
Abstract
Background
Co-crystal of tramadol–celecoxib (CTC), containing equimolar quantities of the active pharmaceutical ingredients (APIs) tramadol and celecoxib (100 mg CTC = 44 mg rac–tramadol hydrochloride and 56 mg celecoxib), is a novel API-API co-crystal for the treatment of pain. We aimed to establish the effective dose of CTC for treating acute pain following oral surgery.
Methods
A dose-finding, double-blind, randomised, placebo- and active-controlled, multicentre (nine Spanish hospitals), phase II study (EudraCT number: 2011-002778-21) was performed in male and female patients aged ≥ 18 years experiencing moderate to severe pain following extraction of two or more impacted third molars requiring bone removal. Eligible patients were randomised via a computer-generated list to receive one of six single-dose treatments (CTC 50, 100, 150, 200 mg; tramadol 100 mg; and placebo). The primary efficacy endpoint was the sum of pain intensity difference (SPID) over 8 h assessed in the per-protocol population.
Results
Between 10 February 2012 and 13 February 2013, 334 patients were randomised and received study treatment: 50 mg (n = 55), 100 mg (n = 53), 150 mg (n = 57), or 200 mg (n = 57) of CTC, 100 mg tramadol (n = 58), or placebo (n = 54). CTC 100, 150, and 200 mg showed significantly higher efficacy compared with placebo and/or tramadol in all measures: SPID (0–8 h) (mean [standard deviation]): − 90 (234), − 139 (227), − 173 (224), 71 (213), and 22 (228), respectively. The proportion of patients experiencing treatment-emergent adverse events was lower in the 50 (12.7% [n = 7]), 100 (11.3% [n = 6]), and 150 (15.8% [n = 9]) mg CTC groups, and similar in the 200 mg (29.8% [n = 17]) CTC group, compared with the tramadol group (29.3% [n = 17]), with nausea, dizziness, and vomiting the most frequent events.
Conclusion
Significant improvement in the benefit–risk ratio was observed for CTC (doses ≥ 100 mg) over tramadol and placebo in the treatment of acute pain following oral surgery.
Funding
Laboratorios del Dr. Esteve, S.A.U.
Similar content being viewed by others

This is the first time that efficacy and safety results of a new medical entity, a co-crystal containing well-known active moieties and therapeutic entities (tramadol and celecoxib), have been presented to the scientific community. |
Findings from this study support the concept that the unique molecular structure of two active pharmaceutical ingredients’ co-crystals can confer altered physicochemical properties to the component drugs, which can ultimately translate into clinical benefits. |
The co-crystal mechanism conferred by co-crystal of tramadol–celecoxib also translates into synergy, through which efficacy is achieved with low amounts of each active moiety, while also resulting in improved safety and tolerability, with an overall enhanced benefit-to-risk ratio. |
1 Introduction
Pain is the most common symptom for which patients seek medical attention, but often pain relief (PAR) is not achieved with the administration of a single drug [1]. Furthermore, studies have shown that many patients with acute pain will not achieve adequate analgesia [1,2,3,4]. Strategies to address this unmet medical need include multimodal analgesia, achieved via the use of multiple classes of pain-relieving drugs that have different mechanisms of action, with the aim of improving PAR [5].
A co-crystal is a solid form consisting of two or more dissociable components in a crystal lattice. Co-crystals containing more than one active pharmaceutical ingredient (API) represent unique molecular structures that offer a novel approach to polypharmacology [6]. Due to weak intermolecular interactions between drugs within the crystalline structure, API-API co-crystals have the potential for improved physicochemical properties compared with their constituent drugs, which may translate into clinical benefits. These may be apparent as enhanced solubility or dissolution characteristics, which in turn may improve pharmacokinetics (PK) compared with open or traditional fixed dose combinations (FDCs) [6]. Furthermore, co-crystals have relatively simple preparation methods and are not associated with many of the formulation issues that can be encountered with FDCs [7].
Co-crystal of tramadol–celecoxib (CTC) is a medical product in development that is based on a co-crystal molecule of two drugs with complementary mechanisms of action (tramadol and celecoxib) in a 1:1 molecular ratio (1:1.27 weight ratio), conferred by the intrinsic stoichiometry of the co-crystal structure. A 100-mg dose of this co-crystal contains 44 mg of racemic tramadol hydrochloride (rac-tramadol.HCl) and 56 mg of celecoxib. Tramadol is a centrally acting, weak mu-opioid receptor agonist and an inhibitor of the neuronal reuptake of noradrenaline and serotonin [8], and is indicated for the treatment of moderate to severe pain worldwide. Celecoxib is a nonsteroidal anti-inflammatory drug (NSAID) that primarily acts via inhibition of cyclooxygenase-2. In Europe and elsewhere, celecoxib is indicated and authorised for the treatment of chronic inflammatory conditions, such as osteoarthritis and rheumatoid arthritis, which are often painful [9].
The unique molecular structure of CTC enables changes in the physicochemical properties of each API. In vitro analysis of a formulation of the co-crystal without additives (ctc) demonstrated intrinsic dissolution profiles for both tramadol and celecoxib from ctc that differed from tramadol and celecoxib administered individually or in open combination [10]. Preclinical pharmacological pain models have shown that the analgesic activity of ctc in suspension (ctcsusp) is greater than the individual activities of tramadol and celecoxib and greater than the activity predicted by the sum of the individual components [11]. This increased analgesic activity is consistent with pharmacodynamic synergism (i.e. supra-additive or synergic effects), resulting from the complementary recruitment of multiple mechanisms of action relevant to pain in central and peripheral targets.
Single- and multiple-dose phase I studies have compared the PK profile of CTC with that of the individual APIs given alone or in open combination in healthy volunteers. These studies demonstrate that CTC exhibits several favourable changes in PK parameters relative to conventional dosage forms. Specifically, tramadol from CTC had a similar area under the concentration–time curve (AUC), with a slightly delayed absorption, compared with tramadol alone or in combination with celecoxib. This was associated with a lower maximum serum concentration (Cmax), and a slightly prolonged time to reach Cmax (Tmax) [12, 13]. Furthermore, celecoxib from CTC displayed a reduced AUC, a lower Cmax and a faster Tmax compared with celecoxib alone [12, 13]. These differences in PK parameters were consistent after single and multiple doses of treatment [12, 13] and, together with the complementary molecular PAR mechanisms of each API, may have implications for both clinical efficacy and safety.
Based on the preclinical efficacy synergism and human PK profile mentioned above, our hypothesis was that 200 mg of CTC (corresponding to 88 mg rac-tramadol.HCl plus 112 mg celecoxib) has better efficacy relative to tramadol 100 mg, and a similar safety profile, while lower doses of CTC have better efficacy and safety relative to tramadol 100 mg, hence resulting in an improved overall benefit–risk ratio. Therefore, the primary objective of this clinical trial was to establish the effective dose among four doses of CTC for moderate to severe acute pain following oral surgery involving the extraction of two or more impacted third molars, requiring bone removal. The secondary objectives were to assess the efficacy and safety of CTC.
2 Patients and Methods
2.1 Study Design
This was a dose-finding, double-blind, randomised, placebo- and active-controlled, parallel-group, phase II study (a regulatory clinical trial: a clinical trial included in the clinical development programme for obtaining marketing authorization) that was performed in nine Spanish hospitals. The study recruited patients with moderate to severe acute pain following oral surgery involving extraction of two or more impacted third molars, requiring bone removal. The ethics committee of each study centre approved the study protocol. The study was conducted in agreement with the updated Declaration of Helsinki, the guidelines for Good Clinical Practice, and applicable Spanish regulatory requirements. The study protocol can be accessed at https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-002778-21/ES.
2.2 Study Participants
Male and female patients aged ≥ 18 years with a body weight of less than 110 kg who were experiencing moderate or severe pain (score of at least 50 mm on the visual analogue scale [VAS], where 0 mm = ‘no pain’ and 100 mm = ‘the worst imaginable pain’) after an oral surgical procedure were eligible for the study. Exclusion criteria included receipt of the following: any analgesic medication other than short-acting preoperative or intraoperative anaesthetic agents within 12 h before taking trial medication; any analgesic medication other than the study drug immediately after the oral surgical procedure; a long-acting NSAID within 3 days prior to dosing; any anti-depressive medication, selective serotonin reuptake inhibitors (SSRIs) (e.g. paroxetine, fluoxetine), diet pills (including fenfluramine and phentermine), or methylphenidate within 4 weeks of study entry; monoamine oxidase inhibitors, tricyclic antidepressants, neuroleptics, or other drugs that reduce the seizure threshold, within 4 weeks of study entry. Patients were also excluded if they experienced any complications during surgery, had evidence of renal or hepatic dysfunction or peptic ulcer disease, or had a history of seizures or drug or alcohol abuse within 6 months of study entry. In addition, patients were excluded if they were sensitive or allergic to tramadol, celecoxib or other NSAIDs, or aspirin, or deemed at risk in terms of precautions, warnings, and contraindications in the package insert for Adolonta® (tramadol hydrochloride) or Celebrex® (celecoxib). All patients provided informed written consent.
2.3 Randomisation and Masking
A computer-generated randomisation schedule was prepared at the start of the study and was balanced by the use of permuted blocks of six. Access to this schedule was limited to the staff who generated it and the staff who manufactured the products. To maintain the double-blind, each medication bottle was labelled with a medication code number and packaged with two identical capsules of study medication according to the randomisation sequence. The investigator assigned medication code numbers to eligible patients in ascending sequential order. Treatment allocation information was contained in a sealed envelope that was only to be opened if needed in an emergency.
2.4 Study Procedure
The study comprised three on-site visits and one telephone interview: screening (visit 1), oral surgery (day 1, visit 2), 24-h post-surgery follow-up telephone interview (day 2, visit 3), and final examination (day 7, visit 4).
The acute pain model used in this study consisted of extraction of at least two impacted third molars (at least one mandibular), requiring bone removal. If only two impacted third molars were extracted, they were required to be ipsilateral and require bone removal (at least one mandibular). Local anaesthesia (lidocaine plus adrenaline 1:100,000 and nitrous oxide) and/or sedation (opioids were not permitted) were used during molar extractions. After the molar extractions, the pain intensity (PI) of the patient was assessed on a 100-mm VAS, scored by the patients themselves, at 15-min intervals until a PI of 50 mm was reached, or up to a maximum of 4 h after extraction. Patients who reached a PI ≥ 50 mm were considered eligible for the study, randomly assigned to one of six treatments, and immediately administered study medication. If a patient did not reach the required PI within 4 h of extraction he/she was excluded (not randomised) from the study and standard hospital practice care was initiated.
Randomised patients received a single dose of study medication consisting of two over-encapsulated tablets administered orally: CTC 50 mg (one tablet of CTC [22 mg tramadol + 28 mg celecoxib] and one tablet of placebo); CTC 100 mg (one tablet of CTC [44 mg tramadol + 56 mg celecoxib] and one tablet of placebo); CTC 150 mg (one tablet of CTC [22 mg tramadol + 28 mg celecoxib] and one tablet of CTC [44 mg tramadol + 56 mg celecoxib]; total dose: 66 mg tramadol + 84 mg celecoxib); CTC 200 mg (two tablets of CTC [44 mg tramadol + 56 mg celecoxib]; total dose: 88 mg tramadol + 112 mg celecoxib); tramadol 100 mg (two tablets of 50 mg Adolonta®, Grünenthal Pharma, S.A., Madrid, Spain); or placebo (two tablets). Patients who received study medication were required to remain at the study site for 12 h after randomisation (i.e. for a maximum of 16 h after oral surgery), after which they could leave the clinic. At any time during the 12-h observation period patients could take a supplementary analgesic medication (rescue medication), but were encouraged to wait at least 1 h after taking the study medication and to wait until pain returned to baseline level. Patients administered rescue medication discontinued the trial. Current PI and PAR were assessed prior to the intake of rescue medication and stopped thereafter. Patients taking rescue medication were asked to remain at the study site for 8 h after randomisation. The administration of rescue medication in terms of substance and amount was based on the investigator’s discretion.
Evaluation of analgesic efficacy included the assessment of PI, PAR, use of rescue medication, time to perceptible and meaningful PAR, and an overall assessment of the study medication. PI was assessed by 100-mm VAS at time points (min): 0 (prior to taking the study medication), 10, 20, 30, 45, 60 (1 h), 75, 90, 105, 120 (2 h), 135, 150, 165, 180 (3 h), 195, 210, 225, 240 (4 h), 270, 300 (5 h), 330, 360 (6 h), 420 (7 h), 480 (8 h), 600 (10 h), 720 (12 h), and 1440 (24 h) (or until rescue medication was taken). PAR was assessed using a 5-point ordinal scale (1 = no PAR; 2 = little PAR; 3 = some PAR; 4 = a lot of PAR; 5 = maximum PAR) at the same time points as PI (excluding baseline). Completion of the VAS was observed by trained study staff. Stopwatches were used to assess time to PAR. Patients started timing upon intake of study medication and stopped one stopwatch at the onset of perceptible PAR and another upon achieving meaningful PAR. Alternatively, stopwatches were stopped at the end of the 12-h observational period, at the time of withdrawal, or at the time of taking rescue medication (whichever occurred first). Patients also made an overall assessment of the study medication using a verbal rating scale (excellent, very good, good, fair, poor) at the time point the stopwatch was stopped.
2.5 Study Outcomes
The primary efficacy endpoint was the sum of pain intensity difference (SPID) from 0 to 8 h. Pain intensity difference (PID) was defined as PID t = PI t − PI0, where PI0 is the baseline PI at t = 0 h and PI t is the PI at specific time points. Therefore, positive values correspond to an increase in pain, while negative values correspond to a decrease in pain. Consequently, SPID was defined as the sum of PID values obtained between time 0 and time t, with each individual PID value weighted according to the relevant time interval (i.e. the time between two consecutive PI measurements).
The secondary efficacy endpoints were SPID from 0 to 12 h, PID at each time point, PAR at 8 and 12 h, total pain relief (TOTPAR) from 0 to 8 h, TOTPAR from 0 to 12 h, overall assessment of the study medication, rescue medication (rate of patients with intake of at least one dose of rescue medication up to 8 h and up to 12 h after study drug administration, and time to first intake of rescue medication), time to perceptible and meaningful PAR, and responder rates. TOTPAR was defined as the sum of PAR between time 0 and time t, with individual PAR t values weighted according to the time interval since the previous evaluation. Five definitions of responder were used for analysis, as set out in the protocol and statistical analysis plan: (1) a 50% reduction in pain compared with baseline; (2) a 30% reduction in pain compared with baseline; (3) a 50% reduction in pain as compared with baseline and a PI below 40 mm VAS; (4) a 30% reduction in pain compared with baseline and a PI below 40 mm VAS; and (5) a PI below 40 mm VAS.
Evaluation of safety included the assessment of adverse events (AEs) spontaneously reported by the patients, safety laboratory tests, dental evaluation, general medical examination, vital signs (pulse rate and blood pressure in sitting position, body temperature), and 12-lead electrocardiogram (ECG). A treatment-emergent AE (TEAE) was defined as an AE with onset on or after the first administration of study treatment, or an AE that worsened even if it was present before first administration.
2.6 Statistical Analysis
Due to the exploratory nature of this study, no confirmatory hypothesis was set. Sample size was determined according to clinical, not statistical, criteria. For the primary efficacy analysis, 40 patients per treatment group were considered the minimum sample size necessary to properly evaluate the results of the study. Based on the sponsor’s previous experience, it was assumed that around 35% of patients would not be evaluable in this analysis, primarily as a result of failing to reach the required PI (VAS ≥ 50 mm) during the first 4 h post-intervention, or as a result of requiring rescue medication during the first hour after study drug administration. Therefore, to compensate for this expected loss, 60 patients per treatment group (360 patients in total) were recruited.
The primary population for efficacy analysis was the per-protocol (PP) analysis set (all randomised and treated patients who had no relevant protocol deviations, provided three or more valid VAS measurements within 8 h of study treatment, and did not take rescue medication during the first hour after study treatment). The primary efficacy endpoint was analysed using an analysis of variance (ANOVA) model, including treatment and centre effects. For SPID from 0–12 h, PID at each time point, PAR at each time point, TOTPAR from 0 to 8 h, TOTPAR from 0 to 12 h, and post hoc analysis carried out to investigate the effect of baseline PI on SPID from 0 to 8 h, the same ANOVA model was applied as for the primary efficacy endpoint. Missing efficacy measurements (in case of drop-outs or administration of rescue medication) were imputed using the last observation carried forward (LOCF) method. The overall assessment of the study medication, response rates, and the number of patients requiring rescue medication were analysed using the Cochran–Mantel–Haenszel test stratified by centre, in order to detect any treatment differences. The time to first intake of rescue medication, time to perceptible PAR, and time to meaningful PAR were analysed using Kaplan–Meier estimates and log-rank tests. Time to response was analysed as time-to-event data, taking the first time at which the response criterion was reached by each patient. Safety variables were assessed in the safety analysis set (all randomised patients who received study treatment) and analysed descriptively. This trial was registered in EudraCT (number: 2011-002778-21).
2.7 Role of the Funding Source
The study’s funder was involved in study design, data interpretation, and revision of the final report, but did not take part in data collection and analysis. The corresponding author had full access to all study data and had final responsibility for the decision to submit the paper for publication.
3 Results
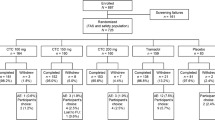
Between 10 February 2012 and 29 January 2013, a total of 420 patients were screened for inclusion. Of these, 335 patients experienced moderate to severe pain within 4 h of oral surgery and were randomised. All but one patient (n = 334) received study medication (Fig. 1).

Study flow chart. CTC co-crystal of tramadol–celecoxib, FAS full analysis set, PP per-protocol, SAS safety analysis set, VAS visual analogue scale
Patient baseline characteristics are shown in Table 1. Patients were predominantly young (mean age 24.5 years), Caucasian (98%), and female (57%). Mean values for demographic variables in the PP analysis set (n = 288) were not markedly different to those in the full analysis set.
The primary efficacy endpoint (SPID up to 8 h post-dose; Fig. 2; Table 2) was lower with CTC 100 mg, 150 mg, and 200 mg compared with placebo (p < 0.05) and tramadol 100 mg (p < 0.05). CTC 50 mg was also numerically lower than placebo and tramadol, although this difference did not reach statistical significance. The effect of CTC on SPID (0–8 h) was dose-dependent, and the test for dose linearity showed a quadratic trend (p < 0.01).

Sum of pain intensity difference (mean + SEM) up to 8 h post-dose (LOCF; PP analysis set). *p < 0.05, significantly better vs. placebo. #p < 0.05, significantly better vs. tramadol (p values from ANOVA with treatment and centre as factors). ANOVA analysis of variance, CTC co-crystal of tramadol–celecoxib, LOCF last observation carried forward, PP per-protocol, SEM standard error of mean, SPID sum of pain intensity difference
CTC doses ≥ 100 mg were more efficacious than placebo (p < 0.05) with respect to PID from 1 h after study drug administration, regardless of the time interval examined (Fig. 3; Table 2) and for all other efficacy endpoints, including TOTPAR (Table 2), PAR (Table 2), median time to first intake of rescue medication, responder rates, and overall assessment of study medication (Table 3). CTC 150 mg and 200 mg were also more efficacious than tramadol 100 mg for most of these efficacy endpoints (p < 0.05; Tables 2, 3). In terms of efficacy, CTC 50 mg was comparable to tramadol, and numerically better than placebo (although not statistically different for any of the efficacy endpoints). No differences between treatment groups were observed for time to perceptible PAR and time to meaningful PAR. When primary and secondary efficacy endpoints were analysed up to 12 h post-dose, results did not change substantially from those seen when analysing up to 8 h post-dose (Tables 2, 3).

Pain intensity difference from baseline, mean over time by treatment group (LOCF; PP analysis set). CTC doses ≥ 100 mg were more efficacious than placebo (p < 0.05) with respect to PID from 1 h after study drug administration, regardless of the time interval examined. CTC co-crystal of tramadol–celecoxib, LOCF last observation carried forward, PI pain intensity, PID pain intensity difference, PP per-protocol
A post hoc analysis of SPID was performed for patients with moderate (50–60 mm VAS, n = 186 [65%]) or severe (> 60 mm VAS, n = 102 [35%]) pain at baseline (Fig. 4). With all doses of CTC, a greater reduction in pain was observed in patients with severe pain at baseline than in those with moderate pain at baseline. A key difference between patients with moderate versus severe baseline pain was observed in patients who received 100 mg tramadol (mean SPID [0–8 h] 101.29 vs. −74.95 h·mm).

Post hoc analysis of SPID (mean + SEM) up to 8 h post-dose in patients with a moderate or b severe pain at baseline (LOCF; PP analysis set). *p < 0.05, significantly better vs. placebo. #p < 0.05, significantly better vs. tramadol (p values from ANOVA with treatment and centre as factors). ANOVA analysis of variance, CTC co-crystal of tramadol–celecoxib, LOCF last observation carried forward, PP per-protocol, SEM standard error of mean, SPID sum of pain intensity difference
A total of 80 TEAEs in 61 patients were reported in this study: 29.8% (n = 17) of patients in the 200 mg CTC group and 29.3% (n = 17) of the patients in the 100 mg tramadol group experienced at least one TEAE. TEAEs occurred in markedly fewer patients after administration of 50 mg (12.7%), 100 mg (11.3%), and 150 mg (15.8%) CTC; there was no marked difference in frequencies between the two lowest dose groups and the placebo group, where an overall TEAE frequency of 9.3% was observed (Fig. 5). The most common side effects were vomiting, nausea, and dizziness (Table 4). TEAEs were mostly mild or moderate, with the exception of one severe drug-related TEAE (an incident of vomiting in a patient who had received 100 mg tramadol).

Percentage of patients with at least one treatment-emergent adverse event (safety analysis set). CTC co-crystal of tramadol–celecoxib
Twenty-nine TEAEs in 23 patients were considered to be related to study treatment. In the 50, 100, 150, and 200 mg CTC groups, 1.8% (n = 1), 5.7% (n = 3), 3.5% (n = 2), and 14.0% (n = 8) of patients, respectively, experienced at least one drug-related TEAE, compared with 13.8% (n = 8) of patients in the tramadol group and 1.9% (n = 1) of patients in the placebo group. Gastrointestinal disorders were the most frequent drug-related TEAE and occurred with similar frequency in the 200 mg CTC (12.3%) and 100 mg tramadol (12.1%) groups, but were markedly less common in the 50 mg (1.8%), 100 mg (5.7%), and 150 mg (3.5%) CTC groups. This included 14 cases of vomiting and ten cases of nausea.
Five serious AEs (SAEs) were reported in four patients. Two SAEs were considered to be drug related: nausea in a patient who received 200 mg CTC, and vomiting in a patient administered 100 mg tramadol. Both patients recovered the next day. No AEs led to patient discontinuation from the study. There were no marked effects on laboratory parameters. No clinically significant vital sign and ECG abnormalities were reported, and there was no obvious trend in these parameters over time.
4 Discussion
To our knowledge, this is the first clinical trial of a co-crystal molecule with two well-known analgesic APIs. This study has demonstrated that CTC (100, 150, and 200 mg) displays improved efficacy compared with tramadol (100 mg) in the management of moderate to severe acute pain, with a comparable (CTC 200 mg) or better (CTC 50 mg, 100 mg, and 150 mg) safety profile. Together with preclinical [11] and phase I data [12, 13], these results suggest that the molecular structure of the co-crystal modifies the physicochemical properties of tramadol and celecoxib [10] in such a way that the PK profile of each API is enhanced. This, together with the complementary analgesic mechanisms of action, translates into improved efficacy and safety. In the present study, the efficacy of CTC improved linearly as the dose increased. Consequently, the 1:1 molecular ratio of tramadol and celecoxib in the co-crystal appears optimal to obtain an enhanced therapeutic effect, as also seen in preclinical studies [11]. Compared with tramadol, the PAR from CTC was associated with a faster onset of action, a longer duration, higher responder rates, and reduced need for rescue medication. Therefore, the potential advantages of this co-crystal molecule include an improvement of the benefit–risk ratio. On the basis of these data, we suggest that CTC 100 mg twice daily (BID) may be an appropriate dose for moderate acute pain, rising to CTC 200 mg BID for severe acute pain.
The extraction of two or more impacted third molars requiring bone removal is considered a gold standard model for measuring the efficacy of new medications in moderate to severe acute pain [14]. Although tramadol has been demonstrated to be more effective than placebo in acute moderate to severe pain models in several clinical trials [15, 16], it has not always been so [17]. In our study, tramadol 100 mg (the active control) was better than placebo in terms of efficacy, but this finding was not statistically significant, which may put into question the assay sensitivity of this clinical trial. Nevertheless, the results obtained in the tramadol 100 mg group are not inconsistent with literature findings. Recently, an increasing number of randomised clinical trials of various analgesic medications have failed to show statistically significant differences compared with placebo in conditions in which their efficacy was previously demonstrated, and for which they have been approved by regulatory agencies [18, 19]. A potential explanation for tramadol not showing statistically significant differences to placebo in this study could be the severity of PI at baseline. In a post hoc analysis, tramadol 100 mg was statistically better than placebo in some efficacy variables in the subgroup of patients with severe pain (VAS > 60 mm) at baseline; this is in agreement with other acute pain studies [20, 21].
Moreover, and in favour of the assay sensitivity of this clinical trial, the results show (1) a clear and statistically significant dose-dependent response to CTC; (2) a consistent response to CTC in all secondary analyses; (3) a reduction in AEs with decreasing CTC dose; and (4) importantly, tramadol 100 mg exhibited a safety profile that was differentiated from placebo and was consistent with the literature and the approved label of tramadol [22]. These findings are similar to profiles demonstrated in other studies of acute pain profiles, such as those evaluating the FDC of dexketoprofen/tramadol [16, 23].
Celecoxib was not included as an active comparator despite the fact that it is a component of the co-crystal. According to the European Summary of Product Characteristics for this drug, celecoxib is not indicated for the treatment of acute pain [9]. Furthermore, literature evidence suggests that the minimum effective dose of celecoxib in dental pain is 400 mg [1, 24, 25]. The maximum dose tested with CTC corresponds to 112 mg of celecoxib, which is, therefore, a subtherapeutic dose. Taking this into account, our results suggest that the efficacy observed with CTC in this study is mainly due to tramadol, and that the presence of celecoxib allows reduction in the dose of tramadol in this oral surgical pain model, leading to improved tolerability. Nevertheless, we cannot neglect the possibility that the observed enhanced therapeutic effect may be bidirectional; that is, tramadol could allow reduction of the dose of celecoxib. If true, this could have a remarkable impact on the management of chronic pain caused by osteoarthritis, as tramadol could reduce the required celecoxib dose and, consequently, reduce the potential risk associated with chronic administration of cyclooxygenase-2 inhibitors. Clinical trials are needed to test this hypothesis.
A potential methodological limitation to this study is the use of the LOCF approach to handle missing data, which includes all observations after patients had received rescue analgesia. However, given that patients included in the study were required to have a post-surgery PI > 50 mm VAS and presented with a higher PI score when requesting rescue medication (treatment failure), this approximation penalises an ineffective treatment and seems to be more conservative than using the alternative ‘baseline observation carried forward’ method. Furthermore, the trial involved a single administration of study drug and may not have been long enough to show duration of effect. These factors will need to be addressed in future clinical trials.
In this study, CTC 100 and 150 mg were associated with fewer AEs than tramadol 100 mg, while CTC 200 mg (tramadol 88 mg plus celecoxib 112 mg) presented a similar safety profile to tramadol 100 mg. The dose-sparing effect of adding celecoxib to the co-crystal (56% less tramadol in CTC 100 mg and 34% less tramadol in CTC 150 mg, vs. tramadol alone) improved tolerability at the same time as enhancing efficacy. Furthermore, CTC 200 mg (12% less tramadol) demonstrated dramatically enhanced efficacy, with a similar safety profile, compared with tramadol 100 mg. These safety findings support the concept that it is possible to improve safety while maintaining or improving efficacy by using lower levels of individual analgesics via an API-API co-crystal approach.
Another methodological limitation to take into account, if we want to estimate the benefit–risk relationship, is the sample size. The sample size (above 50 patients per arm) could be enough for the aim of the study (phase II, dose-finding, clinical trial), but it could be insufficient to estimate this relationship accurately. Therefore, it will need to be addressed in the future phase III clinical trials or in a meta-analysis of the CTC at the end of the clinical development programme.
In conclusion, and in spite of the methodological limitations described above, the data from this phase II clinical trial suggest that the potential clinical benefits of CTC may outweigh the risks. This study has demonstrated that CTC 100, 150, and 200 mg were more efficacious than tramadol 100 mg and placebo in the treatment of acute pain following oral surgery, and that CTC presents a dose-dependent effect on pain efficacy associated with a better (CTC 100 and 150 mg) or similar (CTC 200 mg) safety profile compared with tramadol 100 mg. Other confirmatory clinical trials in moderate to severe post-surgical pain will be needed to confirm this significant benefit-to-risk ratio of CTC.
References
Moore RA, Derry S, Aldington D, Wiffen PJ. Single dose oral analgesics for acute postoperative pain in adults—an overview of Cochrane reviews. Cochrane Database Syst Rev. 2015;9:CD008659.
Correll DJ, Vlassakov KV, Kissin I. No evidence of real progress in treatment of acute pain, 1993–2012: scientometric analysis. J Pain Res. 2014;7:199–210.
Sommer M, de Rijke JM, van Kleef M, et al. The prevalence of postoperative pain in a sample of 1490 surgical inpatients. Eur J Anaesthesiol. 2008;25(4):267–74.
Wu CL, Raja SN. Treatment of acute postoperative pain. Lancet. 2011;377(9784):2215–25.
Chou R, Gordon DB, de Leon-Casasola OA, et al. Management of postoperative pain: a Clinical practice guideline from the American Pain Society, the American Society of Regional Anesthesia and Pain Medicine, and the American Society of Anesthesiologists’ Committee on Regional Anesthesia, Executive Committee, and Administrative Council. J Pain. 2016;17(2):131–57.
Thipparaboina R, Kumar D, Chavan RB, Shastri NR. Multidrug co-crystals: towards the development of effective therapeutic hybrids. Drug Discov Today. 2016;21(3):481–90.
Desai D, Wang J, Wen H, Li X, Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm Dev Technol. 2013;18(6):1265–76.
Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879–923.
European Medicines Agency. Summary of product characteristics for Celebrex 100 mg capsule 2016. http://www.medicines.org.uk/emc/medicine/14534. Accessed 9 March 2017.
Almansa C, Mercè R, Tesson N, Farran J, Tomàs J, Plata-Salamán CR. Co-crystal of tramadol hydrochloride-celecoxib (ctc): a novel API-API co-crystal for the treatment of pain. Cryst Growth Des. 2017;17:1884–92.
Merlos M, Portillo-Salido E, Brenchat B, et al. Co-crystal of tramadol-celecoxib produced synergistic antinociceptive effects in postoperative pain models: preclinical rationale for clinical development. In: Presented at IASP 16th World congress of pain, 26–30 Sept 2016, Yokohama, Toyko.
Videla S, Lahjou M, Vaqué A, Sust M, Encabo M, Soler L, Sans A, Sicard E, Gascón N, Encina G, Plata-Salamán C. Single-dose pharmacokinetics of co-crystal of tramadol-celecoxib: results of a four-way randomized open-label phase I clinical trial in healthy subjects. Br J Clin Pharmacol. 2017;83(12):2718–28. https://doi.org/10.1111/bcp.13395 (Epub 2017 Sep 20).
Videla S, Lahjou M, Vaqué A, Sust M, Escriche M, Soler L, Sans A, Sicard E, Gascón N, Encina G, Plata-Salamán C. Pharmacokinetics of multiple doses of co-crystal of tramadol-celecoxib: findings from a four-way randomized open-label phase I clinical trial. Br J Clin Pharmacol. 2018;84(1):64–78. https://doi.org/10.1111/bcp.13428.
O’Muircheartaigh J, Marquand A, Hodkinson DJ, et al. Multivariate decoding of cerebral blood flow measures in a clinical model of on-going postsurgical pain. Hum Brain Mapp. 2015;36(2):633–42.
Mishra H, Khan FA. A double-blind, placebo-controlled randomized comparison of pre and postoperative administration of ketorolac and tramadol for dental extraction pain. J Anaesthesiol Clin Pharmacol. 2012;28(2):221–5.
Moore RA, McQuay HJ, Tomaszewski J, et al. Dexketoprofen/tramadol 25 mg/75 mg: randomised double-blind trial in moderate-to-severe acute pain after abdominal hysterectomy. BMC Anesthesiol. 2016;16(1):9.
Fricke JR Jr, Hewitt DJ, Jordan DM, Fisher A, Rosenthal NR. A double-blind placebo-controlled comparison of tramadol/acetaminophen and tramadol in patients with postoperative dental pain. Pain. 2004;109(3):250–7.
Dworkin RH, Thakur R, Griesing T, Sharma U, Young JP. Randomized clinical trials of pregabalin for neuropathic pain: methods, results, and implications. Prog Neurother Neuropsychopharmacol. 2008;3(1):167–87.
Katz N. Methodological issues in clinical trials of opioids for chronic pain. Neurology. 2005;65(12 Suppl 4):S32–49.
Averbuch M, Katzper M. Baseline pain and response to analgesic medications in the postsurgery dental pain model. J Clin Pharmacol. 2000;40(2):133–7.
Averbuch M, Katzper M. Severity of baseline pain and degree of analgesia in the third molar post-extraction dental pain model. Anesth Analg. 2003;97(1):163–7.
European Medicines Agency. Summary of product characteristics for tramadol 50 mg capsule 2015. http://www.medicines.org.uk/EMC/medicine/24186/SPC. Accessed 9 March 2017.
Moore RA, Gay-Escoda C, Figueiredo R, Tóth-Bagi Z, Dietrich T, Milleri S. Dexketoprofen/tramadol: randomised double-blind trial and confirmation of empirical theory of combination analgesics in acute pain. J Headache Pain. 2015;16:541.
U.S. Food and Drug Administration. Celebrex® prescribing information 2008. http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020998s027lbl.pdf. Accessed 1 Nov 2016.
Derry S, Moore RA. Single dose oral celecoxib for acute postoperative pain in adults. Cochrane Database Syst Rev. 2012;3:CD004233.
Acknowledgements
Editing support was provided by Alice Wareham, PhD (Aspire Scientific Ltd., Bollington, UK) and funded by Mundipharma Research GmbH & Co. KG (Limburg, Germany). Harrison Clinical Research Deutschland GmbH was the Contract Clinical Research Organisation that provided clinical trial management support, funded by Laboratorios del Dr. Esteve, S.A.U. Co-crystal of Tramadol–Celecoxib Team: The authors present this work on behalf of the ‘CTC phase II dose-finding study team’; in alphabetical order: Soledad Casals, BSc, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; Jesús Cebrecos, MD, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; José-Luis Cebrián, MD, Hospital Universitario La Paz, Madrid, Spain; Jordi García-Linares, MD, Hospital General de Granollers, Granollers, Spain; Neus Gascón, MD, Medical Science, Laboratorios del Dr. Esteve, Barcelona, Spain; Aranxa González-Corchón, MD, Hospital Clínico San Carlos, Madrid, Spain; Mariano Marqués, MD, Hospital Clínico de Valencia, Valencia, Spain; María-José Morán, MD, Hospital Universitario La Paz, Madrid, Spain; Adelaida Morte, MD, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; Ana Otero, MD, Complexo Hospitalario Universitario A Coruña, A Coruña, Spain; Esther Ortiz, BSc, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; María Pombo, MD, Complexo Hospitalario Universitario A Coruña, A Coruña, Spain; Albert Puyada, BSc, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; Artur Sans, RN, Clinical Investigation, Laboratorios del Dr. Esteve, Barcelona, Spain; Jesús Sastre, MD, Hospital Universitario La Princesa, Madrid, Spain; and Juan-Manuel Velázquez, MD, Hospital General de Granollers, Granollers, Spain.
Author information
Authors and Affiliations
Consortia
Contributions
SV, AM, MS, AV, and CP-S designed and wrote the study protocol; JL-C, MB, IJ, SA-H, RM-G, JG, MP, JG-D, and JH approved the final version of the protocol and visited and treated the patients; MS and SV were responsible for data management and the statistical analysis; SV, AM, MS, AV, and CP-S wrote the manuscript. All authors have revised and approved the final manuscript and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Funding
Laboratorios del Dr. Esteve, S.A.U. provided funding for this study. Role of the funding source: The study’s funder was involved in study design, data interpretation, and revision of the final report, but did not take part in data collection and analysis. The corresponding author had full access to all study data and had final responsibility for the decision to submit the paper for publication.
Conflict of interest
AV, MS, and CP are employees of Laboratorios del Dr. Esteve, S.A.U. SV was employee of Laboratorios del Dr. Esteve during clinical trial execution. The remaining authors declare that they have no conflicts of interest.
Additional information
Study group members are listed at the Acknowledegements.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
López-Cedrún, J., Videla, S., Burgueño, M. et al. Co-crystal of Tramadol–Celecoxib in Patients with Moderate to Severe Acute Post-surgical Oral Pain: A Dose-Finding, Randomised, Double-Blind, Placebo- and Active-Controlled, Multicentre, Phase II Trial. Drugs R D 18, 137–148 (2018). https://doi.org/10.1007/s40268-018-0235-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-018-0235-y