
Abstract
Over the past 15 years, single-cell RNA sequencing (scRNA-seq) technology, in combination with other omics, has revealed the mechanisms of human development, tumors, and complex diseases at the genome, transcriptome, and proteome levels. However, this approach fails to directly reflect relevant spatial information, such as cell location and interactions. This limitation has been addressed with the advancement of the combination of high-resolution scRNA-seq and spatial transcriptomics (ST), which enables the identification of cell composition, intercellular and intermolecular interaction, and unravels the mechanisms of disease phenotypes. This review explores two types of ST - imaging-based ST (iST) and sequencing-based ST (sST) - and demonstrates how ST analysis can follow disease pathogenesis in a spatiotemporal manner, searching for disease-specific biomarkers. ST technology is an effective tool for resolving major biomedical and clinical problems, including tumor research, brain science, embryonic development, organ atlas construction and other pathological analysis. Looking towards the future, despite its limitations, ST has the potential to address these problems in conjunction with “dynamics, multi-omics, and resolution”. Ultimately, the development of ST technology, improvement of algorithms, utilization of deep learning, and refinement of the analysis process and interpretation will determine the key to transforming ST from bench to bedside.
Similar content being viewed by others

1 Introduction
Over the course of the past 15 years, scRNA-seq has emerged as an essential tool in scientific research, particularly in the fields of cancer research, brain science, developmental biology, and other pathologies. But scRNA-seq is limited by its requirement for enzymolysis and destruction of the spatial structure of tissues and cells (Williams et al. 2022), while most biological functions in the human body, such as tumorigenesis, brain disease, embryonic development, and other pathological diseases, depend on the spatial structure and properties of associated cells (Moses and Pachter 2022). The high-throughput ST approach has the capability to capture gene expression information in situ, offering valuable insight into regulatory effects of spatial location on gene expression and cellular interactions in biological problems. The construction of the online Spatial Omics DataBase (SODB) platform has provided a driving force for the development of ST (Yuan et al. 2023). Combining ST and scRNA-seq is vital in closely linking pathological phenomena, spatial structure, and molecular changes, revealing molecular communication between cells in situ (Wang et al. 2023; Zhang et al. 2022a; Shen et al. 2022b; Fang et al. 2023b; Vandereyken et al. 2023) and allowing for improved biomedical and clinical applications through the use of sensitive biomarkers, accurate disease classification, and precise risk prediction (Walker et al. 2022; Moffitt et al. 2022; Elmentaite et al. 2022).
2 Development of ST technology and its analysis modules
2.1 Development of ST technology
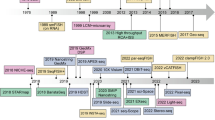
Since its establishment as a technique by Ståhl et al. in 2016 (Stahl et al. 2016), ST has been widely applied across different fields, with numerous new ST technologies emerging. ST technology can be categorized into two types, i.e., iST and sST, with the former detecting spatial gene expression through immunofluorescence images and the latter through sequencing to measure spatial gene expression (Fig. 1a). To date, various ST methods and analysis protocols have been developed, such as 10X Visium, DSP, Stereo-seq (BGI), MERFISH, Slide-seqV2, and STARmap PLUS (Stahl et al. 2016; Merritt et al. 2020; Chen et al. 2022a), alongside the spatial atlas analysis technology of chromatin accessibility (spatial-ATAC-seq) (Deng et al. 2022).

Development timeline of ST technologies. The development timeline of ST has witnessed the emergence of various technologies, including 10X Visium, DSP, Stereo-seq (BGI), MERFISH, Slide-seqV2, and STARmap PLUS, among others
2.2 ST analysis modules
The applications of ST analysis modules are continuously expanding, with applications resolving three fundamental biological problems. Firstly, ST technology enables the clarification of tissue cellular composition, allowing for the observation of specific cell types in cellular spatial structure. Secondly, it facilitates the analysis of interactions between cells in tissues by examining spatial positions and contacts between cells. Lastly, ST technology analyzes molecular interaction by examining ligand-receptor pairs transcription between cells. In tumor research, for instance, ST is particularly well-suited for exploring tumor/vascular/immune/metabolic microenvironments and analyzing spatial immune niche, as well as analyzing the tumor invasion front (TIF) and further constructing the gene networks. ST has demonstrated significant potential for applications in human disease and development.
3 Application potentials of ST in disease and development
3.1 ST in tumor research
Misdiagnosis and complex prognosis of tumors are due to the spatial heterogeneity of tumors and the complexity of defining the TIF (Ahmed et al. 2022). Consequently, there is an urgent need for accurate analysis of the molecular mechanism underlying tumor occurrence and development to improve clinical diagnosis and treatment. The tumor immune microenvironment (TIME) is an incredibly complex ecosystem (Binnewies et al. 2018) where tumor-infiltrating T cells destroy tumor cells based on the signal regulation of bone marrow cells and recognized tumor-specific antigens (TSA). TIME is composed of monocytes, macrophages - especially tumor-associated macrophages (TAM) - as well as immune regulation of cancer-associated fibroblasts (CAF) and nutritional supply through endothelial cells (EC). During the development of anti-PD-1/PD-L1 antibodies and other immune checkpoint blockers (ICBs), understanding the distribution and function of immune cells in detail becomes necessary as TIME strongly affects the effectiveness of ICB therapy. Although scRNA-seq partially addresses questions in cancer research, understanding how different immune cells interact with tumor cells and whether tumor mass is evenly infiltrated requires scRNA-seq data sets embedded with histological structure information, i.e., ST technology (Hsieh et al. 2022).
ST technology is widely utilized in tumor research, combining scRNA-seq to determine cell type-specific gene sets alongside morphological analysis (i.e., HE staining) and ST to determine tissue region-specific gene sets (Moncada et al. 2020). The integration of multi-omics method is then employed to analyze the relationship between cell state and different cell groups and tissue regions in the tumor microenvironment. The former includes tumor immune microenvironment (TIM), vascular microenvironment, and metabolic microenvironment, while the latter encompasses the communication and interaction between tumor cells, immune cells, CAFs, vascular cells, and cell proliferation and death (Fig. 2).

Integrating scRNA-seq, ST technology, and morphological analysis to study tumors. This figure shows the analysis process of multi omics in tumor research
The application of ST technology in tumor research has proven superior to traditional methods, enabling the extraction of more information for clinical diagnosis and treatment than ever before (Lewis et al. 2021). Recently, Seferbekova et al. reviewed the complex landscape of the tumor ecosystem revealed by ST and proteomics, highlighting the necessity of interaction with the tumor microenvironment (TME) in cancer evolution, as well as potential clinical applications and development directions (Seferbekova et al. 2022). Meanwhile, Akhoundova and Rubin focused on the latest research progress in multi-omics analysis of tumor atlas, summarizing the in vitro model of the spatial genome and chromatin conformation, ST and proteome, liquid biopsy, and drug effectiveness evaluation (Akhoundova and Rubin 2022). Novel ST technologies, such as base-specific in situ sequencing (BaSISS), have also emerged to better map the structure, nature, and evolution of cancer clones (Lomakin et al. 2022). Overall, ST is widely utilized in tumor research, with applications spanning various areas (Fig. 3):

The application of ST in tumor research. a Display the cell compositions and communications in the TME, showing TIF, TLS, intratumoral microbiota, etc. b Demonstrate the ST application in the progression of CRC liver metastasis (CRLM), showing prognostic cancer markers
3.1.1 Spatial characteristics of TIME
Solid tumors are primarily composed of tumor cells, immune cells, stromal cells, vascular ECs, and smooth muscle cells (SMC). As one of the most complex tumor microenvironments, ST can effectively interpret TIME by revealing the immune cell types, status, and interaction with other cell types. For instance, Breast cancer (BC) serves as an excellent model for ST application due to its complex histopathological characteristics (Wu et al. 2021; Andersson et al. 2021). Spatial cell labeling has revealed that T cells are co-located in lymphocyte-rich regions (Wu et al. 2021), whereas ST analysis of HER2+ BC has found immune cells and tumor cells compartmentalized, mixed with, or aggregated into the structure of lymphatic organs surrounding the area of ductal carcinoma in situ (DCIS) (Andersson et al. 2021). Furthermore, the analysis of TIME is a crucial focus in tumor research with spatial multi-omics analysis applied to its study (Hsieh et al. 2022). The tumor immune barrier (TIB) formed by the interaction of SPP1+ macrophages and CAF in the TME of hepatocellular carcinoma (HCC) proved related to the efficacy of immunotherapy (Liu et al. 2023d). By blocking SPP1 to destruct the TIB structure could improve the curative effects of immune checkpoint blockade on HCC (Liu et al. 2023d). Real-time imaging via ST has also provided valuable insights into the behavior and function of CAR T cells, demonstrating the theoretical basis for cancer immunotherapy (Ahmed et al. 2022). Additionally, there exists a strong spatial interaction between immune cells and metastatic malignant tumor cells. A CRLM study has identified a specific macrophage group (MRC1+ and CCL18+) scattered around the metastatic tumors and peritumoral boundary with extensive cell-cell interaction (CCI) detected between metastatic tumor cells and macrophages (Wu et al. 2022). Furthermore, SpaCET, a computer framework developed by Ru et al., can infer cell identity in tumor ST data and reveal how CCI at the tumor-immune interface promotes cancer progression (Ru et al. 2023) (Fig. 3a).
3.1.2 Analysis of TIF
TIF, as the junction between tumor and immune cells, generally elicits a fierce immune response. Ji et al. (2020) and Anderssen et al. (2021) conducted high-dimensional and multi-omics analyses to describe the characteristics of human squamous cell carcinoma (cSCC) and HER2+ BC respectively, finding TSK as the center for cell-cell communication (CCC) located in the vascular tissue, and identifying significant cell interaction at TIF. Patients with rich immune cell infiltration at TIF exhibited a better prognosis (Andersson et al. 2021). Meanwhile, ST revealed malignant crosstalk between co-located cells in CRC patients, with CRC cells at TIF inducing human leucocyte antigen G (HLA-G) to produce SPP1+ macrophages, enhancing tumor immunity, proliferation, and invasion (Ozato et al. 2023) (Fig. 3a). The utilization of ST enables the acquisition of high-resolution and high-throughput molecular expression and interaction networks in the TIF, facilitating the observation of changes in a gradient from cancerous to adjacent regions. Furthermore, ST can be employed to investigate alterations in cell types, cellular functions, energy metabolism, and other factors on both sides of the TIF.
3.1.3 Spatial analysis of TLS
Tumor-associated tertiary lymphoid structure (TLS) are complex microenvironments that contain various immune cells with distinct functions, as revealed by ST analysis that TLS are composed of B cells, T cells, and dendritic cells (DC). A BC study showed that TLS was rich in B cells in TNBC patients, while HER2+ BC patients exhibited a TLS highly co-located by B cells and T cells (Keren et al. 2018; Andersson et al. 2021). Bassiouni et al. have investigated the differences in cell composition within TNBC among different racial cohorts (Bassiouni et al. 2023). In human CRC samples, ST can describe the molecular and structural changes that occur during the cancer state transition process and the variations in cell composition and TLS structural pattern (Lin et al. 2023a) (Fig. 3a). The application of ST in the spatial analysis of TLS enables the investigation of dynamic changes and ligand-receptor interactions among immune cells, stromal cells, and cancer cells within the TLS over time.
3.1.4 Intratumoral microbiota on spatial heterogeneity
Recent studies have revealed that microorganisms can influence tumor cells and exhibit intratumoral spatial characteristics. ST and scRNA-seq analysis showed that microbiota distribution in oral squamous cell carcinoma (OSCC) and CRC tumors was not random but highly concentrated in the microenvironment with immune and epithelial cell functions, facilitating cancer progression and highlighting the interactions between space, cell, and host-microorganism (Galeano Nino et al. 2022) (Fig. 3a). The utilization of ST in the analysis of intratumoral microbiota and tumor spatial heterogeneity enables the investigation of interactions between microbiota and tumor cells within the tumor microenvironment, leading to a better understanding of the role of microbiota in tumor development and progression. However, the technology requires further improvement to enhance ST’s ability to capture microbial RNA. Future research may explore the distribution and interaction of microorganisms in tumors through the implementation of a capture strategy such as utilizing random prime capture.
3.1.5 Reveal tumor progression
ST can significantly advance tumor characteristic study, elucidating subpopulation spatial distribution, and interaction and metabolism between different compartments during cancer progression. Through ST analysis, Karras et al. obtained a high-resolution spatial landscape of melanoma cell subpopulations, mapped the phenotypic diversity of melanoma, identified tumorigenic cell groups, and provided insight into tumor growth origins (Karras et al. 2022). In CRC patients, the increased expression of ASCT2 and CD98 (Fig. 3b), prognostic cancer markers in humans, can monitor the immune cell metabolism polarization (Hartmann et al. 2021). Moreover, multi-omics analysis integrated by Heide et al. characterized the CRC phenotypic heterogeneity evolution and constructed the tumor heterogeneity atlas, enlightening CRC biology (Heide et al. 2022; Househam et al. 2022). Multidimensional ST analysis data can identify essential information in tumor progression as shown by Zhu et al. constructing a multi-group invasive lung adenocarcinoma (LUAD) atlas, revealing LUAD-specific cell information and interaction that bolsters early diagnosis and surgical intervention (Zhu et al. 2022). Meanwhile, Cui Zhou et al.’s ST work identified transitional cell groups between normal pancreas and pancreatic ductal adenocarcinoma (PDAC), highlighting differences in chemotherapy’s effect on tumor and interstitial cell abundance and transcription processes (Cui Zhou et al. 2022). In addition to the above research on coding RNA, ST analysis can also unveil tissue-specificity and functional roles of long non-coding RNA (lncRNA)’s in cancer (Xu et al. 2023).
3.1.6 TME related to prognoses
The spatial structure of TME is strongly associated with overall patient survival. Patients with a well-defined TME often exhibit a better disease prognosis. For instance, in PDAC, the immune-related genes and immunophenotypes of the TME influence recurrence patterns and patients with well-defined TME tend to have better survival (Karamitopoulou et al. 2023). Furthermore, structural subtypes of diffuse large B-cell lymphoma (DLBCL) have been found to have better overall survival compared to scattered subtypes (Colombo et al. 2022). In pan-cancer analysis, it has been suggested that the interferon response module varied with tumor location and weakened after lymphocyte elimination, indicating the interaction between cancer recurrence and the TME (Barkley et al. 2022).
3.1.7 Exploration of tumor biomarkers and therapies
The integration of scRNA-seq with ST enables the identification of new tumor biomarkers. For instance, in bladder cancer, a new marker N-Cadherin 2 was discovered through single-cell clustering and was associated with poor prognosis. T cells expressed more depletion markers near tumor cells expressing N-Cadherin 2, indicating that tumor cells with more dedifferentiation, proliferation, and invasiveness were usually co-located with depleted T cells (Sautes-Fridman et al. 2019). ST can also be used to construct molecular and immune atlases of early CRC tumorigenesis (Roelands et al. 2023), identify prognostic markers for CRLM (Cortese et al. 2023), determine multicellular dynamics related to neoadjuvant therapy in PDAC (Hwang et al. 2022), and reveal regulatory pathways for arginine metabolism in human neuroblastoma mouse models (Van de Velde et al. 2021). CAFs, acting as immunomodulatory cells in tumors, can serve as important biomarkers and therapeutic targets. In BC, ST data analysis revealed strong crosstalk between iCAF and adjacent T cells (Wu et al. 2021). Further, Akiyama et al. used integrated scRNA-seq and ST to explore the effect of matrix reprogramming on immune reactivation and potential treatment methods (Akiyama et al. 2023). Exploring tumors using ST to construct a tumor-targeted regulatory network has the potential to facilitate the identification of tumor biomarkers and their subsequent implementation in personalized treatment strategies.
3.2 ST in brain science
The diversity of brain cell types and their communication relationship shall be analyzed to analysis of brain function (Zeng and Sanes 2017). ST plays a crucial role in revealing the relationship between brain cell composition, spatial location, cell communication, organ function, and disease. ST links scRNA-seq characteristics with spatial distribution and is regarded as an advantageous tool in brain analysis. ST-based in situ capture and heterotopic sequencing technologies have enabled spatial transcriptome analysis for regions in human and mouse brains (Maynard et al. 2020; Rodriques et al. 2019; Lei et al. 2022). Though sensitivity is sometimes lower than IHC or IF, high-multiple, high-resolution, and high-throughput ST technologies will continue to improve capture sensitivity, providing a powerful tool for exploring brain science. ST can explore cell type heterogeneity, transformation, and migration during neural development, and reveal specific types of pathological changes in brain diseases and animal models (Fig. 4).

The application of ST in brain science. a ST analysis of the whole brain to construct the brain spatial atlas. Abbreviations: Cerebral cortex (CTX), Hippocampal formation (HPF), Thalamus (TH), Hypothalamus (HY), Corpus callosum (CP); b Spatial analysis of AD and healthy brain demonstrating molecular mechanisms and therapeutic targets (inhibition of Inpp5d)
3.2.1 Construction of brain spatial atlas
ST has been widely used in multiple studies to explore the cellular and spatial characteristics of different brain regions. For instance, Chen et al. used ST (Stereo-seq) to describe the spatial structure of the adult mouse brain (Chen et al. 2022a). Lei et al. used single nuclei RNA sequencing (snRNA-seq), snATAC-seq, and high-resolution ST (Stereo-seq) to identify the regulatory networks of excitatory neurons in different cortex and brain regions of cynomolgus monkeys, highlighting cell types susceptible to neurological diseases (Lei et al. 2022). Lin et al. used ST to map the central nervous system (CNS) region of marmosets (Lin et al. 2022), while Uzquiano et al. used multimodal scRNA-seq, epigenetic sequencing, and ST to define the fate specialization process of the human cortex (Uzquiano et al. 2022). Additionally, Zhang’s team used ST (MERFISH) to identify different cell types and spatial maps in the mouse primary motor cortex, revealing the complexity of different cell types (Zhang et al. 2021) (Fig. 4a).
3.2.2 ST analysis of brain diseases
ST is widely used in Alzheimer’s disease (AD) research, providing insight into the spatiotemporal changes in AD pathology. For example, studies using ST have constructed cellular and spatial atlases of AD progression, identified new markers of plaque like CST7 (cystatin F), and revealed changes in cell type during AD development (Zeng et al. 2023; Chen et al. 2022c; Castranio et al. 2023; Luquez et al. 2022) (Fig. 4b). Additionally, haploid defect models, such as the Inpp5d model constructed by Lin et al., have suggested potential treatment strategies, enhancing microglial function and plaque clearance (Lin et al. 2023b). Other studies have drawn atlases of non-immune and non-neuronal cells in the brain of wild-type and AD model mice, identifying disease-associated oligodendrocytes (DOLs) that respond to the CNS’s steady state (Kenigsbuch et al. 2022). Further multi-omics analysis, as performed by Iturria-Medina et al., could classify AD and predict neuropathy severity, defining three molecular subtypes that explained AD’s pathology and clinical heterogeneity (Iturria-Medina et al. 2022). The combination of ST with radiography or pathology, as well as improved integration methods, can help reveal the formation mechanism and treatment methods of amyloid-beta plaque in this field.
ST is a valuable tool for exploring various brain diseases. For instance, Kathe et al. used snRNA-seq and ST to identify an important subgroup of neurons in post-paralysis recovery tissue necessary for the recovery of walking (Kathe et al. 2022). Allen et al. used ST to study the changes in cell molecular characteristics and spatial organization in the aging process of the mouse brain (Allen et al. 2023), while Fang et al. used ST to analyze the transcriptional substrate of brain structure and function damage in first-episode major depression patients (Fang et al. 2023a). Zhang et al. used ST to study the transcriptional changes in specific brain regions in a mouse model of epilepsy, identifying potential signal targets for immune intervention of epilepsy activities (Zhang et al. 2023b). Additionally, Batiuk et al. used ST to analyze changes in gene expression and neuron composition in the influence circuit of schizophrenia, revealing the network damage mechanism driven by the upper cerebral cortex (Batiuk et al. 2022).
In exploring brain metastasis of cancer, for example, Zhang et al. used ST to obtain complete transcriptional histograms of tumor core, TIME, and the tumor microenvironment in the brains of non-small cell lung cancer (NSCLC) patients, providing insights into the molecular and cellular mechanisms of brain metastasis (Zhang et al. 2022b). Similarly, Biermann et al. used ST to study melanoma brain metastasis (MBM) and identified its rich genome, adaptability, and TME characteristics (Biermann et al. 2022). Furthermore, ST technology has been applied to analyze biopsies of primary tumors of CNS lymphoma, revealing intertumoral heterogeneity and T cell exhaustion (Heming et al. 2022). In the context of infectious diseases such as trypanosoma brucei infection, Quintana et al. employed scRNA-seq and ST analysis to reveal the crosstalk between microglia and plasma cells in the brain (Quintana et al. 2022). These studies shed light on the molecular mechanism underlying various brain diseases.
The utilization of ST omics in the construction of brain atlases and the investigation of brain diseases presents an opportunity to gain insight into the temporal dynamics and intercellular interactions of various brain cell types, as well as to identify potential biomarkers and elucidate the molecular mechanisms underlying these diseases. For instance, ST technology can be employed to partition the brain and examine the spatial distribution of brain regions, as well as changes in the surrounding microenvironment, such as amyloid-beta plaque. Moreover, this approach can facilitate the identification of novel therapeutic targets and the development of personalized treatment modalities.
3.3 ST in development
Developmental biology researchers have traditionally focused on the formation of complex adult organisms from fertilized zygotes. Although scRNA-seq has been utilized to describe gene expression and regulation in mouse embryo development, it cannot accurately depict spatial location and cell interaction. Therefore, the integration of ST techniques is crucial for creating a more complete development atlas. Nevertheless, research on mammalian embryo development is currently limited to mouse and monkey models due to the challenges associated with directly studying human development (Fig. 5).

The application of ST in development. a ST analysis of embryonic development. b ST analysis of organ (heart, lung, intestine, pancreas, and brain) development
3.3.1 Analysis of embryonic development
Rabbani et al. have highlighted the benefits of employing ST analysis in understanding the molecular and cellular mechanisms involved in spermatogenesis and development from newborn to adult (Rabbani et al. 2022) (Fig. 5a). Similarly, Cui et al. used ST to construct a high-precision anatomical atlas of the gastrulating cynomolgus monkey embryo, revealing important information about the regulatory network involved in embryogenesis (Cui et al. 2022). Bergmann et al. studied the early gastrula of Callithrix jacchus using ST and stem cells to depict the spatiotemporal characteristics of embryonic models (Bergmann et al. 2022). Furthermore, a three-dimensional ST spectrum was constructed to provide a panoramic view of mouse embryo development at 16.5 d (Chen et al. 2022a).
3.3.2 Analysis of organ development
Chen et al. created the mouse organogenesis spatiotemporal transcriptomic atlas (MOSTA, https://db.cngb.org/stomics/mosta/) using high-resolution ST, which mapped the dynamics and directionality of mouse organogenesis with high sensitivity at single-cell resolution (Chen et al. 2022a). Similarly, Asp et al. employed ST to map the spatiotemporal gene expression and cell atlas of human heart development, enabling the identification of a unique gene spectrum for each developmental stage and anatomical region (Asp et al. 2019). Sountoulidis et al. combined ST and scRNA-seq to construct a comprehensive atlas of human lung early development, identifying 83 lung cell states and the developmental tracks leading to significant heterogeneity of lung cells (Sountoulidis et al. 2023). Besides, using ST on human fetal lung tissue samples, He et al. established the most detailed and precise multi-group cell atlas of human lung development, revealing cell lineage tracks and providing new insights for understanding the development of adult lung diseases (He et al. 2022). Fawkner-Corbett et al. established an atlas of human intestinal development, providing a detailed account of the formation of the crypt-villus axis and the morphogenesis of nerves, blood vessels, mesenchyme, and intestinal immune population (Fawkner-Corbett et al. 2021). In a study on human fetal pancreas using a combination of scRNA-seq and ST analysis at multiple developmental stages, Olaniru et al. identified novel candidate genes involved in regulating the differentiation of progenitor cells. They also highlighted the potential importance of Schwann cells and mesenchymal cells in the differentiation of human endocrine progenitor cells and acinar cells (Olaniru et al. 2023). Garcia-Alonso et al. constructed a comprehensive spatiotemporal atlas of human and mouse gonadal differentiation using ST (Garcia-Alonso et al. 2022). Finally, Wei et al. utilized ST to draw the first three-dimensional ST spectrum of the development and regeneration of the Ambystoma mexicanum Brain, identifying important neural stem cell subtypes involved in the regeneration process (Wei et al. 2022) (Fig. 5b).
By using ST omics in the study of development, it is possible to learn about the dynamic changes and interactions between varies cell types during embryonic development and organogenesis. This approach can facilitate the identification of key signaling pathways and gene regulatory networks that underlie normal development. Moreover, by comparing healthy and abnormal development, ST can identify transcriptional expression differences of regulatory factors, and subsequently aid in the identification of novel regulators of development and potential therapeutic targets for developmental disorders.
3.4 ST in other pathologies
In addition to the above, the combination of ST and scRNA-seq is a powerful tool for exploring other human complex pathological diseases:
3.4.1 Analysis of key factors of cardiovascular disease (CVD)
The latest review by Miranda et al. provided a comprehensive overview of the use of single-cell transcriptomics in cardiac biology, development, and disease, explaining how to maximize the implementation of these technologies and discusses the potential for multimodal integration with ST and epigenetics (Miranda et al. 2023). Researchers have used ST in multiple studies to explore different aspects of heart disease. Kuppe et al. drew a comprehensive spatial and multi-omics atlas of the human heart after myocardial infarction (Kuppe et al. 2022), while Jung et al. identified an anti-inflammatory macrophage subgroup with Trem2hi characteristics (Jung et al. 2022). Hu et al. explored the origin and function of activated fibroblasts in zebrafish heart regeneration and determined that Wnt signal regulates endocardium fibroblast response (Hu et al. 2022). Additionally, ST analysis has been used in studies of hypertrophic cardiomyopathy (HCM) (Liu et al. 2023b), pigs treated with IGF-1 (Zeng et al. 2023), and human cardiac sarcoidosis (Liu et al. 2022) to reveal transcriptional changes and provide new insights into the pathology of these diseases. Finally, ST has been used by Bondareva et al. to reveal specific molecular changes of organ vascular endothelium in a diabetes mouse model driven by obesity (Bondareva et al. 2022) (Fig. 6a).

The application of ST in other pathologies. a Analysis of key factors of CVD. b Analyze the influencing factors of skin diseases. c Role of virus and inflammation in disease progression. d Construct a normal/ disease atlas
3.4.2 Analyze the influencing factors of skin diseases
The use of ST has helped analyze the diversity of skin cells in different cell types and states (normal/sick), enabling the exploration of biomarkers and disease treatment (Pineiro et al. 2022). Studies of non-communicable inflammatory skin diseases (ncISD) through ST have revealed that a few immune cells promote these diseases (Schabitz et al. 2022). Similarly, ST has advanced our understanding of immune diseases such as leprosy (Ma et al. 2021) and sarcoidosis (Krausgruber et al. 2023). For instance, studies of leprosy and sarcoidosis have utilized ST to explore the spatial structure of granulomas and their immune influencing factors. In a mouse model of wound healing, Foster et al. located four subpopulations of fibroblasts through ST and integrated scRNA-seq data to estimate changes in chromatin accessibility during the three classic wound healing stages. Additionally, Foster et al. found that macrophages play a crucial role in wound healing at the center of the wound after 1 week (Foster et al. 2021) (Fig. 6b).
3.4.3 Role of virus and inflammation in disease progression
ST has proven to be useful in analyzing the response of lung tissue to SARS-COV-2, providing a more accurate description of the heterogeneity and locations of related cells compared to bulk and scRNA-seq (Nienhold et al. 2020; Staines et al. 2021; Cross et al. 2023). Studies of SARS-COV-2 patients have revealed upregulation of gene modules related to angiogenesis, type I interferon production, inflammation, and coagulation (Kulasinghe et al. 2022). Additionally, ST has been utilized to investigate the immunopathology of severe COVID-19 and its relationship to different tissue niches of CCL18 and CCL21 (Mothes et al. 2023). ST is also suitable for examining the immune cell mechanism and tissue diversity associated with chronic inflammatory diseases such as rheumatoid arthritis and spinal arthritis, which involve overexpression of certain genes (Carlberg et al. 2019). Furthermore, ST has been employed to reveal the role of CD4 T cells in the differentiation of effector CD8 T cells during chronic viral infection (Topchyan et al. 2022) and identify the activity and basic subgroup of intestinal eosinophils and their role in colitis (Gurtner et al. 2023) (Fig. 6c).
3.4.4 Construct normal/ disease atlas
ST has the potential to establish a normal/disease human cell atlas of organs, aiding in disease re-classification and the identification of therapeutic targets (Melo Ferreira et al. 2021, 2022; Noel et al. 2021). In the field of lung research, it is crucial to analyze the cellular and microenvironment components. The integration of scRNA-seq and ST has redefined the tissue structure of the lung and airway, revealing that glandular epithelial cells recruited B cells and plasma cells and promoted antibody secretion through the expression of CCL28, APRIL, and IL-6 (Madissoon et al. 2023). Additionally, studies of pulmonary macrophage subpopulations using scRNA-seq and ST have helped clarify the complexity of macrophage biology in homeostasis and disease (Aegerter et al. 2022) (Fig. 6d).
By using ST omics in the study of other pathologies, it is possible to analyze different cell states, modules and networks during disease progression. This approach can facilitate the identification of novel biomarkers for diagnosis, prognosis, and drug response prediction. Furthermore, ST can aid in the identification of potential therapeutic targets and the development of personalized treatment strategies tailored to the individual pathology. Additionally, this approach can provide insights into the molecular mechanisms underlying the pathogenesis of diseases, leading to a better understanding of the disease and the development of new therapeutic approaches.
4 Outlook: future perspectives for ST in clinical application
Precision medicine combines bioinformatics, histomorphology, and molecular biology to achieve personalized treatment. The approach often utilizes multi-omics to decode tumor heterogeneity, brain atlas, transcriptional regulation in development, and the pathogenesis and prognosis of complex diseases. Despite its potential, ST technology still faces certain limitations that need to be addressed to further advance its implementation in precision medicine. The following provides an overview of the limitations and future improvement directions of ST technology to obtain a comprehensive understanding of cellular heterogeneity and spatial organization.
4.1 ST limitations
ST has limitations that need to be addressed for better application in human research and scientific problems (Lee et al. 2022) (Fig. 7). These challenges include correctly locating mRNA transcripts in space to a single cell, improving applicability and performance, capturing transcripts with lower expression levels, accurately tracking immune cell receptor libraries, difficulty in working with FFPE samples, and high cost. The methods for in situ capture and ectopic sequencing, such as 10X Visium, BGI Stereo-seq, and Slide-seq, require spatial transcripts to be located into cells using cell segmentation and the deconvolution algorithm (Chen et al. 2022b). ST is not as suitable as scRNA-seq for capturing transcripts with lower expression levels (Asp et al. 2020), leading to the loss of detection of key genes with lower expression. ST also captures only single-terminal transcripts rather than full-length transcripts, making it difficult to track immune cell receptors (BCR/TCR) library and selective splicing events. Additionally, most ST technologies are well-suited to frozen tissue sections but not ideal for FFPE samples, which are the gold standard for tissue preservation despite being susceptible to RNA degradation (Yu et al. 2022; Li et al. 2022a). The high cost of ST also limits its large-scale application, highlighting the need for a new, lower-cost ST technology in the future.

ST limitations and improvement directions
4.2 ST improvement directions
Given the limitations of the ST above, the improvements are mainly reflected in three aspects (Fig. 7).
4.2.1 Development of new ST technology and platform
New ST technologies are being developed to address previous limitations. Pixel-seq, a new ST technology, uses repeated printing technology on the polony gel to achieve a resolution of 1 μm’s high-density DNA chip and greatly reduces the cost (Fu et al. 2022). The latest progress of ST in FFPE tissue detection will also significantly improve the applicability of spatiotemporal genomics in biomedical and clinical research (Meylan et al. 2022). McKellar et al. captured RNAs missed by traditional workflow through spatial total RNA sequencing (STRS), including non-coding RNA, newly transcribed RNA, and viral RNA, which broadened the application scope of ST (McKellar et al. 2023). While long-read sequencing by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) can improve the accuracy of read sequencing, their use is limited by the high cost and low read counts (Marx 2023). Ultra-sensitive sequential fluorescence in situ hybridization (USeqFISH) could be used to realize the ST analysis of endogenous and viral RNA in the whole tissue volume, such as exploring the different cell subtype deviations of adeno-associated viral vectors (AAV) variants in different brain regions of mice (Jang et al. 2023). Other developments include Image-seq, which preserves spatial information and allows for the recording and analysis of the spatiotemporal history of cells (Haase et al. 2022), and Light-Seq, which indexes biomolecules in cells and tissues for spatial analysis (Kishi et al. 2022). These advancements allow for more sensitive, accurate, and comprehensive analysis of cells and tissues, expanding the potential impact of ST in biomedical and clinical research. Finally, the development of 10X Visium “CytAssist”, BGI “Go Spatial” and cloud computing platform has made ST more accessible to a wider range of researchers, as they do not require specialized equipment or expertise, making them accessible to a wider range of researchers (Watanabe et al. 2023). These developments have the potential to democratize ST technology and enable more researchers to utilize its benefits in their research.
4.2.2 Research and development of new algorithm
The development of ST technology has led to advancements in multiple areas including cell typing, noise reduction, and analyzing cellular interaction. Techniques such as RRST (RNA-Resue Spatial Transcriptions) (Mirzazadeh et al. 2023) and Sprod (Wang et al. 2022) allow for analysis of tissue samples with medium to low RIN scores and the denoising of ST data through combining image and position information. The PRECAST model can integrate ST data by evaluating the probability embedding, clustering, and alignment (Liu et al. 2023a), while Spatial ID serves as a supervised cell typing method (Shen et al. 2022a). The deconvolution algorithm has also been improved to infer the cell type composition from ST data (Zhang et al. 2023a; Liao et al. 2022). In terms of cellular interaction, new exploration of CCC through the COMMOT (COMMUNICATION ANALYSIS BY OPTIMAL TRANSPORT) (Cang et al. 2023) and C-SIDE (Cable et al. 2022) methods help infer competition between different ligand and receptor types and analyze cellular interactions between immune cells and tumors in space. Finally, spatial-CITE-seq technology, developed by Liu et al., facilitates spatially resolved protein and full transcriptome collaborative analysis (Liu et al. 2023c). These advancements open up new avenues for ST analysis and offer valuable insights into the complexity of cellular interactions in a spatial context.
4.2.3 Application of deep learning
Deep learning has played a crucial role in improving the application of ST technology and allowed for joint analysis of scRNA-seq, ST, and image data, providing independent prognostic factors for related diseases (Kleino et al. 2022; Li et al. 2022b; Holscher et al. 2023). The scMGCA method is used to identify specific cell types and their different gene expression levels related to cell and tumor-related signaling pathways (Yu et al. 2023). DIST improves gene expression accuracy using deep learning to infer gene expression in low-quality data (Zhao et al. 2023). TIST-net extracts histopathological features by Markov random fields (MRF) and integrates them with location information and transcriptome data to deal with technical noise in analysis tasks (Shan et al. 2022). These advancements in ST have enabled more accurate and comprehensive analysis of cellular and molecular biology, leading to the development of more effective treatments and therapies for various diseases.
4.3 Summary
In summary, ST marks the new beginning of genome measurement, facilitating the exploration of individuals’ dynamic development and disease processes. With the integration of multi-omics technology and the continuous improvement of resolution, ST development will continue to advance along three main directions: dynamics, multi-omics, and resolution (Tian et al. 2023). These innovations in spatiotemporal genomics will provide powerful tools for exploring human organizational structure and functions, expanding the potential applications of ST beyond biology. The future development direction of ST will focus on improving measurement resolution, enhancing multi-omics integration, and exploring dynamics to better understand complex biological phenomena. These advancements hold promise for uncovering new insights into the spatiotemporal context of gene expression, cell proliferation, and interactions, and may ultimately lead to the development of new diagnostic and therapeutic approaches for various diseases.
Abbreviations
- AAV:
-
Adeno-associated viral vectors
- AD:
-
Alzheimer’s disease
- BaSISS:
-
Base-specific in situ sequencing
- BC:
-
Breast cancer
- CAF:
-
Cancer-associated fibroblasts
- CCC:
-
Cell-cell communication
- CCI:
-
Cell-cell interaction
- CNS:
-
Central nervous system
- COMMOT:
-
COMMUNICATION ANALYSIS BY OPTIMAL TRANSPORT
- CRC:
-
Colorectal cancer
- CRLM:
-
CRC liver metastasis
- cSCC:
-
Squamous cell carcinoma
- CST7:
-
Cystatin F
- CVD:
-
Cardiovascular disease
- DCIS:
-
Ductal carcinoma in situ
- DCs:
-
Dendritic cells
- DLBCL:
-
Diffuse large B-cell lymphoma
- DOLs:
-
Disease-associated oligodendrocytes
- EC:
-
Endothelial cell
- GPNMB:
-
Glycoprotein nonmetastatic melanoma protein B
- HCC:
-
Hepatocellular carcinoma
- HCM:
-
Hypertrophic cardiomyopathy
- HLA-G:
-
Human leucocyte antigen G
- iCAF:
-
Inflammatory CAF
- ICBs:
-
Immune checkpoint blockers
- iST:
-
Imaging-based ST
- lncRNA:
-
Long-non-coding RNA
- LUAD:
-
Lung adenocarcinoma
- MBM:
-
Melanoma brain metastasis
- mCAF:
-
Myofibrolast CAF
- MOSTA:
-
Mouse organogenesis spatiotemporal transcriptomic atlas
- MRF:
-
Markov random field
- ncISD:
-
Non-communicable inflammatory skin diseases
- NSCLC:
-
Non-small cell lung cancer
- OSCC:
-
Oral squamous cell carcinoma
- PDAC:
-
Pancreatic ductal adenocarcinoma
- RR:
-
Reverse reaction
- RRST:
-
RNA-Resue Spatial Transcriptions
- scRNA-seq:
-
Single cell RNA sequencing
- SMC:
-
Smooth muscle cell
- snRNA-seq:
-
Single nuclei RNA sequencing
- SODB:
-
Spatial Omics DataBase
- SpaCET:
-
Spatial Cellular Estimator for Tumors
- sST:
-
Sequencing based ST
- ST:
-
Spatial transcriptomics
- STRS:
-
Spatial total RNA sequencing
- TAM:
-
Tumor-associated macrophages
- TIB:
-
Tumor immune barrier
- TIF:
-
Tumor invasion front
- TIME:
-
Tumor immune microenvironment
- TLS:
-
Tertiary lymphoid structure
- TME:
-
Tumor microenvironment
- TSA:
-
Tumor-specific antigen
- TSK:
-
Tumor-specific keratinocyte
- USeqFISH:
-
Ultra-sensitive sequential fluorescence in situ hybridization
References
Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55:1564–80.
Ahmed R, Zaman T, Chowdhury F, Mraiche F, Tariq M, Ahmad IS, Hasan A. Single-cell RNA sequencing with spatial transcriptomics of cancer tissues. Int J Mol Sci. 2022;23(6):3042.
Akhoundova D, Rubin MA. Clinical application of advanced multi-omics tumor profiling: Shaping precision oncology of the future. Cancer Cell. 2022;40:920–38.
Akiyama T, Yasuda T, Uchihara T, Yasuda-Yoshihara N, Tan BJY, Yonemura A, Semba T, Yamasaki J, Komohara Y, Ohnishi K, Wei F, Fu L, Zhang J, Kitamura F, Yamashita K, Eto K, Iwagami S, Tsukamoto H, Umemoto T, Masuda M, Nagano O, Satou Y, Saya H, Tan P, Baba H, Ishimoto T. Stromal reprogramming through dual PDGFRalpha/beta blockade boosts the efficacy of anti-PD-1 immunotherapy in fibrotic tumors. Cancer Res. 2023;83(5):753–70.
Allen WE, Blosser TR, Sullivan ZA, Dulac C, Zhuang X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell. 2023;186:194–208.e18.
Andersson A, Larsson L, Stenbeck L, Salmen F, Ehinger A, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Borg A, Frisen J, Engblom C, Lundeberg J. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun. 2021;12:6012.
Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, Wardell E, Custodio J, Reimegard J, Salmen F, Osterholm C, Stahl PL, Sundstrom E, Akesson E, Bergmann O, Bienko M, Mansson-Broberg A, Nilsson M, Sylven C, Lundeberg J. A Spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179:1647–60.e19.
Asp M, Bergenstrahle J, Lundeberg J. Spatially resolved transcriptomes-next generation tools for tissue exploration. BioEssays. 2020;42:e1900221.
Barkley D, Moncada R, Pour M, Liberman DA, Dryg I, Werba G, Wang W, Baron M, Rao A, Xia B, Franca GS, Weil A, Delair DF, Hajdu C, Lund AW, Osman I, Yanai I. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat Genet. 2022;54:1192–201.
Bassiouni R, Idowu MO, Gibbs LD, Robila V, Grizzard PJ, Webb MG, Song J, Noriega A, Craig DW, Carpten JD. Spatial transcriptomic analysis of a diverse patient cohort reveals a conserved architecture in triple-negative breast cancer. Cancer Res. 2023;83:34–48.
Batiuk MY, Tyler T, Dragicevic K, Mei S, Rydbirk R, Petukhov V, Deviatiiarov R, Sedmak D, Frank E, Feher V, Habek N, Hu Q, Igolkina A, Roszik L, Pfisterer U, Garcia-Gonzalez D, Petanjek Z, Adorjan I, Kharchenko PV, Khodosevich K. Upper cortical layer-driven network impairment in schizophrenia. Sci Adv. 2022;8:eabn8367.
Bergmann S, Penfold CA, Slatery E, Siriwardena D, Drummer C, Clark S, Strawbridge SE, Kishimoto K, Vickers A, Tewary M, Kohler TN, Hollfelder F, Reik W, Sasaki E, Behr R, Boroviak TE. Spatial profiling of early primate gastrulation in utero. Nature. 2022;609:136–43.
Biermann J, Melms JC, Amin AD, Wang Y, Caprio LA, Karz A, Tagore S, Barrera I, Ibarra-Arellano MA, Andreatta M, Fullerton BT, Gretarsson KH, Sahu V, Mangipudy VS, Nguyen TTT, Nair A, Rogava M, Ho P, Koch PD, Banu M, Humala N, Mahajan A, Walsh ZH, Shah SB, Vaccaro DH, Caldwell B, Mu M, Wunnemann F, Chazotte M, Berhe S, Luoma AM, Driver J, Ingham M, Khan SA, Rapisuwon S, Slingluff CL Jr, Eigentler T, Rocken M, Carvajal R, Atkins MB, Davies MA, Agustinus A, Bakhoum SF, Azizi E, Siegelin M, Lu C, Carmona SJ, Hibshoosh H, Ribas A, Canoll P, Bruce JN, Bi WL, Agrawal P, Schapiro D, Hernando E, Macosko EZ, Chen F, Schwartz GK, Izar B. Dissecting the treatment-naive ecosystem of human melanoma brain metastasis. Cell. 2022;185:2591–608.e30.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50.
Bondareva O, Rodriguez-Aguilera JR, Oliveira F, Liao L, Rose A, Gupta A, Singh K, Geier F, Schuster J, Boeckel JN, Buescher JM, Kohli S, Kloting N, Isermann B, Bluher M, Sheikh BN. Single-cell profiling of vascular endothelial cells reveals progressive organ-specific vulnerabilities during obesity. Nat Metab. 2022;4:1591–610.
Cable DM, Murray E, Shanmugam V, Zhang S, Zou LS, Diao M, Chen H, Macosko EZ, Irizarry RA, Chen F. Cell type-specific inference of differential expression in spatial transcriptomics. Nat Methods. 2022;19:1076–87.
Cang Z, Zhao Y, Almet AA, Stabell A, Ramos R, Plikus MV, Atwood SX, Nie Q. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat Methods. 2023;20(2):218–28.
Carlberg K, Korotkova M, Larsson L, Catrina AI, Stahl PL, Malmstrom V. Exploring inflammatory signatures in arthritic joint biopsies with Spatial Transcriptomics. Sci Rep. 2019;9:18975.
Castranio EL, Hasel P, Haure-Mirande JV, Ramirez Jimenez AV, Hamilton BW, Kim RD, Glabe CG, Wang M, Zhang B, Gandy S, Liddelow SA, Ehrlich ME. Microglial INPP5D limits plaque formation and glial reactivity in the PSAPP mouse model of Alzheimer’s disease. Alzheimers Dement. 2023;19(6):2239–52.
Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, Qiu X, Yang J, Xu J, Hao S, Wang X, Lu H, Chen X, Liu X, Huang X, Li Z, Hong Y, Jiang Y, Peng J, Liu S, Shen M, Liu C, Li Q, Yuan Y, Wei X, Zheng H, Feng W, Wang Z, Liu Y, Wang Z, Yang Y, Xiang H, Han L, Qin B, Guo P, Lai G, Munoz-Canoves P, Maxwell PH, Thiery JP, Wu QF, Zhao F, Chen B, Li M, Dai X, Wang S, Kuang H, Hui J, Wang L, Fei JF, Wang O, Wei X, Lu H, Wang B, Liu S, Gu Y, Ni M, Zhang W, Mu F, Yin Y, Yang H, Lisby M, Cornall RJ, Mulder J, Uhlen M, Esteban MA, Li Y, Liu L, Xu X, Wang J. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022a;185:1777–92.e21.
Chen J, Liu W, Luo T, Yu Z, Jiang M, Wen J, Gupta GP, Giusti P, Zhu H, Yang Y, Li Y. A comprehensive comparison on cell-type composition inference for spatial transcriptomics data. Brief Bioinform. 2022b;23(4):bbac245.
Chen S, Chang Y, Li L, Acosta D, Li Y, Guo Q, Wang C, Turkes E, Morrison C, Julian D, Hester ME, Scharre DW, Santiskulvong C, Song SX, Plummer JT, Serrano GE, Beach TG, Duff KE, Ma Q, Fu H. Spatially resolved transcriptomics reveals genes associated with the vulnerability of middle temporal gyrus in Alzheimer’s disease. Acta Neuropathol Commun. 2022c;10:188.
Colombo AR, Hav M, Singh M, Xu A, Gamboa A, Lemos T, Gerdtsson E, Chen D, Houldsworth J, Shaknovich R, Aoki T, Chong L, Takata K, Chavez EA, Steidl C, Hicks J, Kuhn P, Siddiqi I, Merchant A. Single-cell spatial analysis of tumor immune architecture in diffuse large B-cell lymphoma. Blood Adv. 2022;6:4675–90.
Cortese N, Carriero R, Barbagallo M, Putignano AR, Costa G, Giavazzi F, Grizzi F, Pasqualini F, Peano C, Basso G, Marchini S, Colombo FS, Soldani C, Franceschini B, Di Tommaso L, Terracciano L, Donadon M, Torzilli G, Kunderfranco P, Mantovani A, Marchesi F. High-resolution analysis of mononuclear phagocytes reveals GPNMB as a prognostic marker in human colorectal liver metastasis. Cancer Immunol Res. 2023;11(4):405–20.
Cross AR, de Andrea CE, Villalba-Esparza M, Landecho MF, Cerundolo L, Weeratunga P, Etherington RE, Denney L, Ogg G, Ho LP, Roberts IS, Hester J, Klenerman P, Melero I, Sansom SN, Issa F. Spatial transcriptomic characterization of COVID-19 pneumonitis identifies immune circuits related to tissue injury. JCI Insight. 2023;8(2):e157837.
Cui G, Feng S, Yan Y, Wang L, He X, Li X, Duan Y, Chen J, Tang K, Zheng P, Tam PPL, Si W, Jing N, Peng G. Spatial molecular anatomy of germ layers in the gastrulating cynomolgus monkey embryo. Cell Rep. 2022;40:111285.
Cui Zhou D, Jayasinghe RG, Chen S, Herndon JM, Iglesia MD, Navale P, Wendl MC, Caravan W, Sato K, Storrs E, Mo CK, Liu J, Southard-Smith AN, Wu Y, Naser Al Deen N, Baer JM, Fulton RS, Wyczalkowski MA, Liu R, Fronick CC, Fulton LA, Shinkle A, Thammavong L, Zhu H, Sun H, Wang LB, Li Y, Zuo C, McMichael JF, Davies SR, Appelbaum EL, Robbins KJ, Chasnoff SE, Yang X, Reeb AN, Oh C, Serasanambati M, Lal P, Varghese R, Mashl JR, Ponce J, Terekhanova NV, Yao L, Wang F, Chen L, Schnaubelt M, Lu RJ, Schwarz JK, Puram SV, Kim AH, Song SK, Shoghi KI, Lau KS, Ju T, Chen K, Chatterjee D, Hawkins WG, Zhang H, Achilefu S, Chheda MG, Oh ST, Gillanders WE, Chen F, DeNardo DG, Fields RC, Ding L. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat Genet. 2022;54:1390–405.
Deng Y, Bartosovic M, Ma S, Zhang D, Kukanja P, Xiao Y, Su G, Liu Y, Qin X, Rosoklija GB, Dwork AJ, Mann JJ, Xu ML, Halene S, Craft JE, Leong KW, Boldrini M, Castelo-Branco G, Fan R. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature. 2022;609:375–83.
Elmentaite R, Dominguez Conde C, Yang L, Teichmann SA. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat Rev Genet. 2022;23:395–410.
Fang Q, Cai H, Jiang P, Zhao H, Song Y, Zhao W, Yu Y, Zhu J. Transcriptional substrates of brain structural and functional impairments in drug-naive first-episode patients with major depressive disorder. J Affect Disord. 2023a;325:522–33.
Fang S, Chen B, Zhang Y, Sun H, Liu L, Liu S, Li Y, Xu X. Computational approaches and challenges in spatial transcriptomics. Genomics Proteomics Bioinformatics.2023b;21(1):24–47.
Fawkner-Corbett D, Antanaviciute A, Parikh K, Jagielowicz M, Geros AS, Gupta T, Ashley N, Khamis D, Fowler D, Morrissey E, Cunningham C, Johnson PRV, Koohy H, Simmons A. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell. 2021;184:810–26.e23.
Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, Burcham AR, Salhotra A, Ransom RC, Henn D, Chen K, Mascharak S, Tolentino K, Titan AL, Jones RE, da Silva O, Leavitt WT, Marshall CD, des Jardins-Park HE, Hu MS, Wan DC, Wernig G, Wagh D, Coller J, Norton JA, Gurtner GC, Newman AM, Chang HY, Longaker MT. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc Natl Acad Sci U S A. 2021;118(41):e2110025118.
Fu X, Sun L, Dong R, Chen JY, Silakit R, Condon LF, Lin Y, Lin S, Palmiter RD, Gu L. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell. 2022;185:4621–33.e17.
Galeano Nino JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, Futran N, Houlton J, Sather C, Sicinska E, Taylor A, Minot SS, Johnston CD, Bullman S. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. 2022;611:810–7.
Garcia-Alonso L, Lorenzi V, Mazzeo CI, Alves-Lopes JP, Roberts K, Sancho-Serra C, Engelbert J, Mareckova M, Gruhn WH, Botting RA, Li T, Crespo B, van Dongen S, Kiselev VY, Prigmore E, Herbert M, Moffett A, Chedotal A, Bayraktar OA, Surani A, Haniffa M, Vento-Tormo R. Single-cell roadmap of human gonadal development. Nature. 2022;607:540–7.
Gurtner A, Borrelli C, Gonzalez-Perez I, Bach K, Acar IE, Nunez NG, Crepaz D, Handler K, Vu VP, Lafzi A, Stirm K, Raju D, Gschwend J, Basler K, Schneider C, Slack E, Valenta T, Becher B, Krebs P, Moor AE, Arnold IC. Active eosinophils regulate host defence and immune responses in colitis. Nature. 2023;615(7950):151–7.
Haase C, Gustafsson K, Mei S, Yeh SC, Richter D, Milosevic J, Turcotte R, Kharchenko PV, Sykes DB, Scadden DT, Lin CP. Image-seq: spatially resolved single-cell sequencing guided by in situ and in vivo imaging. Nat Methods. 2022;19:1622–33.
Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, Khair Z, Verberk SGS, Baranski A, Baskar R, Graf W, Van Valen D, Van den Bossche J, Angelo M, Bendall SC. Single-cell metabolic profiling of human cytotoxic T cells. Nat Biotechnol. 2021;39:186–97.
He P, Lim K, Sun D, Pett JP, Jeng Q, Polanski K, Dong Z, Bolt L, Richardson L, Mamanova L, Dabrowska M, Wilbrey-Clark A, Madissoon E, Tuong ZK, Dann E, Suo C, Goh I, Yoshida M, Nikolic MZ, Janes SM, He X, Barker RA, Teichmann SA, Marioni JC, Meyer KB, Rawlins EL. A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell. 2022;185:4841–60.e25.
Heide T, Househam J, Cresswell GD, Spiteri I, Lynn C, Mossner M, Kimberley C, Fernandez-Mateos J, Chen B, Zapata L, James C, Barozzi I, Chkhaidze K, Nichol D, Gunasri V, Berner A, Schmidt M, Lakatos E, Baker AM, Costa H, Mitchinson M, Piazza R, Jansen M, Caravagna G, Ramazzotti D, Shibata D, Bridgewater J, Rodriguez-Justo M, Magnani L, Graham TA, Sottoriva A. The co-evolution of the genome and epigenome in colorectal cancer. Nature. 2022;611:733–43.
Heming M, Haessner S, Wolbert J, Lu IN, Li X, Brokinkel B, Muther M, Holling M, Stummer W, Thomas C, Schulte-Mecklenbeck A, de Faria F, Stoeckius M, Hailfinger S, Lenz G, Kerl K, Wiendl H, Meyer Zu Horste G, Grauer OM. Intratumor heterogeneity and T cell exhaustion in primary CNS lymphoma. Genome Med. 2022;14:109.
Holscher DL, Bouteldja N, Joodaki M, Russo ML, Lan YC, Sadr AV, Cheng M, Tesar V, Stillfried SV, Klinkhammer BM, Barratt J, Floege J, Roberts ISD, Coppo R, Costa IG, Bulow RD, Boor P. Next-Generation Morphometry for pathomics-data mining in histopathology. Nat Commun. 2023;14:470.
Househam J, Heide T, Cresswell GD, Spiteri I, Kimberley C, Zapata L, Lynn C, James C, Mossner M, Fernandez-Mateos J, Vinceti A, Baker AM, Gabbutt C, Berner A, Schmidt M, Chen B, Lakatos E, Gunasri V, Nichol D, Costa H, Mitchinson M, Ramazzotti D, Werner B, Iorio F, Jansen M, Caravagna G, Barnes CP, Shibata D, Bridgewater J, Rodriguez-Justo M, Magnani L, Sottoriva A, Graham TA. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature. 2022;611:744–53.
Hsieh WC, Budiarto BR, Wang YF, Lin CY, Gwo MC, So DK, Tzeng YS, Chen SY. Spatial multi-omics analyses of the tumor immune microenvironment. J Biomed Sci. 2022;29:96.
Hu B, Lelek S, Spanjaard B, El-Sammak H, Simoes MG, Mintcheva J, Aliee H, Schafer R, Meyer AM, Theis F, Stainier DYR, Panakova D, Junker JP. Origin and function of activated fibroblast states during zebrafish heart regeneration. Nat Genet. 2022;54:1227–37.
Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, Mohan R, Drokhlyansky E, Van Wittenberghe N, Ashenberg O, Farhi SL, Schapiro D, Divakar P, Miller E, Zollinger DR, Eng G, Schenkel JM, Su J, Shiau C, Yu P, Freed-Pastor WA, Abbondanza D, Mehta A, Gould J, Lambden C, Porter CBM, Tsankov A, Dionne D, Waldman J, Cuoco MS, Nguyen L, Delorey T, Phillips D, Barth JL, Kem M, Rodrigues C, Ciprani D, Roldan J, Zelga P, Jorgji V, Chen JH, Ely Z, Zhao D, Fuhrman K, Fropf R, Beechem JM, Loeffler JS, Ryan DP, Weekes CD, Ferrone CR, Qadan M, Aryee MJ, Jain RK, Neuberg DS, Wo JY, Hong TS, Xavier R, Aguirre AJ, Rozenblatt-Rosen O, Mino-Kenudson M, Castillo CF, Liss AS, Ting DT, Jacks T, Regev A. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet. 2022;54:1178–91.
Iturria-Medina Y, Adewale Q, Khan AF, Ducharme S, Rosa-Neto P, O'Donnell K, Petyuk VA, Gauthier S, De Jager PL, Breitner J, Bennett DA. Unified epigenomic, transcriptomic, proteomic, and metabolomic taxonomy of Alzheimer's disease progression and heterogeneity. Sci Adv. 2022;8(46):eabo6764.
Jang MJ, Coughlin GM, Jackson CR, Chen X, Chuapoco MR, Vendemiatti JL, Wang AZ, Gradinaru V. Spatial transcriptomics for profiling the tropism of viral vectors in tissues. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-022-01648-w.
Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstrahle J, Larsson L, Bai Y, Zhu B, Bhaduri A, Meyers JM, Rovira-Clave X, Hollmig ST, Aasi SZ, Nolan GP, Lundeberg J, Khavari PA. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell. 2020;182:497–514.e22.
Jung SH, Hwang BH, Shin S, Park EH, Park SH, Kim CW, Kim E, Choo E, Choi IJ, Swirski FK, Chang K, Chung YJ. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2(hi) macrophages in infarcted hearts. Nat Commun. 2022;13:4580.
Karamitopoulou E, Wenning AS, Acharjee A, Zlobec I, Aeschbacher P, Perren A, Gloor B. Spatially restricted tumour-associated and host-associated immune drivers correlate with the recurrence sites of pancreatic cancer. Gut. 2023;72(8):1523–153.
Karras P, Bordeu I, Pozniak J, Nowosad A, Pazzi C, Van Raemdonck N, Landeloos E, Van Herck Y, Pedri D, Bervoets G, Makhzami S, Khoo JH, Pavie B, Lamote J, Marin-Bejar O, Dewaele M, Liang H, Zhang X, Hua Y, Wouters J, Browaeys R, Bergers G, Saeys Y, Bosisio F, van den Oord J, Lambrechts D, Rustgi AK, Bechter O, Blanpain C, Simons BD, Rambow F, Marine JC. A cellular hierarchy in melanoma uncouples growth and metastasis. Nature. 2022;610:190–8.
Kathe C, Skinnider MA, Hutson TH, Regazzi N, Gautier M, Demesmaeker R, Komi S, Ceto S, James ND, Cho N, Baud L, Galan K, Matson KJE, Rowald A, Kim K, Wang R, Minassian K, Prior JO, Asboth L, Barraud Q, Lacour SP, Levine AJ, Wagner F, Bloch J, Squair JW, Courtine G. The neurons that restore walking after paralysis. Nature. 2022;611:540–7.
Kenigsbuch M, Bost P, Halevi S, Chang Y, Chen S, Ma Q, Hajbi R, Schwikowski B, Bodenmiller B, Fu H, Schwartz M, Amit I. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat Neurosci. 2022;25:876–86.
Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R, Bendall SC, Angelo M. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174:1373–87.e19.
Kishi JY, Liu N, West ER, Sheng K, Jordanides JJ, Serrata M, Cepko CL, Saka SK, Yin P. Light-Seq: light-directed in situ barcoding of biomolecules in fixed cells and tissues for spatially indexed sequencing. Nat Methods. 2022;19:1393–402.
Kleino I, Frolovaite P, Suomi T, Elo LL. Computational solutions for spatial transcriptomics. Comput Struct Biotechnol J. 2022;20:4870–84.
Krausgruber T, Redl A, Barreca D, Doberer K, Romanovskaia D, Dobnikar L, Guarini M, Unterluggauer L, Kleissl L, Atzmuller D, Mayerhofer C, Kopf A, Saluzzo S, Lim CX, Rexie P, Weichhart T, Bock C, Stary G. Single-cell and spatial transcriptomics reveal aberrant lymphoid developmental programs driving granuloma formation. Immunity. 2023;56:289–306.e7.
Kulasinghe A, Tan CW, Ribeiro Dos Santos Miggiolaro AF, Monkman J, SadeghiRad H, Bhuva DD, Motta Junior JDS, Busatta Vaz de Paula C, Nagashima S, Baena CP, Souza-Fonseca-Guimaraes P, de Noronha L, McCulloch T, Rossi GR, Cooper C, Tang B, Short KR, Davis MJ, Souza-Fonseca-Guimaraes F, Belz GT, O’Byrne K. Profiling of lung SARS-CoV-2 and influenza virus infection dissects virus-specific host responses and gene signatures. Eur Respir J. 2022;59(6):2101881.
Kuppe C, Ramirez Flores RO, Li Z, Hayat S, Levinson RT, Liao X, Hannani MT, Tanevski J, Wunnemann F, Nagai JS, Halder M, Schumacher D, Menzel S, Schafer G, Hoeft K, Cheng M, Ziegler S, Zhang X, Peisker F, Kaesler N, Saritas T, Xu Y, Kassner A, Gummert J, Morshuis M, Amrute J, Veltrop RJA, Boor P, Klingel K, Van Laake LW, Vink A, Hoogenboezem RM, Bindels EMJ, Schurgers L, Sattler S, Schapiro D, Schneider RK, Lavine K, Milting H, Costa IG, Saez-Rodriguez J, Kramann R. Spatial multi-omic map of human myocardial infarction. Nature. 2022;608:766–77.
Lee J, Yoo M, Choi J. Recent advances in spatially resolved transcriptomics: challenges and opportunities. BMB Rep. 2022;55:113–24.
Lei Y, Cheng M, Li Z, Zhuang Z, Wu L, Sun Y, Han L, Huang Z, Wang Y, Wang Z, Xu L, Yuan Y, Liu S, Pan T, Xie J, Liu C, Volpe G, Ward C, Lai Y, Xu J, Wang M, Yu H, Sun H, Yu Q, Wu L, Wang C, Wong CW, Liu W, Xu L, Wei J, Chen D, Shang Z, Li G, Ma K, Cheng L, Ling F, Tan T, Chen K, Tasic B, Dean M, Ji W, Yang H, Gu Y, Esteban MA, Li Y, Chen A, Niu Y, Zeng H, Hou Y, Liu L, Liu S, Xu X. Spatially resolved gene regulatory and disease-related vulnerability map of the adult Macaque cortex. Nat Commun. 2022;13:6747.
Lewis SM, Asselin-Labat ML, Nguyen Q, Berthelet J, Tan X, Wimmer VC, Merino D, Rogers KL, Naik SH. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods. 2021;18:997–1012.
Li Q, Zhang X, Ke R. Spatial transcriptomics for tumor heterogeneity analysis. Front Genet. 2022a;13:906158.
Li Y, Stanojevic S, Garmire LX. Emerging artificial intelligence applications in Spatial Transcriptomics analysis. Comput Struct Biotechnol J. 2022b;20:2895–908.
Liao J, Qian J, Fang Y, Chen Z, Zhuang X, Zhang N, Shao X, Hu Y, Yang P, Cheng J, Hu Y, Yu L, Yang H, Zhang J, Lu X, Shao L, Wu D, Gao Y, Chen H, Fan X. De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolution. Nat Commun. 2022;13:6498.
Lin JP, Kelly HM, Song Y, Kawaguchi R, Geschwind DH, Jacobson S, Reich DS. Transcriptomic architecture of nuclei in the marmoset CNS. Nat Commun. 2022;13:5531.
Lin JR, Wang S, Coy S, Chen YA, Yapp C, Tyler M, Nariya MK, Heiser CN, Lau KS, Santagata S, Sorger PK. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell. 2023a;186:363–81.e19.
Lin PB, Tsai AP, Soni D, Lee-Gosselin A, Moutinho M, Puntambekar SS, Landreth GE, Lamb BT, Oblak AL. INPP5D deficiency attenuates amyloid pathology in a mouse model of Alzheimer’s disease. Alzheimers Dement. 2023b;19(6):2528–37.
Liu J, Ma P, Lai L, Villanueva A, Koenig A, Bean GR, Bowles DE, Glass C, Watson M, Lavine KJ, Lin CY. Transcriptional and immune landscape of cardiac sarcoidosis. Circ Res. 2022;131:654–69.
Liu W, Liao X, Luo Z, Yang Y, Lau MC, Jiao Y, Shi X, Zhai W, Ji H, Yeong J, Liu J. Probabilistic embedding, clustering, and alignment for integrating spatial transcriptomics data with PRECAST. Nat Commun. 2023a;14:296.
Liu X, Yin K, Chen L, Chen W, Li W, Zhang T, Sun Y, Yuan M, Wang H, Song Y, Wang S, Hu S, Zhou Z. Lineage-specific regulatory changes in hypertrophic cardiomyopathy unraveled by single-nucleus RNA-seq and spatial transcriptomics. Cell Discov. 2023b;9:6.
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, Sun L, Liu Y, Du Y, Guo X, Cui T, Zhou H, Wang J, Yin D, Song R, Zhang S, Cai W, Meng F, Guo H, Zhang B, Yang D, Bao R, Hu Q, Wang J, Ye Y, Liu L. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023d;78(4):770–82.
Liu Y, DiStasio M, Su G, Asashima H, Enninful A, Qin X, Deng Y, Nam J, Gao F, Bordignon P, Cassano M, Tomayko M, Xu M, Halene S, Craft JE, Hafler D, Fan R. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat Biotechnol. 2023c. https://doi.org/10.1038/s41587-023-01676-0.
Lomakin A, Svedlund J, Strell C, Gataric M, Shmatko A, Rukhovich G, Park JS, Ju YS, Dentro S, Kleshchevnikov V, Vaskivskyi V, Li T, Bayraktar OA, Pinder S, Richardson AL, Santagata S, Campbell PJ, Russnes H, Gerstung M, Nilsson M, Yates LR. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature. 2022;611:594–602.
Luquez T, Gaur P, Kosater IM, Lam M, Lee DI, Mares J, Paryani F, Yadav A, Menon V. Cell type-specific changes identified by single-cell transcriptomics in Alzheimer’s disease. Genome Med. 2022;14:136.
Ma F, Hughes TK, Teles RMB, Andrade PR, de Andrade Silva BJ, Plazyo O, Tsoi LC, Do T, Wadsworth MH 2nd, Oulee A, Ochoa MT, Sarno EN, Iruela-Arispe ML, Klechevsky E, Bryson B, Shalek AK, Bloom BR, Gudjonsson JE, Pellegrini M, Modlin RL. The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat Immunol. 2021;22:839–50.
Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N, Ribeiro Orsi A, Mamanova L, Bolt L, Elmentaite R, Pett JP, Huang N, Xu C, He P, Dabrowska M, Pritchard S, Tuck L, Prigmore E, Perera S, Knights A, Oszlanczi A, Hunter A, Vieira SF, Patel M, Lindeboom RGH, Campos LS, Matsuo K, Nakayama T, Yoshida M, Worlock KB, Nikolic MZ, Georgakopoulos N, Mahbubani KT, Saeb-Parsy K, Bayraktar OA, Clatworthy MR, Stegle O, Kumasaka N, Teichmann SA, Meyer KB. A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat Genet. 2023;55(1):66–77.
Marx V. Method of the year: long-read sequencing. Nat Methods. 2023;20:6–11.
Maynard KR, Jaffe AE, Martinowich K. Spatial transcriptomics: putting genome-wide expression on the map. Neuropsychopharmacology. 2020;45:232–3.
McKellar DW, Mantri M, Hinchman MM, Parker JSL, Sethupathy P, Cosgrove BD, De Vlaminck I. Spatial mapping of the total transcriptome by in situ polyadenylation. Nat Biotechnol. 2023;41(4):513–20.
Melo Ferreira R, Freije BJ, Eadon MT. Deconvolution tactics and normalization in renal spatial transcriptomics. Front Physiol. 2021;12:812947.
Melo Ferreira R, Gisch DL, Eadon MT. Spatial transcriptomics and the kidney. Curr Opin Nephrol Hypertens. 2022;31:244–50.
Merritt CR, Ong GT, Church SE, Barker K, Danaher P, Geiss G, Hoang M, Jung J, Liang Y, McKay-Fleisch J, Nguyen K, Norgaard Z, Sorg K, Sprague I, Warren C, Warren S, Webster PJ, Zhou Z, Zollinger DR, Dunaway DL, Mills GB, Beechem JM. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol. 2020;38:586–99.
Meylan M, Petitprez F, Becht E, Bougouin A, Pupier G, Calvez A, Giglioli I, Verkarre V, Lacroix G, Verneau J, Sun CM, Laurent-Puig P, Vano YA, Elaidi R, Mejean A, Sanchez-Salas R, Barret E, Cathelineau X, Oudard S, Reynaud CA, de Reynies A, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55:527–41.e5.
Miranda AMA, Janbandhu V, Maatz H, Kanemaru K, Cranley J, Teichmann SA, Hubner N, Schneider MD, Harvey RP, Noseda M. Single-cell transcriptomics for the assessment of cardiac disease. Nat Rev Cardiol. 2023;20(5):289–308.
Mirzazadeh R, Andrusivova Z, Larsson L, Newton PT, Galicia LA, Abalo XM, Avijgan M, Kvastad L, Denadai-Souza A, Stakenborg N, Firsova AB, Shamikh A, Jurek A, Schultz N, Nister M, Samakovlis C, Boeckxstaens G, Lundeberg J. Spatially resolved transcriptomic profiling of degraded and challenging fresh frozen samples. Nat Commun. 2023;14:509.
Moffitt JR, Lundberg E, Heyn H. The emerging landscape of spatial profiling technologies. Nat Rev Genet. 2022;23:741–59.
Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, Hajdu CH, Simeone DM, Yanai I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38:333–42.
Moses L, Pachter L. Museum of spatial transcriptomics. Nat Methods. 2022;19:534–46.
Mothes R, Pascual-Reguant A, Koehler R, Liebeskind J, Liebheit A, Bauherr S, Philipsen L, Dittmayer C, Laue M, von Manitius R, Elezkurtaj S, Durek P, Heinrich F, Heinz GA, Guerra GM, Obermayer B, Meinhardt J, Ihlow J, Radke J, Heppner FL, Enghard P, Stockmann H, Aschman T, Schneider J, Corman VM, Sander LE, Mashreghi MF, Conrad T, Hocke AC, Niesner RA, Radbruch H, Hauser AE. Distinct tissue niches direct lung immunopathology via CCL18 and CCL21 in severe COVID-19. Nat Commun. 2023;14:791.
Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, Schwab N, Henkel M, Frank A, Zsikla V, Willi N, Kempf W, Hoyler T, Barbareschi M, Moch H, Tolnay M, Cathomas G, Demichelis F, Junt T, Mertz KD. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat Commun. 2020;11:5086.
Noel T, Wang QS, Greka A, Marshall JL. Principles of spatial transcriptomics analysis: a practical walk-through in kidney tissue. Front Physiol. 2021;12:809346.
Olaniru OE, Kadolsky U, Kannambath S, Vaikkinen H, Fung K, Dhami P, Persaud SJ. Single-cell transcriptomic and spatial landscapes of the developing human pancreas. Cell Metab. 2023;35:184–99.e5.
Ozato Y, Kojima Y, Kobayashi Y, Hisamatsu Y, Toshima T, Yonemura Y, Masuda T, Kagawa K, Goto Y, Utou M, Fukunaga M, Gamachi A, Imamura K, Kuze Y, Zenkoh J, Suzuki A, Niida A, Hirose H, Hayashi S, Koseki J, Oki E, Fukuchi S, Murakami K, Tobo T, Nagayama S, Uemura M, Sakamoto T, Oshima M, Doki Y, Eguchi H, Mori M, Iwasaki T, Oda Y, Shibata T, Suzuki Y, Shimamura T, Mimori K. Spatial and single-cell transcriptomics decipher the cellular environment containing HLA-G+ cancer cells and SPP1+ macrophages in colorectal cancer. Cell Rep. 2023;42:111929.
Pineiro AJ, Houser AE, Ji AL. Research techniques made simple: spatial transcriptomics. J Invest Dermatol. 2022;142:993–1001.e1.
Quintana JF, Chandrasegaran P, Sinton MC, Briggs EM, Otto TD, Heslop R, Bentley-Abbot C, Loney C, de Lecea L, Mabbott NA, MacLeod A. Single cell and spatial transcriptomic analyses reveal microglia-plasma cell crosstalk in the brain during Trypanosoma brucei infection. Nat Commun. 2022;13:5752.
Rabbani M, Zheng X, Manske GL, Vargo A, Shami AN, Li JZ, Hammoud SS. Decoding the spermatogenesis program: new insights from transcriptomic analyses. Annu Rev Genet. 2022;56:339–68.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, Macosko EZ. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7.
Roelands J, van der Ploeg M, Ijsselsteijn ME, Dang H, Boonstra JJ, Hardwick JCH, Hawinkels L, Morreau H, de Miranda NF. Transcriptomic and immunophenotypic profiling reveals molecular and immunological hallmarks of colorectal cancer tumourigenesis. Gut. 2023;72(7):1326–39.
Ru B, Huang J, Zhang Y, Aldape K, Jiang P. Estimation of cell lineages in tumors from spatial transcriptomics data. Nat Commun. 2023;14:568.
Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–25.
Schabitz A, Hillig C, Mubarak M, Jargosch M, Farnoud A, Scala E, Kurzen N, Pilz AC, Bhalla N, Thomas J, Stahle M, Biedermann T, Schmidt-Weber CB, Theis F, Garzorz-Stark N, Eyerich K, Menden MP, Eyerich S. Spatial transcriptomics landscape of lesions from non-communicable inflammatory skin diseases. Nat Commun. 2022;13:7729.
Seferbekova Z, Lomakin A, Yates LR, Gerstung M. Spatial biology of cancer evolution. Nat Rev Genet. 2022.
Shan Y, Zhang Q, Guo W, Wu Y, Miao Y, Xin H, Lian Q, Gu J. TIST: transcriptome and histopathological image integrative analysis for spatial transcriptomics. Genomics Proteomics Bioinformatics. 2022;20(5):974–88.
Shen R, Liu L, Wu Z, Zhang Y, Yuan Z, Guo J, Yang F, Zhang C, Chen B, Feng W, Liu C, Guo J, Fan G, Zhang Y, Li Y, Xu X, Yao J. Spatial-ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding. Nat Commun. 2022a;13:7640.
Shen X, Zhao Y, Wang Z, Shi Q. Recent advances in high-throughput single-cell transcriptomics and spatial transcriptomics. Lab Chip. 2022b;22:4774–91.
Sountoulidis A, Marco Salas S, Braun E, Avenel C, Bergenstrahle J, Theelke J, Vicari M, Czarnewski P, Liontos A, Abalo X, Andrusivova Z, Mirzazadeh R, Asp M, Li X, Hu L, Sariyar S, Martinez Casals A, Ayoglu B, Firsova A, Michaelsson J, Lundberg E, Wahlby C, Sundstrom E, Linnarsson S, Lundeberg J, Nilsson M, Samakovlis C. A topographic atlas defines developmental origins of cell heterogeneity in the human embryonic lung. Nat Cell Biol. 2023;25(2):351–65.
Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg A, Ponten F, Costea PI, Sahlen P, Mulder J, Bergmann O, Lundeberg J, Frisen J. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82.
Staines HM, Kirwan DE, Clark DJ, Adams ER, Augustin Y, Byrne RL, Cocozza M, Cubas-Atienzar AI, Cuevas LE, Cusinato M, Davies BMO, Davis M, Davis P, Duvoix A, Eckersley NM, Forton D, Fraser AJ, Garrod G, Hadcocks L, Hu Q, Johnson M, Kay GA, Klekotko K, Lewis Z, Macallan DC, Mensah-Kane J, Menzies S, Monahan I, Moore CM, Nebe-von-Caron G, Owen SI, Sainter C, Sall AA, Schouten J, Williams CT, Wilkins J, Woolston K, Fitchett JRA, Krishna S, Planche T. IgG seroconversion and pathophysiology in severe acute respiratory syndrome coronavirus 2 infection. Emerg Infect Dis. 2021;27:85–91.
Tian L, Chen F, Macosko EZ. The expanding vistas of spatial transcriptomics. Nat Biotechnol. 2023;41(6):773–82.
Topchyan P, Zander R, Kasmani MY, Nguyen C, Brown A, Lin S, Burns R, Cui W. Spatial transcriptomics demonstrates the role of CD4 T cells in effector CD8 T cell differentiation during chronic viral infection. Cell Rep. 2022;41:111736.
Uzquiano A, Kedaigle AJ, Pigoni M, Paulsen B, Adiconis X, Kim K, Faits T, Nagaraja S, Anton-Bolanos N, Gerhardinger C, Tucewicz A, Murray E, Jin X, Buenrostro J, Chen F, Velasco S, Regev A, Levin JZ, Arlotta P. Proper acquisition of cell class identity in organoids allows definition of fate specification programs of the human cerebral cortex. Cell. 2022;185:3770–88.e27.
Van de Velde LA, Allen EK, Crawford JC, Wilson TL, Guy CS, Russier M, Zeitler L, Bahrami A, Finkelstein D, Pelletier S, Schultz-Cherry S, Thomas PG, Murray PJ. Neuroblastoma formation requires unconventional CD4 T cells and arginase-1-dependent myeloid cells. Cancer Res. 2021;81:5047–59.
Vandereyken K, Sifrim A, Thienpont B, Voet T. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet. 2023;24(8):494–515.
Walker BL, Cang Z, Ren H, Bourgain-Chang E, Nie Q. Deciphering tissue structure and function using spatial transcriptomics. Commun Biol. 2022;5:220.
Wang Y, Song B, Wang S, Chen M, Xie Y, Xiao G, Wang L, Wang T. Sprod for de-noising spatially resolved transcriptomics data based on position and image information. Nat Methods. 2022;19:950–8.
Wang R, Peng G, Tam PPL, Jing N. Integration of computational analysis and spatial transcriptomics in single-cell study. Genomics Proteomics Bioinformatics. 2023;21(1):13–23.
Watanabe R, Miura N, Kurata M, Kitazawa R, Kikugawa T, Saika T. Spatial gene expression analysis reveals characteristic gene expression patterns of de novo neuroendocrine prostate cancer coexisting with androgen receptor pathway prostate cancer. Int J Mol Sci. 2023;24(10):8955.
Wei X, Fu S, Li H, Liu Y, Wang S, Feng W, Yang Y, Liu X, Zeng YY, Cheng M, Lai Y, Qiu X, Wu L, Zhang N, Jiang Y, Xu J, Su X, Peng C, Han L, Lou WP, Liu C, Yuan Y, Ma K, Yang T, Pan X, Gao S, Chen A, Esteban MA, Yang H, Wang J, Fan G, Liu L, Chen L, Xu X, Fei JF, Gu Y. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science. 2022;377:eabp9444.
Williams CG, Lee HJ, Asatsuma T, Vento-Tormo R, Haque A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022;14:68.
Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N, Wang T, Larsson L, Kaczorowski D, Weisenfeld NI, Uytingco CR, Chew JG, Bent ZW, Chan CL, Gnanasambandapillai V, Dutertre CA, Gluch L, Hui MN, Beith J, Parker A, Robbins E, Segara D, Cooper C, Mak C, Chan B, Warrier S, Ginhoux F, Millar E, Powell JE, Williams SR, Liu XS, O’Toole S, Lim E, Lundeberg J, Perou CM, Swarbrick A. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53:1334–47.
Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, Cheng Y, Huang S, Liu Y, Jiang S, Liu J, Huang X, Wang X, Qiu S, Xu J, Xi R, Bai F, Zhou J, Fan J, Zhang X, Gao Q. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. 2022;12:134–53.
Xu K, Jin X, Luo Y, Zou H, Lv D, Wang L, Fu L, Cai Y, Shao T, Li Y, Xu J. Spatial transcriptome analysis of long non-coding RNAs reveals tissue specificity and functional roles in cancer. J Zhejiang Univ Sci B. 2023;24:15–31.
Yu Q, Jiang M, Wu L. Spatial transcriptomics technology in cancer research. Front Oncol. 2022;12:1019111.
Yu Z, Su Y, Lu Y, Yang Y, Wang F, Zhang S, Chang Y, Wong KC, Li X. Topological identification and interpretation for single-cell gene regulation elucidation across multiple platforms using scMGCA. Nat Commun. 2023;14:400.
Yuan Z, Pan W, Zhao X, Zhao F, Xu Z, Li X, Zhao Y, Zhang MQ, Yao J. SODB facilitates comprehensive exploration of spatial omics data. Nat Methods. 2023;20(3):387–99.
Zeng H, Sanes JR. Neuronal cell-type classification: challenges, opportunities and the path forward. Nat Rev Neurosci. 2017;18:530–46.
Zeng H, Huang J, Zhou H, Meilandt WJ, Dejanovic B, Zhou Y, Bohlen CJ, Lee SH, Ren J, Liu A, Tang Z, Sheng H, Liu J, Sheng M, Wang X. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat Neurosci. 2023;26(3):430–46.
Zhang M, Eichhorn SW, Zingg B, Yao Z, Cotter K, Zeng H, Dong H, Zhuang X. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature. 2021;598:137–43.
Zhang L, Chen D, Song D, Liu X, Zhang Y, Xu X, Wang X. Clinical and translational values of spatial transcriptomics. Signal Transduct Target Ther. 2022a;7:111.
Zhang Q, Abdo R, Iosef C, Kaneko T, Cecchini M, Han VK, Li SS. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat Commun. 2022b;13:5983.
Zhang Y, Lin X, Yao Z, Sun D, Lin X, Wang X, Yang C, Song J. Deconvolution algorithms for inference of the cell-type composition of the spatial transcriptome. Comput Struct Biotechnol J. 2023a;21:176–84.
Zhang Z, Li Y, Jiang S, Shi FD, Shi K, Jin WN. Targeting CCL5 signaling attenuates neuroinflammation after seizure. CNS Neurosci Ther. 2023b;29:317–30.
Zhao Y, Wang K, Hu G. DIST: spatial transcriptomics enhancement using deep learning. Brief Bioinform. 2023;24(2):bbad013.
Zhu J, Fan Y, Xiong Y, Wang W, Chen J, Xia Y, Lei J, Gong L, Sun S, Jiang T. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp Mol Med. 2022;54:2060–76.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (81700436). Thanks for the support of the China National GeneBank. Besides, we would like to thank X.F for critical feedback on the manuscript. Figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
W.H conducted the literature research. W.H and Y.Z drew the initial figures based on discussions with all authors. J.M, X.F conceived and supervised the study with W.H. W.H wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, W., Zhang, Y., Mei, J. et al. Spatial transcriptomics in human biomedical research and clinical application. Curr Med 2, 6 (2023). https://doi.org/10.1007/s44194-023-00023-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44194-023-00023-4