
Abstract
G protein-coupled receptors (GPCRs) play important roles in inflammation. Inflammatory cells such as polymorphonuclear leukocytes (PMN), monocytes and macrophages express a large number of GPCRs for classic chemoattractants and chemokines. These receptors are critical to the migration of phagocytes and their accumulation at sites of inflammation, where these cells can exacerbate inflammation but also contribute to its resolution. Besides chemoattractant GPCRs, protease activated receptors (PARs) such as PAR1 are involved in the regulation of vascular endothelial permeability. Prostaglandin receptors play different roles in inflammatory cell activation, and can mediate both proinflammatory and anti-inflammatory functions. Many GPCRs present in inflammatory cells also mediate transcription factor activation, resulting in the synthesis and secretion of inflammatory factors and, in some cases, molecules that suppress inflammation. An understanding of the signaling paradigms of GPCRs in inflammatory cells is likely to facilitate translational research and development of improved anti-inflammatory therapies.
Similar content being viewed by others
Introduction
Inflammation is characterized by the cardinal signs of rubor (redness), calor (heat), dolor (pain), tumor (swelling) and function laesa (loss of function). These signs reflect tissue response to inflammatory factors that are either external (eg, bacterial endotoxin) or host-derived (eg, TNFα). GPCRs contribute directly to these clinical manifestations due to their wide presence and diverse functions. Inflammation is shaped not only by leukocytes that accumulate at the site of inflammation, but also by cells in specific tissues and organs such as microglial cells in the brain and synovial fibroblasts in the joints. Endothelial cells in all tissues are actively involved in the process of inflammation and interact closely with leukocytes. GPCRs expressed in these cells play important roles in sensing the presence of chemoattractants, transducing signals that lead to the production of inflammatory cytokines, nociception, and regulation of intracellular and intercellular communications associated with increased blood flow and increased vascular endothelial permeability. Functions mediated by GPCRs can both exacerbate inflammation and promote its resolution.
GPCRs that mediate cell migration and phagocyte activation
It was long observed that phagocytes have the abilities to chase, capture and eventually eliminate invading bacteria. Based on observations made in the last century, it was reported that phagocytes could respond to small molecules derived from invading bacteria and fungi1. In addition, these cells also respond to substances produced by the host during the course of sterile inflammation. A number of small molecules was discovered in the 60's and 70's, including activated complement C5a and N-formylated peptides of bacterial origin2, 3, 4. Evidence that these “classic chemoattractants” act on GPCRs first came from the observation that pertussis toxin (PTX) could alter the binding affinity of chemotactic formyl peptides5, a characteristic feature of certain GPCR ligands6. Working independently, two laboratories reported that PTX could block formyl peptide (eg, fMet-Leu-Phe, fMLF)-induced neutrophil functions through ADP-ribosylation of the Gi class of heterotrimeric G proteins7, 8. These early studies demonstrate that the chemotactic peptide receptor functionally couples to a heterotrimeric G protein that is a substrate for ADP-ribosylation by PTX. Further characterization of the PTX substrate, also termed “islet-activating protein”, found it to be a member of the Gi family of G proteins9 that is critical to a variety of activities downstream of chemoattractant receptor signaling. The identity of the receptors for fMLF and C5a was first revealed, among all chemoattractant receptors, through molecular cloning of their cDNAs and analysis of the deduced protein sequence10, 11. It was confirmed that these receptors belong to the rhodopsin-like, 7-transmembrane (TM) receptor superfamily12, 13. Following these initial cloning efforts, other classic chemoattractant receptors including those for platelet-activating factor (PAF) and leukotriene B4 (LTB4), were sub-sequently identified as GPCRs14, 15.
Chemokines (chemotactic cytokines) are small proteins with cysteine residues located at fixed positions. A large number of chemokines have been identified in the mid-80's and early 90's. These chemokines bind to rhodopsin-like GPCRs, although not all of them are signaling receptors16, 17. Studies of patients with inflammatory disorders found induced expression of many chemokines, indicating that these small proteins play important roles in the development and progression of inflammatory diseases. Published reports have also shown that genetic deletion of selected chemokine receptors causes reduction in the severity of inflammation in various animal models. For instance, deletion of the CCR2 gene markedly reduced lesion in athrosclerosis-prone ApoE-null mice18. Atherosclerosis is an inflammatory disorder and the expression of CCL2, the ligand for CCR2, is upregulated in the atherosclerotic plaque and contributes to local accumulation of monocytes. In addition, CCR2 also contributes to the development of multiple sclerosis, rheumatoid arthritis, scleroderma and ischemia-reperfusion injury. Receptors for the CXCL class of chemokines are found in neutrophils. Among these GPCRs, CXCR1, and CXCR2 are involved in the pathology of myocardial infarction. CXCR1 and CXCR2 interact with CXCL1, CXCL2, and CXCL8, which are present during acute inflammation and acute injury. These chemokines are primarily responsible for the recruitment of neutrophils to the site of inflammation and tissue injury, where these professional phagocytes can affect granule release and reactive oxidant production.
Chemotaxis
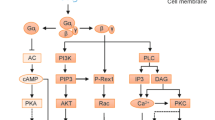
All chemoattractant receptors have the ability to mediate cell migration. In inflammatory disorders such as rheumatoid arthritis and atherosclerosis, the presence of leukocytes is crucial to disease progression. FPR1 is also responsible for sensing mitochondrial N-formyl peptides released from damaged cells19, 20. Activation of the Gi family of G proteins is critical to chemotaxis, which involves a complex network of intracellular signaling and cytoskeleton reorganization. Phagocytes polarize upon chemoattractant stimulation, forming a leading edge and a trailing edge which is characteristic of a migrating cell. G protein signaling initiates at the leading edge, as evidenced by the production of PIP3 and translocation of proteins with the PIP3-binding PH domain such as Akt and guanine nucleotide exchange factors such as P-Rex121 (Figure 1). Cytoskeletal reorganization also requires activation of the small GTPase Rac, whereas RhoA, another small GTPase, is believed to play a role at the trailing edge of a migrating cell22. Not all chemotaxis-mediating receptors are 7-TM receptors; other potent chemoattractants such as TGF-β act on other receptors. However, the fact that all chemokines bind to 7-TM receptors illustrates the importance of this class of receptors in cell migration. Until recently, only Gi proteins were implicated in leukocyte chemotaxis. The fact that several chemoattractant receptors could also couple to Gq proteins prompted a study that identified its role in chemotaxis23. Using mice lacking Gq proteins, it was observed that chemotaxis of dendritic cells to selected chemokines requires both Gi and Gq proteins. This alternative pathway uses a CD38-dependent mechanism for regulation of chemotaxis. It is presently unclear why in these cells Gq is necessary for chemotaxis whereas in other cells Gi is sufficient, but studies of this sort make it clear that the study of chemotaxis will also expand the understanding of GPCR biology.

Signaling pathways of GPCRs involved in inflammation. Signaling by GPCRs is initiated by a specific ligand that binds and activates the receptor inducing conformational changes in the receptor. A partial list of the relevant ligands is given in the top panel. All 4 classes of G proteins are involved in the regulation of events that lead to inflammatory cell activation or inactivation. Major effectors, including second messengers and other signaling molecules involved in the regulation of inflammation are shown. These include Ca2+, cAMP, protein kinases, lipid kinases, lipases, phosphoinositides, small GTPases and the relevant guanine nucleotide exchange factors, and transcriptional factors. The final output is manifested as nuclear, cytoplasmic and extracellular activities such as expression of proinflammatory genes, production of oxidants, and generation of tissue damaging and repairing factors.
Degranulation
A large number of GPCR agonists, including chemotactic peptides, activated complement fragments and histamine, are able to stimulate granule release24. In neutrophils, binding of fMLF to the receptor FPR1 or C5a to C5aR, triggers strong degranulation. In fact, exogenous expression of FPR1 or C5aR in a rat basophilic leukemia cell line renders these cells capable of releasing β-hexosaminidase upon stimulation, demonstrates the sufficiency of these receptors to trigger degranulation25. Simultaneous activation of several signaling pathways is required for degranulation26. As with other secretory cells, fusion of intracellular granules or vesicles in phagocytes requires calcium influx, which is triggered by GPCR signaling (Figure 1). G protein signaling also leads to the activation of several protein kinases, including PKC, cGMP-dependent kinase, and the serine/threonine kinase Akt. Exactly how these kinases promote granule release remains incompletely understood, but it is reported that vesicular fusion-related proteins such as the SNAP proteins are phosphorylated upon cell stimulation and these phosphorylation events precede vesicular fusion27. In addition to the protein kinases, small GTPases are activated downstream of GPCRs and are required for fusion of intracellular vesicles.
Superoxide generation
Although the production of reactive oxygen species (ROS) is widespread among different types of cells, phagocytes can produce large amounts of ROS, a requirement for killing phagocytosed bacteria28. A number of chemoattractants, including fMLF and C5a, potently stimulate ROS production in neutrophils, although production is extracellular rather than intra-phagosome when induced by these soluble mediators. As a result, these oxygen radicals cause damage to endothelial cells that form the lining of vascular wall. Exposure to even low doses of these chemoattractants also “primes” the phagocytes for more robust oxidant production when stimulated with other inflammatory factors such as LPS29. Chemoattractant-induced ROS production requires Gi protein, as it is effectively blocked by PTX treatment. Research conducted thus far has found that chemoattractant-induced ROS production shares basic mechanisms with particle-induced ROS production, in that both require membrane translocation of cytosolic components and assembly of a functional NADPH oxidase at membrane. There are, however, major differences in the upstream signaling mechanisms involved. Whereas phagocytosis-induced oxidant production requires primarily tyrosine kinase activation, chemoattractant-induced ROS production relies mostly on the activation of serine/threonine kinases such as Akt and dual-specificity protein kinases such as p38 MAPK. These different signaling pathways then converge at activation of downstream targets such as PKC (Figure 1). Published reports show that upon activation of chemoattractant GPCRs, the released G protein βγ subunits triggers PI3K activation and the production of PIP330. The membrane-bound PIP3 is required for the subsequent activation of Akt and the small GTPase Rac, which is also required for the assembly of a functional NADPH oxidase complex31, 32. Thus, chemoattractant-induced ROS production requires simultaneous and sustained activation of multiple signaling pathways, which are also negatively regulated by phosphatases33. How chemoattractant GPCRs convert a single binding event to a cascade of signaling events remains incompletely understood. Recent studies have shown that both G protein-dependent and –independent pathways are involved in chemoattractant signaling.
GPCRs and inflammatory pain
Inflammation caused by trauma, infection and other forms of insult to the tissue is often accompanied by the uncomfortable sensation of pain. Inflammatory pain may result from enhanced nociception due to the interaction of inflammatory mediators with neurons and the resulting state of hypersensitivity. Inflammation may also cause damage to neurons and produce neuropathic pain34. Being the largest group of sensory receptors, GPCRs play important roles in inflammatory nociception. At the periphery, a number of GPCR agonists produced during inflammation, including bradykinin (BK) and selected prostaglandins (PGs), participate in inflammatory hyperalgesia. BK and kallidin both activate and sensitize primary afferent neurons. Two classes of BK receptors are present. Experimental data show that B1 BK receptor agonists produce pain only during inflammation, in accordance with the inducible nature of the B1 BK receptor. The B2 BK receptor is constitutively expressed and its blockade reduces inflammatory hyperalgesia in animal models35.
Prostaglandins (PGs) are lipid-derived autacoids generated through the sequential actions of cyclooxygenase and PG synthase. These metabolites of arachidonic acid include thromboxanes (TXA2), PGD2, PGE2, PGI2, and PGF2a. Collectively, they interact with 9 prostanoid receptors that couple to a variety of G proteins and are responsible for several features of inflammation including pain and edema (Table 1). Non-steroidal anti-inflammatory drugs (NSAIDs), including acetylsalicylic acid (aspirin) and the more selective COX-2 specific inhibitors, are effective anti-inflammatory and pain-relieving agents primarily because they block the synthesis of PGs36, 37. Among the various arachdonic acid metabolites, PGE2 interacts with several GPCRs. The 4 EP receptors are individual gene products but in the case of EP3, there are splice variants as well38. The 4 subclasses of EP receptors couple to various G proteins and are responsible for the variety of PGE2 effects. PGE2 is synthesized during the course of inflammation and contributes to tissue edema and hyperalgesia. In the nociceptive primary afferent nerve terminals, PGE2 modulates voltage-gated sodium currents39, a function mediated through the EP3 receptor40. PGE2 is also synthesized in the CNS during peripheral inflammation and contributes to increased pain hypersensitivity41.
Prostacyclin (PGI), which acts on G protein-coupled IP receptor, is another arachdonic metabolite produced during inflammation that plays a central nociceptive role42. Mice lacking the IP receptor display altered pain perception as well as inflammatory responses43. Studies have shown that Gs-coupled PG receptors may enhance nociceptor sensitization by reducing the activation threshold for selected sodium channels through a cAMP and PKA-dependent mechanism44. GPCRs present in dorsal horn neurons are also involved in transmission of nociception and are responsible for the development of central sensitization. The NK1 receptor for tachykinin is a GPCR that responds to substance P and mediates PKC activation, leading to the phosphorylation and potentiation of N-methyl-D-aspartic acid (NMDA) receptors45. In addition to the NK1 receptor, the neuromedin U2 receptor (NMU2) plays a central role in nociception. NMU2 knockout mice display reduced sensitivity to pain induced by capsaicin and formalin46.
Besides functions of GPCRs in inflammatory nociception, there are instances in which GPCRs are antinociceptive. Loperamide, an opioid agonist developed for peripheral use, displays antinociceptive activity in experimental arthritis47. Cannabinoids, which are agonists for the CB1 and CB2 receptors, inhibit peripheral sensitization when used topically48. Exploration of the roles for GPCRs in inflammatory nociception is bound to provide novel therapeutics for effective control of pain associated with inflammatory diseases.
GPCRs and regulation of vascular endothelial permeability
Edema, an abnormal accumulation of interstitial fluid, often accompanies inflammation. The production of inflammatory factors, many of them GPCR agonists, is largely responsible for increased vascular endothelial permeability, which contributes to edema during inflammation. BK and PAF are well known for their roles in the regulation of vascular wall permeability. It has long been recognized that BK reproduces cardinal signs of inflammation, and the BK effect on vascular permeability is direct rather than dependent on histamine49. More recent studies have shown that both the B1 BK receptor and B2 BK receptor are involved in local edema during inflammation, for instance, paw edema and protein extravasation leading to joint swelling50. PAF profoundly affects microvascular permeability, allowing extravasation of plasma contents such as albumin51, 52. Like BK, PAF acts on endothelial cells directly, increasing gap formation between endothelial cells through the actions of eNOS53, tyrosine phosphorylation of VE-cadherin54, and the Rho family small GTPases55.
Histamine and thrombin are also major regulators of vascular permeability. Histamine is released by basophils and mast cells during the allergic response. It binds to one or more of the four G protein-coupled histamine receptors, stimulating vasodilation and increasing vascular permeability56. Histamine also participates in allergic inflammatory diseases such as asthma by inducing chemotaxis and bronchoconstriction57. Thrombin is one of the best studied regulators of vascular endothelial permeability58. The thrombin receptor was initially identified as a GPCR with a unique mechanism for activation. Upon binding of alpha thrombin, the receptor's N-terminus was cleaved, generating a tethered peptide that becomes an agonist for the receptor59. This receptor is the first member of the protease-activated receptor (PAR) subfamily, of which 3 of the 4 PARs bind thrombin (Table 1). Although initially known as a thrombotic agent that catalyzes the conversion of fibrinogen to fibrin, thrombin is also a potent agonist for platelets and its receptors are expressed in human endothelial cells as well as smooth muscle cells (SMC). Its vascular functions include cellular differentiation, migration and proliferation of SMC, angiogenesis and vascular development60. PAR1, which is widely studied in this subfamily of GPCRs, couples to multiple G proteins including Gi, Gq, and G12/13. The selectivity for activation of these G proteins depends on the agonists that bind to the receptor61. In endothelial cells, thrombin binding to PAR1 leads to activation of p115 RhoGEF, which provides a functional link between G13 and RhoA62. Activation of RhoA is responsible for stress fiber formation, and increased calcium flux triggers other signaling pathways which ultimately lead to myosin light chain-dependent contraction of endothelial cells63. This process is reversible, and focal adhesion kinase plays a role in the reversal of the increased vascular permeability64. Thrombin-induced increase in vascular endothelial permeability contributes to the edema seen in inflammatory disorders such as acute lung injury65, 66. Both the receptor and its downstream signaling pathways are targets for therapeutic intervention67, 68. In contrast to thrombin, another GPCR ligand sphingosine 1-phosphate (S1P) promotes stabilization of the endothelial barrier69. This effect of S1P is mediated through its receptor S1P1, which promotes adherens junction assembly in endothelial cells, thus fortifying vascular endothelial barrier70, 71.
GPCRs and regulation of inflammatory gene expression
A large number of GPCRs have been found to participate in transcriptional regulation72. G protein signaling leads to activation of transcription factors including CREB, c-Jun, NF-κB, and STAT3, among others (Figure 1). These transcription factors, particularly NF-κB, are closely associated with the expression of genes that encode inflammatory factors. It was first reported that the receptor for PAF could stimulate NF-κB activation through a PTX-insensitive pathway, suggesting that the Gq protein which couples to the PAF receptor is responsible for this function73. Subsequent studies identified both PTX-sensitive and PTX-insensitive mechanisms by which GPCRs activate NF-κB74, 75, 76, 77, 78. The Gi-dependent pathway requires the Gβγ subunits whereas Gq directly activates PLCβ, thus triggering PKC. In addition to PKC, protein kinases that are found to be involved in GPCR activation of NF-κB include the Ser/Thr kinase Akt77 and tyrosine kinase Pyk279. The β-arrestin pathway was also involved in regulating NF-κB activation. In resting cells, β-arrestins bind to IκBα and protect it from phosphorylation and proteosome degradation80, 81. Upon GPCR activation, β-arrestins participate in intracellular signaling leading to activation of multiple pathways that favor NF-κB activation82, 83, 84. Several intracellular signaling molecules that were initially identified for immune cell functions, including CARMA3 and Bcl10, were found to regulate GPCR signaling leading to NF-κB activation85, 86. These latter findings provide evidence for the complexity of signaling pathways downstream of GPCRs.
In addition to agonist-induced, GPCR-mediated NF-κB activation, a number of constitutively activated GPCRs are able to activate NF-κΒ even in the absence of agonists. Viruses such as KSHV, HHV8, and RCMV encode GPCRs that are constitutivelyactive and, when expressed in mammalian cells, activate NF-κB87, 88, 89, 90. Like agonist-induced GPCRs, these viral GPCRs couples to more than one G proteins for signaling88. Transcriptional activation by these GPCRs is not restricted to NF-κB87. These receptors are believed to contribute to their pathological functions including stimulation of angiogenesis in the case of KSHV GPCR, and are potential therapeutic targets91, 92.
Summary
The diverse actions of inflammatory factors are reflected in the diversity of receptors with which they interact. It is therefore not surprising that inflammatory factors of different nature and composition, including arachidonic acid metabolites, peptides, protein fragments and proteases, are found to partner with the 7-TM GPCRs. The diversity in ligand binding and transmembrane signaling by GPCRs are primarily responsible for the mediation of complex inflammatory (and anti-inflammatory) responses. There is no doubt that GPCRs play important roles in inflammation, as they do in other vital organ functions. A better understanding of these receptors as well as their signaling pathways will help to develop new therapeutic agents with higher specificity than traditional anti-inflammatory agents such as NSAIDs.
References
Harris H . Role of chemotaxis in inflammation. Physiol Rev 1954; 34: 529–62.
Ward PA, Cochrane CG, Muller-eberhard HJ . The role of serum complement in chemotaxis of leukocytes in vitro. J Exp Med 1965; 122: 327–46.
Ward PA, Lepow IH, Newman LJ . Bacterial factors chemotactic for polymorphonuclear leukocytes. Am J Pathol 1968; 52: 725–36.
Schiffmann E, Corcoran BA, Wahl SM . N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci U S A 1975; 72: 1059–62.
Koo C, Lefkowitz RJ, Snyderman R . Guanine nucleotides modulate the binding affinity of the oligopeptide chemoattractant receptor on human polymorphonuclear leukocytes. J Clin Invest 1983; 72: 748–53.
De Lean A, Stadel JM, Lefkowitz RJ . A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 1980; 255: 7108–17.
Bokoch GM, Gilman AG . Inhibition of receptor-mediated release of arachidonic acid by pertussis toxin. Cell 1984; 39: 301–8.
Lad PM, Olson CV, Smiley PA . Association of the N-formyl-Met-Leu-Phe receptor in human neutrophils with a GTP-binding protein sensitive to pertussis toxin. Proc Natl Acad Sci U S A 1985; 82: 869–73.
Simon MI, Strathmann MP, Gautam N . Diversity of G proteins in signal transduction. Science 1991; 252: 802–8.
Boulay F, Tardif M, Brouchon L, Vignais P . The human N-formylpeptide receptor: characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry 1990; 29: 11123–33.
Gerard NP, Gerard C . The chemotactic receptor for human C5a anaphylatoxin. Nature 1991; 349: 614–7.
Gerard C, Gerard NP . C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol 1994; 12: 775–808.
Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 2009; 61: 119–61.
Honda Z, Nakamura M, Miki I, Minami M, Watanabe T, Seyama Y, et al. Cloning by functional expression of platelet-activating factor receptor from guinea-pig lung. Nature 1991; 349: 342–6.
Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T . A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 1997; 387: 620–4.
Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International Union of Pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 2000; 52: 145–76.
Zlotnik A, Yoshie O . Chemokines: a new classification system and their role in immunity. Immunity 2000; 12: 121–7.
Boring L, Gosling J, Cleary M, Charo IF . Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998; 394: 894–7.
Carp H . Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med 1982; 155: 264–75.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010; 330: 362–6.
Chung CY, Potikyan G, Firtel RA . Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol Cell 2001; 7: 937–47.
Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, et al. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 2003; 114: 201–14.
Shi G, Partida-Sanchez S, Misra RS, Tighe M, Borchers MT, Lee JJ, et al. Identification of an alternative G{alpha}q-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med 2007; 204: 2705–18.
Lacy P . Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol 2006; 2: 98–108.
Ahamed J, Haribabu B, Ali H . Cutting edge: Differential regulation of chemoattractant receptor-induced degranulation and chemokine production by receptor phosphorylation. J Immunol 2001; 167: 3559–63.
Nanamori M, Chen J, Du X, Ye RD . Regulation of leukocyte degranulation by cGMP-dependent protein kinase and phosphoinositide 3-kinase: potential roles in phosphorylation of target membrane SNARE complex proteins in rat mast cells. J Immunol 2007; 178: 416–27.
Woska JR Jr, Gillespie ME . SNARE complex-mediated degranulation in mast cells. J Cell Mol Med 2011. doi: 10.1111/j.1582-4934.2011.01443.x.
Nauseef WM . Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 2004; 122: 277–91.
Shapira L, Champagne C, Gordon B, Amar S, Van Dyke TE . Lipopolysaccharide priming of superoxide release by human neutrophils: role of membrane CD14 and serum LPS binding protein. Inflammation 1995; 19: 289–95.
Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D . Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 2000; 287: 1046–9.
Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW . Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991; 353: 668–70.
Knaus UG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM . Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 1991; 254: 1512–5.
Qian F, Deng J, Cheng N, Welch EJ, Zhang Y, Malik AB, et al. A non-redundant role for MKP5 in limiting ROS production and preventing LPS-induced vascular injury. Embo J 2009; 28: 2896–907.
Kidd BL, Urban LA . Mechanisms of inflammatory pain. Br J Anaesth 2001; 87: 3–11.
Burgess GM, Perkins MN, Rang HP, Campbell EA, Brown MC, McIntyre P, et al. Bradyzide, a potent non-peptide B(2) bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br J Pharmacol 2000; 129: 77–86.
Vane JR . Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 1971; 231: 232–5.
Vane JR, Botting RM . Mechanism of action of aspirin-like drugs. Semin Arthritis Rheum 1997; 26: 2–10.
Breyer RM, Bagdassarian CK, Myers SA, Breyer MD . Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 2001; 41: 661–90.
Khasar SG, Gold MS, Levine JD . A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett 1998; 256: 17–20.
Minami T, Nakano H, Kobayashi T, Sugimoto Y, Ushikubi F, Ichikawa A, et al. Characterization of EP receptor subtypes responsible for prostaglandin E2-induced pain responses by use of EP1 and EP3 receptor knockout mice. Br J Pharmacol 2001; 133: 438–44.
Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 2001; 410: 471–5.
Doi Y, Minami T, Nishizawa M, Mabuchi T, Mori H, Ito S . Central nociceptive role of prostacyclin (IP) receptor induced by peripheral inflammation. Neuroreport 2002; 13: 93–6.
Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 1997; 388: 678–82.
England S, Bevan S, Docherty RJ . PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol 1996; 495: 429–40.
Thompson SW, Dray A, Urban L . Injury-induced plasticity of spinal reflex activity: NK1 neurokinin receptor activation and enhanced A- and C-fiber mediated responses in the rat spinal cord in vitro. J Neurosci 1994; 14: 3672–87.
Zeng H, Gragerov A, Hohmann JG, Pavlova MN, Schimpf BA, Xu H, et al. Neuromedin U receptor 2-deficient mice display differential responses in sensory perception, stress, and feeding. Mol Cell Biol 2006; 26: 9352–63.
DeHaven-Hudkins DL, Burgos LC, Cassel JA, Daubert JD, DeHaven RN, Mansson E, et al. Loperamide (ADL 2-1294), an opioid antihyperalgesic agent with peripheral selectivity. J Pharmacol Exp Ther 1999; 289: 494–502.
Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F . The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci 2000; 21: 218–24.
Gaponiuk P, Oivin VI . Direct effect of bradykinin on vascular permeability. Biull Eksp Biol Med 1971; 71: 23–5.
Bhoola K, Ramsaroop R, Plendl J, Cassim B, Dlamini Z, Naicker S . Kallikrein and kinin receptor expression in inflammation and cancer. Biol Chem 2001; 382: 77–89.
Handley DA, Arbeeny CM, Lee ML, Van Valen RG, Saunders RN . Effect of platelet activating factor on endothelial permeability to plasma macromolecules. Immunopharmacology 1984; 8: 137–42.
Burhop KE, Garcia JG, Selig WM, Lo SK, van der Zee H, Kaplan JE, et al. Platelet-activating factor increases lung vascular permeability to protein. J Appl Physiol 1986; 61: 2210–7.
Hatakeyama T, Pappas PJ, Hobson RW 2nd, Boric MP, Sessa WC, Duran WN . Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol 2006; 574: 275–81.
Hudry-Clergeon H, Stengel D, Ninio E, Vilgrain I . Platelet-activating factor increases VE-cadherin tyrosine phosphorylation in mouse endothelial cells and its association with the PtdIns3'-kinase. FASEB J 2005; 19: 512–20.
Knezevic II, Predescu SA, Neamu RF, Gorovoy MS, Knezevic NM, Easington C, et al. Tiam1 and Rac1 are required for platelet-activating factor-induced endothelial junctional disassembly and increase in vascular permeability. J Biol Chem 2009; 284: 5381–94.
Bakker RA, Timmerman H, Leurs R . Histamine receptors: specific ligands, receptor biochemistry, and signal transduction. Clin Allergy Immunol 2002; 17: 27–64.
Hamid Q, Tulic M . Immunobiology of asthma. Annu Rev Physiol 2009; 71: 489–507.
Mehta D, Malik AB . Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006; 86: 279–367.
Vu TK, Hung DT, Wheaton VI, Coughlin SR . Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991; 64: 1057–68.
Patterson C, Stouffer GA, Madamanchi N, Runge MS . New tricks for old dogs: nonthrombotic effects of thrombin in vessel wall biology. Circ Res 2001; 88: 987–97.
McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE . Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 2005; 280: 25048–59
Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 1998; 280: 2109–11.
Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB . Role of Ca2+signaling in the regulation of endothelial permeability. Vascul Pharmacol 2002; 39: 173–85.
Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D . Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem 2006; 281: 2296–305.
Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW 2nd, Malik AB . Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol 1986; 128: 96–104.
Lum H, Aschner JL, Phillips PG, Fletcher PW, Malik AB . Time course of thrombin-induced increase in endothelial permeability: relationship to [Ca2+]i and inositol polyphosphates. Am J Physiol 1992; 263: L219–25.
Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. 'Role reversal' for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol 2007; 8: 1303–12.
Cirino G, Severino B . Thrombin receptors and their antagonists: an update on the patent literature. Expert Opin Ther Pat 2010; 20: 875–84.
English D, Kovala AT, Welch Z, Harvey KA, Siddiqui RA, Brindley DN, et al. Induction of endothelial cell chemotaxis by sphingosine 1-phosphate and stabilization of endothelial monolayer barrier function by lysophosphatidic acid, potential mediators of hematopoietic angiogenesis. J Hematother Stem Cell Res 1999; 8: 627–34.
McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG . Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 2004; 170: 987–93.
Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB . Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 2005; 280: 17320–8.
Ho MK, Su Y, Yeung WW, Wong YH . Regulation of transcription factors by heterotrimeric G proteins. Curr Mol Pharmacol 2009; 2: 19–31.
Kravchenko VV, Pan Z, Han J, Herbert JM, Ulevitch RJ, Ye RD . Platelet-activating factor induces NF-kappa B activation through a G protein-coupled pathway. J Biol Chem 1995; 270: 14928–34.
Cowen DS, Molinoff PB, Manning DR . 5-hydroxytryptamine1A receptor-mediated increases in receptor expression and activation of nuclear factor-kappaB in transfected Chinese hamster ovary cells. Mol Pharmacol 1997; 52: 221–6.
Hsu MH, Wang M, Browning DD, Mukaida N, Ye RD . NF-kappaB activation is required for C5a-induced interleukin-8 gene expression in mononuclear cells. Blood 1999; 93: 3241–9.
Shahrestanifar M, Fan X, Manning DR . Lysophosphatidic acid activates NF-kappaB in fibroblasts. A requirement for multiple inputs. J Biol Chem 1999; 274: 3828–33.
Xie P, Browning DD, Hay N, Mackman N, Ye RD . Activation of NF-kappa B by bradykinin through a Galpha(q)- and Gbeta gamma-dependent pathway that involves phosphoinositide 3-kinase and Akt. J Biol Chem 2000; 275: 24907–14.
Yang M, Sang H, Rahman A, Wu D, Malik AB, Ye RD . G alpha 16 couples chemoattractant receptors to NF-kappa B activation. J Immunol 2001; 166: 6885–92.
Shi CS, Kehrl JH . PYK2 links G(q)alpha and G(13)alpha signaling to NF-kappa B activation. J Biol Chem 2001; 276: 31845–50.
Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell 2004; 14: 303–17.
Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ . beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci U S A 2004; 101: 8603–7.
Yang M, Zhang H, Voyno-Yasenetskaya T, Ye RD . Requirement of Gbetagamma and c-Src in D2 dopamine receptor-mediated nuclear factor-kappaB activation. Mol Pharmacol 2003; 64: 447–55.
Sun J, Lin X . Beta-arrestin 2 is required for lysophosphatidic acid-induced NF-kappaB activation. Proc Natl Acad Sci U S A 2008; 105: 17085–90.
Yang M, He RL, Benovic JL, Ye RD . beta-Arrestin1 interacts with the G-protein subunits beta1gamma2 and promotes beta1gamma2-dependent Akt signalling for NF-kappaB activation. Biochem J 2009; 417: 287–96.
Grabiner BC, Blonska M, Lin PC, You Y, Wang D, Sun J, et al. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev 2007; 21: 984–96.
Wang D, You Y, Lin PC, Xue L, Morris SW, Zeng H, et al. Bcl10 plays a critical role in NF-kappaB activation induced by G protein-coupled receptors. Proc Natl Acad Sci U S A 2007; 104: 145–50.
Schwarz M, Murphy PM . Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol 2001; 167: 505–13.
Shepard LW, Yang M, Xie P, Browning DD, Voyno-Yasenetskaya T, Kozasa T, et al. Constitutive activation of NF-kappa B and secretion of interleukin-8 induced by the G protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus involve G alpha(13) and RhoA. J Biol Chem 2001; 276: 45979–87.
Couty JP, Geras-Raaka E, Weksler BB, Gershengorn MC . Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor signals through multiple pathways in endothelial cells. J Biol Chem 2001; 276: 33805–11.
Gruijthuijsen YK, Casarosa P, Kaptein SJ, Broers JL, Leurs R, Bruggeman CA, et al. The rat cytomegalovirus R33-encoded G protein-coupled receptor signals in a constitutive fashion. J Virol 2002; 76: 1328–38.
Cannon M . The KSHV and other human herpesviral G protein-coupled receptors. Curr Top Microbiol Immunol 2007; 312: 137–56.
Vischer HF, Hulshof JW, de Esch IJ, Smit MJ, Leurs R . Virus-encoded G-protein-coupled receptors: constitutively active (dys)regulators of cell function and their potential as drug target. Ernst Schering Found Symp Proc 2006; 2: 187–209.
Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature 1998; 394: 690–4.
Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG . Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med 2000; 191: 455–62.
Hata AN, Breyer RM . Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 2004; 103: 147–66.
Boie Y, Rushmore TH, Darmon-Goodwin A, Grygorczyk R, Slipetz DM, Metters KM, et al. Cloning and expression of a cDNA for the human prostanoid IP receptor. J Biol Chem 1994; 269: 12173–8.
Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M . Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem 1995; 270: 18910–6.
Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000; 287: 2013–7.
Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL . International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev 2005; 57: 27–77.
Kuduk SD, Bock MG . Bradykinin B1 receptor antagonists as novel analgesics: a retrospective of selected medicinal chemistry developments. Curr Top Med Chem 2008; 8: 1420–30.
Duchene J, Ahluwalia A . The kinin B(1) receptor and inflammation: new therapeutic target for cardiovascular disease. Curr Opin Pharmacol 2009; 9: 125–31.
Duffy RA . Potential therapeutic targets for neurokinin-1 receptor antagonists. Expert Opin Emerg Drugs 2004; 9: 9–21.
Torres R, Croll SD, Vercollone J, Reinhardt J, Griffiths J, Zabski S, et al. Mice genetically deficient in neuromedin U receptor 2, but not neuromedin U receptor 1, have impaired nociceptive responses. Pain 2007; 130: 267–78.
Kaminski NE . Immune regulation by cannabinoid compounds through the inhibition of the cyclic AMP signaling cascade and altered gene expression. Biochem Pharmacol 1996; 52: 1133–40.
Yost CC, Weyrich AS, Zimmerman GA . The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie 2010; 92: 692–7.
Acknowledgements
Due to page limitations, we are unable to cite as many references on GPCRs and inflammation as we wished. We apologize to those whose important work was not mentioned in this article. This work was supported in part by National Basic Research Program of China (973 Program) Grant 2012CB518001 (to Richard D YE).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, L., Ye, R. Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin 33, 342–350 (2012). https://doi.org/10.1038/aps.2011.200
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.200
Keywords
This article is cited by
-
Cell type specific gene expression profiling reveals a role for complement component C3 in neutrophil responses to tissue damage
Scientific Reports (2020)
-
Analysis of gene expression profiles and protein-protein interaction networks in multiple tissues of systemic sclerosis
BMC Medical Genomics (2019)
-
Genome-wide analysis of genetic predisposition to Alzheimer’s disease and related sex disparities
Alzheimer's Research & Therapy (2019)
-
GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine
Journal of Neuroinflammation (2019)
-
The Effect of Recombinant Undercarboxylated Osteocalcin on Endothelial Dysfunction
Calcified Tissue International (2019)
