
Abstract
The inositol 1,4,5-trisphosphate receptor (IP3R) is a major regulator of apoptotic signaling. Through interactions with members of the Bcl-2 family of proteins, it drives calcium (Ca2+) transients from the endoplasmic reticulum (ER) to mitochondria, thereby establishing a functional and physical link between these organelles. Importantly, the IP3R also regulates autophagy, and in particular, its inhibition/depletion strongly induces macroautophagy. Here, we show that the IP3R antagonist xestospongin B induces autophagy by disrupting a molecular complex formed by the IP3R and Beclin 1, an interaction that is increased or inhibited by overexpression or knockdown of Bcl-2, respectively. An effect of Beclin 1 on Ca2+ homeostasis was discarded as siRNA-mediated knockdown of Beclin 1 did not affect cytosolic or luminal ER Ca2+ levels. Xestospongin B- or starvation-induced autophagy was inhibited by overexpression of the IP3R ligand-binding domain, which coimmunoprecipitated with Beclin 1. These results identify IP3R as a new regulator of the Beclin 1 complex that may bridge signals converging on the ER and initial phagophore formation.
Similar content being viewed by others


Main
Macroautophagy (herein referred to as autophagy) is the major catabolic pathway for entire organelles, long-lived/aberrant proteins and superfluous portions of the cytosol. It consists of the stepwise engulfment of substrate elements into distinctive multimembraned autophagosomes, which after fusion with lysosomes form single-membraned autolysosomes. Within the lumen of autolysosomes, macromolecules are enzymatically broken down into metabolites that cope with the bioenergetic and biosynthetic demands of the cell.1, 2
The molecular activation of autophagy is a complex process (reviewed in Yorimitsu and Klionsky3) that is regulated by the mammalian target of rapamycin (mTOR), a protein kinase essential for nutrient-sensing signal transduction.4 Downstream of mTOR, a series of chain reactions are executed by several autophagy-related (Atg) proteins, allowing the preautophagosomal membrane structure (phagophore) to engulf a substrate material, to complete and close the sequestering vacuole in physical association with the microtubular network, and finally fuse autophagosomes with lysosomes to execute degradation. The autophagic functions of the phylogenetically conserved family of atg genes have extensively been studied in yeast.5, 6 One of the most important members of this family is atg6, whose mammalian ortholog is represented by the haploinsufficient tumor suppressor gene beclin 1.7, 8 In mammalian cells, Beclin 1 acts concertedly with Vps34, Vps15, UVRAG, Bif1, Ambra1 and perhaps other proteins to form a multiprotein complex with class III phosphatidylinositol 3-kinase (PI3K) activity that generates phosphatidylinositol-3-phosphate (PI3P). PI3P determines the curvature of the nascent phagophore and promotes the recruitment of other Atg proteins (including Atg5, Atg12, Atg10, Atg4, Atg3, Atg7, Atg8 and Atg16), which catalyze vesicle elongation and phagophore nucleation.6, 9
Although classically viewed as an essential mechanism of adaptation to stress, in particular to that imposed by the lack of nutrients, excessive or inefficient autophagy may be involved in autophagic cell death (ACD).9, 10, 11 At a molecular level, the cross-talk between apoptosis and autophagy is beginning to be understood, and some factors have recently been identified as common regulators of both pathways.9, 12, 13 For instance, the activity of Beclin 1 is inhibited by the antiapoptotic proteins Bcl-2 and Bcl-XL due to the interaction between the BH3 domain present in Beclin 1 and the BH3 receptor cleft of Bcl-2/XL. This binding is competitively disrupted by proapoptotic BH3-only proteins, which therefore can promote autophagy.14, 15, 16
Autophagy can also be induced through an mTOR-independent pathway by lowering myo-inositol 1,4,5-trisphosphate (IP3) levels.17 This effect can be achieved pharmacologically with drugs such as lithium or L-690 330, which disrupt inositol metabolism by inhibiting inositol monophosphatase (IMP).18 As IP3 is a second messenger that mediates Ca2+ release from the endoplasmic reticulum (ER), autophagy might also be regulated by Ca2+. This hypothesis has been addressed by a pioneering study, which concluded that Ca2+ fluxes are not necessary for autophagic stimulation and that autophagy would rather depend on the presence of Ca2+ within an intracellular storage compartment.19 Accordingly, cytosolic Ca2+ may even inhibit autophagy in a cyclical mTOR-independent pathway.18 Three recent studies have also explored the contribution of Ca2+ to autophagy,20, 21, 22 establishing that Ca2+ is required for autophagic induction, but showing some incongruities in whether increases in cytosolic [Ca2+],21, 22 or modifications of ER Ca2+ levels,20 are responsible for the induction of autophagy. Irrespective of such discrepancies, all these studies showed that a Bcl-2 variant that is specifically targeted to the ER inhibits autophagy.20, 22
Ca2+ present in the ER lumen is released through specific channels. Two types of ER-resident Ca2+ release channels exist, namely the ryanodine receptors and the IP3 receptors (IP3R).23 Diverse physiological processes, including the mitochondrial (or intrinsic) pathway of apoptosis, are controlled by Ca2+ fluxes from the ER to mitochondria, which occur in the context of specific microdomains allowing for the functional and physical interaction between these organelles.24, 25 The IP3R plays a critical role in this cross-talk and is currently considered as a major regulator of apoptotic signaling, which is also modulated by the members of the Bcl-2 family of proteins.24, 26, 27, 28 The IP3R has also been shown to regulate autophagy. Indeed, its pharmacological inhibition with xestospongins and its depletion by specific siRNAs, represents a strong stimulus for the induction of autophagy, an effect that can be reverted by ER-targeted Bcl-2 overexpression.29, 30 Moreover, it has been shown that the IP3R gene is required for the induction of ACD in Dyctiostelium discoideum.31, 32
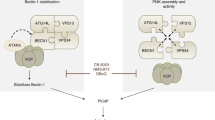
On the basis of these pieces of evidence, IP3R emerges as a possible key integrator of the cross-talk between apoptosis and autophagy. Here, we report that xestospongin B and nutrient starvation disrupt a molecular complex formed by the IP3R, Beclin 1 and Bcl-2, and present evidence that the IP3R represses autophagy through Bcl-2-mediated sequestration of Beclin 1.
Results and Discussion
Xestospongin B induces autophagy by binding to the IP3R in a Bcl-2-inhibitable fashion
The natural compound xestospongin B (purified from the marine sponge Xestospongia exigua) is an IP3R antagonist that induces autophagy when added to a variety of animal cell lines, incuding human cervical adenocarcinoma HeLa cells (Figure 1a–d and Supplementary videos), Rat-1 fibroblasts (from Rattus norvegicus; data not shown) and DT40 B-cell lymphoma cells (from Gallus gallus; Figure 1d). This was assessed by determining the distribution of green fluorescent protein-coupled microtubule-associated protein light chain 3 (GFP-LC3) to cytoplasmic puncta (Figure 1a and b) or the lipidation of LC3, which leads to an increase in its electrophoretic mobility (from LC3-I to LC3-II; Figure 1c and d). Importantly, these effects were inhibited by overexpression of Bcl-2 (Figure 1a–d) and were lost in DT40 cells depleted for all three IP3R isoforms through homologous recombination (Figure 1e and f). As a note, DT40 triple knockout cells remained responsive to nutrient starvation as their wild-type counterparts (Figure 1f), suggesting that at least two independent signaling pathways to autophagy – which are activated by distinct triggers – coexist in these cells. As described earlier,29 xestospongin B caused the dissociation of the Beclin 1/Bcl-2 complex that normally inhibits autophagy. This effect could be detected in coimmunoprecipitation experiments when xestospongin B was added to control DT40 cells, yet was undetectable in IP3R-deficient DT40 cells (Figure 1g and h).

Induction of autophagy by xestospongin B is dependent on the IP3R and Bcl-2. Bcl-2 inhibits xestospongin B-induced autophagy (a–d). Wild-type (WT) or Bcl-2 overexpressing HeLa cells transfected with GFP-LC3 were treated with xestospongin B (2 μM) for 4 h. Autophagy was monitored by assessing the intracellular redistribution of GFP-LC3 (a), and the percentage of vacuolizated cells was quantified (b). LC3 lipidation was compared between WT and Bcl-2 overexpressing HeLa cells by immunoblotting (c), and quantified (d) at the times indicated. The IP3R was necessary for xestospongin B-(4 h), but not starvation-mediated (2 h) LC3 lipidation, as observed in WT DT40 chicken B lymphocytes versus a triple knockout (TKO) DT40 cell line deficient for all IP3R isoforms (e and f). Bcl-2 and Beclin 1 interaction was monitored after 6 h of xestospongin B treatment in WT versus TKO DT40 cells (g), and the coimmunoprecipitated levels of Beclin 1 were normalized to input levels (h). Results are representative of at least three independent experiments. *P<0.05 and **P<0.01 as indicated for WT compared with genetically modified cells. The colour reproduction of this figure is available on the html full version of the manuscript
The redistribution of GFP-LC3 to dots or the accumulation of LC3-II may be a sign of an enhanced formation of autophagosomes or a reduced removal of autophagosomes.33, 34 To discriminate between these two possibilities, we blocked the fusion between autophagosomes and lysosomes (with bafilomycin A1) and/or inhibited lysosomal proteases (with pepstatin A). As an internal control of its efficacy, we ensured that bafilomycin A1 inhibited the colocalization of GFP-LC3 with the lysosomal marker Lamp 2A, which is an indicator of the autophagosome–lysosome fusion (Figure 2a and b). In these conditions, the addition of xestospongin B continued to induce an increase in GFP-LC3 puncta (Figure 2a and c) and in the generation of LC3-II (Figure 2d and e), which strengthens the notion that xestospongin B stimulates the initiation, rather than the late stages, of the autophagic flux.

On-rate autophagic flux induced by xestospongin B. HeLa cells transiently expressing GFP-LC3 were preincubated (30 min) with bafilomycin A1 and then subjected to xestospongin B treatment (4 h) or to nutrient starvation (2 h). Confocal images were acquired, the degree of colocalization between GFP-LC3 and the lysosomal marker Lamp 2a was quantified (a and b), and the percentage of cells with GFP-LC3 puncta was determined (c). A mixture of bafilomycin A1 (1 nM) and pepstatin A1 (10 μg/ml) was employed to inhibit the lysosomal degradation of LC3-II (d). LC3-II relative to GAPDH levels were quantified (e). *P<0.05; **P<0.01 as indicated. The colour reproduction of this figure is available on the html full version of the manuscript
Xestospongin B neither affected the basal levels nor the histamine-induced increase of IP3 (Figure 3a). In conditions in which the IMP inhibitor L-690 330 reduced IP3 levels, non-toxic doses of myo-inositol (which lead to an increase in IP3 concentrations) inhibited L-690 330-induced autophagy, but failed to suppress autophagy induced by xestospongin B (Figure 3b). From these results, we conclude that xestospongin B induces autophagosome formation through a Bcl-2-inhibitable effect on IP3R that does not involve major perturbations of inositol metabolism.

Autophagy induced by xestospongin B is IP3-independent. Intracellular levels of the transient messenger IP3 were measured with a radioreceptor assay under basal or histamine-induced peak conditions (a). The inset graphic depicts a typical time-response to identify the time of IP3 peak concentrations, as induced by histamine (100 μM) stimulation. Treatment with myo-inositol (10 μM, 10 min), L-690 330 (100 μM) and xestospongin B (2 μM) were compared under basal as well as peak conditions evoked by histamine (a). HeLa cells transiently expressing GFP-LC3 were treated with xestospongin B in the presence or absence of myo-inositol. The percentage of vacuolizated cells was determined at the indicated times (b). Data are presented as mean±S.E.M. of three independent triplicates. *P<0.05 and **P<0.01 versus unstimulated control (a) or as indicated (b)
A molecular complex involving the IP3R and Beclin 1 regulates autophagy in response to xestospongin B or starvation
Driven by the observation that Bcl-2 overexpression can inhibit xestospongin B-induced autophagy, we monitored the molecular interaction between the IP3R and Beclin 1, which so far has been indicated as the main target for Bcl-2-mediated autophagy inhibition.14, 15 In unstimulated cells, IP3R could be immunoprecipitated with Beclin 1 and vice versa. Shortly after addition of xestospongin B (usually within 3 h), this interaction was reduced (Figure 4a and b). As described earlier by our group, xestospongin B disrupted the interaction between Beclin 1 and Bcl-2,29 whereas it did not affect the co-immunoprecipitation between the IP3R and Bcl-2 (Figure 4c). This suggests that xestospongin B disrupts the interaction between Beclin 1 and the IP3R/Bcl-2 complex. Similar results were found when xestospongin B treatment was replaced by the most physiological inducer of autophagy, nutrient starvation (Figure 4d), and could also be detected in different cell lines, including human colon cancer HCT116 cells and Rat-1 fibroblasts (not shown). In cells that overexpress Bcl-2, the capacity of xestospongin B to disrupt the IP3R/Beclin 1 complex was reduced and delayed (Figure 4e). Importantly, when Bcl-2 was depleted by specific siRNAs (Figure 4f), the IP3R/Beclin 1 complex was disrupted in resting conditions (Figure 4g), further corroborating the notion that the interaction between IP3R and Beclin 1 is mediated by Bcl-2. Although mTOR phosphorylation and mTOR-mediated phosphorylation of p70S6K were inhibited by rapamycin, xestospongin B did not affect mTOR phosphorylation or p70S6K phosphorylation (Figure 5a). Moreover, mTOR inhibition with rapamycin, which induces autophagy, failed to disrupt the IP3R/Beclin 1 complex (Figure 5b). Altogether, these results strongly suggest that the inhibition of mTOR and that of the IP3R induce autophagy through mechanisms that can be fully separated.

A Bcl-2-dependent molecular complex comprising the IP3R and Beclin 1 regulates autophagy. Immunoprecipitation assays were carried out as indicated in Materials and Methods using wild-type (WT) (a–d) and Bcl-2 overexpressing HeLa cells (e), as well as HeLa cells in which Bcl-2 was depleted by specific siRNAs (g). Xestospongin B induces the dissociation of Beclin 1 from the IP3R (a) and vice versa (b), as well as the separation of Beclin-1 from Bcl-2, without affecting the interaction between the IP3R and Bcl-2 (c). Under nutrient starvation conditions, the interaction between Beclin 1 and the IP3R was also lost (d). In HeLa cells, stably overexpressing Bcl-2, the xestospongin B-mediated dissociation of Beclin 1 from the IP3R was reduced, compared with WT HeLa cells (e). Conversely, in HeLa cells depleted of Bcl-2 (f), the interaction between Beclin 1 and the IP3R was already lost in unstimulated conditions (g). In all experiments, coimmunoprecipitated protein levels were quantified relative to input protein levels. Data are presented as mean±S.E.M. of three independent triplicate assessments. *P<0.05 and **P<0.01 versus untreated controls

Xestospongin B effects are mTOR-independent. HeLa cells were treated with xestospongin B (2 μM) or rapamycin (1 μM) for the indicated periods and the levels of mTOR and p70S6K phosphorylation were assessed by immunoblotting (a). The Beclin 1/IP3R interaction was not modified by rapamycin treatment, in conditions in which xestospongin B caused its dissociation (b). Results are presented as mean±S.E.M. of three independent triplicate assessments. **P<0.01 versus untreated controls
As several IP3R-interacting proteins, including Bcl-2, have major effects on Ca2+ signaling,27 we tested whether Beclin 1 might also affect Ca2+ homeostasis, which reportedly has an important impact on autophagy.18, 19, 20, 21, 22 However, we did not detect any tangible effect of Beclin 1 depletion on IP3R agonist-induced Ca2+ fluxes (Figure 6a and b), steady state levels of Ca2+ in the ER lumen, thapsigargin-induced Ca2+ depletion from the ER (Figure 6c), or increases in cytosolic [Ca2+] upon inhibition of the sarco–ER Ca2+ ATPase (SERCA) with 2,5-di(ter-butyl)-1,4 benzohydroquinone (tBHQ; Figure 6d and e). This was assessed by using state-of-the-art aequorin-based Ca2+ sensors (Figure 6a and b), ERD1 cameleon Ca2+ sensor (Figure 6c) or a chemical Ca2+ probe (Figure 6d and e). Collectively, these data indicate that the IP3R can affect the autophagy-inducing function of Beclin 1, yet suggest that Beclin 1 itself does not play a major role in regulating Ca2+ fluxes governed by the IP3R.

Beclin 1 depletion does not affect Ca2+ homeostasis. HeLa cells were transfected with specific siRNAs targeting Beclin 1 or the unrelated protein emerin. After 48 h, cells were subjected to [Ca2+] measurements (a–e) or immunoblotting to check Beclin 1 levels (f). Histamine-evoked (1 and 100 μM) cytosolic [Ca2+] increases were measured using cytAEQ. Representative traces (a) and mean±S.E.M. peak values are shown for each condition (b). Steady-state ER Ca2+ levels and thapsigargin-induced ER Ca2+ depletion levels were measured with the FRET probe ERD1 and mean±S.E.M. values are shown (c). Cytosolic [Ca2+] after ER depletion with tBHQ (d), as well as steady-state cytosolic Ca2+ levels (e) were quantified using Fura-2. Representative traces and mean values±S.E.M. are shown, respectively. *P<0.05 versus culture medium with Ca2+. **P<0.01
The IP3R ligand-binding domain (IP3R-LBD) inhibits autophagy through an effect on the Beclin 1/Bcl-2 complex
A target for xestospongin B is the IP3R-LBD (aa 224–604), which is engaged in several protein–protein interactions;35, 36 and has a major effect on the regulation of ER-mitochondrial microdomains that affect Ca2+ signaling and the physical contact between ER and mitochondria.24 The transfection-enforced expression of the IP3R-LBD coupled to red fluorescent protein (RFP) strongly inhibited autophagy induced by xestospongin B or nutrient starvation (Figure 7a and b). This held so for either ER-targeted, outer mitochondrial membrane (OMM)-targeted and cytosolic (Cyto) variants of the IP3R-LBD coupled to RFP, all of which affect the ER–mitochondrial cross-talk.24 As negative controls, equally targeted versions of RFP (namely Cyto-ER- and OMM-RFP) failed to inhibit autophagy in this system, as assessed by measuring GFP-LC3 puncta (Figure 7a and b) or the accumulation of LC3-II (Figure 7e). Moreover, an inactive IP3R-LBD mutant (IP3R-LBD-9aaER-RFP) in which the insertion of a nonapeptide linker abolishes the interaction with endogenous IP3R,37 failed to affect xestospongin B-induced autophagy (Figure 7b). All the functional variants of the IP3R-LBD-RFP (Cyto-, OMM- and ER-targeted) but not IP3R-LBD-9aaER-RFP, interacted with Beclin 1 (not shown), and this interaction was reduced in the ER by depletion of Bcl-2 (Figure 7c and d). Altogether, these results indicate that IP3R-LBD is the moiety of the IP3R that mediates its interaction with Beclin 1 (through Bcl-2), and accounts for IP3R-mediated inhibition of autophagy.

Overexpression of the IP3R-LBD inhibits xestospongin B-induced autophagy through interaction with Beclin 1. HeLa cells were cotransfected with GFP-LC3 and three IP3R-LBD-RFP chimeric proteins targeted to different subcellular compartments, namely the cytosol (Cyto), the outer mitochondrial membrane (OMM) and the endoplasmic reticulum (ER). An inactive ER-targeted IP3R-LBD-RFP, including a non-apeptide linker (9aaER) as well as equally targeted variants of RFP alone were used as controls. Twenty-four hours after transfection, cells were subjected to treatment with xestospongin B or nutrient starvation. Representative pictures of Hoechst 33342-counterstained cells were taken 4 h after treatment (a) and the percentage of adherent cells exhibiting GFP-LC3 vacuolization into cytoplasmic puncta was determined (b). The interaction between the IP3R-LBD and Beclin 1 was assessed by immunoprecipitation experiments in HeLa cells that were transiently expressing IP3R-LBD-ER-RFP and concomitantly subjected to siRNA-mediated emerin or Bcl-2 knockdown as indicated (c). Coimmunoprecipitated Beclin 1 levels were normalized relative to input protein levels (d). LC3-II levels from the input lysates were normalized relative to GAPDH levels (e). Data are reported as mean±S.E.M. of three independent experiments carried out in triplicates. *P<0.05 and **P<0.01. The colour reproduction of this figure is available on the html full version of the manuscript
Conclusions
The results from this and several earlier studies,17, 18, 29, 30 indicate that IP3R agonists (such as IP3 itself) and IP3R antagonists (such as xestospongins) act as inhibitors and inducers of autophagy, respectively, through an effect on the IP3R. Thus, xestospongin B, which reportedly competes with IP3 for IP3R binding,38, 39 induces autophagy through the IP3R (in the sense that this effect is lost in IP3R knockout cells) rather than through an off-target effect. Although acute depletion of the IP3R by RNA interference is sufficient to induce autophagy in HeLa cells,29 permanent IP3R knockout by homologous recombination does not induce autophagy, at least in DT40 cells, presumably because these cells have adapted to the absence of the IP3R in a long-term selection process and express a truncated version of IP3R.40 Conversely, plasmid-driven overexpression of the IP3R-LBD inhibits autophagic vacuolization induced by both xestospongin and nutrient starvation, further confirming the major impact of the IP3R on the regulation of autophagy.
Although accumulating evidence point to the modulation of autophagy by the IP3R, the underlying mechanisms are still unclear. IP3R controls the agonist-induced release of Ca2+ from the ER lumen to the cytosol,35 and elevations of cytosolic Ca2+ have been involved in pharmacologically induced autophagy, as occurred in response to vitamin D analogs.22 Hence, the finding that IP3R inhibition with xestospongins induces autophagy (although it should reduce39 cytosolic Ca2+) suggests that the effects of the IP3R and/or IP3R ligands on autophagy cannot be explained only by a modulation of Ca2+ levels. Here, we show that the IP3R coimmunoprecipitates with Beclin 1, suggesting another mechanism through which the IP3R might act as an endogenous inhibitor of autophagy. Indeed, we found that IP3R binding by its antagonist xestospongin B, as well as physiological induction of autophagy by nutrient starvation, disrupted the interaction between the IP3R and Beclin 1. As this interaction was abolished by the knockdown of Bcl-2, it is likely that the IP3R and Beclin 1 interact through an indirect link established by Bcl-2.
In the triangular game among the IP3R, Beclin 1 and Bcl-2, the IP3R-LBD (which is the site of interaction with IP3 and xestospongins) appears as the most important domain for the regulation of autophagy. Thus, the IP3R-LBD (which itself has no function as a Ca2+ channel)41 can inhibit autophagy irrespective of its precise subcellular localization or topology, once more arguing against the idea that modified Ca2+ fluxes account for the effects of IP3R on autophagy. We were unable to detect any effects of Beclin 1 knockdown on Ca2+ fluxes at the ER membrane, which suggests that Beclin 1 affects autophagy regulation mainly as an allosteric regulator of Vps34 (and perhaps other yet-to-be-discovered enzymatic activities and molecular motors), rather than through gross effects on Ca2+ handling.
In synthesis, the results contained in this study identify the IP3R as a new, unsuspected regulator of the Beclin 1 complex that bridges signals converging on the ER and initial phagophore formation. Although the molecular details of this cross-talk require further exploration, our data point to the presence of an intriguing regulatory network in which the conformation of the IP3R, as influenced by agonists and/or antagonists acting on its LBD, regulates the initiation of autophagy through an effect on Beclin 1.
Materials and Methods
Cells and treatments
Wild-type, Bcl-2 overexpressing and GFP-LC3 overexpressing derivatives of the human cervix adenocarcinoma HeLa cell line, as well as the rat fibroblast cell line Rat-1, were grown in Glutamax-containing Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10 mM HEPES buffer. Wild-type and IP3R-triple deficient chicken lymphoma DT40 cells were maintained in Glutamax-containing RPMI 1640 medium supplemented with 10% FBS, 1% chicken serum, 10 mM HEPES buffer and 10 μM 2-mercaptoethanol. All cell lines were cultured in the presence of 100 U/ml penicillin G and 100 μg/ml streptomycin, at 37 °C (5%) CO2. All media and supplements for cell culture were purchased from Gibco-Invitrogen (Carlsbad, USA). For serum and amino-acid starvation, cells were cultured in serum-free Earle's Balanced Salt Solution medium (Sigma-Aldrich, St. Louis, MO, USA), a condition that we refer to as ‘nutrient starvation’. All cell lines were seeded in 6-, 12- or 24-well plates and grown for 24 h before treatments. Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich. Bafilomycin A1 (1 nM), histamine (1–100 μM), L-690 330 (100 μM; Tocris, Bristol, UK), myo-inositol (10 μM; Calbiochem, Darmstadt, Germany), pepstatin A (10 μg/ml) and xestospongin B (2 μM, extracted from the marine sponge Xestospongia exigua as described earlier38, 42) were added for 0–8 h, as indicated.
Dynamic in vivo [Ca2+] measurements
Basal and 2,5-di(ter-butyl)-1,4 benzohydroquinone (tBHQ)- or histamine-induced cytosolic Ca2+ signals were measured using either Fura-2 or the cytosolic version of the recombinant Ca2+ sensor aequorin (cytAEQ). All measurements were carried out in Krebs–Ringer modified buffer (KRB): 135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 5.5 mM glucose, 20 mM HEPES (pH=7.4), supplemented with 1 mM CaCl2. HeLa cells were loaded with 3 μM Fura-2/AM in KRB for 20 min at 37 °C and 10 min at RT. Cells were then perfused with KRB followed by 100 μM tBHQ, and dye calibration was performed using ionomycin (1 μM, the highest peak value) and ionomycin/EDTA (1 μM/1 mM, the lowest peak value). Changes in emission at 520 nm after sequential excitation at 340 and 380 nm were recorded and calibrated into [Ca2+]c from the ratio of emitted fluorescence, on the basis of an earlier described procedure that used a dissociation constant of 224 nM for Fura-2.43 HeLa cells transiently expressing cytAEQ were administrated with coelenterazine and transferred to a perfusion chamber. The light signal was collected in a purpose-built luminometer and calibrated into [Ca2+]c values as described earlier.44 To quantify the Ca2+ content of the ER, the fluorescence resonance energy transfer (FRET)-based ER-targeted ERD1 probe45 was imaged using a Zeiss LSM 510 Meta laser confocal system (Carl Zeiss AG, Oberkochen, Germany). The probe was excited by a 405 nm laser diode, emission spectra were acquired at 420–600 nm and the yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) signals were obtained by unmixing the spectrum on the basis of previously registered spectra of separate CFP and YFP proteins, as well as the autofluorescence of non-transfected cells. FRET efficiency, which is a function of ER luminal [Ca2+], was quantified using the acceptor bleaching method.45 Briefly, after five acquisitions, YFP was bleached (at both 488 and 514 nm excitation wavelengths, typically by about 80–90%), followed by acquisition of further five image spectra. Reduction of the YFP signal leads to an increase in the CFP signal, which is normalized to the decrease of YFP intensity during bleaching. The normalized increase of CFP intensity is presented as FRET efficiency.
Immunoblots and immunoprecipitation
All cell lines (4 × 106 cells) were washed with PBS and lysed as describe earlier.46 For immunoblotting, 50 μg of protein were separated onto NuPAGE gels (Invitrogen) and transferred to Immobilion-PSQ PVDF membranes (Millipore Corporation, Billerica, MA, USA). Membranes were incubated for 1 h in PBS-Tween 20 (0.05%) containing 5% BSA. Primary antibodies specific for Bcl-2, Beclin 1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), IP3R-I (Calbiochem), IP3R types I-II-III (Santa Cruz), LC3B, mTOR, p70S6K, phospho-mTOR or phospho-p70S6K (Cell Signaling, Danvers, MA, USA) were incubated overnight at 4 °C and revealed with the appropriate horseradish peroxidase-labeled secondary antibodies (SouthernBiotech, Birmingham, AL, USA) by means of the SuperSignal West Pico chemoluminiscent substrate (Pierce, Rockford, IL, USA). An antibody recognizing GAPDH (Chemicon) was used to control equal loading of lanes. For immunoprecipitation, extracts from HeLa cells (8 × 106 cells) were lysed and 400 μg of protein were precleared for 1 h with 15 μl of Protein G Sepharose 4 Fast Flow (GE Healthcare, Piscataway, NJ, USA), and subsequently incubated for 3 h in the presence of anti-Beclin 1 antibody, anti-IP3R-I, anti-RFP (Abcam, Cambridge, UK) and anti-Bcl-2 or immunoglobulin control. Immunoprecipitation immunoblotting was carried out using TrueBlot-HRP (eBioscience, San Diego, CA, USA) secondary antibodies. Quantitative analysis of immunoblots was carried out by using the open source ImageJ software (freely available at http://rsbweb.nih.gov/ij/index.html). Results are presented as mean±S.E.M. Three independent experiments were carried out in triplicate, and statistical significance was evaluated by Student's t-test.
Immunofluorescence microscopy
For immunofluorescence staining, cells were fixed with paraformaldehyde (4% w/v, 20 min), permeabilized with Triton X-100 (0.3% in PBS, 30 min), blocked with 3% BSA and incubated overnight at 4°C with a specific antibody against Lamp 2a (200 ng/ml; Santa Cruz). Nuclei were counterstained with 10 μg/ml Hoechst 33342 (Molecular Probes-Invitrogen, Carlsbad, CA, USA). Conventional fluorescence microscopy was performed with a Leica IRE2 microscope equipped with a DC300F camera (Leica Microsystems, Wetzlar, Germany). Confocal microscopy was carried out with a Leica TSC-SPE microscope equipped with a 63 × /1.15 Olympus objective by using LAS software (Leica Microsystems). Cells presenting a diffuse distribution of GFP-LC3 in the cytoplasm and nucleus were considered as non-autophagic, whereas cells exhibiting both several intense punctuate GFP-LC3 aggregates and nuclear GFP-LC3 exclusion were classified as autophagic (GFP-LC3vac). Each GFP-LC3 staining was independently quantified by two investigators (JMV and CO).
Measurement of intracellular IP3 levels
HeLa cells (4 × 106) were seeded in 100 mm culture dishes and were incubated after 24 h with L-690 330 (100 μM), myo-inositol (10 μM) and/or xestospongin B (2 μM) for 0–100 min. Immediately afterwards, they were stimulated with histamine (100 μM) for 0–3 min and lysed with 0.2 M ice-cold trichloroacetic acid. Cell extracts were then subjected to a radioreceptor assay (Perkin Elmer no. NEK064, Waltham, MA, USA). IP3 levels were quantified in a liquid scintillation counter (Packard Tri-carb 2100TR, Packard Instrument, Meriden, CT, USA) and normalized to the amount of protein contained in each sample. Results are presented as mean±S.E.M. Three independent experiments were carried out in triplicate and statistical significance was evaluated by Student's t-test.
Plasmids, transfection and RNA interference
Cells were cultured in 6-well plates and transfected at 80% confluence by using Oligofectamine reagent (Invitrogen), with small interfering RNAs (siRNAs) specifically targeting human Beclin 1 (sense 5′-GAUUGAAGACACAGGAGGC-3),47, 48 Bcl-2 (sense 5′-GCUGCACCUGACGCCCUUCTT-3′)47, 49 or the unrelated protein emerin (sense 5′-TATGTCCTCCTCATCATCTTCCT-3′).50 All siRNAs were purchased from Sigma-Proligo. Transient transfections with plasmids were carried out with Lipofectamine 2000 reagent (Invitrogen) and cells were used 24 h after transfection. Cells were transfected with an empty control vector or with a plasmid encoding for GFP-LC3.51 Cotransfection was carried out using a mixture of the GFP-LC3-encoding plasmid and plasmids encoding for variants of the IP3R-LBD coupled to RFP targeted to different subcellular compartments, namely the cytosol (IP3R-LBD-RFP-Cyto), the OMM (IP3R-LBD-RFP-OMM) and the ER (IP3R-LBD-RFP-ER). As controls, a plasmid coding for an ER-targeted IP3R-LBD that is rendered inactive by the incorporation of a nonapeptide linker sequence (IP3R-9aa-LBD-RFP-ER), as well as plasmids encoding RFP alone targeted to the cytosol (RFP-Cyto), the OMM (RFP-OMM) or the ER (RFP-ER) were used.24, 37, 52
Abbreviations
- ACD:
-
autophagic cell death
- Ambra:
-
activating molecule in Beclin 1-regulated autophagy
- Atg:
-
autophagy-related gene
- Ca2+:
-
calcium
- [Ca2+]:
-
Ca2+ concentration
- [Ca2+]c:
-
cytosolic [Ca2+]
- CFP:
-
cyan fluorescent protein
- cytAEQ:
-
cytosolic aequorin
- Cyto:
-
cytosolic
- ER:
-
endoplasmic reticulum
- FBS:
-
fetal bovine serum
- FRET:
-
fluorescence resonance energy transfer
- GFP:
-
green fluorescent protein
- GFP-LC3:
-
GFP-coupled microtubule-associated protein light chain 3
- IMP:
-
inositol monophosphatase
- IP3:
-
myo-inositol 1,4,5-trisphosphate
- IP3R:
-
IP3 receptor
- LBD:
-
ligand-binding domain
- mTOR:
-
mammalian target of rapamycin
- OMM:
-
outer mitochondrial membrane
- PI3K:
-
phosphatidylinositol 3-kinase
- PI3P:
-
phosphatidylinositol-3-phosphate
- RFP:
-
red fluorescent protein
- SERCA:
-
sarco–endoplasmic reticulum Ca2+ ATPase
- tBHQ:
-
2,5-di(ter-butyl)-1,4 benzohydroquinone
- UVRAG:
-
UV irradiation resistance-associated tumor suppressor gene
- Vps:
-
vacuolar protein sorting
- YFP:
-
yellow fluorescent protein
References
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721.
Yorimitsu T, Klionsky DJ . Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12 (Suppl 2): 1542–1552.
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ . Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6: 304–312.
Klionsky DJ, Cregg JM, Dunn Jr WA, Emr SD, Sakai Y, Sandoval IV et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5: 539–545.
Xie Z, Klionsky DJ . Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9: 1102–1109.
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999; 59: 59–65.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N . Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100: 15077–15082.
Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G . To die or not to die: that is the autophagic question. Curr Mol Med 2008; 8: 78–91.
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14: 1237–1243.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11.
Levine B, Sinha S, Kroemer G . Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4: 600–606.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26: 2527–2539.
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B . JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30: 678–688.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–1111.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305.
Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO . Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993; 268: 26107–26112.
Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA . The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J 2007; 274: 3184–3197.
Gao W, Ding WX, Stolz DB, Yin XM . Induction of macroautophagy by exogenously introduced calcium. Autophagy 2008; 4: 754–761.
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25: 193–205.
Berridge MJ, Bootman MD, Roderick HL . Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003; 4: 517–529.
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006; 175: 901–911.
Szabadkai G, Rizzuto R . Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett 2004; 567: 111–115.
Berridge MJ . The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 2002; 32: 235–249.
Rong Y, Distelhorst CW . Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 2008; 70: 73–91.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14: 1029–1039.
Criollo A, Vicencio JM, Tasdemir E, Maiuri MC, Lavandero S, Kroemer G . The inositol trisphosphate receptor in the control of autophagy. Autophagy 2007; 3: 350–353.
Lam D, Golstein P . A specific pathway inducing autophagic cell death is marked by an IP3R mutation. Autophagy 2008; 4: 349–350.
Lam D, Kosta A, Luciani MF, Golstein P . The Inositol 1,4,5-Trisphosphate Receptor Is Required to Signal Autophagic Cell Death. Mol Biol Cell 2008; 19: 691–700.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–175.
Mizushima N, Yoshimori T . How to interpret LC3 immunoblotting. Autophagy 2007; 3: 542–545.
Mikoshiba K . IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 2007; 102: 1426–1446.
Mikoshiba K . The IP3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp 2007: 74: 9–22.
Varnai P, Balla A, Hunyady L, Balla T . Targeted expression of the inositol 1,4,5-triphosphate receptor (IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R channels. Proc Natl Acad Sci USA 2005; 102: 7859–7864.
Jaimovich E, Mattei C, Liberona JL, Cardenas C, Estrada M, Barbier J et al. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108-15) cells. FEBS Lett 2005; 579: 2051–2057.
Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF et al. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 1997; 19: 723–733.
Guillemette J, Caron AZ, Regimbald-Dumas Y, Arguin G, Mignery GA, Boulay G et al. Expression of a truncated form of inositol 1,4,5-trisphosphate receptor type III in the cytosol of DT40 triple inositol 1,4,5-trisphosphate receptor-knockout cells. Cell Calcium 2005; 37: 97–104.
Devogelaere B, Verbert L, Parys JB, Missiaen L, De Smedt H . The complex regulatory function of the ligand-binding domain of the inositol 1,4,5-trisphosphate receptor. Cell Calcium 2008; 43: 17–27.
Quirion JC, Sevenet T, Husson HP, Weniger B, Debitus C . Two new alkaloids from Xestospongia sp., a New Caledonian sponge. J Nat Prod 1992; 55: 1505–1508.
Grynkiewicz G, Poenie M, Tsien RY . A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985; 260: 3440–3450.
Chiesa A, Rapizzi E, Tosello V, Pinton P, de Virgilio M, Fogarty KE et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem J 2001; 355: 1–12.
Palmer AE, Jin C, Reed JC, Tsien RY . Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA 2004; 101: 17404–17409.
Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 2005; 118: 3091–3102.
Criollo A, Galluzzi L, Maiuri MC, Tasdemir E, Lavandero S, Kroemer G . Mitochondrial control of cell death induced by hyperosmotic stress. Apoptosis 2007; 12: 3–18.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
Jiang M, Milner J . Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev 2003; 17: 832–837.
Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K . Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 2001; 114: 4557–4565.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Lin X, Varnai P, Csordas G, Balla A, Nagai T, Miyawaki A et al. Control of calcium signal propagation to the mitochondria by inositol 1,4,5-trisphosphate-binding proteins. J Biol Chem 2005; 280: 12820–12832.
Acknowledgements
GK is supported by the Ligue Nationale contre le Cancer (équipe labellisée), European Commission (RIGHT, ChemoRes, Apop-Train, Apo-SYs), Agence Nationale pour la Recherche, Cancéropôle Ile-de-France and Institut National contre le Cancer (INCa). We thank the International Collaboration Program ECOS-CONICYT, project C08S01. JMV is supported by Fondation pour la Recherche Médicale and CONICYT. CO holds a PhD scholarship from CONICYT, Chile. OK is supported by EMBO. AWEJ is a Medical Research Council student. We also thank Michael R Duchen (University College London) for the use of confocal facility (Zeiss LSM 510 Meta) and discussions, as well as to Dr. Roger Y Tsien (University of California at San Diego) for the donation of ERD1.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by D Rubinsztein
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Vicencio, J., Ortiz, C., Criollo, A. et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006–1017 (2009). https://doi.org/10.1038/cdd.2009.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.34
Keywords
This article is cited by
-
Autophagy and autophagy signaling in Epilepsy: possible role of autophagy activator
Molecular Medicine (2023)
-
The dual roles of autophagy and the GPCRs-mediating autophagy signaling pathway after cerebral ischemic stroke
Molecular Brain (2022)
-
Pitavastatin activates mitophagy to protect EPC proliferation through a calcium-dependent CAMK1-PINK1 pathway in atherosclerotic mice
Communications Biology (2022)
-
Palmitate reduces starvation-induced ER stress by inhibiting ER-phagy in hypothalamic cells
Molecular Brain (2021)
-
IL-1β receptor antagonist (IL-1Ra) combined with autophagy inducer (TAT-Beclin1) is an effective alternative for attenuating extracellular matrix degradation in rat and human osteoarthritis chondrocytes
Arthritis Research & Therapy (2019)
