
Abstract
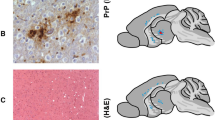
THE cellular prion protein (PrPc) is a sialoglycoprotein of Mr 33–35K that is expressed predominantly in neurons1–3. In transmissible and genetic neurodegenerative disorders such as scrapie of sheep, spongiform encephalopathy of cattle and Creutzfeldt–Jakob or Gerstmann–Sträussler–Scheinker diseases of humans4,5, PrPc is converted into an altered form (termed PrPSc) which is distinguishable from its normal homologue by its relative resistance to protease digestion6–8. PrPSc accumulates in the central nervous system of affected individuals8,9, and its protease-resistant core aggregates extracellularly into amyloid fibrils10–12. The process is accompanied by nerve cell loss, whose pathogenesis and molecular basis are not understood. We report here that neuronal death results from chronic exposure of primary rat hippocampal cultures to micromolar concentrations of a peptide corresponding to residues 106–126 of the amino-acid sequence deduced from human PrP complementary DNA. DNA fragmentation of degenerating neurons indicates that cell death occurred by apoptosis. The PrP peptide 106–126 has a high intrinsic ability to polymerize into amyloid-like fibrils in vitro. These findings indicate that cerebral accumulation of PrPSc and its degradation products may play a role in the nerve cell degeneration that occurs in prion-related encephalopathies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
Oesch, B. et al. Cell 40, 735–746 (1985).
Chesebro, B. et al. Nature 315, 331–333 (1985).
Kretzschmar, H. A., Prusiner, S. B., Stowring, L. E. & DeArmond, S. J. Am. J. Path. 122, 1–5 (1986).
Gajdusek, D. C. in Virology (eds Field, B. N. et al.) 2289–2324 (Raven, New York, 1990).
Prusiner, S. B. Science 252, 1515–1522 (1991).
Bolton, D. C., McKinley, M. P. & Prusiner, S. B. Science 218, 1309–1311 (1982).
Prusiner, S. B. et al. Biochemistry 21, 6942–6950 (1982).
McKinley, M. P., Bolton, D. C. & Prusiner, S. B. Cell 35, 57–62 (1983).
Meyer, R. K. et al. Proc. natn. Acad. Sci. U.S.A. 83, 2310–2314 (1986).
Prusiner, S. B. et al. Cell 35, 349–358 (1983).
DeArmond, S. J. et al. Cell 41, 221–235 (1985).
Tagliavini, F. et al. EMBO J. 10, 513–519 (1991).
Yankner, B. A., Duffy, L. K. & Kirschner, D. A. Science 250, 279–282 (1990).
Kowall, N. W. et al. Proc. natn. Acad. Sci. U.S.A. 88, 7247–7251 (1991).
Wyllie, A. H., Kerr, F. F. R. & Currie, A. R. Int. Rev. Cytol. 68, 251–306 (1980).
Server, A. C. & Mobley, W. C. in: The Molecular Basis of Cell Death (eds Tomei, L. D. & Cope, F. O.) 263–277 (Cold Spring Harbor Laboratory Press, New York, 1991).
Martin, S. J., Lennon, S. V., Bonham, A. M. & Cotter, T. G. J. Immun. 145, 1859–1867 (1990).
Wyllie, A. H. Nature 284, 555–556 (1980).
Clemens, J. A. & Stephenson, D. T. Neurobiol. Aging 13, 581–586 (1992).
Emre, M., Geula, C., Ransil, B. J. & Mesulam, M. M. Neurobiol. Aging 13, 553–559 (1992).
Price, D., Borchelt, D. R., Walker, L. C. & Sisodia, S. Neurobiol. Aging 13, 623–625 (1992).
Pike, V. C. J., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. Brain Res. 563, 311–314 (1991).
Tagliavini, F. et al. Soc. Neurosci. Abs. 18, 520.13 (1992).
Bruce, M. E., McBride, P. A. & Farquhar, C. F. Neurosci. Lett. 102, 1–6 (1989).
De Armond, S. J. et al. Neurology 37, 1271–1280 (1987).
Giaccone, G. et al. Proc. natn. Acad. Sci. U.S.A. 89, 9349–9353 (1992).
Montpetit, M. K., Lawless, K. R. & K. R. & Tenniswood, P. R. Prostrate 8, 25–31 (1986).
Leger, J., Le Guellec, R. & Tenniswood, P. R. Prostrate 13, 131–142 (1988).
Bettuzzi, S., Hilpakka, R. A., Gilna, P. & Liao, S. Biochem. J. 257, 293–296 (1989).
Buttyan, R. in The Molecular Basis of Cell Death (eds Tomei, L. D. & Cope, F. O.) 157–173 (Cold Spring Harbor Laboratory Press, New York, 1991).
Duguid, J. R., Bohmont, C. W., Liu, N. & Tourtellote, W. W. Proc. natn. Acad Sci. U.S.A. 86, 7260–7264 (1989).
Forloni, G., Chiesa, R., Angeretti, N. & Smiroldo, S. Soc. Neurosci. Abs. 18, 601.16 (1992).
Forloni, G. et al. Molec. Brain Res. 16, 128–134 (1992).
Aggarwal, B. B., Moffat, B. & Harkins, R. N. J. biol. Chem. 259, 686–691 (1984).
Fraser, P. E., Nguyen, W. K., Surewicz, W. D. & Kirschner, D. A. Biophys. J. 60, 1190–1201 (1991).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Forloni, G., Angeretti, N., Chiesa, R. et al. Neurotoxicity of a prion protein fragment. Nature 362, 543–546 (1993). https://doi.org/10.1038/362543a0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/362543a0
This article is cited by
-
Prion strains: shining new light on old concepts
Cell and Tissue Research (2023)
-
PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease
Cell Death & Disease (2022)
-
Non-cell autonomous astrocyte-mediated neuronal toxicity in prion diseases
Acta Neuropathologica Communications (2021)
-
Prion peptide-mediated calcium level alteration governs neuronal cell damage through AMPK-autophagy flux
Cell Communication and Signaling (2020)
-
OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases
Cell Death & Disease (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
