
Abstract
The formation of small crystallites is governed by two competing factors: the free energy gained upon transferring constituent atoms, molecules or colloidal particles from the metastable liquid to the more stable solid, and the free energy needed to create the surface area of the crystallite1. Because the ratio of surface area to bulk is large for small particles, small crystallites dissolve spontaneously under conditions where larger crystallites are stable and macroscopic crystal growth occurs only if spontaneously formed crystallites exceed a critical minimum size. On theoretical grounds1, the probability of forming such critical crystal nuclei is expected to increase rapidly with supersaturation. However, experiments show1,2 that the rate of crystal nucleation in many systems goes through a maximum as the supersaturation is increased. It is commonly assumed that the nucleation rate peaks because, even though the probability of forming critical nuclei increases with increasing concentration, the rate of growth of such nuclei decreases. Here we report simulations of crystal nucleation in suspensions of colloidal spheres with varying size distributions that show that the probability that critical nuclei will form itself goes through a maximum as the supersaturation is increased. We find that this effect, which is strongest for systems with the broadest particle size distribution, results from an increase with supersaturation of the solid–liquid interfacial free energy. The magnitude of this effect suggests that vitrification at high supersaturations should yield colloidal glasses that are truly amorphous, rather than nano-crystalline.
Similar content being viewed by others

Main
Colloidal suspensions of identical, hard, spherical particles can be either fluid or crystalline. At low densities, the fluid state is stable, but when the colloids occupy more than 49.4% of the volume, a crystalline phase should form2. In practice, several factors influence the crystallization of hard-sphere colloids. First of all, synthetic colloids have a distribution of particle radii with a width that is rarely less than 2–3% of the average radius. This non-uniformity of size (“polydispersity”) is known to affect the location of the freezing curve. Simulations3 show that higher compressions are needed to freeze a polydisperse suspension. Irrespective of the composition of the coexisting fluid, the polydispersity of the crystal never exceeds 5.7%. Experiments on crystal formation in hard-sphere colloids indicate that crystallization is suppressed in suspensions with a polydispersity exceeding 12% (ref. 2). This must be due to kinetic factors, as crystallization of strongly polydisperse suspensions is not excluded on thermodynamic grounds.
Classical nucleation theory (CNT)1 offers a simple thermodynamic explanation for why small crystal nuclei are less stable (that is, they have a higher free energy) than the supersaturated parent phase. CNT uses macroscopic arguments to estimate the free energy required to form a crystallite. The decrease in free energy due to the transfer of N particles from the metastable liquid to the solid state is approximated as NΔμ, where Δμ = μsolid-μliquid is the difference in chemical potential between the solid and the liquid state. The CNT estimate for the free-energy cost involved in the creation of the surface area A of the nucleus is γA, where γ is the surface free energy of the solid–liquid interface.
Owing to the competition between bulk and surface terms, the Gibbs free energy ΔG(N) required to form an N-particle nucleus goes through a maximum at a value of N called the critical nucleus size. For a spherical nucleus, the maximum value of ΔG(N) is:

where ρ is the number density of the crystal phase. The rate I at which nuclei are formed depends exponentially on ΔG*:

where T is the absolute temperature, k is Boltzmann's constant and κ is a kinetic prefactor that is proportional to the short-time self-diffusion constant of the colloids. The form of equation (2) does not rely on the validity of CNT.
In the CNT picture, increasing the supersaturation (that is, increasing |Δμ|) lowers the nucleation barrier. If γ were independent of |Δμ|, then ΔG* would always decrease with increasing supersaturation. In experiments4,5,6 the rate of colloidal crystal nucleation starts to decrease again for large supersaturations. This effect is attributed to the decrease in the kinetic prefactor κ: in order to crystallize, colloidal fluids must be compressed beyond the freezing curve. But eventually, the suspension will vitrify under compression. This vitrification slows down the particle motion and presumably reduces κ in equation (2). A problem with this interpretation is that recent experiments on colloidal crystallization in micro-gravity have found evidence for crystallization at densities that are well beyond the glass transition point7.
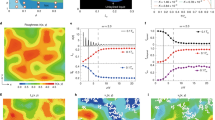
We performed Monte Carlo simulations to study the crystal-nucleation barrier and the structure of the critical nucleus, as a function of both polydispersity and supersaturation. As in the case of monodisperse suspensions8, we find that all critical nuclei have a randomly stacked close-packed structure. During crystallization, size-fractionation occurs3: the particles that make up the critical nucleus are on average larger than those in the metastable liquid. At fixed |Δμ| we find that ΔG*, the height of the nucleation barrier, does not depend on the polydispersity for polydispersities ≤5% (see Fig. 1; Table 1). As the polydispersity is increased beyond 5%, ΔG* increases rapidly. This implies that the probability of a critical nucleus forming is suppressed in polydisperse suspensions. It follows from equation (1) (or actually, from its polydisperse equivalent) that, at constant |Δμ|, the variation of ΔG* with polydispersity is due to an increase of the interfacial free energy γ. The increase of γ with polydispersity runs counter to Turnbull's suggestion that the interfacial free energy should be proportional to ΔH, the latent heat of fusion1. For the systems that we studied, ΔH crosses zero at a polydispersity of about 9%, where the liquid becomes denser than the coexisting solid3. Yet, γ clearly remains non-zero.

The free energy ΔG* is expressed in terms of kT, where k is Boltzmann's constant and T is the absolute temperature. |Δμ| (also in terms of kT) is the absolute difference between the chemical potential of the liquid and the solid. It is a measure for the degree of supersaturation. The curves are fits that have been drawn as a guide to the eye. To facilitate comparison with experiment, we show, in Table 1, the relationship between |Δμ| and the volume fraction ϕ of the liquid for the different systems that we studied.
Surprisingly, the variation of ΔG* with |Δμ| is non-monotonic. As |Δμ| is increased, the nucleation barrier goes through a minimum (Fig. 1). This non-monotonic behaviour of ΔG* is due to the increase of γ with |Δμ| (Fig. 2). To illustrate this, let us approximate the |Δμ|-dependence of γ by γ ≈ γ0 (1 + α|Δμ|). If we disregard the slight |Δμ|-dependence of the solid density, it then follows from equation (1) that ΔG* must go through a minimum when |Δμ| = 2/α. The nucleation theorem9 suggests that the minimum in ΔG* is due to the inversion of the number densities of the polydisperse fluid and the crystal nucleus. In CNT it is usually assumed that γ is constant. A linear variation of γ with |Δμ| has been observed in inorganic glasses1, but there the constant α is negative and hence there is no minimum in ΔG*. In other systems10,11, non-monotonic behaviour of ΔG* is induced by a hidden phase transition in the metastable phase.

The interfacial free energy γ is expressed in terms of kT/σ2, where σ is the average hard-sphere diameter. The curves are fits that have been drawn as a guide to the eye.
The minimum value of ΔG* increases rapidly with polydispersity. Using kinetic Monte Carlo simulations, we can estimate the value of the kinetic prefactor8. We find that, over the range of supersaturations studied, the kinetic prefactors vary by at most an order of magnitude (S.A. and D.F., manuscript in preparation). This means that the variation in the rate of nucleation is dominated by the behaviour of ΔG*. We estimate that, for colloidal particles with a radius ≥500 nm, homogeneous nucleation will be effectively suppressed (less than one nucleus per cubic centimetre per day) when the polydispersity exceeds 10%. This finding has important implications for the morphology of polycrystalline colloidal materials. Using a simplified version of the analysis proposed in ref. 10 to estimate the size of crystallites in a polycrystalline sample, it is easy to derive that Rc, the average crystallite size at the end of a nucleation experiment, should scale as exp(ΔG*/4kT). Our observation of a minimum in ΔG* thus implies the existence of a minimum in the typical crystallite size. This should be experimentally observable.
We could only compute ΔG* if spontaneous nucleation did not occur in the course of a simulation. In practice, this implied that we could not study barriers lower than 15kT. As a result, we could not test whether ΔG* in systems with a low polydispersity (less than 8.5%) also has a minimum. If we assume that at lower polydispersities we can extrapolate the increase of γ with |Δμ| to large supersaturations, then we predict that a minimum in ΔG* should occur even in nearly monodisperse systems. Again, this should be experimentally observable, because we should expect to see the formation of larger crystallites if the solution can be compressed rapidly through the region where ΔG* is small.
There are two ways to interpret the experimental finding that crystallization is not observed in suspensions with a polydispersity greater than 12%: either crystals do not form, or they are too small to be observed. Our simulations support the first interpretation. In a suspension of colloids with a 500-nm radius, we would expect10 to see less than one crystallite per cubic centimetre, once ΔG* > 32kT. In other words, under those conditions the colloidal glass is truly amorphous.
Our predictions concerning the structure and free energy of colloidal crystal nuclei can be tested experimentally. Recently, the technique of confocal scanning laser microscopy has been used12 to study the structure and size of critical crystal nuclei in dense colloidal suspensions and would thus be perfectly suited to test our predictions. Our prediction concerning the minimum in ΔG* is even easier to verify. By visual inspection, one could verify whether the crystallites that nucleate in strongly supersaturated solutions are larger than those that form at lower supersaturations. Over a decade ago, Pusey and van Megen produced images of the morphology of polycrystalline hard-sphere colloids13 and similar morphologies have recently been observed in a study of colloidal crystallization in micro-gravity (Z. D. Cheng, W. B. Russel and P. M. Chaikin, unpublished data). The observed increase of the crystallite size at large supersaturations was attributed to heterogeneous nucleation13, so a direct test of our prediction is still lacking.
Methods
The simulation techniques that are required to compute the free energy of small crystal nuclei have been described in earlier publications8,14. In the present work, we used constant-pressure, semi-grand-canonical Monte Carlo (SGMC) simulations of the type described in ref. 3. To eliminate possible finite-size effects, we used systems of 3,375 particles. Very long runs (up to 1.6 × 107 trial moves per particle), in combination with parallel tempering8, were needed to ensure equilibration of the dense, polydisperse fluid.
References
Kelton, K. F. Solid State Physics Vol. 45 (eds Ehrenreich, H. & Turnbull, D.) 75–178 (Academic, New York, 1991).
Pusey, P. in Liquids, Freezing and Glass Transition (eds Hansen, J. P., Levesque, D. & Zinn-Justin, J.) 763–931 (North-Holland, Amsterdam, 1991).
Kofke, D. A. & Bolhuis, P. G. Freezing of polydisperse hard spheres. Phys. Rev. E 59, 618–622 (1999).
Palberg, T. Crystallization kinetics of repulsive colloidal spheres. J. Phys. Condens. Matter 11, 323–360 (1999).
Van Duijneveldt, J. S. & Lekkerkerker, H. N. W. in Science and Technology of Crystal Growth (eds van der Eerden, J. P. & Bruinsma, O. S. L.) 279–290 (Kluwer Academic, Dordrecht, 1995).
Harland, J. L. & Van Megen, W. Crystallization kinetics of suspensions of hard colloidal spheres. Phys. Rev. E 55, 3054–3067 (1997).
Zhu, J. et al. Crystallization of hard-sphere colloids under microgravity. Nature 387, 883–885 (1997).
Auer, S. & Frenkel, D. Prediction of absolute crystal-nucleation rate in hard-sphere colloids. Nature 409, 1020–1023 (2001).
Oxtoby, D. W. & Kashchiev, D. A general relation between the nucleation work and the size of the nucleus in multicomponent nucleation. J. Chem. Phys. 100, 7665–7671 (1994).
Shi, F. G., Tong, H. Y. & Ayers, J. D. Free energy barrier to nucleation of amorphous-to-crystalline transformation selects the scale of microsctructure of crystallized materials. Appl. Phys. Lett. 67(3), 350–352 (1995).
Ten Wolde, P. R. & Frenkel, D. Enhancement of protein crystal nucleation by critical density fluctuations. Science 277, 1975–1978 (1997).
Gasser, U., Weeks, E. R., Schofield, A., Pusey, P. N. & Weitz, D. A. Real-space imaging of nucleation and growth in colloidal crystallization. Science 292, 258–262 (2001).
Pusey, P. N. & Van Megen, W. Phase behaviour of concentrated suspensions of nearly hard colloidal spheres. Nature 320, 340–342 (1986).
Ten Wolde, P. R., Ruiz-Montero, M. J. & Frenkel, D. Numerical evidence for bcc ordering at the surface of a critical fcc nucleus. Phys. Rev. Lett. 75, 2714–2717 (1995).
Acknowledgements
We thank A. van Blaaderen, P. M. Chaikin, H. N. W. Lekkerkerker, D. Oxtoby, W. B. Russel and F. Shi for comments and suggestions. This work was supported by the division of Chemical Sciences of the Netherlands Organization for Scientific Research (NWO). The work of the FOM Institute is part of the research programme of FOM and is made possible by financial support from the NWO. Access to the TERAS supercomputer was made possible through a grant by the NCF.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Auer, S., Frenkel, D. Suppression of crystal nucleation in polydisperse colloids due to increase of the surface free energy. Nature 413, 711–713 (2001). https://doi.org/10.1038/35099513
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/35099513
This article is cited by
-
Emerging exotic compositional order on approaching low-temperature equilibrium glasses
Nature Communications (2023)
-
Bifunctional interface modification for efficient and UV-robust α-Fe2O3-based planar organic–inorganic hybrid perovskite solar cells
Advanced Composites and Hybrid Materials (2022)
-
Energy polydisperse 2d Lennard–Jones fluid in the presence of flow field
Pramana (2022)
-
A conceptual change in crystallisation mechanisms of oxide materials from solutions in closed systems
Scientific Reports (2020)
-
Instantaneous dimensionless numbers for transient nonlinear rheology
Rheologica Acta (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
