglobulins are highly polymorphic (known as allotypes), and each therapeutic is made from only one variant, which, therefore, may be recognised as non-self in individuals who lack that polymorphism [40, 41, 127,128,129]. Therefore, tolerance to an originator biologic does not necessarily predict immunological tolerance to its biosimilar and vice versa.
Significant effects of ADAs can be divided into those that neutralise the therapeutic action of the drug and/or its endogenous analogue and those that mediate hypersensitivity reactions to the drug. For example, EPO-induced ADA that neutralises endogenous EPO has been implicated in rare cases of pure red cell aplasia [130]. Unsurprisingly, there is also some evidence that ADAs arising against the reference product can also react to its biosimilars [114]. For example, in a retrospective analysis of 250 patients with arthritis who received either a reference biologic (infliximab) or a biosimilar, none of whom had undergone switching, ADAs against infliximab (detected in 126 patients) cross-reacted in in vitro antibody assays with either of two biosimilar products [114]. In the current review, no studies reported any treatment failure related to the development of ADAs. Comparative data on neutralising drug antibodies were reported in only three studies.
4.4 Regulatory Considerations
Ongoing post-marketing pharmacovigilance and accurate documentation of the specific product used by each patient are important. The current lack of standardisation of naming conventions for biosimilars poses several potential concerns. A unique name for the reference product and its biosimilar supports traceability in relation to pharmacovigilance and adverse event reporting [11, 131, 132]. Prescribers need to be able to distinguish between the reference product and its biosimilar to avoid prescribing errors. Naming conventions vary across regions, with the EU licensing biosimilars under the same international non-proprietary name (INN) as the originator and the FDA using a non-specific four-letter suffix (suggested by the sponsor) added to the non-proprietary name. The Australian TGA currently uses the locally approved name without any unique identifier for a biologic and its biosimilar, but is considering various options [11]; hence, at present, traceability may be difficult when multiple switches occur. A study on the traceability of biopharmaceuticals in spontaneous reporting systems over the period 2004–2010 found that, of six biosimilars approved in Europe at the time (sold under 12 different trade names), five contained the same INN as the innovator [133]. The product name was clearly identifiable for 90–96% of biopharmaceuticals for which biosimilars were available in the EU, although batch traceability was poorly maintained [133]. Given the evidence gaps and the difficulty of conducting randomised trials, particularly in the post-marketing setting, there is a need to establish or use pre-existing registries and administrative data, and to develop novel approaches to understanding the effectiveness data being generated by what are, in effect, natural, real-world experiments.
5 Conclusions
In summary, results from this systematic literature review show important evidence gaps around the safety of switching between originator biologics and biosimilars or between biosimilars. While emergent evidence from the EGALITY and NOR-SWITCH studies suggests that switching between TNF-α biologics and their corresponding biosimilars can be safe and efficacy is not compromised, we believe that sufficiently powered and appropriately statistically analysed clinical trials and pharmacovigilance studies, with long-term follow-ups and multiple switching sequences, are needed to support decision-making around biosimilar interchangeability. In the interim, switching should remain a clinical decision made by the treating physician and the patient based on available evidence and individual patient circumstances. Global harmonisation of the regulatory approach to these agents would facilitate an improved understanding of the safety and efficacy of biosimilars.
References
Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25(12):1325–34.
Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363–72.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 5 Sep 2016.
European Medicines Agency (EMA). Guideline on similar biological medicinal products. 2014. http://www.ema.europaeu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed 5 Sep 2015.
Australian Therapeutic Goods Administration (TGA). Regulation of biosimilar medicines (Version 2.0, December 2015). https://www.tga.gov.au/sites/default/files/evaluation-biosimilars-151217_0.pdf. Accessed 5 Sep 2016.
US Food and Drug Administration (FDA). Guidance for industry: immunogenicity assessment for therapeutic protein products. 2014. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338856.pdf. Accessed 6 Sep 2016.
Australian Government Department of Health. Biosimilar medicines—factsheet for healthcare professionals. 2016. http://www.pbs.gov.au/publication/factsheets/biosimilars/biosimilars-factsheet-healthcare-professionals.pdf. Accessed 8 Sep 2016.
Australian Pharmaceutical Benefits Scheme (PBS) Pharmaceutical Benefit Advisory Committee (PBAC). PBAC special meeting minutes. 2015. http://www.pbs.gov.au/industry/listing/elements/pbac-meetings/pbac-outcomes/2015-04/2015-04-biosimilars.pdf. Accessed 8 Sep 2016.
US Food and Drug Administration (FDA). Biosimilars: questions and answers regarding implementation of the biologics price competition and innovation act 2009: guidance for industry. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm444661.pdf. Accessed 8 Sep 2016.
European Medicines Agency (EMA). Biosimilars in the EU: information guide for healthcare professionals. 2017. http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf. Accessed 5 Oct 2017.
Australian Therapeutic Goods Administration (TGA). Consultation: nomenclature of biological medicines (Version 1.0, July 2017). https://www.tga.gov.au/sites/default/files/consultation-nomenclature-biological-medicines.pdf. Accessed 24 Oct 2017.
Papamichael K, Van Stappen T, Jairath V, Gecse K, Khanna R, D’Haens G, et al. Review article: pharmacological aspects of anti-TNF biosimilars in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;42(10):1158–69.
Dörner T, Strand V, Cornes P, Goncalves J, Gulacsi L, Kay J, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–82.
Heinemann L, Home PD, Hompesch M. Biosimilar insulins: guidance for data interpretation by clinicians and users. Diabetes Obes Metab. 2015;17(10):911–8.
European Commission. Consensus information paper: what you need to know about biosimilar medicinal products. 2013. http://ec.europa.eu/DocsRoom/documents/8242. Accessed 8 Sep 2016.
Declerck P, Endrenyi L, Chow SC. Interchangeability, switchability and substitution of biosimilar products. In: Endrenyi LDPJ, Chow SC, editors. Biosimilar drug product development. London and New York: CRC Press/Taylor & Francis; 2017.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31(2):83–91.
US Food and Drug Administration. Therapeutic biologic applications (BLA): information for industry (biosimilars). 2016. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241720.htm. Accessed 5 Sep 2016.
US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product: guidance for industry (draft). 2017. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed 25 Jan 2017.
Allens Linklaters. Costs before caution—Australia’s unique approach to the interchangeability of biosimilars. 2016. http://www.allens.com.au/pubs/pdf/healthcare/BiosimilarsWhitePaper-CostsbeforeCaution.pdf. Accessed 8 Sep 2016.
Kay J, Isaacs JD. Clinical trials of biosimilars should become more similar. Ann Rheum Dis. 2017;76(1):4–6.
American College of Rheumatology (ACR). Position statement: biosimilars. 2016. http://www.rheumatology.org/Portals/0/Files/Biosimilars-Position-Statement.pdf. Accessed 11 Aug 2016.
Australian Diabetes Society (ADS) and Diabetes Australia and Australian Diabetes Educators Association (ADEA). 2015. Position statement: use of ‘biosimilar’ insulins for diabetes. https://diabetessociety.com.au/documents/BiosimilarSubstitutionPositionStatementDAADSADEATBFV20150911.pdf. Accessed 5 Sep 2016.
American Society of Clinical Oncology. ASCO policy brief: biosimilars. 2015. https://www.asco.org/sites/new-wwwascoorg/files/content-files/advocacy-and-policy/documents/2013_biosilimars%20New.pdf. Accessed 5 Sep 2016.
National Rheumatoid Arthritis Society (NRAS). NRAS position paper on biosimilars—revised June 2016. http://www.nras.org.uk/data/files/About%20RA/How%20is%20RA%20managed/NRAS%20Biosimilars%20Position%20Paper%20Final.pdf. Accessed 5 Sep 2016.
American Academy of Dermatology. Position paper on generic therapeutic & biosimilar substitution. 2013. https://www.aad.org/Forms/Policies/Uploads/PS/PS-Generic%20Therapeutic%20and%20%20Biosimilar%20Substitution.pdf. Accessed 6 Oct 2016.
Tabernero J, Vyas M, Giuliani R, Arnold D, Cardoso F, Casali PG, et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open. 2017;1(6):e000142.
Annese V, Vecchi M. Use of biosimilars in inflammatory bowel disease: statements of the Italian Group for Inflammatory Bowel Disease. Dig Liver Dis. 2014;46(11):963–8.
Fiorino G, Girolomoni G, Lapadula G, Orlando A, Danese S, Olivieri I. The use of biosimilars in immune-mediated disease: a joint Italian Society of Rheumatology (SIR), Italian Society of Dermatology (SIDeMaST), and Italian Group of Inflammatory Bowel Disease (IG-IBD) position paper. Autoimmun Rev. 2014;13(7):751–5.
Abad Hernández MA, Andreu JL, Caracuel Ruiz MA, Belmonte Serrano MA, Diaz-Gonzalez F, Moreno Muelas JV. Position paper from the Spanish Society of Rheumatology on biosimilar drugs. Reumatol Clin. 2015;11(5):269–78.
Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. GaBi J. 2014;3(3):150–4.
Devlin SM, Bressler B, Bernstein CN, Fedorak RN, Bitton A, Singh H, et al. Overview of subsequent entry biologics for the management of inflammatory bowel disease and Canadian Association of Gastroenterology position statement on subsequent entry biologics. Can J Gastroenterol. 2013;27(10):567–71.
Gastroenterological Society of Australia (GESA) and Australian Inflammatory Bowel Disease Association (AIBDA). GESA-AIBDA position on biosimilars substitution. 2015. http://www.gesa.org.au/files/editor_upload/File/NEWS/Position%20on%20Biosimilars%20GESA_AIBDA.pdf. Accessed 6 Oct 2016.
Arthritis Australia. Biosimilars and biologics: what you need to know. 2015. http://arthritisaustralia.com.au/indexphp/63-news/301-biosimilars-and-biologics-what-you-need-to-know.html. Accessed 6 Oct 2016.
Australian Rheumatology Association (ARA). ARA position on the introduction of biosimilars for the treatment of rheumatic diseases. 2016. http://rheumatology.org.au/rheumatologists/documents/20161124%20ARA%20position%20statement%20biosimilars%20Nov%2016%20final.pdf. Accessed 13 Dec 2016.
Danese S, Gomollon F. ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis. 2013;7(7):586–9.
European League Against Rheumatism (EULAR). Biosimilars: what do patients need to consider? 2015. http://www.eular.org/myUploadData/files/Biosimilars_2015.pdf. Accessed 5 Sep 2016.
Pharmaceutical Society of Australia. Biosimilar medicines: position statement. 2015. http://www.psa.org.au/downloads/ent/uploads/filebase/policies/biosimilar-medicines-position-statement.pdf. Accessed 6 Oct 2016.
The Pharmacy Guild of Australia. Policy: biosimilar medicines. 2015. https://www.guild.org.au/docs/default-source/public-documents/issues-and-resources/Policy-and-Position-Statements/20151026-guild-biosimilar-medicines.pdf?sfvrsn=0. Accessed 6 Oct 2016.
Hausberger A, Lamanna WC, Hartinger M, Seidl A, Toll H, Holzmann J. Identification of low-level product-related variants in filgrastim products presently available in highly regulated markets. BioDrugs. 2016;30(3):233–42.
Mastrangeli R, Satwekar A, Cutillo F, Ciampolillo C, Palinsky W, Longobardi S. In-vivo biological activity and glycosylation analysis of a biosimilar recombinant human follicle-stimulating hormone product (Bemfola) compared with its reference medicinal product (GONAL-f). PLoS One. 2017;12(9):e0184139.
Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol Bioeng. 2015;112(9):1727–37.
Abdalla A, Byrne N, Conway R, Walsh T, Mannion G, Hanly M, et al. Long-term safety and efficacy of biosimilar infliximab among patients with inflammatory arthritis switched from reference product. Open Access Rheumatol. 2017;9:29–35.
Ala K, Avery P, Wilson R, Prowse A, Shutt J, Jupp J, et al. Early experience with biosimilar infliximab at a district general hospital for an entire Crohns disease patient cohort switch from Remicade to Inflectra. Gut. 2016;65:A81 (abstract).
Arguelles-Arias F, Guerra Veloz MF, Perea Amarillo R, Vilches-Arenas A, Castro Laria L, Maldonado Perez B, et al. Effectiveness and safety of CT-P13 (biosimilar infliximab) in patients with inflammatory bowel disease in real life at 6 months. Dig Dis Sci. 2017;62(5):1305–12.
Bennett KJ, Heap GA, Hawkins S, Ahmad T. Prospective evaluation of the safety and efficacy of switching stable patients with inflammatory bowel disease from Remicade™ to biosimilar infliximab (IFX). Gut. 2016;65:A146 (abstract).
Benucci M, Gobbi FL, Bandinelli F, Damiani A, Infantino M, Grossi V, et al. Safety, efficacy and immunogenicity of switching from innovator to biosimilar infliximab in patients with spondyloarthritis: a 6-month real-life observational study. Immunol Res. 2017;65(1):419–22.
Buer LC, Moum BA, Cvancarova M, Warren DJ, Medhus AW, Hoivik ML. Switching from Remicade® to Remsima® is well tolerated and feasible: a prospective, open-label study. J Crohns Colitis. 2017;11(3):297–304.
Dapavo P, Vujic I, Fierro MT, Quaglino P, Sanlorenzo M. The infliximab biosimilar in the treatment of moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75(4):736–9.
Fiorino G, Manetti N, Armuzzi A, Orlando A, Variola A, Bonovas S, et al. The PROSIT-BIO cohort: a prospective observational study of patients with inflammatory bowel disease treated with infliximab biosimilar. Inflamm Bowel Dis. 2017;23(2):233–43.
Gentileschi S, Barreca C, Bellisai F, Biasi G, Brizi MG, De Stefano R, et al. Switch from infliximab to infliximab biosimilar: efficacy and safety in a cohort of patients with different rheumatic diseases. Response to: Nikiphorou E, Kautiainen H, Hannonen P, et al. Clinical effectiveness of CT-P13 (infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther. 2015;15:1677–1683. Expert Opin Biol Ther. 2016:1–2.
Glintborg B, Sorensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76:1426–31.
Jorgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304–16.
Jung YS, Park DI, Kim YH, Lee JH, Seo PJ, Cheon JH, et al. Efficacy and safety of CT-P13, a biosimilar of infliximab, in patients with inflammatory bowel disease: a retrospective multicenter study. J Gastroenterol Hepatol. 2015;30(12):1705–12.
Kay J, Chopra A, Lassen C, Shneyer L, Wyand M. BOW015, a biosimilar infliximab: disease activity and disability outcomes from a phase 3 active comparator study in patients with active rheumatoid arthritis on stable methotrexate doses. Ann Rheum Dis. 2015;74(Suppl 2):462–3 (abstract).
Kay J, Lassen C, Trokan L, Wyand M. Safety profile of BOW015, a biosimilar infliximab, in healthy subjects and patients with active rheumatoid arthritis. Ann Rheum Dis. 2015;74(Suppl 2):706 (abstract).
Kolar M, Duricova D, Bortlik M, Hruba V, Machkova N, Mitrova K, et al. Infliximab biosimilar (Remsima) in therapy of inflammatory bowel diseases patients: experience from one tertiary inflammatory bowel diseases centre. Dig Dis. 2017;35(1–2):91–100.
Nikiphorou E, Kautiainen H, Hannonen P, Asikainen J, Kokko A, Rannio T, et al. Clinical effectiveness of CT-P13 (infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther. 2015;15(12):1677–83.
Park SH, Kim YH, Lee JH, Kwon HJ, Lee SH, Park DI, et al. Post-marketing study of biosimilar infliximab (CT-P13) to evaluate its safety and efficacy in Korea. Expert Rev Gastroenterol Hepatol. 2015;9(Suppl 1):35–44.
Park W, Suh CH, Shim SC, Molina FFC, Jeka S, Medina-Rodriguez FG, et al. Efficacy and safety of switching from innovator rituximab to biosimilar CT-P10 compared with continued treatment with CT-P10: results of a 56-week open-label study in patients with rheumatoid arthritis. BioDrugs. 2017;31(4):369–77.
Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Urena S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76:346–54.
Rahmany S, Cotton S, Garnish S, Brown M, Saich R, Lloyd D, et al. In patients with IBD switching from originator infliximab (Remicade) to biosimilar infliximab (CT-P13) is safe and effective. Gut. 2016;65:A89 (abstract).
Razanskaite V, Bettey M, Downey L, Wright J, Callaghan J, Rush M, et al. Biosimilar infliximab in inflammatory bowel disease: outcomes of a managed switching programme. J Crohns Colitis. 2017;11(6):690–6.
Rubio E, Ruiz A, Lopez J, Lopez-Chozas JM, Bermudez L, Aguilera C, et al. Prospective study of 78 patients treated with infliximab biosimilar Remsima®. Ann Rheum Dis. 2016;75(Suppl 2):1006 (abstract).
Sheppard M, Hadavi S, Hayes F, Kent J, Dasgupta B. Preliminary data on the introduction of the infliximab biosimilar (CT-P13) to a real world cohort of rheumatology patients. Ann Rheum Dis. 2016;75(Suppl 2):1011 (abstract).
Sieczkowska J, Jarzebicka D, Banaszkiewicz A, Plocek A, Gawronska A, Toporowska-Kowalska E, et al. Switching between infliximab originator and biosimilar in paediatric patients with inflammatory bowel disease. Preliminary observations. J Crohns Colitis. 2016;10(2):127–32.
Schmitz EMH, Benoy-De Keuster S, Meier AJL, Scharnhorst V, Traksel RAM, Broeren MAC, et al. Therapeutic drug monitoring (TDM) as a tool in the switch from infliximab innovator to biosimilar in rheumatic patients: results of a 12-month observational prospective cohort study. Clin Rheumatol. 2017;36(9):2129–34.
Smits LJ, Derikx LA, de Jong DJ, Boshuizen RS, van Esch AA, Drenth JP, et al. Clinical outcomes following a switch from Remicade® to the biosimilar CT-P13 in inflammatory bowel disease patients: a prospective observational cohort study. J Crohns Colitis. 2016;10:1287–93.
Smolen JS, Choe J-Y, Prodanovic N, Niebrzydowski J, Staykov I, Dokoupilova E, et al. Comparable safety and immunogenicity and sustained efficacy after transition to SB2 (an infliximab biosimilar) vs ongoing infliximab reference product in patients with rheumatoid arthritis: results of phase III transition study. Ann Rheum Dis. 2016;75(Suppl 2):488 (abstract).
Tanaka Y, Yamanaka H, Takeuchi T, Inoue M, Saito K, Saeki Y, et al. Safety and efficacy of CT-P13 in Japanese patients with rheumatoid arthritis in an extension phase or after switching from infliximab. Mod Rheumatol. 2017;27(2):237–45.
Tweehuysen L, Van Den Bemt BJF, Van Ingen IL, De Jong AJL, Van Der Laan WH, Van Den Hoogen FHJ, et al. Clinical and immunogenicity outcomes after switching treatment from innovator infliximab to biosimilar infliximab in rheumatic diseases in daily clinical practice. Arthritis Rheumatol. 2016;68:821–3 (abstract).
Yazici Y, Xie L, Ogbomo A, Parenti D, Goyal K, Teeple A, et al. A descriptive analysis of real-world treatment patterns in a Turkish rheumatology population that continued innovator infliximab (Remicade) therapy or switched to biosimilar infliximab. Arthritis Rheumatol. 2016;68:2903–6 (abstract).
Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76:355–63.
Haag-Weber M, Vetter A, Thyroff-Friesinger U. Therapeutic equivalence, long-term efficacy and safety of HX575 in the treatment of anemia in chronic renal failure patients receiving hemodialysis. Clin Nephrol. 2009;72(5):380–90.
Harzallah A, Zouaghi K, Dridi A, Boubaker K, Beji S, Ayari M, et al. Therapeutic efficacy of a biosimilar epoetin alfa in hemodialysis patients. Saudi J Kidney Dis Transpl. 2015;26(1):78–82.
Hörbrand F, Bramlage P, Fischaleck J, Hasford J, Brunkhorst R. A population-based study comparing biosimilar versus originator erythropoiesis-stimulating agent consumption in 6,117 patients with renal anaemia. Eur J Clin Pharmacol. 2013;69(4):929–36.
Lopez MJ, del García Busto N, Carrascosa O, Mejía L, Antonino G, De La Vega I, et al. Biosimilar epoetin zeta in the treatment of chemotherapy-induced anaemia. Eur J Hosp Pharm. 2014;21:A38–9.
Minutolo R, Borzumati M, Sposini S, Abaterusso C, Carraro G, Santoboni A, et al. Dosing penalty of erythropoiesis-stimulating agents after switching from originator to biosimilar preparations in stable hemodialysis patients. Am J Kidney Dis. 2016;68(1):170–2.
Morosetti M, Dominijanni S, Calzona AB, Zappalà L, Nicolais R, Di Turi R. Switch to biosimilars in hemodialysis patients: efficacy, safety and cost analysis in a single centre. Ricerca e Pratica. 2017;33(1):5–12.
Ohta S, Yasuno N, Inomoto Y, Matsuda K, Nakagawa Y, Sasagawa I, et al. Efficacy of once or twice weekly administration of epoetin kappa in patients receiving hemodialysis: a retrospective study. Exp Ther Med. 2014;7(1):27–30.
Wiecek A, Ahmed I, Scigalla P, Koytchev R. Switching epoetin alfa and epoetin zeta in patients with renal anemia on dialysis: posthoc analysis. Adv Ther. 2010;27(12):941–52.
Wizemann V, Rutkowski B, Baldamus C, Scigalla P, Koytchev R. Comparison of the therapeutic effects of epoetin zeta to epoetin alfa in the maintenance phase of renal anaemia treatment. Curr Med Res Opin. 2008;24(3):625–37.
Balili CAV, Mendoza ESR, Mercado-Asis LB. Shifting to biosimilar insulin preparation: impact on glycemic control and cost. Endocrine Reviews. 2015;36:abstract THR-659.
Hadjiyianni I, Dahl D, Lacaya LB, Pollom RK, Chang CL, Ilag LL. Efficacy and safety of LY2963016 insulin glargine in patients with type 1 and type 2 diabetes previously treated with insulin glargine. Diabetes Obes Metab. 2016;18(4):425–9.
Ilag LL, Deeg MA, Costigan T, Hollander P, Blevins TC, Edelman SV, et al. Evaluation of immunogenicity of LY2963016 insulin glargine compared with Lantus(R) insulin glargine in patients with type 1 or type 2 diabetes mellitus. Diabetes Obes Metab. 2016;18(2):159–68.
Rosenstock J, Hollander P, Bhargava A, Ilag LL, Pollom RK, Zielonka JS, et al. Similar efficacy and safety of LY2963016 insulin glargine and insulin glargine (Lantus(R)) in patients with type 2 diabetes who were insulin-naive or previously treated with insulin glargine: a randomized, double-blind controlled trial (the ELEMENT 2 study). Diabetes Obes Metab. 2015;17(8):734–41.
Segal D, Tupy D, Distiller L. The Biosulin equivalence in standard therapy (BEST) study—a multicentre, open-label, non-randomised, interventional, observational study in subjects using Biosulin 30/70 for the treatment of insulin-dependent type 1 and type 2 diabetes mellitus. S Afr Med J. 2013;103(7):458–60.
Flodmark CE, Lilja K, Woehling H, Jarvholm K. Switching from originator to biosimilar human growth hormone using dialogue teamwork: single-center experience from Sweden. Biol Ther. 2013;3:35–43.
Gila AG, Garcia MP. Switching from the original to the biosimilar recombinant human GH-Omnitrope®: an experience of a single paediatric centre in Spain. Horm Res Paediatr. 2014;82:414 (abstract).
Rashid N, Saenger P, Wu YL, Woehling H, Frankel M, Lifshitz F, et al. Switching to Omnitrope® from other recombinant human growth hormone therapies: a retrospective study in an integrated healthcare system. Biol Ther. 2014;4(1–2):27–39.
Belleli R, Fisch R, Renard D, Woehling H, Gsteiger S. Assessing switchability for biosimilar products: modelling approaches applied to children’s growth. Pharm Stat. 2015;14(4):341–9.
Romer T, Zabransky M, Walczak M, Szalecki M, Balser S. Effect of switching recombinant human growth hormone: comparative analysis of phase 3 clinical data. Biol Ther. 2011;1:5.
Engert A, Griskevicius L, Zyuzgin Y, Lubenau H, del Giglio A. XM02, the first granulocyte colony-stimulating factor biosimilar, is safe and effective in reducing the duration of severe neutropenia and incidence of febrile neutropenia in patients with non-Hodgkin lymphoma receiving chemotherapy. Leuk Lymphoma. 2009;50(3):374–9.
Gatzemeier U, Ciuleanu T, Dediu M, Ganea-Motan E, Lubenau H, Del Giglio A. XM02, the first biosimilar G-CSF, is safe and effective in reducing the duration of severe neutropenia and incidence of febrile neutropenia in patients with small cell or non-small cell lung cancer receiving platinum-based chemotherapy. J Thorac Oncol. 2009;4(6):736–40.
Krendyukov A, Harbeck N, Gascon P, Gattu S, Li Y, Blackwell KL. Safety and efficacy of alternating treatment with EP2006, a filgrastim biosimilar, and reference filgrastim for the prevention of severe neutropenia, in patients with breast cancer receiving myelosuppressive chemotherapy. J Clin Oncol. 2017;35:abstr10116 (abstract).
Gooderham M, Spelman L, Kaliaperumal A, Costanzo A, Narbutt J, Zhang N, et al. Single transition from adalimumab to ABP 501: evaluation of immunogenicity in a phase 3 study in subjects with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;74(5):AB275 (abstract).
Papp K, Bachelez H, Costanzo A, Foley P, Gooderham M, Kaur P, et al. Clinical similarity of biosimilar ABP 501 to adalimumab in the treatment of patients with moderate to severe plaque psoriasis: a randomized, double-blind, multicenter, phase III study. J Am Acad Dermatol. 2017;76(6):1093–102.
Weinblatt M, Baranauskaite A, Niebrzydowski J, Dokoupilova E, Zielinska A, Sitek-Ziolkowska K, et al. Sustained efficacy and comparable safety and immunogenicity after transition to SB5 (an adalimumab biosimilar) vs continuation of the adalimumab reference product in patients with rheumatoid arthritis: results of phase III study EUCLAR 2016; 8–11 June 2016; London UK (abstract FRI0161). Arthritis Rheumatol. 2016;68(suppl 10):abstract 604.
Nasonov E, Mazurov V, Plaksina T, Nesmeyanova O, Knyazeva L, Eremeeva A, et al. Interchangeability of innovator rituximab and its biosimilar: results from international controlled comparative 1-year study in patients with active rheumatoid arthritis. Arthritis Rheumatol. 2016;68:2046–7.
Roy PS, John S, Karankal S, Kannan S, Pawaskar P, Gawande J, et al. Comparison of the efficacy and safety of rituximab (Mabthera) and its biosimilar (Reditux) in diffuse large B-cell lymphoma patients treated with chemo-immunotherapy: a retrospective analysis. Indian J Med Paediatr Oncol. 2013;34(4):292–8.
Emery P, Vencovský J, Sylwestrzak A, Leszczyñski P, Porawska W, Stasiuk B, et al. Additional efficacy results of SB4 (etanercept biosimilar) up to week 100: comparison between continuing SB4 and switching from reference etanercept (Enbrel®) to SB4. Arthritis Rheumatol. 2016;68(suppl 10):789–90 (abstract).
Emery P, Vencovsky J, Sylwestrzak P, Leszczynski P, Porawska W, Stasiuk B, et al. Long-term safety and efficacy of SB4 (etanercept biosimilar) in patients with rheumatoid arthritis: comparison between continuing SB4 and switching from etanercept reference product to SB4. Ann Rheum Dis. 2016;75(Suppl 2):236 (abstract).
Griffiths CEM, Thaci D, Gerdes S, Arenberger P, Pulka G, Kingo K, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176(4):928–38.
Strowitzki T, Kuczynski W, Mueller A, Bias P. Safety and efficacy of Ovaleap® (recombinant human follicle-stimulating hormone) for up to 3 cycles in infertile women using assisted reproductive technology: a phase 3 open-label follow-up to main study. Reprod Biol Endocrinol. 2016;14(1):31.
Glintborg B, Kringelbach T, Hogdall E, Sorensen IJ, Jensen DV, Loft AG, et al. Non-medical switch from originator to biosimilar infliximab among patients with inflammatory rheumatic disease—impact on S-infliximab and antidrug-antibodies. Results from the National Danish Rheumatologic Biobank and the Danbio Registry. Ann Rheum Dis. 2016;75(Suppl 2):224 (abstract).
Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–8.
Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology (Oxford). 2011;50(8):1445–52.
Thomas SS, Borazan N, Barroso N, Duan L, Taroumian S, Kretzmann B, et al. Comparative immunogenicity of TNF inhibitors: impact on clinical efficacy and tolerability in the management of autoimmune diseases. A systematic review and meta-analysis. BioDrugs. 2015;29(4):241–58.
Dowlat HA, Kuhlmann MK, Khatami H, Ampudia-Blasco FJ. Interchangeability among reference insulin analogues and their biosimilars: regulatory framework, study design and clinical implications. Diabetes Obes Metab. 2016;18(8):737–46.
Goll GL, Olsen IC, Jorgensen KK, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Biosimilar infliximab (CT-P13) is not inferior to originator infliximab: results from a 52-week randomized switch trial in Norway Arthritis Rheumatol. 2016;68 (suppl 10) abstract 19L.
Gautam A. Market watch: strategies for biosimilars in emerging markets. Nat Rev Drug Discov. 2017;16(8):520–1.
Marciano I, Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, et al. How did the introduction of biosimilar filgrastim influence the prescribing pattern of granulocyte colony-stimulating factors? Results from a multicentre, population-based study, from five Italian centres in the years 2009–2014. BioDrugs. 2016;30(4):295–306.
Ingrasciotta Y, Giorgianni F, Bolcato J, Chinellato A, Pirolo R, Tari DU, et al. How much are biosimilars used in clinical practice? A retrospective Italian population-based study of erythropoiesis-stimulating agents in the years 2009–2013. BioDrugs. 2015;29(4):275–84.
Ruiz-Arguello MB, Maguregui A, Ruiz Del Agua A, Pascual-Salcedo D, Martinez-Feito A, Jurado T, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75(9):1693–6.
Li E, Ramanan S, Green L. Pharmacist substitution of biological products: issues and considerations. J Manag Care Spec Pharm. 2015;21(7):532–9.
DePalma A. Glycosylation affirmed as quality metric. GEN Select. 2014;34(13):5263.
Chung S, Quarmby V, Gao X, Ying Y, Lin L, Reed C, et al. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcgamma receptor binding and antibody-dependent cell-mediated cytotoxicity activities. MAbs. 2012;4(3):326–40.
Kanda Y, Yamada T, Mori K, Okazaki A, Inoue M, Kitajima-Miyama K, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. 2007;17(1):104–18.
Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9(8):1716–28.
Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, et al. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs. 2012;4(4):475–87.
Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant. 2001;16(Suppl 3):3–13.
Murakami M, Kiuchi T, Nishihara M, Tezuka K, Okamoto R, Izumi M, et al. Chemical synthesis of erythropoietin glycoforms for insights into the relationship between glycosylation pattern and bioactivity. Sci Adv. 2016;2(1):e1500678.
Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358(11):1109–17.
Burska AN, Hunt L, Boissinot M, Strollo R, Ryan BJ, Vital E, et al. Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediators Inflamm. 2014;492873(10):492873.
Zhong X, Wright JF. Biological insights into therapeutic protein modifications throughout trafficking and their biopharmaceutical applications. Int J Cell Biol. 2013;273086(10):273086.
Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72(2):165–78.
Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–3.
Magnenat L, Palmese A, Fremaux C, D’Amici F, Terlizzese M, Rossi M, et al. Demonstration of physicochemical and functional similarity between the proposed biosimilar adalimumab MSB11022 and Humira(R). MAbs. 2017;9(1):127–39.
Jefferis R, Lefranc MP. Human immunoglobulin allotypes: possible implications for immunogenicity. MAbs. 2009;1(4):332–8.
Locatelli F, Del Vecchio L, Pozzoni P. Pure red-cell aplasia “epidemic”—mystery completely revealed? Perit Dial Int. 2007;27(2):S303–7.
US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. 2017. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Accessed 17 Oct 2017.
Grampp G, Felix T. Pharmacovigilance considerations for biosimilars in the USA. BioDrugs. 2015;29(5):309–21.
Vermeer NS, Straus SM, Mantel-Teeuwisse AK, Domergue F, Egberts TC, Leufkens HG, et al. Traceability of biopharmaceuticals in spontaneous reporting systems: a cross-sectional study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance databases. Drug Saf. 2013;36(8):617–25.
Ekelund M, Bidad C, Gomez R. To the editor; a commentary on “Switching from originator to biosimilar human growth hormone using a dialogue teamwork: single-center experience from Sweden”. Biol Ther. 2014;4(1–2):69–71.
Funding
Administrative and medical writing support was provided by Ms Donna Bartlett (Allori Pty Ltd, Australia) and Dr. Anja Becher (Sydney, Australia), and was funded through Medicines Australia with specific support from the Australian affiliates of AbbVie, Amgen, Eli Lilly, Janssen, Novo Nordisk, Roche and Sanofi. The sponsors were given the opportunity to review the final version of the article for accuracy pertaining to sponsored products and compliance with individual processes; however, they did not influence the content. The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ross McKinnon: Education and advisory service fees in relation to biosimilars for AbbVie and Sanofi Australia. Matthew Cook: Advisory board for Roche and Baxter. Winston Liauw: Advisory board for NPS MedicineWise. Mona Marabani: Consultancy, speaker fees or conference support from AbbVie, BMS, UCB, MSD, Pfizer, Janssen, Novartis and Sanofi. Ian Marschner: Research grant and consulting fees from Janssen-Cilag and consulting fees from Pfizer, AbbVie and Generic Health. Nicolle Packer reports having no conflicts of interest. John Prins holds clinical and scientific advisory board positions and/or has received speaking fees from Pfizer, Merck and Novo Nordisk.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McKinnon, R.A., Cook, M., Liauw, W. et al. Biosimilarity and Interchangeability: Principles and Evidence: A Systematic Review. BioDrugs 32, 27–52 (2018). https://doi.org/10.1007/s40259-017-0256-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-017-0256-z