
Abstract
Experiments in rodents revealed neuropeptide S (NPS) to constitute a potential novel treatment option for anxiety diseases such as panic and post-traumatic stress disorder. However, both its cerebral target sites and the molecular underpinnings of NPS-mediated effects still remain elusive. By administration of fluorophore-conjugated NPS, we pinpointed NPS target neurons in distinct regions throughout the entire brain. We demonstrated their functional relevance in the hippocampus. In the CA1 region, NPS modulates synaptic transmission and plasticity. NPS is taken up into NPS receptor-expressing neurons by internalization of the receptor–ligand complex as we confirmed by subsequent cell culture studies. Furthermore, we tracked internalization of intranasally applied NPS at the single-neuron level and additionally demonstrate that it is delivered into the mouse brain without losing its anxiolytic properties. Finally, we show that NPS differentially modulates the expression of proteins of the glutamatergic system involved inter alia in synaptic plasticity. These results not only enlighten the path of NPS in the brain, but also establish a non-invasive method for NPS administration in mice, thus strongly encouraging translation into a novel therapeutic approach for pathological anxiety in humans.
Similar content being viewed by others


INTRODUCTION
The novel neuropeptide S (NPS) has been shown to elicit strong anxiolytic effects in rodents upon intracerebral injection (Leonard et al, 2008; Xu et al, 2004). NPS treatment facilitates fear extinction in mice and reduces fear memory retrieval after extinction training (Jüngling et al, 2008), thus becoming interesting especially for post-traumatic stress disorder (PTSD) treatment. This peptide is also known to influence the HPA axis (Smith et al, 2006), which is involved in the pathogenesis of anxiety and other affective disorders (de Kloet et al, 2005). NPS interacts with many neurotransmitter systems in the brain, including the glutamatergic network (Han et al, 2009; Raiteri et al, 2009; Si et al, 2010; Okamura et al, 2010), which is involved in the pathology of disorders such as PTSD (Rossi et al, 2009; Ravindran and Stein, 2009). The anxiolytic properties of NPS are not mediated by the benzodiazepine-binding site of the GABAA receptor (Leonard et al, 2008), thus suggesting that its administration would not cause benzodiazepine-like side effects. However, the intracerebral NPS administration techniques employed in animal experiments reported so far are hardly transferable to human clinical use.
NPS exerts its function through the NPS receptor (NPSR), a G-protein-coupled receptor (GPCR) bound to either Gq or Gs (Reinscheid et al, 2005). In NPSR-KO mice, NPS no longer exerts its previously described effects, proving that NPS actions are mediated exclusively by NPSR (Zhu et al, 2010). Interestingly, single-nucleotide polymorphisms (SNPs) of the NPSR gene have been associated with panic disorder (Domschke et al, 2010). Although GPCR internalization upon ligand binding is a known desensitization mechanism (Hökfelt et al, 2003), this process has not yet been explicitly demonstrated for the NPS/NPSR complex.
Even though, in rats, NPS expression is limited to three brain regions (the peri-locus coeruleus, the lateral parabrachial nucleus, and the principle sensory 5 nucleus of the trigeminus), NPSR mRNA (Xu et al, 2007) and protein (Leonard and Ring, 2011) are expressed throughout the rat brain. However, NPSR protein and mRNA expression patterns do not overlap completely, as in the case of the hippocampal CA1–3 regions and the dentate gyrus, leaving the ultimate identity of NPS target sites open.
Here, we identified NPS target neurons and target regions in the mouse brain and validated their functional relevance in the ventral hippocampus. Furthermore, we show that NPS is internalized by an NPSR-dependent mechanism. Finally, we developed an NPS administration technique that has the potential to facilitate animal studies with NPS and, more important, is most likely also applicable to humans.
MATERIALS AND METHODS
Animals
For behavioral experiments, C57BL/6N males were purchased from Charles River Germany GmbH (Sulzfeld, Germany). Male HAB mice were obtained from the animal facility of the Max Planck Institute (MPI) of Biochemistry, Martinsried, Germany. For all other animal experiments, we used C57BL/6N males bred in the animal facility of the MPI of Biochemistry, Martinsried, Germany. Experiments were performed on 10-week-old animals. All procedures were approved by the Government of Upper Bavaria and were in accordance with European Union Directive 86/609/EEC.
Administration of Fluorophore-Labeled NPS
For ICV injection, a guide cannula was implanted into the right lateral ventricle using a stereotaxic frame (coordinates: 0.3 mm caudal and 1.1 mm lateral from the bregma; 1.3 mm ventral from the skull surface). Mice were injected with 2 μl of Cy3-NPS or rhodamine-NPS (10 μM; Phoenix Pharmaceuticals, Karisruhe, Germany) or pure rhodamine (1 g/ml; Sigma-Aldrich, Taufkirchen, Germany). The mice were killed at 2, 10, or 30 min after injection. To additionally clarify the specificity of the NPS internalization mechanism, 2 μl of native NPS (50 μM or 100 μM, rat; Bachem, Weil am Rhein, Germany) in Ringer solution were pre-injected 10 min before injection of Cy3-NPS. Additionally, to investigate whether NPS is internalized through the already described NPSR, we co-injected Cy3-NPS with NPSR-specific antagonists: 1.5 mM [D-Cys(tBu)5]Neuropeptide S (Camarda et al, 2009) and the active enantiomer of SHA 68, (R)-SHA 68 (Okamura et al, 2008; Trapella et al, 2011), both at 150-fold concentration of Cy3-NPS. (R)-SHA 68 was a gift from A Sailer, Novartis (Basel, Switzerland). The mice were killed 30 min after injection.
For intranasal application, mice were anesthetized with ketamine–rompun at 0.1 ml/10 g (2% rompun 2%, xylazin—xylazin hydrochloride (Bayer Vital GmbH, Lederhosen, Germany); 5% ketamine 10%, ketamine hydrochloride (Essex Pharma GmbH, Munich, Germany); in NaCl) and placed in a supine position, with the head supported at a 45-degree angle to the body (van den Berg et al, 2002). A 7-μl volume of Cy3-NPS (10 μM) or pure rhodamine (10 g/ml) was applied alternatingly to each nostril; after 5 min, the procedure was repeated. The effect of the anesthetic lasted for approximately 20 min. The mice were killed at 15, 30 min, and 4 h after the first application.
For comparison of Cy3-NPS uptake between ICV and intranasal administration, a four-point scale was used to score both signal intensity and number of labeled cells as follows: very strong, +++; strong, ++; moderate, +; weak/scattered, −/+; no signal, −.
Immunohistochemistry
Brains were removed, post-fixed in 4% paraformaldehyde, and cryoprotected in 20% sucrose, then shock-frozen in methylbutane. Immunohistochemistry was performed on free-floating cryosections (40 μm). The primary antibodies used were specific for the neurofilament (1 : 1000; Abcam, Cambridge, UK), glial fibrillary acidic protein (GFAP) (1 : 250; Dako, Glostrup, Denmark), and Iba-1 (1 : 1000; Wako, Richmond, VA, USA). The secondary antibodies used were specific for mouse (Alexa-488 goat anti-mouse IgG) and rabbit (Alexa-488 donkey anti-rabbit IgG) (1 : 300; Invitrogen, Darmstadt, Germany). The sections were counterstained with 4′,6-diamidin-2-phenylindole (DAPI, 200 ng/ml; Carl Roth, Karlsruhe, Germany) and mounted with a fluorescence-preserving mounting medium (Shandon Immu-Mount; Thermo Scientific, Schwerte, Germany).
Behavioral Experiments
Behavioral tests (open field, dark–light test and EPM) following intranasal NPS application were performed sequentially in the order mentioned. Each test lasted for 5 min, with an inter-test interval of 5 min, as described previously (Bunck et al, 2009; Krömer et al, 2005). The animals’ behavior during testing was videotaped and relevant parameters were analyzed with the tracking software ANY-maze version 4.30 (Stoelting, Wood Dale, IL, USA). Three different doses of native NPS (7, 14, and 28 nmol) or vehicle (Ringer solution) were administered alternatingly into each nostril of the alert mouse using a 10-μl pipette. A total volume of 14 μl was administered in two steps of 7 μl per step, with a 5-min rest period between the administration steps in order to avoid overfilling of the nostrils and loss of the substance by exhalation, as well as reduced or delayed absorption. The alert mice were restrained manually during the administration procedure in a supine position and the head was held immobile at an angle of approximately 45 degrees to the body. This position was used to avoid loss of the substance into the sinus cavities, as it has been described as being the most effective for nasal delivery to the CNS in rodents (van den Berg et al, 2002). Mice were held immobile in this position until all the substance had been absorbed (approximately 10 s). Mice were tested 30 min and 4 h after the beginning of intranasal administration. For protein isolation, animals were killed 4 and 24 h after treatment, and the prefrontal cortex and bilateral hippocampi were prepared immediately and shock-frozen in methylbutane. For RNA isolation, animals were killed 4 h after treatment and the prepared brain regions were shock-frozen in liquid nitrogen.
Immunoblotting
For immunoblotting, proteins were extracted from the prefrontal cortex and hippocampus. Quantitative blot analysis was performed using the ImageJ software (US National Institutes of Health; http://rsbweb.nih.gov/ij/). The primary antibodies used were as follows: Glt-1, GluR1, GluR2 (1 : 100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), synapsin (1 : 2000; Synaptic Systems, Göttingen, Germany), and GAPDH (1 : 2000; Santa Cruz Biotechnology). The secondary antibodies were as follows: donkey anti-goat IgG-HRP (1 : 10 000; Santa Cruz Biotechnology), goat anti-rabbit IgG-HRP (1 : 7500; Sigma-Aldrich), and goat anti-mouse IgG-HRP (1 : 25 000; Sigma-Aldrich).
Real-Time PCR
Total RNA was extracted using the Trizol protocol (GibcoBRL, Darmstadt, Germany) and reverse-transcribed using the Omniscript Reverse Transcription kit (Qiagen, Hilden, Germany). For real-time PCR, the QuantiFast SYBR Green PCR kit (Qiagen) was used according to the manufacturer's instructions. Experiments were performed in duplicates with a Lightcycler2.0 instrument (Roche Diagnostics, Mannheim, Germany) under the following PCR conditions: initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation (95 °C for 10 s), and a combined annealing and extension phase (60 °C for 30 s). The primers used were as follows: synapsin I (fwd: 5′-CACCGACTGGGCAAAATACT-3′; rev: 5′-TCCGAAGAACTTCCATGTCC-3′); synapsin II (fwd: 5′-CCTTCATCGACGCCAAGTAT-3′; rev: 5′-GAGCAGGCATCTACCCAGAG-3′); Glt-1 (Perisic et al, 2010); GluR1 and GluR2 (Blanco et al, 2011); and GAPDH (fwd: 5′-CCATCACCATCTTCCAGGAGCGAG-3′; rev: 5′-GATGGCATGGACTGTGGTCATGAG-3′). Relative gene expression was determined by the method (Livak and Schmittgen, 2001). Crossing points were calculated by the LightCycler software 4.0 (Roche Diagnostics GmbH) and normalized to the housekeeping gene GAPDH.
Cloning of EGFP-NPSR, Cell Culture, and Transfection Experiments
The cDNA encoding mNPSR was cloned into the pEGFP-C1 vector (Clontech, Mountain View, CA, USA) to generate NPSR fused to the C-terminus of EGFP (NPSR pEGFP-C1). The primers used were as follows: forward, 5′-AATGCCTCGAGTTATGCCAGCCAACCTCACAGAG-3′ and reverse, 5′-GCCGCGGATCCTCAGCCTAGCACTACTGCCTC-3′. Prior to microscopic analysis, 2.5 × 106 HEK cells per well were seeded on gelatine-coated coverslips in 24-well plates, then transfected with NPSR pEGFP-C1 (Clontech) using ExGen 500 (Fermentas, St Leon-Rot, Germany). To visualize internalization of Cy3-NPS, we adapted a protocol from Grady et al (1995). In brief, we added 100 nM Cy3-NPS to the culture medium and incubated the cells for 60 min at 4 °C. After washing, cells were re-heated to 37 °C for 10–30 min, then fixed in 4% paraformaldehyde, counterstained with DAPI, and mounted.
Electrophysiology
Field potential recordings in horizontal brain slices (350-μm thick) containing the ventral hippocampus were performed as described previously (Schmidt et al, 2011). Only the first two slices from the ventral surface of the brain in which the CA1 region was clearly visible were used for the experiments. After recovery for 30 min at 34 °C, slices were incubated for 2 h either in normal artificial cerebrospinal fluid (ACSF), ACSF containing 1 μM NPS (Bachem), or ACSF containing 1 μM NPS and 10 μM (R)-SHA 68 at room temperature (23–25 °C). During experimentation, slices were superfused with NPS-/(R)-SHA 68-free ACSF.
Image Acquisition and Processing
Images were acquired either with a confocal microscope (Olympus IX81, software, FluoView FV1000 2.1.2.5) or with an epifluorescence microscope (Olympus BX61, software, cell^F 2.8; Olympus Soft Imaging Systems GmbH, Münster, Germany). After acquisition, images were processed using Photoshop (Adobe).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.03 (GraphPad Software, La Jolla, CA, USA). For the behavioral data, we used Grubbs’ test to identify and exclude significant outliers (p<0.05); one animal per group was excluded at the most. For the paired-pulse facilitation analysis, we used two-way ANOVA (Treatment/Interstimulus Interval (ISI)) for dependent measures (ISI), and Newman–Keuls post-hoc test. LTP experiments were analyzed by one-way ANOVA with Bonferroni's post-hoc test. The results of the behavioral assays of the C57BL/6N groups treated with different doses of NPS were normalized to the controls and analyzed by one-way ANOVA with Bonferroni's post-hoc test. For the behavioral data from the HAB mice, we used the two-tailed unpaired Student's t-test. Immunoblotting and real-time PCR data were normalized to the internal control GAPDH and analyzed by two-tailed unpaired Student's t-test. p-values below 0.05 were considered significant, and p-values between 0.05 and 0.1 were considered to represent a trend.
RESULTS
Target Brain Regions and Target Cells of NPS
To identify NPS target cells in the murine brain, we examined the distribution pattern of a fluorescent NPS conjugate (Cy3-NPS) after unilateral ICV injection in C57BL/6N mice. Ten minutes after ICV injection, single cells already showed discrete patterns of fluorescence (data not shown). Thirty minutes after ICV injection, various cell populations in distinct brain regions had already internalized Cy3-NPS (Table 1).
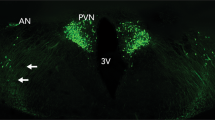
Cells containing Cy3-NPS were present within the basal ganglia (globus pallidus and nucleus accumbens) and also in the amygdaloid nuclei, including the basolateral and central amygdala (Figure 1a). Uptake of the labeled substance was detected even in regions further away from the ventricular system, such as the somatosensory cortex (Figure 1b). Cy3-NPS was additionally found in other regions associated with stress response and learning, such as the lateral habenula and the mediodorsal thalamic nuclei, respectively (Figure 1c), as well as in regions with neuroendocrine functions, such as the arcuate and ventromedial hypothalamic nuclei (Figure 1d). It also targeted single cells within the locus coeruleus, the tegmental nucleus, Barrington's nucleus, and the parabrachial nucleus (Figure 1e).

Representative selection of mouse brain regions targeted by ICV-administered fluorescent Cy3-NPS. (a) Amygdaloid structures (Cy3-NPS: bright white): Central amygdala (CeA), medial amygdala (MeA), basolateral amygdala (BLA), basomedial amygdala (BMA). Cortical structures: Dorsal endopiriform cortex (DEn). Basal ganglia: Globus pallidus (GP). Scale bar, 200 μm. (b–e) The leftmost panels show a schematic overview of murine brain regions (Franklin and Paxinos, 2007). The middle panels show the nuclear counterstain DAPI (blue) (scale bar, 100 μm) and cell populations having taken up Cy3-NPS (red). The images in the red channel are presented in two different magnifications (scale bars, 100 and 10 μm)—white rectangles indicate the area of magnification. The rightmost panels show an overlay of the blue and red channels (scale bar, 100 μm). (b) Cortical structures: Primary somatosensory cortex (S1). (c) Thalamic structures: Paraventricular thalamic nucleus (PV); sporadically in medial habenula (MHb); lateral habenula (LHb); mediodorsal thalamic nucleus (MD): medial (MDM), central (MDC), and lateral (MDL). Third ventricle (3V). (d) Hypothalamic structures: Periventricular hypothalamic nucleus (Pe), dorsomedial hypothalamic nucleus (DM), ventromedial hypothalamic nucleus (VMH), arcuate hypothalamic nucleus (Arc). Third ventricle (3V). (e) Brainstem structures: Central gray of the pons (CGPn), medial vestibular nucleus (MVe), sporadically in posterodorsal tegmental nucleus (PDTg), Barrington's nucleus (Bar), sporadically in locus coeruleus (LC) and in medial parabrachial nucleus (MPB). Fourth ventricle (4V). All images were acquired with a confocal microscope and are representative for a total of 10 mice. See Table 1 for a complete list of brain regions where uptake of Cy3-NPS was detected.
Most notable was the internalization of Cy3-NPS in the hippocampal CA1, CA2, and CA3 regions, mainly in the pyramidal and oriens layers, and sparingly in the radiate and molecular layers (Figure 2a), as well as in the granulate and polymorphic layers of the dentate gyrus (Figure 2b).

Analysis of cell types targeted by Cy3-NPS. (a) A representative overview image of the hippocampus. Scale bar, 100 μm. (b) Morphologically representative cells from the granular dentate gyrus. Granular dentate gyrus (GrDG), molecular dentate gyrus (MoDG). Scale bar, 20 μm. Z-stack of 15 images in 0.59-μm intervals. (c) Co-staining with the neuronal marker NF (green). This representative image was taken from the dentate gyrus. Scale bar, 20 μm. Z-stack of 10 images in 1-μm intervals. (d) The hippocampal CA3 region after co-staining with GFAP (green), an astroglial marker. Z-stack of 18 images in 1-μm intervals. Scale bar, 20 μm. (e) Dentate gyrus after co-staining with the microglial marker Iba-1 (green). Z-stack of 19 slices in 1-μm intervals. Scale bar, 20 μm. Cy3-NPS: Red; nuclear counterstain DAPI: Blue. All images (a–e) were acquired with a confocal microscope and are representative for a total of 10 mice.
To test for the specificity of the distribution pattern of ICV-injected Cy3-NPS, we then compared it to that of rhodamine-NPS and unconjugated rhodamine. Cy3-NPS and rhodamine-NPS were internalized specifically into distinct cells, and showed almost identical intracerebral distribution patterns (Supplementary Figure S1), whereas pure rhodamine dispersed homogenously in the intercellular space throughout the entire brain, forming aggregates not corresponding to any cellular structure (Supplementary Figure S2). These findings indicate that the distribution pattern described here is determined by NPS and not by the fusion of NPS and fluorophore, or by the fluorophore alone.
Cy3-NPS was found in the cytosol and throughout the processes of target cells (Figure 2b). To characterize these cells, we performed immunostainings against the neuronal marker neurofilament (NF), the astroglial marker GFAP, and the microglial marker Iba-1 in brain sections from animals treated with Cy3-NPS. Cy3-NPS colocalized exclusively with the neuronal marker (Figure 2c). Additionally, cells containing Cy3-NPS possessed typical morphological features of neurons, being larger and showing fewer processes than astroglia and microglia (Figure 2d and e). Cells not expressing the neuronal marker did not take up Cy3-NPS. Taken together, these data strongly suggest that NPS is internalized exclusively into neurons after ICV administration.
Intracellular Uptake of Cy3-NPS is Mediated by Internalization of the Receptor–Ligand Complex
To clarify the mechanism of intracellular Cy3-NPS uptake, we injected native, that is, unlabeled, NPS at fivefold concentration 10 min prior to ICV injection of Cy3-NPS. Pre-injection of native NPS drastically reduced Cy3-NPS uptake throughout the brain (Figure 3a and Supplementary Figure S3). This points toward a receptor-mediated uptake mechanism, since, as shown for other neuropeptides (Grady et al, 1995; Hubbard et al, 2009), pre-treatment with unlabeled agonists leads to receptor saturation, thereby antagonizing the uptake of labeled agonist (here Cy3-NPS). To exclude the potential contribution of a putative NPS transporter to intracellular Cy3-NPS uptake, we additionally co-injected Cy3-NPS with NPSR-specific competitive antagonists, [D-Cys(tBu)5]Neuropeptide S (Camarda et al, 2009) and the active enantiomer of SHA 68, (R)-SHA 68 (Okamura et al, 2008; Trapella et al, 2011), both at 150-fold concentration of Cy3-NPS. We saw, as expected, highly reduced uptake of Cy3-NPS throughout the brain after co-injection of both antagonists (Figure 3a and Supplementary Figure S3), which strengthened our assumption that the internalization process described here is largely dependent on NPSR expression at the cell surface.

Analysis of the specificity of Cy3-NPS uptake in vivo and in vitro, and intracerebral distribution of Cy3-NPS after intranasal application. (a) From left to right: Coronal sections through mouse brain (overview (Franklin and Paxinos, 2007)) with and without pre-injection of native NPS at fivefold concentration 10 min before ICV administration of Cy3-NPS, and after co-injection with the NPSR antagonists [D-Cys(tBu)5]Neuropeptide S and (R)-SHA 68 at 150-fold concentration. Posteroventral nucleus of the medial amygdala (MePV), cortical amygdala (ACo). Optic tract (opt). Additional brain regions are depicted in Supplementary Figure S3 for comparison. All images are representative for a total of four mice pre-treated with native NPS before ICV administration of Cy3-NPS and a total of three mice co-treated with the antagonists. (b) HEK cells transiently transfected with EGFP-NPSR (green) after 10 min of incubation with Cy3-NPS (red). Nuclear counterstain: DAPI (blue). The rightmost panel depicts an overlay of all three channels and shows colocalization of Cy3-NPS and EGFP-NPSR (yellow) in cytoplasmic (arrows) and perinuclear (arrowheads) vesicular structures. All images were taken with a confocal microscope. Scale bars, 20 μm. (c) Intraneuronal uptake of Cy3-NPS (red) 30 min after intranasal delivery shown exemplarily in the hippocampus. DAPI (blue). Left: Hippocampal neuron from the oriens layer (CA3 region). Z-stack of 10 images in 1-μm intervals. Right: Hippocampal neuron from the pyramidal layer (CA3 region) after NF staining (green). Scale bars, 20 μm. All images were taken with a confocal microscope and are representative for a total of three mice.
To show that Cy3-NPS internalization is receptor-mediated, we studied the cellular NPS uptake mechanism in cultured HEK cells incubated with Cy3-NPS after transient transfection with EGFP-NPSR. As colocalization of Cy3-NPS and EGFP-NPSR indicates (Figure 3b), the receptor–ligand complex was internalized and subsequently accumulated in cytoplasmic and perinuclear vesicular structures. Together with our finding that Cy3-NPS is not taken up by HEK cells expressing only EGFP but not EGFP-NSPR (data not shown), this indicates that surface expression of NPSR is a requirement for Cy3-NPS internalization. The fact that NPSR was reported to be the only receptor mediating NPS effects (Zhu et al, 2010) allows no other conclusion but that the in vivo uptake of fluorescently labeled NPS observed here (Figures 1, 2, and 3a and c) is also dependent on NPSR expression.
NPS Modulates Synaptic Transmission and Plasticity in the Ventral Hippocampus
Because of the prominent uptake of Cy3-NPS by hippocampal CA1 neurons (Figure 2a) and the importance of the hippocampus in regulating anxiety (Fanselow and Dong, 2010), we next asked whether NPS impacts on synaptic transmission and plasticity in the ventral hippocampus. We therefore performed extracellular recordings of neurotransmission at CA3–CA1 synapses in brain slices (Figure 4a). Treatment of slices with 1 μM NPS decreased paired-pulse facilitation, pointing to an elevated release probability of glutamate at CA3–CA1 synapses (Figure 4b). Two-way ANOVA (Treatment/ISI) revealed both a significant effect of treatment (F2,31=11.286, p=0.0002) and a significant Treatment*ISI interaction (F8,124=7.620, p<0.001). NPS also reduced the magnitude of long-term potentiation (LTP) at these synapses (F2,27=11.199, p<0.001) (Figure 4c). Both NPS-induced effects failed to appear if slices were treated with NPS in the presence of the specific NPSR antagonist (R)-SHA 68 (10 μM; Figure 4b and c) (Jüngling et al, 2008). These findings indicate that NPS, through NPSR, modulates short-term as well as long-term synaptic plasticity in the CA1 output subfield of the ventral hippocampus.

Effects of NPS on synaptic transmission and plasticity in the ventral hippocampus. (a) An overview picture of a horizontal slice of the ventral hippocampus, with the position of the stimulation (Stim) and recording (Rec) electrode. DG, dentate gyrus. (b) Paired-pulse facilitation at CA3–CA1 synapses in control (n=10), NPS-treated (n=13), and NPS+(R)-SHA 68-treated (n=11) slices. Newman–Keuls post-hoc test showed a significant decrease in the paired-pulse ratio after NPS treatment, on the one hand compared with control or NPS+(R)-SHA 68-treated slices and on the other hand for every ISI (400-ms control vs NPS, p=0.0049, and NPS vs NPS+(R)-SHA 68, p=0.006; for every other ISI, p<0.001). (c) NPS decreases LTP at CA3–CA1 synapses through NPSR. fEPSP, field excitatory postsynaptic potential (Bonferroni's post-hoc test: control vs NPS, p=0.049; NPS vs NPS+(R)-SHA 68, p<0.001). HFS, high-frequency stimulation (control, n=8; NPS, n=10; NPS+(R)-SHA 68, n=10). *p<0.05, **p<0.01, ***p<0.001. All data are shown as mean±SEM.
Intranasal Administration Delivers Cy3-NPS to its Target Cells
To establish a non-invasive NPS administration method also applicable in patients, we investigated the effectiveness of intranasal NPS delivery to mice. First, we designed a protocol for intranasal application of liquid substances to alert or anesthetized mice, which considerably reduces the stress the animals are exposed to during the surgery required for intracerebral NPS injection. We then compared the cerebral distribution patterns of intranasally and ICV-administered Cy3-NPS. Although the overall intensity and amount of Cy3-NPS reaching the brain were lower in mice having received Cy3-NPS intranasally, the distribution patterns were almost identical 30 min after application (see Table 1). At this time point after intranasal administration, Cy3-NPS distributes throughout the brain, from the olfactory bulb to caudal subcortical structures such as the hippocampus. There, it becomes internalized into the cytosol and processes of its target neurons, the same as after ICV injection (Figure 3c). A distribution timeline of intranasally applied Cy3-NPS additionally revealed that at 15 min after application, intracellular Cy3-NPS uptake was visible only in the olfactory bulb, and that 4 h after application almost all traces of Cy3-NPS disappeared (data not shown). Furthermore, the intraneuronal distribution pattern of transnasally delivered Cy3-NPS is similar to the one observed in HEK cells having internalized the EGFP-NPSR/Cy3-NPS complex (Figure 3b). In conclusion, we demonstrate that intranasally applied NPS is effectively delivered to its brain target neurons in the living mouse.
Intranasally Administered NPS Exerts Strong Anxiolytic Effects on C57BL/6N and HAB Mice
After having ascertained that intranasally administered NPS reaches its target cells, we established its anxiolytic effects reported in intracerebral administration studies (Xu et al, 2004; Leonard et al, 2008) for this transnasal delivery method. In order to define the most efficient treatment dose, we delivered three different doses of NPS (7, 14, and 28 nmol) intranasally to C57BL/6N mice, a mouse strain reported to show reduced anxiety after ICV administration of NPS (Xu et al, 2004; Jüngling et al, 2008; Fendt et al, 2010). We observed no appreciable exhalation of the substance during or immediately following intranasal application, pointing toward efficient inhalation. To accurately characterize the anxiety-relieving properties of NPS, we performed two standardized behavioral assays measuring anxiolytic effects at two different time points after intranasal NPS administration. First, we performed the open field test in order to obtain a baseline of locomotion-related changes following NPS treatment. We then examined the effects of intranasal NPS vs vehicle treatment on both anxiety- and locomotion-related parameters in the dark–light test and in the elevated plus maze (EPM); both tests have been shown to cover differential facets of anxiety-related behavior (Bailey et al, 2007; van Gaalen and Steckler, 2000).
As it has been shown that effective concentrations of intranasally applied substances can be detected in the CNS up to 4 h after delivery (Thorne et al, 1995; Jansson and Björk, 2002), we chose this time point for behavioral testing.
One-way ANOVA with Bonferroni's post-hoc test of the total distance traveled in the open field did not show any NPS-induced changes in locomotion for either dose (F2, 26=1.364, p=0.2733) (Figure 5a). In the dark–light test, NPS-treated and control animals did not differ significantly in the time spent in the light chamber (F2,24=1.666, p=0.2102) or in the percent distance traveled in the light chamber (F2,24=0.2080, p=0.8136); a trend toward reduced latency to the first entry into the light chamber was observed for animals treated with 14 and 28 nmol of NPS (F2,24=3.382, p=0.0508) (Figure 5a). Among animals challenged in the EPM, individuals treated with NPS (14 nmol) significantly increased their percent time on the open arms (F2,24=4.127, p=0.0288), whereas there was no difference between treatment and control in the total number of entries (F2,24=0.5957, p=0.5591) (Figure 5a). While we cannot exclude that performing the open field first may impact the results of subsequent behavioral assays, we assume that possibility to be minimal in view of the fact that in the open field itself, we see no differences between control and treatment groups.

Behavioral effects of transnasally delivered NPS in C57BL/6N and in HAB mice. (a) Behavioral testing of C57BL/6N mice 4 h after intranasal NPS treatment (7, 14, and 28 nmol) in open field, dark–light test, and EPM (n=10). Statistical analysis: One-way ANOVA with Bonferroni's post-hoc test. The data are shown as % change relative to control. (b) Behavioral testing of HAB mice 4 h after intranasal NPS treatment (14 nmol) in open field, dark–light test, and EPM (n=10). Statistical analysis: Two-tailed unpaired t-test. (c) Behavioral testing of C57BL/6N mice and HAB mice 30 min after intranasal NPS treatment (14 nmol) in open field, dark–light test, and EPM (n=10). Statistical analysis: Two-tailed unpaired t-test. C57BL/6N mice: Total distance traveled in open field: t=0.6222, df=17, p=0.2710; % time spent in light chamber: t=0.4437, df=18, p=0.3313; latency to first entry in light chamber: t=1.190, df=18, p=0.1247; % distance traveled in light chamber: p=0.1007, t=1.326, df=18; % time spent on the open arm: p=0.2109, t=0.8219, df=18; entries in closed arm: p=0.4721, t=0.07093, df=18. HAB mice: Total distance traveled in open field: p=0.2198, t=0.7905, df=18; % time spent in light chamber: p=0.3856, t=0.2953, df=18; latency to first entry in light chamber: p=0.4085, t=0.2348, df=18; % distance traveled in light chamber: p=0.4524, t=0.1214, df=18; % time spent on the open arm: p=0.2459, t=0.7009, df=19; entries in closed arm: p=0.4081, t=0.2356, df=19. tp<0.1, *p<0.05. All data are shown as mean±SEM.
From these results, we conclude that intranasally administered NPS at 14 nmol has the potential to elicit anxiolytic locomotion-independent effects in C57BL/6N mice as shown by the results of the EPM and the dark–light test.
As the EPM results revealed 14 nmol of NPS to be the optimal dose for producing clear-cut anxiolytic effects 4 h after intranasal administration, we employed this protocol to test the efficacy of NPS-mediated anxiolysis in HAB (high-anxiety behavior) mice that have been inbred for >40 generations for pathologically high anxiety (Landgraf et al, 2007). We analyzed both C57BL/6N and HAB mice to assess the influence of NPS on the temporary condition of state anxiety vs the general condition of inborn trait anxiety (Bunck et al, 2009; Krömer et al, 2005).
In HAB mice, the open field test revealed the locomotor activity to be unaffected by NPS treatment (t=0.8610, df=19, p=0.2000) (Figure 5b). However, in the dark–light test, NPS treatment significantly increased the time spent in the light chamber 4 h after intranasal administration (t=2.316, df=18, p=0.0163) and moreover trend-wise reduced the latency to the first entry in the light chamber (t=1.451, df=18, p=0.0820), indicating anxiolytic effects, whereas the percent distance traveled in the light chamber remained unaffected (t=0.8146, df=18, p=0.2130) (Figure 5b). There were no differences on the EPM in any parameter tested in these mice (% time on the open arm: t=0.6839, df=18, p=0.2514; entries in closed arm: t=0.07032, df=18, p=0.4724) (Figure 5b).
We then tested whether intranasally delivered NPS at the effective dose of 14 nmol produces anxiolytic effects at 30 min after administration, as this is the time point when behavioral effects become apparent after intracerebral injection of NPS. Thirty minutes after intranasal administration, there were no behavioral differences between NPS- and vehicle-treated animals among either C57BL/6N or HAB mice in any of the three tests performed (Figure 5c).
In summary, 4 h after intranasal administration, NPS induced anxiolytic effects on the EPM in C57BL/6N mice and in the dark–light test in genetically predisposed HAB mice. In both cases, the anxiolytic effects of intranasal NPS were independent of locomotor activity.
Intranasally Administered NPS Upregulates the Expression Levels of Proteins of the Glutamatergic System Involved Inter Alia in Synaptic Function
Although the behavioral effects of NPS have been well documented, its effects on cerebral protein expression have hitherto not been studied. Therefore, we selected candidate proteins for examination relying on publications linking NPS to the glutamatergic system (Han et al, 2009) and to synaptic function (Jüngling et al, 2008; Raiteri et al, 2009). Expression levels of proteins of the glutamatergic network, such as AMPA receptor subunits and glutamate transporters, and synapsin, a protein involved in synaptic formation and function, were examined 4 and 24 h after intranasal NPS treatment by immunoblotting of lysates from the prefrontal cortex and the hippocampus. Changes in mRNA expression of these candidates were examined by real-time PCR 4 h after treatment.
The behavioral changes elicited by intranasally administered NPS after 4 h were accompanied by a significant increase in prefrontocortical glutamate transporter type-1 (Glt-1) (t=4.562, df=7, p=0.0026) and synapsin II mRNA levels (t=2.776, df=8, p=0.0241) (Figure 6d) in C57BL/6N mice, and by a significant reduction in hippocampal AMPA receptor subunit-1 (GluR1) mRNA levels in HAB mice (t=2.361, df=8, p=0.0459) (Figure 7a). A trend toward decrease in hippocampal Glt-1 mRNA levels after treatment was detectable in both C57BL/6N (t=1.865, df=8, p=0.0992) and HAB mice (t=2.198, df=8, p=0.0591) (Figures 6b and 7a). As expected, 4 h after intranasal NPS treatment of C57BL/6N mice, we did not find any changes in the protein levels of any candidate protein tested (Figure 6a and c), as usually, the cellular protein synthesis machinery is too slow to produce detectable changes in protein expression levels within 4 h. However, at a later time point, that is, 24 h after intranasal NPS treatment, immunoblot analyses revealed a significant upregulation of prefrontocortical GluR2 (t=2.832, df=10, p=0.0178) and a trend in upregulation of GluR1 (t=2.228, df=10, p=0.0500) protein levels in HAB mice (Figure 8d), as well as a significant increase in both GluR1 (t=3.219, df=8, p=0.0123) and Glt-1 protein levels (t=2.561, df=8, p=0.0336) in C57/BL6N mice (Figure 8b). In addition, in the hippocampus of C57BL/6N mice, we detected a significant increase in synapsin Ia–b/IIa protein expression (t=2.561, df=8, p=0.0336) (Figure 8a).

Effects of transnasally delivered NPS (14 nmol) 4 h after application on protein and mRNA levels in the prefrontal cortex and hippocampus of C57BL/6N mice. (a) Immunoblot analysis of hippocampal (Hc) lysate from C57BL/6N mice 4 h after intranasal NPS treatment. GluR1: t=1.192, df=8, p=0.2675; GluR2: t=0.657, df=8, p=0.5552; Glt-1: t=0.02711, df=8, p=0.9790; synapsin Ia–b/IIa: t=1.506, df=6, p=0.1828. (b) Real-time PCR analysis of Hc lysate from C57BL/6N mice 4 h after intranasal NPS treatment. GluR1: t=1.236, df=7, p=0.2562; GluR2: t=1.242, df=8, p=0.2493; synapsin I: t=0.3580, df=4, p=0.7385; synapsin II: t=1.243, df=7, p=0.2540. (c) Immunoblot analysis of prefrontal cortex (Pfc) lysate from C57BL/6N mice 4 h after intranasal NPS treatment. GluR1: t=0.1030, df=8, p=0.9205; GluR2: t=0.8469, df=8, p=0.4217; Glt-1: t=0.03901, df=8, p=0.9698; synapsin Ia–b/IIa: t=1.384, df=6, p=0.2038. (d) Real-time PCR analysis of Pfc lysate from C57BL/6N mice 4 h after intranasal NPS treatment. GluR1: t=0.7166, df=5, p=0.5057; GluR2: t=0.6839, df=8, p=0.5133; synapsin I: t=1.063, df=7, p=0.3230. Internal expression control: GAPDH (35 kDa in immunoblot excerpts). The blot excerpts show three representative adjacent bands of each group. The immunoblot data represent cumulated data from at least three independent experiments. C57BL/6N: n=5 for each group. Statistical analysis: Two-tailed unpaired t-test. tp<0.1, *p<0.05, **p<0.01. All data are shown as mean±SEM.

Effects of transnasally delivered NPS (14 nmol) 4 h after application on mRNA levels in the prefrontal cortex and hippocampus of HAB mice. (a) Real-time PCR analysis of Hc lysate from HAB mice 4 h after intranasal NPS treatment. GluR2: t=1.540, df=8, p=0.1620; synapsin I: t=1.123, df=8, p=0.2940; synapsin II: t=1.339, df=5, p=0.2383. (b) Real-time PCR analysis of Pfc lysate from HAB mice 4 h after intranasal NPS treatment. GluR1: t=0.7371, df=7, p=0.4850; GluR2: t=0.7957, df=8, p=0.4492; Glt-1: t=0.2440, df=7, p=0.8143; synapsin I: t=0.8174, df=8, p=0.4373; synapsin II: t=0.4126, df=8, p=0.6907. HAB: n=5 for each group. Statistical analysis: Two-tailed unpaired t-test. tp<0.1, *p<0.05. All data are shown as mean±SEM.

Effects of transnasally delivered NPS (14 nmol) 24 h after application on protein levels in the prefrontal cortex and hippocampus of C57BL/6N and HAB mice. (a) Immunoblot analysis of hippocampal (Hc) lysate from C57BL/6N mice 24 h after intranasal NPS treatment. GluR1: t=1.271, df=8, p=0.2396; GluR2: t=0.9666, df=8, p=0.3621; Glt-1: t=1.628, df=8, p=0.1421. (b) Immunoblot analysis of prefrontal cortex (Pfc) lysate from C57BL/6N mice 24 h after intranasal NPS treatment. GluR2: t=0.2798, df=8, p=0.7868; synapsin Ia–b/IIa: t=1.549, df=6, p=0.1601. (c) Immunoblot analysis of Hc lysate from HAB mice 24 h after intranasal NPS treatment. GluR1: t=0.2502, df=9, p=0.8081; GluR2: t=0.1124, df=10, p=0.9127; Glt-1: t=1.582, df=10, p=0.1447; synapsin Ia–b/IIa: t=1.197, df=9, p=0.2619. (d) Immunoblot analysis of Pfc lysate from HAB mice 24 h after intranasal NPS treatment. Glt-1: t=0.8817, df=9, p=0.4009; synapsin Ia–b/IIa: t=0.9739, df=10, p=0.3531. Internal expression control: GAPDH (35 kDa in immunoblot excerpts). The blot excerpts show three representative adjacent bands of each group. The immunoblot data represent cumulated data from at least three independent experiments. C57BL/6N: n=5 for each group. HAB: n=5 for each group. Statistical analysis: Two-tailed unpaired t-test. tp<0.1, *p<0.05, **p<0.01. All data are shown as mean±SEM.
Taken together, these results point toward robust and brain region-specific regulatory effects of intranasal NPS treatment both in C57BL/6N mice and in a mouse model of pathological anxiety.
DISCUSSION
The evidence for the identity of NPS target neurons presented here is compelling given the specificity of the method used. Fluorescently labeled neuropeptides are known to be appropriate for studying specific interactions with their receptors in vitro (Grady et al, 1995; Bunnett et al, 1995) and in vivo (Hubbard et al, 2009).
Comparison of the here identified murine NPS target neurons with the published rat NPSR expression patterns (Xu et al, 2007; Leonard and Ring, 2011) revealed that mouse and rat brain NPSR expression sites overlap largely but not completely. We found NPS target neurons in the murine basolateral and central amygdaloid nuclei, where no NPSR expression in the rat brain had been reported. In contrast to rat mRNA expression studies (Xu et al, 2007) but in agreement with rat NPSR protein expression analyses (Leonard and Ring, 2011), we observed NPS target neurons in the murine hippocampus, a region involved in fear memory and anxiety formation. We chose the ventral hippocampus to validate the physiological significance of our results employing electrophysiological measurements. We show here for the first time that NPS, through NPSR, decreases paired-pulse facilitation (pointing to an increased release probability of glutamate) and LTP at CA3–CA1 synapses, two important forms of synaptic plasticity. At first glance, the coincidental occurrence of these two effects appears counterintuitive. One of many possible explanations for this phenomenon is that during high-frequency activation of CA3–CA1 synapses, the available pool of presynaptic glutamate is depleted faster upon enhancement of the release probability. This may result in a shorter depolarization period of the postsynaptic membrane and thus in a decreased magnitude of LTP, which might causally contribute to the reported NPS-mediated reduction of fear memory retrieval after extinction training (Jüngling et al, 2008). Here, we propose an involvement of the ventral hippocampus in the mechanistic model of NPS-mediated regulation of anxiety demonstrated by others who identified the amygdala as a major player in mediating the anxiolytic effects of NPS (Meis et al, 2008; Fendt et al, 2010; Jüngling et al, 2008). As the ventral hippocampus and especially the ventral CA1 region are known to be closely connected to the amygdaloid nuclei by extensive bidirectional connections (Fanselow and Dong, 2010), our results suggest that the ventral hippocampus may, in addition to the well-described direct effects of NPS in the amygdala, modulate the activity of amygdaloid nuclei with a view toward reducing anxiety. The NPS-mediated increase of glutamate release probability 2 h after slice incubation with NPS may continue in the long term, as suggested by the upregulation of hippocampal synapsin Ia–b/IIa 24 h after intranasal NPS application in vivo (Figure 8a). As synapsin is known to be involved in the modulation of neurotransmitter release through regulation of synaptic vesicle availability (Cesca et al, 2010), its upregulation may mirror an increased number of synaptic vesicles at presynaptic terminals.
To our knowledge, this report is the first showing internalization and accumulation of an intranasally administered neuropeptide in single brain neurons. The neuronal populations targeted by intranasally and ICV-administered Cy3-NPS are identical (Table 1). This experimental technique could improve future studies of intranasally applied neuropeptides as the radioactively labeled substances employed so far (Thorne et al, 2008, 2004) did not allow for a comparably high anatomical resolution of their intracerebral distribution patterns. We detected first traces of Cy3-NPS at 15 min after its intranasal instillation followed by a peak at 30 min and almost complete disappearance after 4 h. This timeline coincides with the one reported by others after transnasal delivery of radioactively labeled compounds (Thorne et al, 2008, 2004; Thorne and Frey, 2001) and supports the hypothesis of extraneuronal substance transport from the nasal cavity to the brain (Thorne et al, 2004).
We hypothesized that the observed Cy3-NPS uptake into neurons is mediated by GPCR-dependent internalization of the complex between neuropeptide and specific receptor, a cellular desensitization mechanism described for other neuropeptide receptors both in vitro and in vivo (Hökfelt et al, 2003; Grady et al, 1995; Reyes et al, 2006). The only evidence for ligand-induced internalization of NPSR hitherto was achieved with cell culture transfection experiments not only employing a chimeric construct of vasopressin receptor and NPS receptor elements, but also vasopressin instead of NPS stimulation, resulting in a limited physiological significance of the results obtained (Gupte et al, 2004). We bridge this gap by providing the first in vitro evidence for agonist-induced internalization of wild-type murine NPSR. The fact that NPSR is the only receptor mediating NPS effects in mice (Zhu et al, 2010) allows no other conclusion but that the in vivo uptake of Cy3-NPS observed here is likewise dependent on surface expression of active NPSR. This inference is additionally supported by the striking similarity of intracellular Cy3-NPS distribution patterns in HEK cells and in murine brain neurons (Figure 3b and c).
Treatment of anxiety disorders with antidepressants and/or cognitive behavior therapy, although effective in most patients, often leads only to partial remission and furthermore takes weeks for onset of action (Ravindran and Stein, 2009; Furukawa et al, 2006; Rodrigues et al, 2011). By contrast, benzodiazepines act very fast, but are nevertheless improper as long-term anti-anxiety medications owing to their high abuse potential (Ravindran and Stein, 2009; Tan et al, 2010; Cloos and Ferreira, 2009). Hence, there is need for developing effective anxiolytics with a rapid onset of action and lacking benzodiazepine-like side effects. NPS is an excellent candidate for this purpose, as, although reward-like effects have been reported by others (Cao et al, 2011), NPS does not act as a GABAA receptor agonist.
Our data on successful transnasal NPS delivery to alert mice represent an important step toward developing NPS-based anxiolytics as they provide the basis for intranasal NPS administration to humans and furthermore could facilitate future animal experiments studying NPS effects. A variety of substances, among them neuropeptides, have been shown to cross the nose–brain barrier both in animals and humans (Dufes et al, 2003; Dhuria et al, 2009a; Born et al, 2002). Some of them are used therapeutically as nasal spray formulations (Guastella et al, 2010). Even though several chemical and biological characteristics influencing the nose-to-brain transportability of molecules have been already identified, the mere molecular structure of a substance cannot predict exactly to what extent it reaches the brain after intranasal administration (Dhuria et al, 2010; Ozsoy et al, 2009). The nasal bioavailability of hydrophilic peptides and proteins is usually less than 1% (Ozsoy et al, 2009). However, a significant amount of hydrophilic NPS obviously manages to escape the neuropeptide-degrading enzymes of the murine nasal mucosa (Ohkubo et al, 1994) and to enter the mouse brain where it rapidly distributes before inducing anxiolytic effects. Thus, we were able to prove the feasibility of intranasal NPS delivery and, most important, demonstrate that it elicits similar behavioral effects as ICV administration of NPS. These results represent a starting point for further studies exploring the precise pharmacokinetics of NPS with a view toward implementing intranasal application of NPS as a novel anxiolytic therapy for patients suffering from pathological anxiety.
We show here that intranasally applied NPS elicits anxiolytic actions not only in C57BL/6N mice, but also in HAB mice, thus mitigating the phenotypic consequences of the latters’ genetic predisposition to high-anxiety-related behavior (Krömer et al, 2005; Hambsch et al, 2010). NPS treatment caused anxiolytic behavior in HAB mice in the dark–light test only, but not in the EPM, like in C57BL/6N mice. These differences possibly result from different neurobiological mechanisms underlying trait vs state anxiety and from the fact that HAB mice are bred for >40 generations for extreme inborn anxiety on the EPM (Krömer et al, 2005; Hambsch et al, 2010), making their behavior particularly resistant against changes in this test.
NPS effects become manifest 30 min after ICV injection and 4 h after intranasal application. This different onset of action probably originates from dissimilar pharmacokinetics and targeting efficiencies of intranasally vs ICV-administered drugs (Thorne et al, 1995, 2008; Shi et al, 2010). As at 30 min after intranasal application of Cy3-NPS, a much weaker signal was observed than after ICV injection, it is possible that, although NPS does reach the brain rapidly after transnasal delivery, it may require additional time to reach its brain target cells in the full concentration required to produce behavioral effects. Our finding that NPS effects can last up to several hours after application highlights their robustness. In view of our results describing changes in the expression levels of candidate proteins up to 24 h after application, future studies on an extended timeline of duration of NPS effects will be necessary for developing NPS-based therapeutics. Chronic treatment, fine-tuning of dosage, and possibly also chemical modifications most likely will enhance the anxiolytic effects of transnasally administered NPS, as described by others for intranasal drug delivery (Dhuria et al, 2009b).
Another proof for the effectiveness of intranasal NPS treatment in mice is the differential regulation of hippocampal and prefrontocortical mRNA and proteins induced by transnasal NPS delivery. Here we report that in the prefrontal cortex and the hippocampus of C57BL/6N and HAB mice, intranasally applied NPS differentially regulates the expression levels of proteins of the glutamatergic system, a player in disorders such as PTSD (Ravindran and Stein, 2009; Rossi et al, 2009). Changes in GluR1 : GluR2 ratios are known to mirror an enhancement of AMPA receptor function (Isaac et al, 2007). As potentiation of AMPA receptors in the prefrontal cortex was shown to facilitate fear extinction (Zushida et al, 2007), we speculate that the increase in the GluR1 : GluR2 protein expression ratio observed in the prefrontal cortex of NPS-treated C57BL/6N mice may provide one possible explanatory mechanism for long-term NPS-mediated fear extinction following chronic NPS treatment, especially as this phenomenon has already been described after acute intra-amygdala NPS injection by others (Jüngling et al, 2008). Moreover, NPS regulates the levels of Glt-1, which is expressed exclusively by astrocytes (Huang and Bergles, 2004), showing that NPS, although internalized only into neurons, ultimately impacts the entire cellular brain network. Although changes in protein expression only become apparent 24 h after treatment, regulation of membrane transport and insertion mechanisms may occur already at an earlier time point and thus lead to the electrophysiological and behavioral effects described at 2 and 4 h after NPS administration, respectively. Further studies on protein levels of cellular sub-fractions and on protein dynamics are needed to clarify this aspect. The fact that the expression of the examined candidates is differentially modulated at protein and mRNA levels, as well as the fact that these processes seem to be time-, region-, and strain-dependent, suggest that NPS-mediated regulation of protein expression taps into a variety of post-transcriptional and post-translational mechanisms to exert its effects. In conclusion, these results provide new insights into potential regulatory mechanisms of NPS in vivo, which have hitherto been explored in vitro and at the RNA level only (Vendelin et al, 2006).
In summary, the findings presented here not only improve the understanding of the molecular effects and the path of NPS in the brain, but also establish a non-invasive method for NPS administration in mice. These results represent an important step toward the implementation of NPS as a potential novel treatment option for anxiety disorders.
References
Bailey KR, Pavlova MN, Rohde AD, Hohmann JG, Crawley JN (2007). Galanin receptor subtype 2 (GalR2) null mutant mice display an anxiogenic-like phenotype specific to the elevated plus-maze. Pharmacol Biochem Behav 86: 8–20.
Blanco E, Bilbao A, Luque-Rojas MJ, Palomino A, Bermúdez-Silva FJ, Suárez J et al (2011). Attenuation of cocaine-induced conditioned locomotion is associated with altered expression of hippocampal glutamate receptors in mice lacking LPA1 receptors. Psychopharmacology; e-pub ahead of print 2 September 2011.
Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci 5: 514–516.
Bunck M, Czibere L, Horvath C, Graf C, Frank E, Kessler MS et al (2009). A hypomorphic vasopressin allele prevents anxiety-related behavior. PLoS ONE 4: e5129.
Bunnett NW, Dazin PF, Payan DG, Grady EF (1995). Characterization of receptors using cyanine 3-labeled neuropeptides. Peptides 16: 733–740.
Camarda V, Rizzi A, Ruzza C, Zucchini S, Marzola G, Marzola E et al (2009). In vitro and in vivo pharmacological characterization of the neuropeptide s receptor antagonist [D-Cys(tBu)5]neuropeptide S. J Pharmacol Exp Ther 328: 549–555.
Cao J, de Lecea L, Ikemoto S (2011). Intraventricular administration of neuropeptide S has reward-like effects. Eur J Pharmacol 658: 16–21.
Cesca F, Baldelli P, Valtorta F, Benfenati F (2010). The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91: 313–348.
Cloos J-M, Ferreira V (2009). Current use of benzodiazepines in anxiety disorders. Curr Opin Psychiatry 22: 90–95.
de Kloet ER, Joëls M, Holsboer F (2005). Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6: 463–475.
Dhuria SV, Hanson LR, Frey WH (2009a). Intranasal drug targeting of hypocretin-1 (orexin-A) to the central nervous system. J Pharm Sci 98: 2501–2515.
Dhuria SV, Hanson LR, Frey WH (2009b). Novel vasoconstrictor formulation to enhance intranasal targeting of neuropeptide therapeutics to the central nervous system. J Pharmacol Exp Ther 328: 312–320.
Dhuria SV, Hanson LR, Frey WH (2010). Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99: 1654–1673.
Domschke K, Reif A, Weber H, Richter J, Hohoff C, Ohrmann P et al (2010). Neuropeptide S receptor gene—converging evidence for a role in panic disorder. Mol Psychiatry 16: 938–948.
Dufes C, Olivier J-C, Gaillard F, Gaillard A, Couet W, Muller J-M (2003). Brain delivery of vasoactive intestinal peptide (VIP) following nasal administration to rats. Int J Pharm 255: 87–97.
Fanselow MS, Dong H-W (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65: 7–19.
Fendt M, Imobersteg S, Bürki H, McAllister KH, Sailer AW (2010). Intra-amygdala injections of neuropeptide S block fear-potentiated startle. Neurosci Lett 474: 154–157.
Franklin KBJ, Paxinos G (2007). The Mouse Brain in Stereotaxic Coordinates with CDROM, 3rd edn. Academic Press, New York, NY, USA.
Furukawa TA, Watanabe N, Churchill R (2006). Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry 188: 305–312.
Grady E, Garland A, Gamp P, Lovett M, Payan D, Bunnett N (1995). Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol Biol Cell 6: 509–524.
Guastella AJ, Einfeld SL, Gray KM, Rinehart NJ, Tonge BJ, Lambert TJ et al (2010). Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol Psychiatry 67: 692–694.
Gupte J, Cutler G, Chen J-L, Tian H (2004). Elucidation of signaling properties of vasopressin receptor-related receptor 1 by using the chimeric receptor approach. Proc Natl Acad Sci USA 101: 1508–1513.
Hambsch B, Chen B-G, Brenndörfer J, Meyer M, Avrabos C, Maccarrone G et al (2010). Methylglyoxal-mediated anxiolysis involves increased protein modification and elevated expression of glyoxalase 1 in the brain. J Neurochem 113: 1240–1251.
Han R-W, Yin X-Q, Chang M, Peng Y-L, Li W, Wang R (2009). Neuropeptide S facilitates spatial memory and mitigates spatial memory impairment induced by N-methyl-D-aspartate receptor antagonist in mice. Neurosci Lett 455: 74–77.
Hökfelt T, Bartfai T, Bloom F (2003). Neuropeptides: opportunities for drug discovery. Lancet Neurol 2: 463–472.
Huang YH, Bergles DE (2004). Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol 14: 346–352.
Hubbard CS, Dolence EK, Shires JA, Rose JD (2009). Identification of brain target neurons using a fluorescent conjugate of corticotropin-releasing factor. J Chem Neuroanat 37: 245–253.
Isaac JTR, Ashby M, McBain CJ (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54: 859–871.
Jansson B, Björk E (2002). Visualization of in vivo olfactory uptake and transfer using fluorescein dextran. J Drug Target 10: 379–386.
Jüngling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD et al (2008). Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron 59: 298–310.
Krömer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L et al (2005). Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci 25: 4375–4384.
Landgraf R, Kessler MS, Bunck M, Murgatroyd C, Spengler D, Zimbelmann M et al (2007). Candidate genes of anxiety-related behavior in HAB/LAB rats and mice: focus on vasopressin and glyoxalase-I. Neurosci Biobehav Rev 31: 89–102.
Leonard SK, Dwyer JM, Sukoff Rizzo SJ, Platt B, Logue SF, Neal SJ et al (2008). Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacology (Berl) 197: 601–611.
Leonard SK, Ring RH (2011). Immunohistochemical localization of the neuropeptide S receptor in the rat central nervous system. Neuroscience 172: 153–163.
Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25: 402–408.
Meis S, Bergado-Acosta JR, Yanagawa Y, Obata K, Stork O, Munsch T (2008). Identification of a neuropeptide s responsive circuitry shaping amygdala activity via the endopiriform nucleus. PLoS ONE 3: e2695.
Ohkubo K, Okuda M, Kaliner MA (1994). Immunological localization of neuropeptide-degrading enzymes in the nasal mucosa. Rhinology 32: 130–133.
Okamura N, Garau C, Duangdao DM, Clark SD, Jüngling K, Pape H-C et al (2010). Neuropeptide S enhances memory during the consolidation phase and interacts with noradrenergic systems in the brain. Neuropsychopharmacology 36: 744–752.
Okamura N, Habay SA, Zeng J, Chamberlin AR, Reinscheid RK (2008). Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J Pharmacol Exp Ther 325: 893–901.
Ozsoy Y, Gungor S, Cevher E (2009). Nasal delivery of high molecular weight drugs. Molecules 14: 3754–3779.
Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M et al (2010). Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 35: 792–805.
Raiteri L, Luccini E, Romei C, Salvadori S, Calò G (2009). Neuropeptide S selectively inhibits the release of 5-HT and noradrenaline from mouse frontal cortex nerve endings. Br J Pharmacol 157: 474–481.
Ravindran LN, Stein MB (2009). Pharmacotherapy of PTSD: premises, principles, and priorities. Brain Res 1293: 24–39.
Reinscheid RK, Xu Y-L, Okamura N, Zeng J, Chung S, Pai R et al (2005). Pharmacological characterization of human and murine neuropeptide s receptor variants. J Pharmacol Exp Ther 315: 1338–1345.
Reyes BAS, Fox K, Valentino RJ, Van Bockstaele EJ (2006). Agonist-induced internalization of corticotropin-releasing factor receptors in noradrenergic neurons of the rat locus coeruleus. Eur J Neurosci 23: 2991–2998.
Rodrigues H, Figueira I, Gonçalves R, Mendlowicz M, Macedo T, Ventura P (2011). CBT for pharmacotherapy non-remitters—a systematic review of a next-step strategy. J Affect Disord 129: 219–228.
Rossi S, De Capua A, Tavanti M, Calossi S, Polizzotto NR, Mantovani A et al (2009). Dysfunctions of cortical excitability in drug-naïve posttraumatic stress disorder patients. Biol Psychiatry 66: 54–61.
Schmidt MV, Schülke J-P, Liebl C, Stiess M, Avrabos C, Bock J et al (2011). Tumor suppressor downregulated in renal cell carcinoma 1 (DRR1) is a stress-induced actin bundling factor that modulates synaptic efficacy and cognition. Proc Natl Acad Sci USA 108: 17213–17218.
Shi C-G, Wang L-M, Wu Y, Wang P, Gan Z-J, Lin K et al (2010). Intranasal administration of nerve growth factor produces antidepressant-like effects in animals. Neurochem Res 35: 1302–1314.
Si W, Aluisio L, Okamura N, Clark SD, Fraser I, Sutton SW et al (2010). Neuropeptide S stimulates dopaminergic neurotransmission in the medial prefrontal cortex. J Neurochem 115: 475–482.
Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV et al (2006). Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology 147: 3510–3518.
Tan KR, Brown M, Labouèbe G, Yvon C, Creton C, Fritschy J-M et al (2010). Neural bases for addictive properties of benzodiazepines. Nature 463: 769–774.
Thorne RG, Emory CR, Ala TA, Frey WH (1995). Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res 692: 278–282.
Thorne RG, Frey WH (2001). Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet 40: 907–946.
Thorne RG, Hanson LR, Ross TM, Tung D, Frey WH (2008). Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 152: 785–797.
Thorne RG, Pronk GJ, Padmanabhan V, Frey WH (2004). Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127: 481–496.
Trapella C, Pela M, Del Zoppo L, Calo G, Camarda V, Ruzza C et al (2011). Synthesis and separation of the enantiomers of the neuropeptide S receptor antagonist (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68). J Med Chem 54: 2738–2744.
van den Berg MP, Romeijn SG, Verhoef JC, Merkus FWHM (2002). Serial cerebrospinal fluid sampling in a rat model to study drug uptake from the nasal cavity. J Neurosci Methods 116: 99–107.
van Gaalen MM, Steckler T (2000). Behavioural analysis of four mouse strains in an anxiety test battery. Behav Brain Res 115: 95–106.
Vendelin J, Bruce S, Holopainen P, Pulkkinen V, Rytilä P, Pirskanen A et al (2006). Downstream target genes of the neuropeptide S–NPSR1 pathway. Hum Mol Genet 15: 2923–2935.
Xu Y-L, Gall CM, Jackson VR, Civelli O, Reinscheid RK (2007). Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol 500: 84–102.
Xu Y-L, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH et al (2004). Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43: 487–497.
Zhu H, Mingler MK, McBride ML, Murphy AJ, Valenzuela DM, Yancopoulos GD et al (2010). Abnormal response to stress and impaired NPS-induced hyperlocomotion, anxiolytic effect and corticosterone increase in mice lacking NPSR1. Psychoneuroendocrinology 35: 1119–1132.
Zushida K, Sakurai M, Wada K, Sekiguchi M (2007). Facilitation of extinction learning for contextual fear memory by PEPA: a potentiator of AMPA receptors. J Neurosci 27: 158–166.
Acknowledgements
We thank Carsten T Wotjak for important feedback on the microscopy data and for crucial help with the statistical analysis. We also thank Christoph Thoeringer and Theo Rein for fruitful discussion on the confocal microscopy images; Markus Nussbaumer for technical assistance in performing the behavioral tests; Bozidar Novak for technical assistance in performing the immunoblots, for cloning EGFP-NPSR, and for performing real-time PCRs; Christine Huber for technical assistance in performing the immunoblots; and Nancy Xin Ru Wang for performing the RNA extraction and real-time PCRs. We thank Andreas Sailer from Novartis for providing us with (R)-SHA 68, and Benno Pütz and Tonia Ludwig for most important advice on the statistical analysis. The research was supported in part by the Horst Kübler-Foundation, Bad Ragaz. The Horst Kübler-Foundation had no further role in study design, in the collection, analysis, and interpretation of the data; in the writing of the report; and in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
IA Ionescu, Y-C Yen, F Holsboer, R Landgraf, and U Schmidt declare a conflict of interest due to a patent application on intranasal NPS pending since April 2011. J Dine, DR Buell, L Herrmann, and M Eder declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article
Cite this article
Ionescu, I., Dine, J., Yen, YC. et al. Intranasally Administered Neuropeptide S (NPS) Exerts Anxiolytic Effects Following Internalization Into NPS Receptor-Expressing Neurons. Neuropsychopharmacol 37, 1323–1337 (2012). https://doi.org/10.1038/npp.2011.317
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.317
Keywords
This article is cited by
-
Peroxisome proliferator-activated receptor-α activation facilitates contextual fear extinction and modulates intrinsic excitability of dentate gyrus neurons
Translational Psychiatry (2023)
-
Hippocampus: Molecular, Cellular, and Circuit Features in Anxiety
Neuroscience Bulletin (2023)
-
Brain neuropeptide S: via GPCR activation to a powerful neuromodulator of socio-emotional behaviors
Cell and Tissue Research (2019)
-
Neuropeptide S Induces Acute Anxiolysis by Phospholipase C-Dependent Signaling within the Medial Amygdala
Neuropsychopharmacology (2018)
-
Novel analgesic effects of melanin-concentrating hormone on persistent neuropathic and inflammatory pain in mice
Scientific Reports (2018)
