
Abstract
Asthma is a common allergic disorder involving a complex interplay among multiple genetic and environmental factors. Recent studies identified genetic variants of human GAB1 as a novel asthma susceptibility factor. However, the functions of Gab1 in lung remain largely unexplored. In this study, we first observed an elevation of Gab1 level in peripheral blood mononuclear cells from asthmatic patients during acute exacerbation compared with convalescence. Mice with a selectively disrupted Gab1 in myeloid dendritic cells (mDCs) considerably attenuated allergic inflammation in experimental models of asthma. Further investigations revealed a prominent reduction in CCL19-mediated migration of Gab1-deficient mDCs to draining lymph nodes and subsequent impairment of Th2-driven adaptive activation. Mechanistically, Gab1 is an essential component of the CCL19/CCR7 chemokine axis that regulates mDC migration during asthmatic responses. Together, these findings provide the first evidence for the roles of Gab1 in lung, giving us deeper understanding of asthmatic pathogenesis.
Similar content being viewed by others


Introduction
Bronchial asthma is a common chronic inflammation with well-defined pathological features including intermittent airway hyperresponsiveness (AHR), pulmonary eosinophil infiltration, and excessive mucus secretion1,2. There is currently no proven cure for this allergic lung disease, which is caused primarily by intricate interactions between genetic susceptibility and environmental influences3,4. Numerous studies have demonstrated that allergic asthma is driven predominantly by a Th2 type of immune response in both human and mouse models of asthma5. This suggests that a therapeutic option targeting Th2-driven allergic inflammation would be valuable for treating asthma6. Dendritic cells (DCs), the most potent antigen-presenting cells (APCs), play an essential role in allergic asthma by inducing Th2 immunity to inhaled allergens. Following persistent antigen uptake and recognition, respiratory DCs are recruited and migrate to draining lymph nodes (DLNs), activating naive T cells and initiating the adaptive immune response7,8. Thus, there is an urgent need to elucidate the molecular mechanisms controlling DCs in asthmatic inflammation.
DCs are the most competent myeloid-lineage cells, linking the innate and adaptive immune responses. Conceptually, they are categorized into two major subsets: myeloid DCs (mDCs) and plasmacytoid DCs (pDCs). pDCs are characterized by their ability to produce abundant type-1 interferon, thus contributing to antiviral responses; they play a major role in innate immunity but a limited role in antigen presentation. Conversely, mDCs are important in allergic asthma because they have the ability to phagocytose the antigen and subsequently activate naive T cells in the lymph nodes8,9,10. DCs at mucosal surfaces are one of the first lines of defense to counter inhaled allergens during the early onset of allergic inflammation. In response to a variety of environmental allergens, these immature DCs mature into activated APCs upon antigen capture. Activation of DCs triggers their migration from lung to mediastinal lymph nodes, where they present processed antigen to naive T cells and induce their polarization into Th2 cells, eventually driving allergic inflammation8,11. The fact that mDC function is crucial to the etiology of chronic asthma is well established, and elucidating the signaling events involved in DC function could help guide the development of novel therapeutic options for allergic asthma.
Tyrosine phosphorylation has received considerable attention in asthma research over the past few years12,13,14. In general, this protein modification is post-transcriptionally catalyzed by tyrosine kinases and phosphatases in a highly flexible manner15. More importantly, this reversible phosphorylation is finely orchestrated by various scaffolding proteins which confer signal diversity, specificity and integration. These scaffolding proteins are apparently devoid of catalytic domains, and in most cases, they serve as adaptors by providing multiple tyrosine phosphorylation sites that function as binding sites for kinases and phosphatases16,17. The present study was designed to characterize a novel function of Gab1 in DC-mediated asthmatic inflammation. Gab1 belongs to the Gab family of scaffolding molecules (Gab1, Gab2, and Gab3 in mammals). It has been documented that Gab1 is rapidly phosphorylated upon stimulation by many extracellular stimuli, and subsequently activates Erk/MAPK, PI3K/Akt, and JAK/STAT pathways through direct binding with the phosphatase Shp2, members of the Crk kinase family, or the p85 subunit of PI3K18,19,20. Recently, Gab1 has attracted much interest because genetic variants of human GAB1 have been identified as a risk factor for asthma21. Gab1 is ubiquitously expressed at a relatively low level, except for elevated expression in the heart and blood22. In this study, we found an elevated Gab1 level in peripheral blood mononuclear cells (PBMCs) collected from asthmatic patients during acute exacerbation. PBMCs originating from a common myeloid lineage in the bone marrow are the main source of inflammation mediators. In asthmatic inflammation, PBMCs are recruited and circulate in inflamed peripheral sites, where they further differentiate into macrophages and DCs23. Thus, it has been hypothesized that myeloid Gab1 may participate in the pathogenesis of allergic asthma. Using genetic approaches, we found striking protection from ovalbumin (OVA)- and house dust mite (HDM)-induced asthma in mice lacking Gab1 in their myeloid cells. Further experiments suggested that this effect was a consequence of loss of Gab1 in mDCs rather than in macrophages. We identified Gab1 as a critical signaling component of the CCL19-mediated C-C chemokine receptor 7 (CCR7) signaling pathway, which thereby contributes to DC migration in the development of asthmatic inflammation. This result highlights the importance of Gab1 as a clinically-relevant genetic risk factor for asthma and suggests a novel therapeutic strategy for targeting the pathogenesis of asthma.
Results
Myeloid Gab1-deficient mice are protected from OVA- and HDM-induced allergic asthma
We first conducted a paired comparison study to assess the changes of Gab1 levels in PBMCs from patients suffering from recurrent asthma. It was evident that the level of Gab1 increased during exacerbation compared with convalescence, while the level of Gab2 was unaltered (Figure 1A). This clinical finding implies a functional requirement of Gab1 for asthmatic inflammation. In addition, mice were sensitized and challenged by OVA to induce asthma. We observed an increase in both mRNA and protein levels of Gab1 in PBMCs from OVA-sensitized mice compared with unsensitized mice (Figure 1B and 1C). Taken together, our findings support the potential clinical importance of Gab1 in the myeloid lineage in driving asthmatic inflammation.

Gab1 expression is increased in PBMCs during acute exacerbation in asthmatic patients and mice. (A) Gab1 expression was enhanced in PBMCs from asthmatic patients during acute exacerbation compared with convalescence, while Gab2 expression did not change. Each dot represents one patient. n = 20/group, **P < 0.01. (B) A significant increase in Gab1 expression occurred in PBMCs from asthmatic mice. In contrast, there was no significant change in Gab2 expression. n = 9/group, *P < 0.05. (C) Gab1 and Gab2 protein levels in asthmatic mice were examined by western blot. Each sample represents three mice.
To determine the physiological role of Gab1 in asthma, we generated mice with a conditional Gab1 deletion in myeloid cells (LysMcreGab1fl/fl). Conditional disruption of Gab1 was confirmed by western blot analysis (Supplementary information, Figure S1A). LysMcreGab1fl/fl mice appeared to be normal, with weights and lifespan similar to littermate controls. Histological evaluation also revealed no signs of pathological changes and inflammation in LysMcreGab1fl/fl lungs (Supplementary information, Figure S1B-S1D).
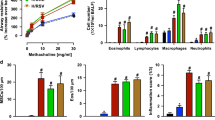
To test our hypothesis, we first examined the effect of Gab1 deficiency on asthmatic injury. Mice were challenged with OVA or HDM to induce asthma and as expected, Th2 responses were augmented: remarkable increases in secreted IL-4, IL-5 and IL-13 were observed in bronchoalveolar lavage fluid (BALF) of OVA- and HDM-sensitized mice compared with saline-treated mice. In contrast, we found that Th2 responses in the LysMcreGab1fl/fl asthmatic model were dramatically reduced (Figure 2A and 2G). Meanwhile, we measured IFN-γ levels in the BALF and the results showed that this protein was increased in LysMcreGab1fl/fl mice challenged with OVA or HDM, which suggest that this altered signaling contributed to the phenotypic switch from predominant Th2 to Th1 (Supplementary information, Figure S2). Furthermore, the total number of leukocytes in BALF was determined by cell counting, and a remarkable reduction was found in LysMcreGab1fl/fl mice (Figure 2B and 2H). Differential cell counts revealed a notable decrease in the number of eosinophils and slight decreases in macrophages, lymphocytes, and neutrophils in LysMcreGab1fl/fl mice compared with Gab1fl/fl mice (Figure 2C and 2I). To further assess the effect of Gab1 on allergic inflammatory responses, the lungs were removed and sectioned for histochemical analysis. Periodic acid-Schiff (PAS) staining showed reduced mucus production in the airways of LysMcreGab1fl/fl mice (Figure 2D, 2E, 2J and 2K). Meantime, immunohistochemical analysis of Muc5ac, which is a prominent component of airway mucus and commonly used to evaluate airway inflammation24, indicated a notable attenuation of mucus production and airway remodeling in LysMcreGab1fl/fl mice compared with controls (Figure 2F and 2L). Collectively, our findings showed that Gab1 positively regulates OVA- and HDM-stimulated allergic asthma and that myeloid Gab1 deficiency may protect mice from asthmatic attacks.

Gab1 deficiency in myeloid-derived cells attenuates OVA- and HDM-stimulated allergic airway inflammation. (A) ELISA results showing that levels of Th2 cytokines (IL-4, IL-5 and IL-13) in BALF were reduced in OVA-challenged LysMcreGab1fl/fl mice. n = 6 or 10/group, **P < 0.01. (B, C) Total and differential leukocyte counts in BALF from saline- or OVA-challenged Gab1fl/fl and LysMcreGab1fl/fl mice. n = 6 or 10/group, **P < 0.01, ***P < 0.001. (D) OVA-induced mucus production (visualized by PAS-positive staining) was decreased in LysMcreGab1fl/fl mice. Scale bar, 100 μm; n = 6 or 10/group. (E) Percentages of PAS-positive cells in airway epithelial cells. n = 6 or 10/group, **P < 0.01. (F) Decreased Muc5ac expression in LysMcreGab1fl/fl mice treated with OVA. Scale bar, 100 μm; n = 6 or 10/group. (G) ELISA results showing that levels of Th2 cytokines (IL-4, IL-5 and IL-13) in BALF were reduced in HDM-challenged LysMcreGab1fl/fl mice. n = 4/group, **P < 0.01. (H, I) Total and differential leukocyte counts in BALF from saline- or HDM-challenged Gab1fl/fl and LysMcreGab1fl/fl mice. n = 4/group, *P < 0.05. (J) HDM-induced mucus production (visualized by PAS-positive staining) was decreased in LysMcreGab1fl/fl mice. Scale bar, 100 μm; n = 4/group. (K) Percentages of PAS-positive cells in airway epithelial cells. n = 4/group, **P < 0.01. (L) Decreased Muc5ac expression in LysMcreGab1fl/fl mice treated with HDM. Scale bar, 100 μm; n = 4/group. Eos, eosinophils; Macs, macrophages; Lymphs, lymphocytes; Neuts, neutrophils.
Adoptive transfer of wild-type BMDCs reconstitutes allergic inflammation in LysMcreGab1fl/fl mice
In OVA-immunized LysMcreGab1fl/fl mice, we also noted a reduction of CD11c+ MHC-II+ cells in the mediastinal lymph nodes (MLN; Supplementary information, Figure S3), where Th2 responses are primed. CD11c+ MHC-II+ cells include macrophages and DCs. Therefore, adoptive transfer experiments were performed to determine which subtypes of myeloid cells contributed to the attenuation of asthmatic inflammation in Gab1-deficient mice. Briefly, BMDCs or BMDMs prepared from Gab1fl/fl and LysMcreGab1fl/fl donor mice were adoptively transferred into Gab1fl/fl and LysMcreGab1fl/fl recipients. LysMcreGab1fl/fl mice that received wild-type BMDCs exhibited Th2 responses and asthmatic injury like Gab1fl/fl mice following OVA challenge (Figure 3A-3C). This phenotypic reconstitution in LysMcreGab1fl/fl mice was demonstrated by Th2 cytokines (Figure 3A), total number of leukocytes (Figure 3B), and differential counts of leukocytes (Figure 3C) in the BALF. In contrast, Gab1fl/fl mice receiving Gab1-deficient BMDCs appeared similar to LysMcreGab1fl/fl mice after OVA administration, with lower Th2 responses and attenuated leukocyte infiltration (Figure 3A-3C). Consistent with these results, histological analysis of asthmatic airway remodeling suggested that the adoptive transfer of wild-type BMDCs restored OVA-induced mucus secretion in LysMcreGab1fl/fl mice (Figure 3D). Meanwhile, BMDMs from Gab1fl/fl and LysMcreGab1fl/fl donor mice were adoptively transferred to LysMcreGab1fl/fl and Gab1fl/fl recipients, respectively, and indicators of allergic asthma were assessed. As shown in Figure 3E-3H, Th2 cytokine secretion (Figure 3E), total leukocytes (Figure 3F) and differential leukocyte counts (Figure 3G) in BALF, as well as mucus production (Figure 3H) remained decreased in OVA-challenged LysMcreGab1fl/fl mice compared with controls after transfer of wild-type BMDMs. Furthermore, transfer of Gab1-deficient BMDMs to control mice had no apparent influence on the development of asthma (Figure 3E-3H). Collectively, this result clearly defines a unique function of Gab1 in mDCs rather than macrophages in the regulation of allergic asthma.

Normal BMDCs, rather than BMDMs, reconstitute allergic inflammation in LysMcreGab1fl/fl mice. (A) ELISA results (IL-4, IL-5 and IL-13 in the BALF) showing that LysMcreGab1fl/fl mice that received wild-type BMDCs exhibited normal Th2 responses and asthmatic injury, while the Th2 cytokine level was lower when control mice received Gab1-deficient BMDCs. n = 4/group, **P < 0.01. (B, C) Total and differential leukocyte counts were unchanged in OVA-challenged LysMcreGab1fl/fl mice compared with controls after transfer of wild-type BMDCs. On the contrary, the number of leukocytes was decreased when control mice received Gab1-deficient BMDCs. n = 4/group, *P < 0.05. (D) OVA-induced mucus production (visualized by PAS-positive staining) was unchanged in OVA-challenged LysMcreGab1fl/fl mice compared with controls after transfer of wild-type BMDCs, while mucus production was reduced when control mice received Gab1-deficient BMDCs. Scale bar, 100 μm; n = 4/group. (E) Th2 cytokine levels (IL-4, IL-5 and IL-13) in BALF were still attenuated in OVA-challenged LysMcreGab1fl/fl mice compared with controls after transfer of wild-type BMDMs, while Th2 cytokine level was unchanged in control mice receiving Gab1-deficient BMDMs compared with those receiving normal BMDMs. n = 4/group, *P < 0.05, ***P < 0.001. (F, G) Total and differential leukocyte counts were lower in OVA-challenged LysMcreGab1fl/fl mice than in controls after transfer of normal BMDMs. On the contrary, the number of leukocytes remained unchanged in control mice receiving Gab1-deficient BMDMs compared with those receiving normal BMDMs. n = 4/group, *P < 0.05, **P < 0.01, ***P < 0.001. (H) OVA-induced mucus production (visualized by PAS-positive staining) was still blocked in OVA-challenged LysMcreGab1fl/fl mice compared with controls after transfer of normal BMDMs, while mucus production was unchanged in control mice receiving Gab1-deficient BMDMs compared with those receiving normal BMDMs. Scale bar, 100 μm; n = 4/group.
Gab1 deficiency in DCs relieves allergic asthma
To further verify our finding, CD11c-CreGab1fl/fl mice were generated by crossing floxed Gab1 (Gab1fl/fl) mice with CD11c-Cre mice, in which Gab1 was conditionally knocked out in CD11c-positive DCs. These mice were treated with OVA and the allergic inflammation was assessed as discribed above. We also found relieved asthmatic injury in CD11c-CreGab1fl/fl mice compared with control mice, including reduced Th2 inflammation factor secretion (Figure 4A), total leukocytes (Figure 4B), and differential leukocyte counts (Figure 4C) in BALF. In addition, the goblet cell metaplasia and mucus production in airways were also blocked (Figure 4D and 4E). These results further proved the unique role of Gab1 in regulating DC-mediated allergic asthma.

Gab1 deficiency in DCs relieves OVA-induced allergic asthma. (A) ELISA results showing that levels of Th2 cytokines (IL-4, IL-5 and IL-13) in BALF were reduced in OVA-challenged CD11c-CreGab1fl/fl mice. n = 3 or 7/group, *P < 0.05, **P < 0.01, ***P < 0.001. (B, C) Total and differential leukocyte counts in BALF from saline- or OVA-challenged Gab1fl/fl and CD11c-CreGab1fl/fl mice. n = 3 or 7/group, *P < 0.05, **P < 0.01. (D) OVA-induced mucus production (visualized by PAS-positive staining) was decreased in CD11c-CreGab1fl/fl mice. Scale bar, 100 μm; n = 3 or 7/group. (E) Percentages of PAS-positive cells in airway epithelial cells. n = 3 or 7/group, **P < 0.01.
Gab1-deficient mDCs exhibit normal maturation, antigen uptake and antigen presentation
The scaffolding protein Gab1 has been reported as a crucial signaling element involved in many aspects of cell physiology25,26,27,28. In allergic asthma, the functions of mDCs vary greatly at different stages of development, from their origin in the bone marrow and migration into the circulation to their full maturation in the lymph nodes.
To further investigate the role of Gab1 in mDC functions, we next explored whether loss of Gab1 influences mDC maturation or antigen uptake and presentation. First, immature DCs were harvested and induced to mature following OVA stimulation in vitro. CD80, CD86, and MHC-II on mDCs as markers of maturation were analyzed by flow cytometry. Our findings revealed that Gab1 deficiency had no effect on the OVA-induced maturation of mDCs (Figure 5A). Further experiments evaluated the ability of mDCs to capture and internalize antigen. Immature DCs were incubated with Alexa Fluor 647-conjugated chicken OVA (Alexa Fluor 647-OVA) and we found no detectable difference in phagocytosis between Gab1-deficient and control BMDCs (Figure 5B). In addition, an in vitro antigen presentation assay indicated that Gab1-deficient BMDCs exhibited normal antigen presentation, characterized by unaltered proliferation of OT-II CD4+ T cells stimulated by OVA-pulsed control and Gab1-deficient BMDCs (Figure 5C and 5D). These results suggest that Gab1 deficiency in BMDCs has no effect on maturation, antigen uptake, or antigen presentation in vitro.

Gab1-deficient DCs exhibit normal maturation, antigen uptake, and antigen presentation compared with control DCs. (A) FACS analysis of cell surface markers showing that Gab1 deficiency in DCs had no influence on their maturation. Data are representative of three independent experiments. (B) Antigen uptake (OVA-Alexa Flour 647) by DCs measured with FACS showed no difference between control and Gab1-deficient DCs. Data are representative of three independent experiments. (C) FACS analysis of naive T cell proliferation showing that Gab1 deficiency in DCs had no influence on antigen presentation. Data are representative of three independent experiments. (D) Mean fluorescence intensity of CD4+ T cell proliferation.
Gab1 is required for mDC migration to lymph nodes in vivo
In vitro evidence suggested a limited involvement of Gab1 in the modulation of DC maturation, antigen uptake, and antigen presentation, thus enabling us to focus more on its role in mDC migration. At times of allergen challenge, immature DCs in the airways migrate into the DLNs, where Th2 responses are initiated. Migratory DCs are known to play a crucial role in the development of allergic asthma. We thus hypothesized that direct alteration of mDC migration might be responsible for the attenuated asthma in LysMcreGab1fl/fl mice. To test this possibility, equal numbers of mDCs from Gab1fl/fl and LysMcreGab1fl/fl mice were labeled with different fluorescent dyes and subcutaneously injected into congenic mice. Then, DLNs were disaggregated for analysis of mDC migration. As shown in Figure 6A and 6B, the loss of Gab1 dramatically reduced mDC migration to DLNs. Similar results were obtained with exchanged dyes, showing that the differences were not due to variation of dyes. Furthermore, reduced migration of antigen-loaded DCs to DLNs due to Gab1 deficiency also led to impaired T cell activation (Figure 6C). Carboxyfluorescein succinimidyl ester (CFSE)-labeled CD4+ T cells from OT-II mice were intravenously injected into recipient mice. After 24 h, these mice were subcutaneously injected at the hind footpads with OVA-loaded mature BMDCs from Gab1fl/fl or LysMcreGab1fl/fl mice. T cell proliferation evaluated by monitoring CFSE dilution of OT-II T cells from DLNs represented DC migration capacity since we had ruled out the possibility that Gab1 contributes to antigen presentation in DCs (Figure 5C and 5D). Altogether, we demonstrated that Gab1 deficiency disturbs mDC migration and ultimately results in defective T cell activation in vivo.

Loss of Gab1 impairs migration of BMDCs in DLNs. (A) Gab1-deficient DCs were unable to migrate to DLNs. Upper panel, more CFSE-labeled control DCs migrated to DLNs than eFluor670-labeled Gab1-deficient DCs. Lower panel, fewer CFSE-labeled Gab1-deficient DCs migrated to DLNs than eFluor670-labeled control DCs. Each plot represents one mouse. n = 3/group. (B) Percentage of labeled-DCs migrating to DLNs. n = 6/group, **P < 0.01. (C) Gab1-deficient DCs failed to migrate to DLNs and prime naive T cells. Each plot represents one mouse. n = 3/group.
Loss of Gab1 impairs the chemotactic motility of mDCs in vitro
mDCs play a major role in the pathogenesis of allergic asthma, and targeting their migration is a promising strategy for curing asthma29,30. CCL19 binding with CCR7 is critical for DC homing to lymph nodes31,32,33,34,35. We thus determined whether Gab1 removal influenced CCL19-dependent mDC migration. Live-cell imaging experiments revealed a similar random migration path (Figure 7A) and velocity (Figure 7B) in control and Gab1-deficient mDCs. However, in a CCL19 concentration gradient, Gab1-deficient mDCs showed disorganized and blocked migration (Figure 7C) compared with the directional and ordered movement of control mDCs, and the velocity of Gab1-deficient mDCs was reduced (Figure 7D). Similarly, impaired CCL19-mediated chemotaxis was also observed in Gab1-deficient DCs using an in vitro transwell assay (Figure 7E). Collectively, these observations clearly define a unique function of Gab1 in CCL19-mediated mDC migration.

Genetic removal of Gab1 results in attenuated CCL19-induced chemotactic motility in BMDCs. (A, B) Normal random migration of Gab1-deficient DCs in a planar under-agarose assay. Migration tracks (A) and cell velocity (B) of randomly-selected cells were shown. (C, D) Impaired directional motility of Gab1-deficient DCs. Migration tracks (C) and velocity (D) of randomly-selected cells were shown. Each dot represented one cell and > 30 cells were tracked. ***P < 0.001. (E) Transwell assay showing that fewer Gab1-deficient DCs migrated toward CCL19. All data represent three independent experiments. **P < 0.01.
Gab1 positively regulates the CCL19-mediated CCR7/Rho/Pyk2 axis by bridging p115-RhoGEF and Rho
CCL19/CCR7 is essential for DC migration to lymph nodes, but it remains largely elusive which intracellular signaling components are involved. To correlate Gab1 function with CCL19-dependent signaling events, mDCs were treated with CCL19 in an in vitro time-course experiment and then analyzed for activation of Pyk2, Cofilin, Akt, JNK, and p38, which have been reported to be involved in DC migration34,36. As shown in Figure 8A, levels of phosphorylated Akt, Pyk2, and its downstream Cofilin were markedly reduced, but there was no evident impairment of Erk, p38, or JNK activation in Gab1-deficient mDCs. In addition, the diminished activation of Pyk2 and Cofilin were found in DCs of MLNs from OVA-induced LysMcreGab1fl/fl mice (Supplementary information, Figure S4). Moreover, activation of JNK and p38 were slightly altered in Gab1-deficient DCs, possibly due to a compromised environment in vivo. Pharmacological suppression of Pyk2 activation notably reduced CCL19-induced DC migration in vitro while inhibition of Akt activation had no detectable influence (Figure 8B), suggesting that the CCL19/CCR7/Akt axis may not be a major factor contributing to mDC migration. Activated Pyk2 plays a pivotal role in controlling Cofilin activity and cytoskeleton rearrangement36,37; accordingly, we observed migratory defects in Gab1-deficient DCs, visualized as less-polarized actin polymerization in response to CCL19 (Figure 8C). These findings indicated a role of Gab1 in modulating CCL19-mediated Rho/Pyk2/Cofilin activation.

Gab1 positively regulates CCL19-mediated CCR7/Pyk2 signaling by mediating the interaction of p115-RhoGEF with Rho. (A) Loss of Gab1 in DCs inhibited the phosphorylation of Pyk2, Cofilin, and Akt in response to CCL19 stimulation, while the phosphorylation of Erk, JNK and p38 was little changed. BMDCs were stimulated with CCL19 (100 ng/ml) for 0, 2, 5, 10 and 30 min. β-Actin was used as loading control. Data are representative of three independent experiments. (B) Transwell assays using control DCs pre-treated with specific inhibitors of Pyk2 (PF-431396) or PI3K (LY294002) demonstrated that Gab1 regulates DC trafficking to lymph nodes via the Pyk2/Cofilin pathway rather than the PI3K/Akt pathway. ***P < 0.001. (C) F-actin staining showing that Gab1 deficiency in DCs impaired the actin cytoskeleton rearrangement in response to CCL19. Scale bar, 10 μm. (D) Co-immunoprecipitation assays indicating that Gab1, CCR7, Rho, and p115-RhoGEF formed a complex upon CCL19 stimulation. Data are representative of three independent experiments. (E) Co-immunoprecipitation assays in 293T cells indicating that CCR7 interacted with Gab1 through its C-terminal domain of 332-378. Data are representative of three independent experiments. (F) Removal of Gab1 in DCs reduced the interaction of Rho with p115-RhoGEF, while had slight influence on the interaction of CCR7 and Rho upon stimulation by CCL19. Whole-cell lysates were immunoprecipitated with anti-Rho or anti-p115-RhoGEF antibody and then immunoblotted with anti-p115-RhoGEF, anti-Rho, or anti-CCR7 antibodies. Data are representative of three independent experiments. (G) The activation of Rho induced by CCL19 was attenuated in Gab1-deficient DCs. Whole-cell lysates were incubated with GST-Rhotekin-RBD and then immunoblotted with anti-Rho antibody. Data are representative of three independent experiments.
As a small GTP-binding protein, Rho has both GDP/GTP-binding and GTPase activities and functions as a binary switch, assisting CCR7 to convey intracellular signals and regulate actin organization, cell polarity and migration. Guanine nucleotide exchange factors (GEFs) are necessary for the activation of Rho by catalyzing the exchange of bound GDP for GTP38,39. Gab1 serves as an essential docking protein for integrating the cytoplasmic elements involved in transmembrane signal amplification and previous studies have suggested a role of Gab1 in the activation of Rho family members40,41. Here, we hypothesized that Gab1 regulates the activation of CCL19 downstream elements by facilitating the binding of p115-RhoGEF to Rho, which is necessary for Rho activation. To test this, we performed co-immunoprecipitation assays and indeed, the results showed that Gab1 interacted with CCR7, Rho, and p115-RhoGEF (Figure 8D), and further study implied that CCR7 bound Gab1 through its C-terminal domain of 332-378 in cytoplasm (Figure 8E). More importantly, Gab1 deficiency disrupted the assembly of Rho and p115-RhoGEF, while had no influences on the interaction of CCR7 and Rho (Figure 8F). Correspondingly, CCL19-induced Rho activation was also inhibited in Gab1-deficient DCs (Figure 8G). Collectively, the findings obtained in this study define a novel role of the docking protein Gab1 in regulating CCL19-dependent mDC migration in the pathogenesis of allergic asthma. It is most likely that Gab1 serves as an essential docking protein that integrates CCL19/CCR7/Rho signals, thus contributing to Rho-induced Pyk2 activity.
Discussion
Asthma is a highly prevalent form of allergic inflammation, but the fundamental causes are still largely unknown. An important pathogenesis of asthma is the development of an allergic inflammatory response to inhaled allergens42,43. In the present study, we defined a unique role of the scaffolding protein Gab1 in regulating DC-mediated asthma. First, overexpression of Gab1 was found in PBMCs from asthma patients during exacerbations. Second, selective deletion of Gab1 in myeloid monocytes significantly attenuated OVA- and HDM-induced asthmatic inflammation. Further experiments revealed the importance of Gab1 in the regulation of mDC migration. Mechanistically, Gab1 acts as a docking protein, linking Rho with p115-RhoGEF, which is required for the activation of Rho- and CCL19-directed cell migration. Our findings add new insight into the pathogenesis of allergic asthma and indicate that Gab1 may be an attractive therapeutic target for this lung disease.
It is becoming increasingly understood that perturbations in tyrosine phosphorylation underlie the development of asthma, and this has prompted great efforts to develop inhibitors targeting the tyrosine kinases and phosphatases implicated in asthma and other inflammatory diseases44,45,46. It has become evident that scaffolding proteins are integral parts of signal modification by controlling signal diversity, specificity and crosstalk47,48. Elucidating the mechanism by which these proteins finely orchestrate phosphorylation would provide not only a better understanding of the fundamental processes in asthma but also desirable specific targets for curing this disease. Although lacking enzymatic activity itself, Gab1 contains multiple tyrosine-based activation motifs and domains for direct binding with kinases, phosphatases, and other signal partners. It is most likely that Gab1 serves as a docking site to amplify more specific signaling in a fine-tuned manner16. In contrast to other GAB family proteins, Gab1 and Gab2 are ubiquitously expressed. It has been suggested that they are functionally redundant, owing to their high homology and similar expression profiles. However, emerging evidence indicates that Gab1 and Gab2 have distinct physiological functions in vivo. Gab1 deficiency in mice leads to embryonic lethality, with profound developmental defects in heart, placenta, and skin, whereas Gab2-deficient mice appear normal and are generally healthy, without obvious developmental defects22,24. In recent years, the Cre-driven conditional knockout system has been used to demonstrate the importance of Gab1 in the liver and cardiovascular system49,50,51. Hepatic Gab1 is required for liver regeneration and hepatoprotection52. Disruption of Gab1 in the liver enhances hepatic insulin sensitivity and glucose tolerance51. Gab1 has crucial functions in coordinating ErbB signaling in regulating the postnatal maintenance of cardiac function53. Endothelial Gab1 is also required for postnatal angiogenesis and vascular inflammation after injury. Endothelial-specific Gab1-knockout mice show exacerbated vascular inflammation and impaired angiogenesis following ischemic injury49. However, the roles of Gab1 in lung and in inflammation remain largely unknown. Previous studies have defined a unique function of Gab2 in allergic reactions, i.e., mediating IgE-induced mast cell activation54. Intriguingly, we found that both Gab1 and Gab2 are involved in allergic asthma but have distinct roles. Gab2 is highly expressed in the airway epithelium with a weak expression in myeloid cells in an OVA-induced asthmatic mouse model. Gab2-knockout mice exhibit a reduction in mucus production and attenuated goblet cell hyperplasia, suggesting a major role of Gab2 in airway epithelium24. Additional evidence has shown that migration of Gab2-deficient mDCs is not affected in response to CCL19 stimulation (Supplementary information, Figure S5). Conversely, Gab1 is mainly elevated in PBMCs in asthmatic patients and asthmatic mouse models. We first introduced LysMcreGab1fl/fl mice in which Gab1 is selectively disrupted in macrophages, neutrophils, eosinophils, and mDCs. Considering the key roles of eosinophils in asthma and a significant reduction of eosinophil infiltration in LysMcreGab1fl/fl mice, eosinophils were isolated and transwell assay showed that the eosinphil migration was unaltered in LysmcreGab1fl/fl mice (Supplementary information, Figure S6). Then adoptive transfer of mDCs in LysMcreGab1fl/fl mice suggests a primary role of Gab1 in DC-mediated asthma. Furthermore, gab1 gene was targeted for deletion with a CD11c-Cre, whose activity is largely restrained in mDCs while relatively limited in alveolar macrophages. Previous studies have reported that DCs, rather than alveolar macrophages, mediated the Th2 airway inflammation55,56. We therefore hypothesized that Gab1 dominates DC functions, which are essential for initiating and maintaining adaptive Th2 cell responses to allergens.
CCL19/CCR7 signaling plays a pivotal role in tissue DC migration. Blockade of CCR7 impairs dermal and ocular DC trafficking to neighboring lymphoid nodes, suggesting that targeting CCR7 would be therapeutically beneficial in the treatment of atopic allergic inflammation57,58. Here, we defined the docking protein Gab1 as a novel component of CCL19/CCR7-induced Rho/Pyk2 pathway. The CCR7/Gab1/p115-RhoGEF/Rho complex is formed in response to CCL19 stimulation, upon which, Gab1 is coupled with CCR7 to facilitate p115-RhoGEF interaction with Rho, finally activating Rho and downstream Pyk2. Our study provides new insight into the molecular mechanism by which Gab1 modulates CCL19-dependent signals for DC trafficking to lymph nodes. Despite previous studies showing that Gab1 mediates cell migration via the PI3K/Akt and MAPK pathways26,27,59, we demonstrated that it acts as a docking protein to facilitate p115-RhoGEF interaction with Rho, thereby contributing to DC migration. First, we found that loss of Gab1 in DCs resulted in impaired CCL19-induced activation of Akt and Pyk2, while the activation of three MAPK family members (Erk1/2, p38, and JNK) was unchanged. Further study using specific inhibitors of the two pathways demonstrated that Gab1 regulates DC trafficking to lymph nodes via the Pyk2/Cofilin pathway rather than the PI3K/Akt pathway. Consistent with this result, attenuated activation of Rho, upstream of Pyk2, was also detected in Gab1-deficient DCs. All these results suggest that Gab1 modulates DC migration by positively regulating Rho activation. As an adaptor protein, Gab1 recruits p115-RhoGEF, and mediates the interaction of Rho with p115-RhoGEF in DCs and finally regulates DC migration.
The pathogenesis of allergic asthma is strongly genetic in origin. Over the past decade, major advances in high-throughput technologies for genome-wide association studies (GWASs) have enabled screening of some important chromosomal regions, including loci that may contain susceptibility genes affecting the development of asthma. To date, over a hundred such genes have been identified in population-based genetic studies, and many of them are thought to be immune-relevant60,61. In fact, asthma is highly atopic, involving a complicated interplay between genetic variants and the environment, highlighting the complexity of these interactions in its etiology62. A recent GWAS identified a novel and narrow region on chromosome 4q31, USP38-GAB1, a potential susceptibility region for asthma21. Here, we used a genetic approach to functionally characterize the unique role of Gab1 in DC-mediated asthmatic inflammation, providing additional information linking genetic evidence with functional relevance for this common atopic inflammation. Germline mutations in gab1 gene have been identified in cancer63, and further studies focusing on genetic analysis of gab1 polymorphisms in asthma may contribute to the understanding of asthma susceptibility.
In conclusion, our findings support a functional requirement for Gab1, a recently identified genetic risk factor for asthma, in allergic inflammation. Overexpression of human GAB1 was found in PBMCs during exacerbation in asthma patients and deleting Gab1 in myeloid monocytes attenuated asthmatic injury and inflammation. Further, our results indicate that Gab1 links Rho and p115-RhoGEF, thereby promoting Pyk2-activated DC migration. Recently, a novel structure-based screening approach, targeting conformational changes of Gab1 upon specific ligand binding, has been reported64. Further studies may shed light on the detailed mechanisms underlying the interplay between the scaffolding protein Gab1 and CCL19/CCR7 signaling in regulation of DC migration during allergic responses. Based on these findings, it may be possible to develop a promising therapeutic option for asthma.
Materials and Methods
Preparation of human peripheral blood monocytes
We collected peripheral blood from children who were diagnosed with asthma in Lishui Children's Hospital. Monocytes were isolated using a human monocyte isolation kit (Haoyang Biological, Tanjin, China) according to the manufacturer's recommendations. This study was approved by the Medical Ethics Committee of Zhejiang University School of Medicine.
OVA- or HDM-induced asthma model
Gab1fl/fl mice and LysMcre mice were generous gifts from Dr Gen-Sheng Feng (University of California, San Diego, CA, USA) and Dr Ximei Wu (Zhejiang University, China). CD11c-Cre mice were bought from Jackson Laboratory. Gab1 conditional knockout mice (LysMcreGab1fl/fl, 129Sv × C57B6/J; CD11c-CreGab1fl/fl, C57B6/J) were generated by crossing Gab1fl/fl mice with LysMcre mice and CD11c-Cre mice. OT-II TCR transgenic mice were gifts from Dr Shumin Duan (Zhejiang University, China). Gab1fl/fl and LysMcreGab1fl/fl littermates or CD11c-CreGab1fl/fl (6-8 weeks old) were challenged with 100 μg OVA (Sigma-Aldrich, St. Louis, MO, USA) adsorbed on 2 mg aluminum hydroxide in the footpad, neck, back, and groin on day 0 and then given with the same amount of OVA by intraperitoneal injection on day 14. Beginning on day 21, the sensitized mice were exposed to aerosolized 1% OVA for 30 min daily for 7 days. Control group mice (Gab1fl/fl and LysMcreGab1fl/fl or CD11c-CreGab1fl/fl mice) were treated with saline using the same protocol. On day 28, mice were sacrificed for pathological examinations. Gab1fl/fl and LysMcreGab1fl/fl littermates were sensitized intratracheally on day 0 with 100 μg HDM (Greer Laboratories, Lenoir, NC, USA) and again on days 7, 8, 9, and 10 with 25 μg HDM, and sacrificed on day 13. All experiments in this study were reviewed and approved by the Animal Care and Use Committee of Zhejiang University School of Medicine.
BALF analysis and cytokine detection
Twenty-four hours after allergen challenge, mice were sacrificed by injection of pentobarbital. BALF from each animal was collected as described previously13,24. All BALF samples were centrifuged at 930× g at 4 °C for 10 min. Cell pellets were resuspended in 100 μl phosphate-buffered saline (PBS), and differential cell counts were performed using standard morphological criteria after Wright-Giemsa staining. The BALF supernatant from each experimental mouse was stored at −80 °C until use. The Th2 cytokine levels in the BALF supernatant were measured using ELISA kits (IL-4, IL-5 and IL-13; eBioscience, San Diego, CA, USA) according to the manufacturer's recommendations.
Histology and immunohistochemistry
Lungs were equivalently inflation-fixed with an intratracheal injection of 4% paraformaldehyde in PBS at 25 cm H2O and immersed in the same fixative to preserve the pulmonary architecture. The left lung of each mouse was embedded in paraffin according to standard procedures. Sections (5 μm) were mounted on slides for histological or immunohistochemistry (IHC) analysis. Hematoxylin and eosin staining was used to evaluate changes in lung morphology. Mucus secretion was assessed by PAS staining. IHC analysis was performed using an indirect method with an antibody against the Muc5ac protein (Thermo Scientific, Bremen, Germany) and 3,3′-diaminobenzidine and hematoxylin for general morphology.
Generation of BMDCs and BMDMs
To generate BMDCs, mouse bone marrow cells were cultured for 7 days at 1 × 106 cells/ml in complete medium (RPMI 1640 with 10% heat-inactivated fetal bovine serum (FBS), 50 U/ml penicillin, and 50 U/ml streptomycin) supplemented with 10 ng/ml mouse GM-CSF (Peprotech, Rocky Hill, NJ, USA) and 10 ng/ml mouse IL-4 (Peprotech). The culture medium was changed every 2 days. On day 7, non-adherent cells were collected and purified using anti-mouse CD11c magnetic beads (Miltenyi Biotec, Auburn, CA, USA) with an AutoMacs device (Miltenyi Biotec). The purity was determined by FACS analysis (> 98% CD11c+). The purified cells were immature DCs. To obtain mature DCs, these cells were treated with 100 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich) for 16 h. Bone marrow cells were cultured in complete medium supplemented with 10 ng/ml mouse M-CSF (Peprotech) for 7 days to obtain BMDMs as described above.
Adoptive transfer of BMDCs and BMDMs
Adoptive transfer of BMDCs and BMDMs was performed according to a previously described method65. BMDCs or BMDMs (1 × 107 cells/mouse) were adoptively transferred to Gab1fl/fl or LysMcreGab1fl/fl mice via tail vein injection on days 0, 6, and 12 when mice were treated with OVA.
RT-PCR and real-time PCR
Total RNA from human peripheral blood monocytes was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The concentration of RNA was measured using a Nanodrop spectrophotometer (Thermo Scientific). Total RNA (1 μg) was reverse-transcribed using the ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). Real-time quantitative PCR was performed using the FastStart Universal SYBR Green Master Kit (Roche, Mannheim, Germany).
FACS analysis
Cells were subjected to FACS analysis using an FC 500 MC system (Beckman Coulter, Fullerton, CA, USA). For DC maturation studies, immature DCs were incubated with 10 μg/ml OVA for 24 h and then stained with anti-CD80-FITC, anti-CD86-APC, anti-CD11c-PE, and anti-MHC-II-PE (eBioscience). For antigen uptake analysis, phagocytosis by DCs was detected using FACS analysis. For the in vitro antigen presentation assay, OT-II T cells were labeled with CFSE (eBioscience) and T cell proliferation was assessed with FACS. For in vivo DC migration studies, mature DCs from Gab1fl/fl and LysMcreGab1fl/fl mice were labeled with CFSE and eFluor 670, respectively, and the migration of DCs was evaluated by FACS. A minimum of 20 000 cells were collected and the data were analyzed using FlowJo software (Tree Star, Eugene, OR, USA) and CXP analysis software (Beckman Coulter).
Analysis of antigen uptake
Alexa Fluor 647-conjugated chicken OVA (Alexa Fluor 647-OVA, Invitrogen) was used to detect antigen uptake by DCs. Immature DCs (1 × 106 cells/ml) were incubated with Alexa Fluor 647-OVA (25, 50, and 100 μg/ml) at 37 °C for 45 min. Control incubations were performed at 4 °C. Cells were then washed three times with cold PBS and analyzed by FACS.
In vitro antigen presentation assay
CD4+ T cells were isolated from the spleen of OT-II Ly5.2+ TCR transgenic mice and enriched by negative selection using a mouse CD4 T Lymphocyte Enrichment Set-DM according to the manufacturer's protocol (BD Biosciences, San Diego, CA, USA). Wild-type and Gab1-deficient BMDCs were incubated with 250 μg/ml OVA (Sigma-Aldrich) for 45 min at 37 °C in culture medium, washed three times, and then co-cultured with CFSE-labeled CD4+ T cells in round-bottom 96-well plates at ratios of 1:5, 1:10, and 1:20 (1 × 105 T cells) for 72 h. T cell proliferation was assessed by FACS.
In vivo T cell proliferation assay
CD4+ T cells isolated from the spleen of OT-II Ly5.2+ TCR mice were purified and then labeled with 1.25 μM CFSE. About 5 × 106 CFSE-labeled OT-II T cells were intravenously injected into control recipient mice. After 24 h, those mice were subcutaneously injected at the hind footpads with 6 × 105 OVA-loaded mature BMDCs from Gab1fl/fl or LysMcreGab1fl/fl mice. After 72 h, popliteal DLNs were digested and T cell proliferation was assessed by FACS.
In vivo migration of DCs
Equal numbers of mature DCs from Gab1fl/fl and LysMcreGab1fl/fl mice were labeled with 1.25 μM CFSE (eBioscience) and 1.25 μM eFluor 670 (eBioscience), respectively, and then mixed. A total of 4 × 106 DCs were suspended in 20 μl PBS and then subcutaneously injected into the hind footpads of control littermates. After 48 h, popliteal DLNs were disaggregated and digested with 1 mg/ml collagenase D (Sigma-Aldrich) and 0.4 mg/ml DNase I (Roche) for 30 min at 37 °C. The enzymatically treated cell suspension was evaluated by FACS.
Chemotaxis assay
Chemotaxis of mature DCs was determined by measuring the number of cells that migrated through a polycarbonate filter (5 μm pore size) in 24-well transwell chambers (Corning Costar, Cambridge, MA, USA). DCs (1 × 105) from Gab1fl/fl or LysMcreGab1fl/fl mice were resuspended in 100 μl RPMI 1640 with 0.1% bovine serum albumin and then added to the upper chamber. The lower chamber contained 400 μl of the same medium with or without the chemokine CCL19 (100 ng/ml and 300 ng/ml; Peprotech). After incubation for 3 h at 37 °C, DCs that migrated to the bottom chamber were counted by a Cedex XS cell analysis system (Roche). To determine which pathway was involved in CCL19-mediated DC migration, wild-type DCs were pre-treated with 0.5, 1 and 2 μM of PF-431396 (inhibitor of Pyk2, Sigma-Aldrich) or 0.5, 1 and 2 μM of LY294002 (inhibitor of PI3K, Cell Signaling Technology, Danvers, MA, USA) for 2 h and the transwell assay was performed.
In vitro migration assay (under-agarose chemotaxis assays)
Under-agarose chemotaxis assays were performed as described previously66. Briefly, 2.5% UltraPure agarose (Invitrogen) in distilled water was heated and mixed with prewarmed (56 °C) RPMI 1640 containing 20% FBS and 2× Hanks' Balanced Salts (Sigma-Aldrich) at a ratio of 1:2:1 to achieve a final agarose concentration of 6.25 mg/ml. Two milliliters of liquid agarose-medium mixture was used to coat 35-mm dishes (Shengyou Biotechnology, Hangzhou, China) and allowed to polymerize at room temperature. Two holes (1 mm) were punched into the agarose: a “responder” hole for DCs and an “attractor” hole for chemokine CCL19. After equilibration at 37 °C for 30 min, 2 × 104 DCs from Gab1fl/fl or LysMcreGab1fl/fl mice were transferred into the responder hole and 1.2 μg/ml CCL19 was transferred into the attractor hole. After culture at 37 °C for 1 h, live-cell imaging on an IX81-FV1000 confocal microscope (Olympus, Tokyo, Japan) was initiated to record the directional migration of DCs. For under-agarose chemotaxis assays with homogenous CCL19, CCL19 was added when the agarose-medium mixture had cooled to about 37 °C, resulting in a final CCL19 concentration of 2.5 μg/ml. One hole (1 mm) was punched into the agarose and 2 × 104 DCs from Gab1fl/fl or LysMcreGab1fl/fl mice were transferred into the hole. After culture at 37 °C for 1 h, live-cell imaging was initiated to record the migration of DCs. The results were analyzed using Imaris 7.6.3 (Bitplane AG, Zurich, Switzerland).
Immunofluorescence
Immunofluorescence of BMDCs was carried out as follows. Mature BMDCs (1 × 104) from Gab1fl/fl or LysMcreGab1fl/fl mice were grown on poly-L-lysine-coated (Sigma-Aldrich, 100 μg/ml) coverslips in 12-well plates for 1 h. CCL19 (100 ng/ml) was added to stimulate the cells for 0, 0.5, 1, or 2 h and then the cells were washed three times with calcium- and magnesium-free PBS, fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 20 min, blocked with 4% normal goat serum for 1 h, and incubated overnight with phalloidin-FITC (Beyotime, 1:200 dilution). 4′,6-diamidino-2-phenylindole (DAPI) was used to stain nuclei (blue). Confocal images of stained BMDCs were analyzed with an inverted microscope (Carl Zeiss, Göttingen, Germany) using the Zeiss LSM program.
Active Rho detection
Rho activity was assessed using a commercially available active Rho detection kit (Cell Signaling Technology). According to the manufacturer's protocol, CCL19 (100 ng/ml) was added to DCs (1 × 107/sample) and incubated for 0, 0.5, and 2 min. Then the cells were washed with ice-cold PBS. PBS was removed and 0.5 ml ice-cold 1× lysis buffer/binding/wash buffer plus 1 mM PMSF was added to each sample, followed by vortexing and incubating on ice for 5 min. Cell lysates were collected by centrifugation at 4 °C. Next the 50% resin slurry was washed once with 1× lysis buffer/binding/wash buffer and mixed with GST-Rhotekin-RBD. The lysates were then transferred. The reaction mixture was incubated at 4 °C for 1 h with gentle rocking. The resin was washed 3 times with 1× lysis buffer/binding/wash buffer (400 μl). The resin was suspended in 50 μl of SDS sample buffer and boiled for 5 min. Bound Rho proteins were detected by western blot using a Rho antibody (Cell Signaling Technology) and the total amount of Rho in cell lysates served as control.
Immunoblotting analysis and co-immunoprecipitation
Primary cells were homogenized in RIPA buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS, supplemented with protease and phosphatase inhibitors) before further analysis. Cell lysates (40 μg) were separated on SDS-PAGE and immunoblotted using antibodies against the following proteins: Gab1, phospho-Akt (Thr308), phospho-Akt (Ser473), Akt, phospho-Erk, Erk, phospho-Pyk2 (Tyr402), Pyk2, P-Cofilin (Ser3), Cofilin, p-p38, p38, P-JNK and JNK (Cell Signaling Technology), CCR7 (Abcam, Cambridge, MA, USA), and β-actin (Sigma-Aldrich). For co-immunoprecipitation assays, cells were lysed in NP-40 buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 1% NP-40) containing protease and phosphatase inhibitors, and the lysates were immunoprecipitated overnight at 4 °C using antibodies against the following proteins: Gab1, CCR7, Rho, and p115-RhoGEF (Cell Signaling Technology) coupled to protein A-agarose (Roche). Immunoprecipitates were washed three times with lysis buffer. Protein levels were determined by western blotting as indicated above. Scanned immunoblots were representative of three independent experiments.
In 293T cells, 4 μg of Flag-Gab1 were co-transfected with HA-CCR7 (4 μg) or GST-CCR7332-378 (4 μg) plasmids for 48 h. Flag co-transfected with HA-CCR7 or GST-CCR7332–378 plasmids were as control. Then cells were lysed in NP-40 buffer containing protease and phosphatase inhibitors, and the lysates were immunoprecipitated overnight at 4 °C using HA or GST antibodies (Cell Signaling Technology) coupled to protein A-agarose. Immunoprecipitates were washed three times with lysis buffer. Protein levels were determined by western blotting as indicated above. Scanned immunoblots were representative of three independent experiments.
Statistical analysis
Data are expressed as mean ± SEM and were assessed for significance by Student's t-test or ANOVA when appropriate. P values < 0.05 were considered significant.
Author Contributions
YZ, YX, LL, YT, HQ, XZ and YK designed the study; YZ, YX, SW, XH, DC, JQ and HY performed experiments, collected, and analyzed data. KJ and CL collected human samples. YZ, LL, HS and YK wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
References
Chang PJ, Michaeloudes C, Zhu J, et al. Impaired nuclear translocation of the glucocorticoid receptor in corticosteroid-insensitive airway smooth muscle in severe asthma. Am J Respir Crit Care Med 2015; 191:54–62.
Barnes PJ . Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008; 8:183–192.
Weiss ST . New approaches to personalized medicine for asthma: where are we? J Allergy Clin Immunol 2012; 129:327–334.
Vercelli D . Gene-environment interactions in asthma and allergy: the end of the beginning? Curr Opin Allergy Clin Immunol 2010; 10:145–148.
Kaiko GE, Foster PS . New insights into the generation of Th2 immunity and potential therapeutic targets for the treatment of asthma. Curr Opin Allergy Clin Immunol 2011; 11:39–45.
Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM . Treatment of allergic asthma: modulation of Th2 cells and their responses. Respir Res 2011; 12:114.
Polte T, Petzold S, Bertrand J, et al. Critical role for syndecan-4 in dendritic cell migration during development of allergic airway inflammation. Nat Commun 2015; 6:7554.
Gill MA . The role of dendritic cells in asthma. J Allergy Clin Immunol 2012; 129:889–901.
Dua B, Smith S, Kinoshita T, Imaoka H, Gauvreau G, O'Byrne P . Myeloid dendritic cells type 2 in allergic asthma. Allergy 2013; 68:1322–1326.
Lambrecht BN, De Veerman M, Coyle AJ, Gutierrez-Ramos JC, Thielemans K, Pauwels RA . Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J Clin Invest 2000; 106:551–559.
van Helden MJ, Lambrecht BN . Dendritic cells in asthma. Curr Opin Immunol 2013; 25:745–754.
Katsumoto TR, Kudo M, Chen C, et al. The phosphatase CD148 promotes airway hyperresponsiveness through SRC family kinases. J Clin Invest 2013; 123:2037–2048.
Qin XJ, Zhang GS, Zhang X, et al. Protein tyrosine phosphatase SHP2 regulates TGF-beta1 production in airway epithelia and asthmatic airway remodeling in mice. Allergy 2012; 67:1547–1556.
Guntur VP, Reinero CR . The potential use of tyrosine kinase inhibitors in severe asthma. Curr Opin Allergy Clin Immunol 2012; 12:68–75.
Hunter T . Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 1995; 80:225–236.
Gu H, Neel BG . The “Gab” in signal transduction. Trends Cell Biol 2003; 13:122–130.
Pawson T, Scott JD . Signaling through scaffold, anchoring, and adaptor proteins. Science 1997; 278:2075–2080.
Rajadurai CV, Havrylov S, Zaoui K, et al. Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J Cell Sci 2012; 125:2940–2953.
Rodrigues GA, Falasca M, Zhang Z, Ong SH, Schlessinger J . A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol Cell Biol 2000; 20:1448–1459.
Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W . Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol 2000; 149:1419–1432.
Hirota T, Takahashi A, Kubo M, et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat Genet 2011; 43:893–896.
Itoh M, Yoshida Y, Nishida K, Narimatsu M, Hibi M, Hirano T . Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol Cell Biol 2000; 20:3695–3704.
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K . Development of monocytes, macrophages, and dendritic cells. Science 2010; 327:656–661.
Zhang X, Zhang Y, Tao B, et al. Docking protein Gab2 regulates mucin expression and goblet cell hyperplasia through TYK2/STAT6 pathway. FASEB J 2012; 26:4603–4613.
Yang Z, Xue B, Umitsu M, Ikura M, Muthuswamy SK, Neel BG . The signaling adaptor GAB1 regulates cell polarity by acting as a PAR protein scaffold. Mol Cell 2012; 47:469–483.
Zhu X, Li Z, Pan W, et al. Participation of Gab1 and Gab2 in IL-22-mediated keratinocyte proliferation, migration, and differentiation. Mol Cell Biochem 2012; 369:255–266.
Caron C, Spring K, Laramee M, et al. Non-redundant roles of the Gab1 and Gab2 scaffolding adapters in VEGF-mediated signalling, migration, and survival of endothelial cells. Cell Signal 2009; 21:943–953.
Laramee M, Chabot C, Cloutier M, et al. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem 2007; 282:7758–7769.
Kool M, Lambrecht BN . Dendritic cells in asthma and COPD: opportunities for drug development. Curr Opin Immunol 2007; 19:701–710.
Idzko M, Hammad H, van Nimwegen M, et al. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest 2006; 116:2935–2944.
Clatworthy MR, Aronin CE, Mathews RJ, Morgan NY, Smith KG, Germain RN . Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat Med 2014; 20:1458–1463.
Holtzman MJ . Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest 2012; 122:2741–2748.
Braun A, Worbs T, Moschovakis GL, et al. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol 2011; 12:879–887.
Forster R, Davalos-Misslitz AC, Rot A . CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 2008; 8:362–371.
Marsland BJ, Battig P, Bauer M, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 2005; 22:493–505.
Riol-Blanco L, Sanchez-Sanchez N, Torres A, et al. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J Immunol 2005; 174:4070–4080.
Freeman SA, Lei V, Dang-Lawson M, Mizuno K, Roskelley CD, Gold MR . Cofilin-mediated F-actin severing is regulated by the Rap GTPase and controls the cytoskeletal dynamics that drive lymphocyte spreading and BCR microcluster formation. J Immunol 2011; 187:5887–5900.
David M, Petit D, Bertoglio J . Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012; 11:3003–3010.
Rossman KL, Der CJ, Sondek J . GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167–180.
Gadepalli R, Kotla S, Heckle MR, Verma SK, Singh NK, Rao GN . Novel role for p21-activated kinase 2 in thrombin-induced monocyte migration. J Biol Chem 2013; 288:30815–30831.
Zhao C, Ma H, Bossy-Wetzel E, Lipton SA, Zhang Z, Feng GS . GC-GAP, a Rho family GTPase-activating protein that interacts with signaling adapters Gab1 and Gab2. J Biol Chem 2003; 278:34641–34653.
Kim HY, DeKruyff RH, Umetsu DT . The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol 2010; 11:577–584.
Lambrecht BN, Hammad H . Taking our breath away: dendritic cells in the pathogenesis of asthma. Nat Rev Immunol 2003; 3:994–1003.
Wong WS . Inhibitors of the tyrosine kinase signaling cascade for asthma. Curr Opin Pharmacol 2005; 5:264–271.
Wong WS, Leong KP . Tyrosine kinase inhibitors: a new approach for asthma. Biochim Biophys Acta 2004; 1697:53–69.
Mueller C, August A . Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J Immunol 2003; 170:5056–5063.
Pawson T . Dynamic control of signaling by modular adaptor proteins. Curr Opin Cell Biol 2007; 19:112–116.
Birge RB, Knudsen BS, Besser D, Hanafusa H . SH2 and SH3-containing adaptor proteins: redundant or independent mediators of intracellular signal transduction. Genes Cells 1996; 1:595–613.
Shioyama W, Nakaoka Y, Higuchi K, et al. Docking protein Gab1 is an essential component of postnatal angiogenesis after ischemia via HGF/c-met signaling. Circ Res 2011; 108:664–675.
Lu Y, Xiong Y, Huo Y, et al. Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc Natl Acad Sci USA 2011; 108:2957–2962.
Bard-Chapeau EA, Hevener AL, Long S, Zhang EE, Olefsky JM, Feng GS . Deletion of Gab1 in the liver leads to enhanced glucose tolerance and improved hepatic insulin action. Nat Med 2005; 11:567–571.
Bard-Chapeau EA, Yuan J, Droin N, et al. Concerted functions of Gab1 and Shp2 in liver regeneration and hepatoprotection. Mol Cell Biol 2006; 26:4664–4674.
Nakaoka Y, Nishida K, Narimatsu M, et al. Gab family proteins are essential for postnatal maintenance of cardiac function via neuregulin-1/ErbB signaling. J Clin Invest 2007; 117:1771–1781.
Gu H, Saito K, Klaman LD, et al. Essential role for Gab2 in the allergic response. Nature 2001; 412:186–190.
Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ . Modulation of dendritic cell trafficking to and from the airways. J Immunol 2006; 176:3578–3584.
van Rijt LS, Jung S, Kleinjan A, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med 2005; 201:981–991.
Schlereth S, Lee HS, Khandelwal P, Saban DR . Blocking CCR7 at the ocular surface impairs the pathogenic contribution of dendritic cells in allergic conjunctivitis. Am J Pathol 2012; 180:2351–2360.
Jin Y, Shen L, Chong EM, et al. The chemokine receptor CCR7 mediates corneal antigen-presenting cell trafficking. Mol Vis 2007; 13:626–634.
Laramee M, Chabot C, Cloutier M, et al. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem 2007; 282:7758–7769.
Torgerson DG, Capurso D, Mathias RA, et al. Resequencing candidate genes implicates rare variants in asthma susceptibility. Am J Hum Genet 2012; 90:273–281.
Vercelli D . Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol 2008; 8:169–182.
Apter AJ . Advances in adult asthma diagnosis and treatment in 2012: potential therapeutics and gene-environment interactions. J Allergy Clin Immunol 2013; 131:47–54.
Ortiz-Padilla C, Gallego-Ortega D, Browne BC, et al. Functional characterization of cancer-associated Gab1 mutations. Oncogene 2013; 32:2696–2702.
Chen L, Du-Cuny L, Moses S, et al. Novel inhibitors induce large conformational changes of GAB1 pleckstrin homology domain and kill breast cancer cells. PLoS Comput Biol 2015; 11:e1004021.
de Heer HJ, Hammad H, Soullie T, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med 2004; 200:89–98.
Frittoli E, Matteoli G, Palamidessi A, et al. The signaling adaptor Eps8 is an essential actin capping protein for dendritic cell migration. Immunity 2011; 35:388–399.
Acknowledgements
We thank Dr Gen-Sheng Feng (University of California, San Diego, CA, USA) for the Gab1fl/fl mice; Dr Ximei Wu (Zhejiang University School of Medicine) for the LysMcre mice and Dr Shumin Duan (Zhejiang University School of Medicine) for the OT-II TCR transgenic mice. This work was supported by the National Natural Science Foundation of China (31371415 to YK, 31370857 to XZ), Key Project of the National Natural Science Foundation of China (81530001 to YK) and Key Research and Development Project of Ministry of Science and Technology of China (2016YFA0501800 to YK).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
LysMcreGab1fl/fl mice exhibited normal lung morphology and leukocytes. (PDF 331 kb)
Supplementary information, Figure S2
IFN-γ secretion was increased in OVA- and HDM-sensitized Gab1-deficient mice. (PDF 84 kb)
Supplementary information, Figure S3
FACS analysis showing a reduction of CD11c+ MHC-II+ cells in the MLNs of OVA-treated LysMcreGab1fl/fl mice. (PDF 167 kb)
Supplementary information, Figure S4
Diminished activation of Pyk2 and Cofilin in DCs of MLNs in OVA-induced LysMcreGab1fl/fl mice. (PDF 56 kb)
Supplementary information, Figure S5
Gab2-deficient DCs showing normal migration. (PDF 39 kb)
Supplementary information, Figure S6
Eosinophil migration is normal in LysmcreGab1fl/fl mice. (PDF 144 kb)
Rights and permissions
About this article

Cite this article
Zhang, Y., Xu, Y., Liu, S. et al. Scaffolding protein Gab1 regulates myeloid dendritic cell migration in allergic asthma. Cell Res 26, 1226–1241 (2016). https://doi.org/10.1038/cr.2016.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.124
