
Abstract
Late-onset Fuchs endothelial corneal dystrophy (FECD) shows genetic heterogeneity. Identification of SLC4A11 as a candidate gene for congenital hereditary endothelial dystrophy with similar corneal endothelial defects as FECD and reduced mRNA expression of SLC4A11 in the endothelium of FECD cases suggested that this gene may also be involved in pathogenesis of FECD. Mutations in SLC4A11 give rise to SLC4A11 protein marked by retention in the endoplasmic reticulum as a result of mis-folding. We screened 45 sporadic late-onset, 4 early-onset FECD patients and an early-onset autosomal dominant FECD family. We identified three previously unreported missense mutations: c.719G>C (p.W240S), c.1519G>A (p.V507I) and c.1304C>T (p.T434I) in unrelated individuals. These SLC4A11 mutants, expressed in HEK293 cells, had defects in either their cell surface expression or functional activity (rate of osmotically driven water flux). SLC4A11 mutations contribute to 11% (5/45) of sporadic late-onset FECD in the cohort studied. COL8A2, which causes some cases of early-onset FECD, was also screened in this cohort. No mutations were identified in COL8A2, in neither the late-onset cohort nor the early-onset family, suggesting genetic heterogeneity in this FECD family.
Similar content being viewed by others

Introduction
Fuchs endothelial corneal dystrophy (FECD; MIM 136800, 613268, 613270, 610158, 613267, 613271, 613269) is a hereditary bilateral degenerative disorder of the cornea, clinically characterized by guttae, which are localized thickenings of the Descemet membrane secreted by the corneal endothelium.1, 2 Predominantly, a late-onset disease, the symptoms (the characteristic guttae) present in the fourth or fifth decade and slowly progress over the next two decades. There is loss of endothelial cell density, which in turn compromises the corneal ion transport and solute barrier functions. This leads to stromal and epithelial edema, finally causing visual impairment.1, 3 In the rare early-onset variant of FECD, the first symptoms present as early as the first decade, but the disease progresses as in the late-onset variant, causing corneal edema and visual impairment by the third or fourth decade of life.2, 3, 4 Penetrating keratoplasty or more recently endothelial keratoplasty is the only treatment modality for the advanced disease.5 In the more common late-onset FECD, 50% of cases are familial, with an autosomal dominant pattern of inheritance, whereas the rest are sporadic.1 Few families with the rare early-onset autosomal dominant FECD have been reported and mutations in COL8A2 gene have been identified in all these families.2, 3, 4 Late-onset familial as well as the sporadic FECD cases, however, show considerable genetic heterogeneity. Linkage studies on large multi-generational, late-onset families have so far identified nine loci; 13pter-13q21.13.23 (FECD1), 18q21.2-q21.32 (FECD2), 5q33.1-q35.2 (FECD3), 9p22.1-p24.1 (FECD4), chromosome 1, 7, 15, 17 and X.1, 6, 7, 8, 9 Recently, LOXHD1 was identified as the causative gene in one of the three families with linkage to 18q21.2-q21.32 (FECD2),10 but the causative gene(s) at the other loci remain unknown.
Mutations in the SLC4A11 gene were first identified in autosomal recessive congenital hereditary endothelial dystrophy (CHED).11 Commonality of features among the corneal endothelial dystrophies (posterior polymorphous corneal dystrophy, CHED and FECD) and reduced expression of SLC4A11 in Fuchs endothelium compared with normal endothelium led us to screen the SLC4A11 gene as a candidate for FECD. An earlier study screened 25 and 64 FECD cases in Indian and Chinese cohorts and identified one (c.719G>C (p.E399K)) and three (c.2126G>A (p.Gly709Glu), c.2261C>T (p.Thr754Met), c.99-100delTC (p.S33SfsX18)) heterozygous mutations, respectively.12 Mutations in SLC4A11 were reported in 2% of FECD cases in one Caucasian cohort.13 Similarly, ZEB1 (TCF8) first identified in posterior polymorphous corneal dystrophy, also contributes to a small proportion of FECD.7
Genome-wide association study of late-onset sporadic FECD identified rs613872 in the intronic region of TCF4 gene to be significantly associated with the disease and also been replicated in various cohorts14, 15, 16, 17 (our cohort—data unpublished). Very recently, TGC trinucleotide repeat expansion in the intron of TCF4 gene has been identified to be associated with the disease and an expansion of >50 repeats being highly specific for the disease compared with the single-nucleotide polymorphism (SNP; rs613872).18
Here we describe results of mutation screening of SLC4A11 in 49 cases (including the 25 cases described earlier) of FECD (predominantly late-onset and sporadic) and an autosomal dominant early-onset FECD family and report the frequency of mutations in SLC4A11 that contribute to FECD in our cohort. We functionally characterized the novel missense mutations identified in SLC4A11, when expressed in HEK293 cells. We also screened COL8A2 gene in this cohort to determine its contribution to late-onset FECD and in an autosomal dominant early-onset family.
Materials and methods
DNA blood mini kit (Qiagen, GmbH, Hilden, Germany), M&N Nucleospin DNA maxi kit (Macherey-Nagel, GmbH, Düren, Germany), primers (Shrimpex Biotech Services, Chennai, India), Taq DNA polymerase (Merck, Mumbai, India), dNTPs (Applied Biosystems, Foster City, CA, USA) Applied Biosystems 2720 and 9700 thermal cyclers, ABI3100 Avant Genetic Analyzer (Applied Biosystems) were used to carry out the experiments.
Subjects and clinical examination
Forty-five unrelated individuals with late-onset sporadic and four early-onset (<40 years of age and no family history of FECD) FECD were recruited. An autosomal dominant early-onset FECD (ADFCD1) family with 15 affected and 18 unaffected members was also enrolled. All the individuals enrolled were of south Asian origin. All patients underwent complete ophthalmic examination, including slit lamp examination, applanation tension and fundus examination. Evaluation included ultrasound corneal pachymetry (Tomey, Pheonix, AZ, USA) and specular microscopy (Noncon, ROBO CA, KONAN Medical, Nishinomiya, Hyogo, Japan) to assess corneal thickness and endothelial morphology quantitatively and qualitatively, respectively. Diagnosis of FECD was based on clinical examination, above investigations and/or histopathologically confirmed FECD after performing a penetrating keratoplasty or endothelial transplantation. The study was conducted in accordance with institutional guidelines and the Declaration of Helsinki and approved by institutional ethics committee. Blood (10 ml) was drawn from individuals, after obtaining informed consent. DNA was extracted using DNA blood mini kit (Qiagen, GmbH) or M&N Nucleospin maxi kit (Macherey-Nagel, GmbH), according to the manufacturer’s protocol. Control individuals, recruited from epidemiological studies on diabetic retinopathy19 and glaucoma20 conducted by our hospital (Medical Research Foundation, Chennai, India), underwent complete ophthalmic screening and subjects with any corneal abnormality were excluded from the study.
SLC4A11 and COL8A2 screening
The 19 coding exons and flanking intronic regions (NCBI reference sequence NM_032034.3) of the SLC4A11 gene were amplified using 11 primer pairs as described previously.12 Two exons and the intermittent intron of COL8A2 gene were amplified using 16 overlapping primers as described.3 Amplified products were ExoSap treated (Escherichia coli exonuclease I and Shrimp Alkaline phosphatase (Fermentas Life Sciences, Waltham, MA, USA)) followed by direct sequencing using BigDye terminator v3.1 kit (Applied Biosystems) in ABI 3100 Avant Genetic Analyser (Applied Biosystems). Sequence chromatograms were analyzed, using Sequence analysis software v5.1.1 (Applied Biosystems) or Bioedit with NM_032034.3 and NM_005202.2 as reference for SLC4A11 and COL8A2 genes, respectively. Mutations/variations were confirmed by reverse sequence and in a second PCR-sequencing reaction as well. Screening was performed on all the sporadic cases and three affected members of the ADFCD1 family.
In the SLC4A11 gene, the c.719G>C (p.W240S) and c.1519G>A (p.V507I) variations were screened by direct sequencing of the corresponding exon fragments and allele-specific PCR was used to screen for c.1304C>T (p.T434I) variation in 140 controls. Synonymous and non-synonymous variations identified in COL8A2 gene were screened by direct sequencing of the respective amplified fragments in 100 controls.
Bioinformatics
Non-synonymous variations observed in SLC4A11 and COL8A2 were analyzed by SIFT21 and Polyohen222 to predict the probable effect of the missense variation(s) in the structure and function of their respective proteins.
cDNA constructs and mutagenesis
Expression constructs encoding amino-terminally hemagglutinin epitope (HA)-tagged human SLC4A11 were engineered by PCR, using human SLC4A11 cDNA (plasmid myc-SLC4A11 in pcDNA3.1, a vector that drives expression from a CMV promoter).23 The forward primer used was 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTAGCCAGGTCGGGGG-3′ (IDT Technologies, Coralville, IA, USA) that contains a 5′ NheI restriction site and codons corresponding to an HA tag (amino-acid sequence YPYDVPDYA). The initiation codon of SLC4A11 was removed and replaced by an ATG 5′ to the HA-tag. The reverse primer 5′-AGCGGCGAAGCA-3′ flanks a unique EcoNI restriction site in the SLC4A11-coding sequence. PCR product and plasmid pMyc-SLC4A11 were digested with Nhe1 and EcoNI. The cut PCR fragment replaced the original 5′ end of the plasmid myc-SLC4A11-coding region, to yield pSKL1. FECD mutants were prepared, using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Stratagene, La Jolla, CA, USA) and pSKL1 as a template. Primers used for mutant construction are given in Supplementary Table 1. Integrity of all the clones was confirmed by sequencing (IBD Core facility, Department of Biochemistry, University of Alberta).
Culture and transfection of HEK293 cells
HEK293 cells were grown in Dulbecco’s modified Eagle’s medium, supplemented with 5% (v/v) fetal bovine serum, 5% (v/v) calf serum, 1% (v/v) penicillin/streptomycin/glutamine and maintained at 37 °C in 5% CO2/95% air environment.
SLC4A11 variants were expressed in HEK293 cells by transient transfection using the calcium phosphate method.24 All experiments involving transfected HEK293 cells were carried out 40–48 h post-transfection. In transfections performed for functional assays (see below), cells were co-transfected with peGFPC1 and cDNA encoding the appropriate SLC4A11 variant at a 1:8 molar ratio.
Cell surface processing assays
HEK293 cells were transiently transfected with cDNAs, as described above. In brief, cells were incubated with the membrane-impermeant, amine-directed biotinylation reagent, Sulpho-NHS-SS-biotin (Thermo Scientific, Waltham, MA, USA). After appropriate washes, cells were lysed with immunoprecipitation buffer (1% (v/v) IGEPAL CA-630, 5 mM EDTA, 150 mM NaCl, 0.5% (w/v) sodium deoxycholate, 10 mM Tris, pH 7.5). Lysates were divided in two, where one half was incubated with streptavidin Sepharose (Thermo Scientific) and the other half was not (total (T) sample). Total protein and the fraction of protein not bound to the resin (unbound (U) sample) were subjected to SDS-polyacrylamide gel electrophoresis and transferred to poly(vinylidene fluoride) membranes and probed as immunoblots with anti-HA antibody. SLC4A11 abundance in each sample was quantified by densitometry. The efficiency of cell surface processing was calculated from the difference in abundance of SLC4A11 in the total and unbound fractions (%biotinylation=T−U/T × 100), as described previously.23
Immunoblots
Immunoblots were performed as described previously,25 using anti-HA epitope antibody (Covance, Princeton, NJ, USA) and anti-glyceraldehyde phosphate dyhydrogenase (Santa Cruz Biotechnology, Dallas, TX, USA).
Fluorescence immunocytochemistry
Transfected HEK293 cells grown on glass coverslips were washed with phosphate-buffered saline (PBS) pH 7.4, fixed with 4% (w/v) paraformaldehyde in PBS for 20 min, washed 5 min with 100 mM glycine in PBS and permeabilized by incubating in 0.1% Triton X-100 for 2 min. Samples were then incubated with 1% BSA in PBS pH 7.4 (PBS) for 30 min at room temperature to block non-specific binding. Cells were combined with mouse anti-HA antibody (Covance) (1:500 dilution) and rabbit anti-Na+-K+ ATPase antibody (Santa Cruz Biotechnology; 1:1000 dilution) in PBS for 1 h at 20 °C to detect HA-tagged SLC4A11 and endogenously expressed Na+/K+-ATPase, respectively. After washing 15 min with PBS, samples were incubated in the dark, for 1 h at room temperature, with 1:1000 dilutions of chicken anti-mouse IgG conjugated with Alexa Fluor-594 and chicken anti-rabbit Alexa Fluor-488 (Invitrogen, Burlington, ON, Canada) in PBS. Cells were washed in the dark for 15 min with PBS to remove excess antibody. Coverslips were mounted in Prolong Gold Antifade Solution (Invitrogen), containing the DNA-specific fluorescent dye 4′,6-diamidino-2-phenylindole. Confocal images were collected using an IX81 motorized inverted microscope (Olympus, Burlington, ON, Canada) with an MS-2000 motorized XY stage harboring a piezo Z insert that has 100 μm travel (Advanced Scientific Instrumentation, Eugene, OR, USA) and a CSU 10 spinning disk confocal scan-head (Yokogawa, Tokyo, Japan). Samples were observed through a × 60 PlanApo oil immersion objective (numerical aperture 1.42) and image capture was performed with a C9100-13 EM-CCD Digital Camera (Hamamatsu, Hamamatsu, Japan), using Volocity software (Perkin-Elmer, Mississauga, ON, Canada) with constant exposure times and gains for every channel.
Water flux assays
HEK293 cells, co-transfected with enhanced green fluorescent protein cDNA (peGFPC1; Clontech, Palo Alto, CA, USA) and SLC4A11 (or empty vector), grown on poly-L-lysine-coated 25 mm glass coverslips, were mounted in a 35-mm diameter Attofluor Cell Chamber (Molecular Probes, Eugene, OR, USA). During experiments, the chamber was perfused at 3.5 ml min−1 with isotonic MBSS buffer (90 mM NaCl, 5.4 mM KCl, 0.4 mM MgCl2, 0.4 mM MgSO4, 3.3 mM NaHCO3, 2 mM CaCl2, 5.5 mM glucose, 100 mM D-mannitol, 10 mM HEPES, pH 7.4, 300 mOsm kg−1) and then with hypotonic (200 mOsm kg−1) MBSS (modified balanced salt solution) buffer, pH 7.4 (same composition as previous, but lacking D-mannitol). The chamber was placed on the stage of a Wave FX Spinning Disc Confocal Microscope (Quorum Technologies, Guelph, ON, Canada) and a live-cell environment chamber (Chamlide, Seoul, Korea), set to 24 °C during the duration of the experiment. Acquisition was performed with a Hamamatsu C9100-13 Digital Camera (EM-CCD) and a × 20 objective during excitation with the FiTC/eGFP laser (491 nm). Enhanced green fluorescent protein (eGFP) fluorescence data were acquired at 1 point/s. Quantitative image analysis was performed with Volocity 4.2 software (Perkin-Elmer, Woodbridge, ON, Canada). Cell swelling rates were determined by measuring the rate of fluorescence decrease in the initial 15 s of the linear fluorescence decrease, following switch to hypo-osmotic medium. The time point where this occurs was determined for WT-SLC4A11 and this time point was used as the time at which slopes were measured for all other transfected cells.
Statistical analysis
Frequency distribution of SNPs in the coding region of COL8A2 gene in cases and controls were analyzed by Fisher’s exact t-test to assess for association using online tool VassarStats. In functional studies, values are expressed as mean±standard error of measurement. Statistical significance analyses were performed using Prism software (Graphpad, La Jolla, CA, USA). Groups were compared with one-way analysis of variance and unpaired t-test with P<0.05 considered significant.
Results
Forty-five late-onset sporadic FECD cases were analyzed, including 26 females and 19 males with age range 42–81 years and average of 57.7 years (standard deviation 11.1 years). The four early-onset cases were females with age range from 22 to 39 years and average of 31.3 years (standard deviation 7.7 years). In ADFCD1 family, the youngest member showing corneal guttae was 9 years of age at presentation.
Variations in the SLC4A11 gene
Four variations in exons were observed in five cases: c.719G>C (p.W240S), c.1195G>A (p.E399K), c.1304C>T (p.T434I) and c.1519G>A (p.V507I). Variant c.1195G>A (p.E399K) was previously described12 in a 59-year-old female and was observed in a 49-year-old female as well. The other three variations have not been previously reported. Screening of 140 control samples for the variations, c.719G>C, c.1519G>A and c.1304C>T, revealed the wild-type (WT) allele in the controls.
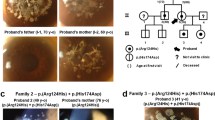
The variants c.[719G>C];[=] and c.[1519G>A];[=] were heterozygous, whereas c.[1304C>T];[1304C>T] was homozygous. The three novel variations are not reported in exome variant server and 1000 genome database. Residues at mutant positions W240 and T434 are conserved across the mammals, but V507 was not conserved (UCSC genome browser; Figure 1). Residues at codons 240, 434 and 507 were not conserved in the closely related SLC4 family of proteins (AE1 (SLC4A1), AE2 (SLC4A1) and AE3 (SLC4A3; data not shown)). Nucleotide variations with their predicted amino-acid change in protein, age of onset of disease and SIFT and Polyphen2 predictions are given in Table 1. All the mutation-positive cases presented to us with advanced disease. The visual acuity, corneal thickness, endothelial concentration and the details of treatment/surgical management of the mutation-positive cases are given in Table 2. We also observed many previously reported SNPs in coding and intronic regions, providing confirmation to the earlier reports.

Mutations identified in SLC4A11. Sequence chromatogram trace (upper) and conservation of the residues across the vertebrates as obtained from the UCSC database (lower; the species listed between mouse_lemur to human represented in blue indicate primates, the species represented in green are non-primate mammals and species listed between zebrafish and opossum in blue are vertebrates. Double line in the alignment region indicates unalignable bases between the species and N represents lack of sequence in the relevent portions between the aligning species). (a) c.719G>C;(p.W240S) heterozygous mutation and the tryptophan residue begin conserved across the vertebrates. (b) c.1304C>T;(p.T434I) homozygous mutation, the threonine residue is conserved across the vertebrates. (c) c.1519G>A;(p.V507I) heterozygous mutation.
Variations in the COL8A2 gene
Two reported non-synonymous variations c.464G>A (p.R155Q) and c.1505C>T (p.T502M) were found in two late-onset and one early-onset cases, respectively. c.464G>A was present in both cases and controls with no statistically significant association with the disease, whereas c.1505C>T was not present in 100 controls screened. SIFT predicts both the non-synonymous variations to ‘affect protein function,’ whereas polyphen2 scores them as ‘benign.’ Reported synonymous variations c.105G>A, c.1485G>A, c.1758C>T were observed in both cases and controls with no statistical significant association of specific genotype or allele with the disease (Supplementary Table 2). No previously unreported or reported pathogenic mutation or variations in COL8A2 gene were observed in the ADFCD1 family.
Although, two other candidate genes, TCF8 and LOXHD1, contribute to small percentage of late-onset FECD, their involvement in the above cohort is currently being studied and not presented here.
Expression and localization of SLC4A11
To begin to assess differences between WT-SLC4A11 and the FECD mutants, we expressed the proteins in HEK293 cells and examined their expression on immunoblots (Figure 2). Specificity of detection is indicated by the absence of immunoreactivity in empty vector-transfected cells. Previously, we determined that SLC4A11, expressed in HEK293 cells, migrates as two bands, an upper mature form predominantly associated with the cell surface and a lower molecular weight form, associated with endoplasmic reticulum retention.23, 25 Consistent with the earlier characterization, WT-SLC4A11 migrates as two bands, with a predominant upper band. Similarly, V507I-SLC4A11 is also found predominantly as a higher molecular weight form, aligned with that of WT-SLC4A11. In contrast, W240S-SLC4A11 is found only as an immature lower molecular weight protein, with no evident mature protein. Although T434I-SLC4A11 appears as both mature and immature forms, the relative abundance of the mature form is much lower than observed for WT-SLC4A11 (Table 3). To correct for the amount of SLC4A11 present/cell, SLC4A11 expression levels were normalized to the abundance of the housekeeping protein, GAPDH, as assessed on immunoblots (not shown). Densitometry of the total amount of SLC4A11 present as both the mature and immature bands (Figure 2b) reveals that W240S and T434I-SLC4A11 accumulate to significantly lower levels than WT, whereas V507I accumulation is indistinguishable from WT-SLC4A11.

Quantification of SLC4A11 expression and cell surface targeting. HEK293 cells were transfected with WT-SLC4A11 (WT), or the indicated SLC4A11 mutants. (a, b) Cell lysates were resolved on 7.5% acrylamide gels, and probed on immunoblots with monoclonal antibody (mAb), anti-hemagglutinin (HA) and anti-GAPDH antibodies. (b) Total SLC4A11 expression per cell was quantified by densitometry of immunoblots and data were normalized to WT-SLC4A11. (c, d) Transfected cells were treated with membrane-impermeant biotinylating reagent, SNSB. Samples of total cell lysates (T) and lysates, following removal of biotinylated protein with streptavidin resin (U), were assayed on immunoblots probed with anti-HA antibody. (d) Fraction of protein present at the cell surface was quantified, normalized to WT-SLC4A11. Dashed line indicates level of background SNSB (sulpho-NHS-SS-biotin)-labeling indicated by cytosolic GAPDH. Data represent the mean±s.e.m. of three or four replicates. *Indicates statistical difference relative to WT-SLC4A11. #Indicates statistical difference relative to all other samples.
As FECD mutants of SLC4A11 are frequently found to have reduced efficiency of processing to the cell surface, we performed cell surface biotinylation assays to measure cell surface abundance in transfected HEK293 cells (Figures 2c and d). About 60% of WT-SLC4A11 was biotinylated and thus present at the cell surface. Interestingly, the fraction of V507I present at the cell surface was indistinguishable from WT. Both T434I and W240S-SLC4A11 had cell surface expression that was less efficient than WT-SLC4A11.
The data need interpretation cautiously because the values (Figure 2d) represent fraction of protein relative to the total for that protein, not relative to the absolute amount present for WT-SLC4A11. The amount of SLC4A11 at the cell surface depends on the relative amount of SLC4A11 expressed and the fraction of this protein that traffics to the cell surface. In this light (Table 3), W240S, T434I and V507I-SLC4A11 all have reduced cell surface abundance compared with WT-SLC4A11 (15%, 40% and 65%, respectively). Disease in W240S, T434I-affected individuals is readily explained by a failure of the protein to process normally to the cell surface. Disease in V507I is not as easily explained by a processing defect.
We also assessed SLC4A11 localization in HEK293 cells by confocal immunofluorescence (Figure 3). Endogenous Na+/K+-ATPase, which serves as a marker of the plasma membrane, has a clear peri-cellular location. No immunofluorescence was detected for the HA-epitope tag in empty vector-transfected cells, indicating specificity of detection. WT-SLC4A11, however, has both intracellular and plasma membrane localization, consistent with the observation from immunoblots and cell surface processing assays (Figure 3). W240S-SLC4A11 has a reticular pattern of localization, consistent with endoplasmic reticulum. T434I-SLC4A11 displays a pattern of expression consistent with a mixture of intracellular and plasma membrane localization, which is especially evident in the merge image, which indicates some degree of co-localization with Na+-K+ ATPase. Finally, V507I-SLC4A11 has clear plasma membrane localization, with some evident intracellular localization.

Assessment of SLC4A11 localization by confocal immunofluorescence. HEK293 cells were transfected with WT-SLC4A11 (WT) or the indicated SLC4A11 mutants. Cells were processed for immunofluorescence and images were collected by confocal microscopy, with scale bars indicated in each set of panels. Nuclei were detected with 4′,6-diamidino-2-phenylindole (DAPI) staining (blue). DAPI staining was included in all panels to provide a reference for cell positioning. The hemagglutinin (HA)-tag on SLC4A11 was detected with anti-HA-antibody and chicken anti-mouse IgG conjugated with Alexa Fluor-594 (red). Plasma membrane marker, Na+/K+-ATPase, was detected with anti-Na+/K+-ATPase and chicken anti-rabbit Alexa Fluor-488 (green). Merge represents merged images.
Functional activity of SLC4A11
Mutations of SLC4A11 can affect either the efficiency of protein processing to the cell surface, functional activity of the protein, or both. Although SLC4A11 has been reported as a borate transporter, this function does not readily explain corneal dystrophies associated with mutations of the protein. We recently observed that SLC4A11 mediates a water flux,26 which is compromised in mutants of the protein. Here we assessed the ability of SLC4A11 and mutants to mediate water movement, in transfected HEK293 cells. The basis for the assay is to transfect cells with SLC4A11, along with eGFP. Cells are exposed to a hypo-osmotic challenge induced by switching between media with the same composition, except for concentration of mannitol. The rate of cell swelling was measured by the dilution of eGFP, monitored by confocal microscopy of a small region of interest in the cytosol.26
All cells swelled in response to a switch from iso-osmotic medium to hypo-osmotic medium (Figure 4a), as evident from the reduction of fluorescence, corresponding to a dilution of cytosolic eGFP as the cell swelled. Rates of cell swelling were corrected for background activity of empty vector-transfected cells (Supplementary Table 3) and normalized to WT-SLC4A11 (Figure 4b). All of the mutants assessed here had functional activity significantly less than WT. V507I-SLC4A11 had transport activity that was significantly higher than the other two mutants, but still much lower than WT (26% of WT level).

Measurement of SLC4A11 functional activity. HEK293 cells were transiently co-transfected with enhanced green fluorescent protein (eGFP) cDNA and wild-type (WT) or mutant SLC4A11, or empty vector (pcDNA3.1), as indicated. (a) Cells were perfused alternately with isotonic (black bar) and hypotonic (white bar) medium. eGFP fluorescence in regions of interest was measured digitally from images captured every second by confocal microscopy. Traces represent WT-SLC4A11 (black), empty vector (grey), W240S-SLC4A11 (red), T434I-SLC4A11 (blue), and V507I-SLC4A11 (brown). (b) Rate of fluorescence change, calculated by linear regression of the initial 15 s of linear fluorescence decrease, following perfusion with hypotonic buffer (Supplementary Table 3). Data were corrected for background activity of empty vector-transfected cells and normalized to WT-SLC4A11. Data represent the mean±s.e.m. of three or four independent swelling experiments with 30–40 cells measured per assay. * and # indicate statistical difference relative to WT-SLC4A11 and the other mutants, respectively.
A summary of expression and functional activity of the mutants clarifies the nature of their defects (Table 3). Cell surface abundance relative to WT, indicates that very low levels of W240S and T434I-SLC4A11 process to the cell surface. In contrast, V507I accumulates at the cell surface at about two-third the level of WT, suggesting that this mutant has a functional defect combined with some degree of intracellular retention. Indeed, cells expressing this mutant displayed only 26% of WT levels of water flux. The functional defect in these mutants is best displayed in the right hand column of Table 3, which represents the relative water flux carried by each molecule of SLC4A11 at the plasma membrane. All of the mutants have some degree of compromised activity relative to WT.
Discussion
We sought to identify mutations responsible for early- and late-onset FECD in the Indian ethnic patients, by screening the genes SLC4A11 and COL8A2. Mutations of SLC4A11 were originally identified in autosomal recessive CHED, where homozygous truncating mutations were reported.11 Heterozygous misssense SLC4A11 mutations in late-onset sporadic FECD subsequently provided examples of allelic heterogeneity in corneal dystrophies.12, 13 Here, we identified three previously unreported mutations, c.719G>C (p.W240S), c.1304C>T (p.T434I) and c.1519G>A (p.V507I), in late-onset sporadic FECD. The frequency of mutations identified in SLC4A11 in FECD in different ethnic populations reported so far is detailed in Table 4.12, 13, 27, 28 This study presents the first characterization of the functional effects of SLC4A11 mutations, measuring activity by an osmotically activated cell swelling assay. Although disease from two of these mutations was readily explained by intracellular retention, we also found that the protein that processed to the cell surface was functionally compromised, with unitary functional activity that was 5–42% that of WT.
SLC4A11 protein was originally characterized as an electrogenic Na+-coupled borate co-transporter that co-transports Na+ and OH− in the absence of borate.29 More recently, SLC4A11 was found not to act as a bicarbonate transporter, unlike other members of the SLC4 family.30 SLC4A11 may have multiple functions, with reports that it does not mediate borate transport,23, 30 but rather functions as a water conductive pathway, or a Na+/OH− co-transporter.30 SLC4A11 is predicted to have 14 transmembrane segments and N- and C-terminal cytosolic domains.25
Here we report three previously unknown mutations in late-onset FECD cases, W240S, T434I andV507I. On the basis of a published topology model,25 amino-acid 240 localizes to the N- terminal cytosolic domain, whereas amino acids 434 and 507 are present in third and fourth trans-membrane segments, respectively.
Ideally, SLC4A11 mutants would be studied in the native context of corneal cells, as the HEK293 cell system used here may not represent the biology present in intact cornea. Corneal endothelial cells form a well-polarized endothelial layer, whereas HEK293 cells are non-polarized. That said, even immortalized corneal endothelial cells might be expected to de-differentiate and have a phenotype differing from that in the cornea. Studies here of SLC4A11 in HEK293 cells are limited, but we note that transfected HEK293 cells express a similar amount of SLC4A11 as corneal endothelial cells (Loganathan and Casey, Submitted).
W240S is expressed in immature form only, indicating that it is retained intracellularly, likely as a result of mis-folding, so it is unavailable for normal function at the plasma membrane. T434I-SLC4A11, expressed in HEK293 cells, is found in mature form, but at a reduced level. This suggests that the protein is mis-folded and degraded and is not processed fully to the cell surface; low steady-state level of the mutant explains the disease. T434I was observed as homozygous in the patient, who was not born to consanguineous parents. Late-onset FECD is considered dominant and all reported mutations thus far have been heterozygous. The unaffected brother and sister of this index patient were homozygous for the WT (data not shown), the father was deceased and mother was not available for testing. Therefore, we unfortunately cannot assess whether the parents are heterozygous for the mutant or homozygous WT. As there is a still low amount of mature form of the protein for the T434I mutant, as indicated by osmotically driven water flux assays, the disease may manifest as late-onset even with homozygous mutation. V507I-SLC4A11 protein is expressed and processed to the cell surface nearly as well as WT. Unlike, the other characterized mutants, disease cannot be explained by a defect in cell surface accumulation of SLC4A11. Instead, V507I has a functional defect, retaining only 26% of WT-SLC4A11 activity.
In the absence of a high-resolution structure for SLC4A11 or any member of the SLC4 family, it is difficult to interpret the potential significance of the identified mutants. Amino-acid sequence alignments across species (Figure 1) reveal complete conservation of W240 and T434, suggesting these are functionally or structurally important. W240S and T434I are not structurally conservative mutations, so significant structural/functional perturbation would be expected, arising from these substitutions. Disease associated with V507I is more difficult to explain, as the valine to isoleucine change is subtle. V507, however, is conserved across species (Figure 1), except for bovine, chicken and Western clawed frog (Xenopus tropicalis). At this position, there is a clear preference for a hydrophobic residue (V, F, L, I). The isoleucine found in bovine SLC4A11 mimics the sequence alteration that we identified in one patient.
Why is isoleucine tolerated at this position in cattle SLC4A11, but not human? Cell surfacing processing and functional defects identified in V507I SLC4A11 are consistent with the attribution of V507I as deleterious and disease causing. Two speculative possibilities arise: (1) Bovine SLC4A11 has a lower functional activity than human SLC4A11 (as V507I human SLC4A11 has only, 42% of WT activity), which is tolerated because of differences between humans and cattle, or (2) Bovine SLC4A11 has another sequence change at a different position that compensates for the isoleucine. That is, this amino-acid position is involved in a buried portion of the protein, such that substitutions give rise to mis-folding. In bovine SLC4A11, a second amino-acid change may allow conservation of protein packing.
Regarding the dominant inheritance of FECD, we note that patients are heterozygous, whereas functional assays were performed under homozygous conditions (that is, mutants were expressed in the absence of WT SLC4A11). In this regard, the functional activity does not represent the level that would be found in patients, but the analysis represents the molecular defects found in the mutants when expressed alone. Previously, SLC4A11 was found to form heterodimers between WT and mutant forms.25 Moreover, the ER-retained phenotype of heterodimers was revealed as dominant in the case of Fuchs heterodimers and recessive for CHED, consistent with the pattern of inheritance of the two diseases.
Mutations in SLC4A11 have been reported in FECD, CHED and in Harboyan syndrome.23 Missense, nonsense, frameshift and splice site mutations leading to truncated protein in CHED, whereas missense and frameshift mutations in FECD and Harboyan syndrome have been described.11, 28, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 These mutations are located throughout the protein in predicted N-terminal, cytosolic domains, transmembrane segments, and extracellular and cytosolic loops. Missense mutations in all three diseases are spread across different domains of the SLC4A11 protein and do not show any genotype–phenotype correlation. Until now, 82 mutations (missense, nonsense, small insertions and deletions, indels and gross deletions) in SLC4A11 have been reported in all three diseases (HGMD), here we have described further three missense mutations identified in late-onset sporadic FECD cases.
COL8A2 gene mutations are reported in all early-onset FECD families described so far.2, 3, 4, 43 Although the first report on COL8A2 gene, describing its role in early-onset FECD, also suggested its involvement in some cases of late-onset disease, later studies have not identified any pathogenic mutation in COL8A2 in late-onset FECD.2, 27, 43, 44 In our cohort, no pathogenic mutations were seen in COL8A2 in the late-onset FECD cases either. The frequencies of synonymous SNPs c.105G>A, c.1485G>A and c.1758C>T and non-synonymous SNP c.464G>A (p.R155Q) did not show any statistical significant difference between the cases and controls.
The c.1505C>T (p.T502M) variation found in an early-onset case (28 years at diagnosis) was not observed in 200 control chromosomes. This variation is predicted as ‘affecting protein function’ by SIFT but ‘benign’ by Polyphen2. This variation was reported in Japanese cohort with no significant difference in frequency between cases and controls.43 The minor allele frequency of the T allele is reported as 0.066 in the dbSNP database. This individual was lost to follow-up and we could not recruit any other family members for segregation analysis. Thus, the pathogenicity of the c.1505C>T (p.T502M) variation remains unclear.
Three individuals of the early-onset FECD family also did not show any mutation in the COL8A2 gene. Whole-genome linkage analysis using Affymetrix 10K XbaI gene chip excluded the COL8A2 loci in chromosome 1 (data not shown). Thus, defects in genes other than SLC4A11 and COL8A2 could be responsible for some cases of early-onset FECD.
In summary, we identified three previously unreported mutations in the SLC4A11 gene, two of them (c.719G>C (p.W240S) and c.1304C>T (p.T434I)) affecting the ability of SLC4A11 protein to localize and accumulate at the plasma membrane. The third newly identified mutation (c.1519G>A (p.V507I)) inactivates the protein, resulting in greatly reduced functional activity (osmotically driven water flux) in spite of near normal cell surface abundance. In this study, 5 of the 45 late-onset sporadic FECD cases were identified to harbor a mutation in SLC4A11 gene, contributing to 11% of the disease pathogenesis. In the cohort studied here, COL8A2 gene was not involved in the pathogenesis of sporadic late-onset FECD. It is, however, interesting to note that mutations were not identified in COL8A2 gene in the early-onset family studied (it was excluded by whole-genome linkage analysis), indicating genetic heterogeneity in the early-onset phenotype as well.
References
Sundin, O. H., Jun, A. S., Broman, K. W., Liu, S. H., Sheehan, S. E., Vito, E. C. et al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest. Ophthalmol. Vis. Sci. 47, 140–145 (2006).
Gottsch, J. D., Sundin, O. H., Liu, S. H., Jun, A. S., Broman, K. W., Stark, W. J. et al. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 46, 1934–1939 (2005).
Biswas, S., Munier, F. L., Yardley, J., Hart-Holden, N., Perveen, R., Cousin, P. et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 10, 2415–2423 (2001).
Mok, J. W., Kim, H. S. & Joo, C. K. Q455V mutation in COL8A2 is associated with Fuchs' corneal dystrophy in Korean patients. Eye (Lond) 23, 895–903 (2009).
Li, J. Y., Terry, M. A., Goshe, J., Davis-Boozer, D. & Shamie, N. Three-year visual acuity outcomes after Descemet's stripping automated endothelial keratoplasty. Ophthalmology 119, 1126–1129 (2012).
Sundin, O. H., Broman, K. W., Chang, H. H., Vito, E. C., Stark, W. J. & Gottsch, J. D. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest. Ophthalmol. Vis. Sci. 47, 3919–3926 (2006).
Riazuddin, S. A., Zaghloul, N. A., Al-Saif, A., Davey, L., Diplas, B. H., Meadows, D. N. et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45–53 (2010).
Riazuddin, S. A., Eghrari, A. O., Al-Saif, A., Davey, L., Meadows, D. N., Katsanis, N. et al. Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35.2. Invest. Ophthalmol. Vis. Sci. 50, 5667–5671 (2009).
Afshari, N. A., Li, Y. J., Pericak-Vance, M. A., Gregory, S. & Klintworth, G. K. Genome-wide linkage scan in fuchs endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 50, 1093–1097 (2009).
Riazuddin, S. A., Parker, D. S., McGlumphy, E. J., Oh, E. C., Iliff, B. W., Schmedt, T. et al. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am. J. Hum. Genet. 90, 533–539 (2012).
Vithana, E. N., Morgan, P., Sundaresan, P., Ebenezer, N. D., Tan, D. T., Mohamed, M. D. et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat. Genet. 38, 755–757 (2006).
Vithana, E. N., Morgan, P. E., Ramprasad, V., Tan, D. T., Yong, V. H., Venkataraman, D. et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum. Mol. Genet. 17, 656–666 (2008).
Riazuddin, S. A., Vithana, E. N., Seet, L. F., Liu, Y., Al-Saif, A., Koh, L. W. et al. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum. Mutat. 31, 1261–1268 (2010).
Baratz, K. H., Tosakulwong, N., Ryu, E., Brown, W. L., Branham, K., Chen, W. et al. E2-2 protein and Fuchs's corneal dystrophy. New Engl. J. Med. 363, 1016–1024 (2010).
Li, Y. J., Minear, M. A., Rimmler, J., Zhao, B., Balajonda, E., Hauser, M. A. et al. Replication of TCF4 through association and linkage studies in late-onset Fuchs endothelial corneal dystrophy. PLoS ONE 6, e18044 (2011).
Riazuddin, S. A., McGlumphy, E. J., Yeo, W. S., Wang, J., Katsanis, N. & Gottsch, J. D. Replication of the TCF4 intronic variant in late-onset Fuchs corneal dystrophy and evidence of independence from the FCD2 locus. Invest. Ophthalmol. Vis. Sci. 52, 2825–2829 (2011).
Thalamuthu, A., Khor, C. C., Venkataraman, D., Koh, L. W., Tan, D. T., Aung, T. et al. Association of TCF4 gene polymorphisms with Fuchs' corneal dystrophy in the Chinese. Invest. Ophthalmol. Vis. Sci. 52, 5573–5578 (2011).
Wieben, E. D., Aleff, R. A., Tosakulwong, N., Butz, M. L., Highsmith, W. E., Edwards, A. O. et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS ONE 7, e49083 (2012).
Saumya Pal, S., Raman, R., Ganesan, S., Sahu, C. & Sharma, T. Sankara Nethralaya diabetic retinopathy epidemiology and molecular genetic study (SN–DREAMS III): study design and research methodology. BMC Ophthalmol. 11, 7 (2011).
George, R., Arvind, H., Baskaran, M., Ramesh, S. V., Raju, P. & Vijaya, L. The Chennai glaucoma study: prevalence and risk factors for glaucoma in cataract operated eyes in urban Chennai. Indian J. Ophthalmol. 58, 243–245 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Vilas, G. L., Morgan, P. E., Loganathan, S. K., Quon, A. & Casey, J. R. A biochemical framework for SLC4A11, the plasma membrane protein defective in corneal dystrophies. Biochemistry 50, 2157–2169 (2011).
Ruetz, S., Lindsey, A. E. & Kopito, R. R. Function and biosynthesis of erythroid and nonerythroid anion exchangers. Soc. Gen. Physiol. Ser. 48, 193–200 (1993).
Vilas, G. L., Loganathan, S. K., Quon, A., Sundaresan, P., Vithana, E. N. & Casey, J. Oligomerization of SLC4A11 protein and the severity of FECD and CHED2 corneal dystrophies caused by SLC4A11 mutations. Hum. Mutat. 33, 419–428 (2012).
Vilas, G. L., Loganathan, S. K., Liu, J., Riau, A. K., Young, J. D., Mehta, J. S. et al. Transmembrane water flux through SLC4A11: a route defective in genetic corneal diseases. Hum. Mol. Genet. 22, 4579–4590 (2013).
Hemadevi, B., Srinivasan, M., Arunkumar, J., Prajna, N. V. & Sundaresan, P. Genetic analysis of patients with Fuchs endothelial corneal dystrophy in India. BMC Ophthalmol 10, 3 (2010).
Minear, M. A., Li, Y. J., Rimmler, J., Balajonda, E., Watson, S., Allingham, R. R. et al. Genetic screen of African Americans with Fuchs endothelial corneal dystrophy. Mol. Vis. 19, 2508–2516 (2013).
Park, M., Li, Q., Shcheynikov, N., Zeng, W. & Muallem, S. NaBC1 is a ubiquitous electrogenic Na+ -coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation. Mol. Cell 16, 331–341 (2004).
Jalimarada, S. S., Ogando, D. G., Vithana, E. N. & Bonanno, J. A. Ion transport function of SLC4A11 in corneal endothelium. Invest. Ophthalmol. Vis. Sci. 54, 4330–4340 (2013).
Ramprasad, V. L., Ebenezer, N. D., Aung, T., Rajagopal, R., Yong, V. H., Tuft, S. J. et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Mutation in brief #958. Online. Hum. Mutat. 28, 522–523 (2007).
Aldave, A. J., Yellore, V. S., Bourla, N., Momi, R. S., Khan, M. A., Salem, A. K. et al. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea 26, 896–900 (2007).
Kumar, A., Bhattacharjee, S., Prakash, D. R. & Sadanand, C. S. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Mol. Vis. 13, 39–46 (2007).
Desir, J., Moya, G., Reish, O., Van Regemorter, N., Deconinck, H., David, K. L. et al. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J. Med. Genet. 44, 322–326 (2007).
Hemadevi, B., Veitia, R. A., Srinivasan, M., Arunkumar, J., Prajna, N. V., Lesaffre, C. et al. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch. Ophthalmol 126, 700–708 (2008).
Shah, S. S., Al-Rajhi, A., Brandt, J. D., Mannis, M. J., Roos, B., Sheffield, V. C. et al. Mutation in the SLC4A11 gene associated with autosomal recessive congenital hereditary endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 29, 41–45 (2008).
Aldahmesh, M. A., Khan, A. O., Meyer, B. F. & Alkuraya, F. S. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest. Ophthalmol. Vis. Sci 50, 4142–4145 (2009).
Paliwal, P., Sharma, A., Tandon, R., Sharma, N., Titiyal, J. S., Sen, S. et al. Congenital hereditary endothelial dystrophy - mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol. Vis. 16, 2955–2963 (2010).
Mehta, J. S., Hemadevi, B., Vithana, E. N., Arunkumar, J., Srinivasan, M., Prajna, V. et al. Absence of phenotype-genotype correlation of patients expressing mutations in the SLC4A11 gene. Cornea 29, 302–306 (2010).
Kodaganur, S. G., Kapoor, S., Veerappa, A. M., Tontanahal, S. J., Sarda, A., Yathish, S. et al. Mutation analysis of the SLC4A11 gene in Indian families with congenital hereditary endothelial dystrophy 2 and a review of the literature. Mol. Vis. 19, 1694–1706 (2013).
Park, S. H., Jeong, H. J., Kim, M. & Kim, M. S. A novel nonsense mutation of the SLC4A11 gene in a Korean patient with autosomal recessive congenital hereditary endothelial dystrophy. Cornea 32, e181–e182 (2013).
Kim, J. H., Ko, J. M. & Tchah, H. Fuchs endothelial corneal dystrophy in a heterozygous carrier of congenital hereditary endothelial dystrophy type 2 with a novel mutation in SLC4A11. Ophthalmic. Genet. (e-pub ahead of print 6 February 2014).
Kobayashi, A., Fujiki, K., Murakami, A., Kato, T., Chen, L. Z., Onoe, H. et al. Analysis of COL8A2 gene mutation in Japanese patients with Fuchs' endothelial dystrophy and posterior polymorphous dystrophy. Jpn J. Ophthalmol. 48, 195–198 (2004).
Aldave, A. J., Rayner, S. A., Salem, A. K., Yoo, G. L., Kim, B. T., Saeedian, M. et al. No pathogenic mutations identified in the COL8A1 and COL8A2 genes in familial Fuchs corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 47, 3787–3790 (2006).
Acknowledgements
The work in SNONGC Department of Genetics and Molecular Biology, Vision Research Foundation was part of the project funded by Indian Council of Medical Research, Government of India (no. 53/11/2006-BMS). We thank all the patients for participating in the study. We also thank late Dr G Sitalakshmi (cornea consultant) for her contribution in patient evaluation. Work in the laboratory of JRC was supported by an operating grant from the Canadian Institutes of Health Research. SKL is supported by a graduate studentship from the International Research Training Group in Membrane Biology, supported by Canada’s Natural Sciences and Engineering Research Council.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Soumittra, N., Loganathan, S., Madhavan, D. et al. Biosynthetic and functional defects in newly identified SLC4A11 mutants and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J Hum Genet 59, 444–453 (2014). https://doi.org/10.1038/jhg.2014.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.55
This article is cited by
-
Genetic mutations and molecular mechanisms of Fuchs endothelial corneal dystrophy
Eye and Vision (2021)
-
Human Corneal Expression of SLC4A11, a Gene Mutated in Endothelial Corneal Dystrophies
Scientific Reports (2019)
-
The Molecular Basis of Fuchs’ Endothelial Corneal Dystrophy
Molecular Diagnosis & Therapy (2019)
