
Key Points
-
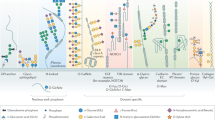
More than 1% of the human genome encodes proteins that make, modify or bind to glycans. In the past decade nearly 30 disease-causing genes in 6 distinct glycan biosynthetic pathways have been discovered. It is likely that many more will be identified in the coming years.
-
There is enormous diversity of clinical phenotypes in patients with glycosylation defects. The disorders cut across many medical subspecialties, making it difficult for physicians to easily recognise and diagnose glycosylation disorders.
-
Identifying the molecular and genetic basis of many glycosylation defects has relied heavily on complementation testing using yeast mutants and mammalian cell lines carrying mutations that affect glycosylation.
-
Although most genetic glycosylation disorders are autosomal recessive, a few are dominant and/or sex linked. Several glycosylation disorders are uniquely associated with mutations that affect a single enzyme. However, recent discoveries point to inherited and somatic mutations in genes that encode cytoplasmic and endoplasmic-reticulum- and Golgi-associated proteins that chaperone multiple components of various glycan biosynthetic pathways.
-
Several well-studied types of muscular dystrophy are now known to result from defects in the glycosylation of α-dystroglycan, one of the vital components of the dystrophin glycoprotein complex.
-
Many mouse gene knockouts for components of glycan biosynthetic pathways are lethal and therefore unsuitable models for the human diseases that result from mutations in the same genes. In the future, better models might be provided by creating mice that carry hypomorphic alleles of these genes.
-
A few glycosylation disorders are remarkably responsive to simple dietary supplements: free sugars — but not the indigestible 'glyconutrients' that are promoted by some companies — can be used to reverse the effect of some glycosylation disorders. However, for most disorders of this type no therapy is available; an increased understanding of the biology of these diseases is needed to gain insights into potential pathways to treatment.
Abstract
The spectrum of all glycan structures — the glycome — is immense. In humans, its size is orders of magnitude greater than the number of proteins that are encoded by the genome, one percent of which encodes proteins that make, modify, localize or bind sugar chains, which are known as glycans. In the past decade, over 30 genetic diseases have been identified that alter glycan synthesis and structure, and ultimately the function of nearly all organ systems. Many of the causal mutations affect key biosynthetic enzymes, but more recent discoveries point to defects in chaperones and Golgi-trafficking complexes that impair several glycosylation pathways. As more glycosylation disorders and patients with these disorders are identified, the functions of the glycome are starting to be revealed.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Raman, R., Raguram, S., Venkataraman, G., Paulson, J. C. & Sasisekharan, R. Glycomics: an integrated systems approach to structure–function relationships of glycans. Nature Methods 2, 817–824 (2005). A 'one-stop shopping' guide that explains how to connect structure–function relationships in glycobiology.
Wopereis, S., Lefeber, D. J., Morava, E. & Wevers, R. A. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin. Chem. 52, 574–600 (2006).
Jaeken, J. Komrower Lecture. Congenital disorders of glycosylation (CDG): it's all in it! J. Inherit. Metab. Dis. 26, 99–118 (2003).
Muntoni, F. Journey into muscular dystrophies caused by abnormal glycosylation. Acta Myol. 23, 79–84 (2004).
Spiro, R. G. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R–56R (2002).
Kornfeld, R. & Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 (1985). A classic description of N -glycan biosynthesis; this review is clear and detailed.
Chavan, M. & Lennarz, W. The molecular basis of coupling of translocation and N-glycosylation. Trends Biochem. Sci. 31, 17–20 (2006).
Kelleher, D. J. & Gilmore, R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R (2006).
Schachter, H., Vajsar, J. & Zhang, W. The role of defective glycosylation in congenital muscular dystrophy. Glycoconj. J. 20, 291–300 (2004).
Chai, W. et al. High prevalence of 2-mono- and 2,6-di-substituted manol-terminating sequences among O-glycans released from brain glycopeptides by reductive alkaline hydrolysis. Eur. J. Biochem. 263, 879–888 (1999).
Marth, J. D. in Essentials of Glycobiology (eds Varki, A. et al.) 101–113 (Cold Spring Harbor Laboratory Press, New York, 1999).
Esko, J. in Essentials of Glycobiology (eds Varki, A. et al.) 145–160 (Cold Spring Harbor Laboratory Press, New York, 1999).
Esko, J. D. & Selleck, S. B. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 (2002).
Varki, A. in Essentials of Glycobiology (eds Varki, A. et al.) 115–129 (Cold Spring Harbor Laboratory Press, New York, 1999).
Hakomori, S. Carbohydrate-to-carbohydrate interaction, through glycosynapse, as a basis of cell recognition and membrane organization. Glycoconj. J. 21, 125–137 (2004).
Hwa, K. Y. Glycosyl phosphatidylinositol-linked glycoconjugates: structure, biosynthesis and function. Adv. Exp. Med. Biol. 491, 207–214 (2001).
Hancock, J. F. GPI-anchor synthesis: Ras takes charge. Dev. Cell 6, 743–745 (2004).
Jaeken, J. & Matthijs, G. Congenital disorders of glycosylation. Annu. Rev. Genomics Hum. Genet. 2, 129–151 (2001).
Aebi, M. et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj. J. 16, 669–671 (1999).
Hagberg, B. A., Blennow, G., Kristiansson, B. & Stibler, H. Carbohydrate-deficient glycoprotein syndromes: peculiar group of new disorders. Pediatr. Neurol. 9, 255–262 (1993).
Freeze, H. H. Update and perspectives on congenital disorders of glycosylation. Glycobiology 11, 129R–143R (2001).
Jaeken, J., Matthijs, G., Carchon, H. & Schaftingen, E. V. in The Metabolic & Molecular Bases of Inherited Diseases (eds Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D.) 1601–1622 (McGraw-Hill Medical Publishing Division, New York, 2001).
Marquardt, T. & Denecke, J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 162, 359–379 (2003).
Grünewald, S., Schollen, E., Van Schaftingen, E., Jaeken, J. & Matthijs, G. High residual activity of PMM2 in patients' fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am. J. Hum. Genet. 68, 347–354. (2001).
van Ommen, C. H. et al. Carbohydrate-deficient glycoprotein syndrome type 1a: a variant phenotype with borderline cognitive dysfunction, cerebellar hypoplasia, and coagulation disturbances. J. Pediatr. 136, 400–403 (2000).
Briones, P. et al. Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J. Inherit. Metab. Dis. 25, 635–646 (2002).
Coman, D., Klingberg, S., Morris, D., McGill, J. & Mercer, H. Congenital disorder of glycosylation type Ia in a 6-year-old girl with a mild intellectual phenotype: two novel PMM2 mutations. J. Inherit. Metab. Dis. 28, 1189–1190 (2005).
Matthijs, G., Schollen, E., Heykants, L. & Grünewald, S. Phosphomannomutase deficiency: the molecular basis of the classical Jaeken syndrome (CDGS type Ia). Mol. Genet. Metab. 68, 220–226 (1999).
Pirard, M. et al. Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Lett. 452, 319–322 (1999).
Kjaergaard, S., Skovby, F. & Schwartz, M. Carbohydrate-deficient glycoprotein syndrome type 1A: expression and characterisation of wild type and mutant PMM2 in E. coli. Eur. J. Hum. Genet. 7, 884–888 (1999).
Westphal, V. et al. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet. Med. 3, 393–398 (2001).
Schollen, E., Kjaergaard, S., Legius, E., Schwartz, M. & Matthijs, G. Lack of Hardy–Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur. J. Hum. Genet. 8, 367–371 (2000).
Westphal, V., Xiao, M., Kwok, P. Y. & Freeze, H. H. Identification of a frequent variant in ALG6, the cause of congenital disorder of glycosylation-Ic. Hum. Mutat. 22, 420–421 (2003).
Niehues, R. et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Invest. 101, 1414–1420 (1998). First description of a novel glycosylation disease, its genetic basis, and a successful 'low-tech' treatment.
Denecke, J. et al. Congenital disorder of glycosylation type Id: clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr. Res. 58, 248–253 (2005).
Tan, J., Dunn, J., Jaeken, J. & Schachter, H. Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Am. J. Hum. Genet. 59, 810–817 (1996).
Jaeken, J. et al. Carbohydrate deficient glycoprotein syndrome type II: a deficiency in Golgi localised N-acetyl-glucosaminyltransferase II. Arch. Dis. Child. 71, 123–127 (1994).
Hansske, B. et al. Deficiency of UDP-galactose:N-acetylglucosamine β-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J. Clin. Invest. 109, 725–733 (2002).
Lübke, T. et al. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nature Genet. 28, 73–76 (2001).
Martinez-Duncker, I. et al. Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood 105, 2671–2676 (2005).
Marquardt, T. et al. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94, 3976–3985 (1999).
Marquardt, T. et al. Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism. J. Pediatr. 134, 681–688 (1999).
Etzioni, A. et al. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N. Engl. J. Med. 327, 1789–1792 (1992).
Willig, T. B. et al. Macrothrombocytopenia with abnormal demarcation membranes in megakaryocytes and neutropenia with a complete lack of sialyl-Lewis-X antigen in leukocytes — a new syndrome? Blood 97, 826–828 (2001).
Spaapen, L. J. et al. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 28, 707–714 (2005).
Wu, X. et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nature Med. 10, 518–523 (2004). First description of a patient with a defect in trafficking of glycosyltransferases and nucleotide sugar transporters.
Steet, R. & Kornfeld, S. COG-7-deficient human fibroblasts exhibit altered recycling of Golgi proteins. Mol. Biol. Cell 17, 2312–2321 (2006).
Ungar, D., Oka, T., Krieger, M. & Hughson, F. M. Retrograde transport on the COG railway. Trends Cell Biol. 16, 113–120 (2006).
Oka, T. et al. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5- and COG7-deficient mammalian cells. J. Biol. Chem. 280, 32736–32745 (2005).
Shestakova, A., Zolov, S. & Lupashin, V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 7, 191–204 (2006).
Kubota, Y., Sano, M., Goda, S., Suzuki, N. & Nishiwaki, K. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development 133, 263–273 (2006).
Wopereis, S. et al. Apolipoprotein C-III isofocusing in the diagnosis of genetic defects in O-glycan biosynthesis. Clin. Chem. 49, 1839–1845 (2003).
Morava, E. et al. Defective protein glycosylation in patients with cutis laxa syndrome. Eur. J. Hum. Genet. 13, 414–421 (2005).
Wopereis, S. et al. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology 15, 1312–1319 (2005).
Matthijs, G. et al. Deficiencies in different subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorder of Glycosylation. Am. Soc. Genet. Annu. Meeting 27 October 2005; available from the ASGH Abstract Search and Personal Itinerary Planner web page, http://www.ashg.org/genetics/ashg05s.
Foulquier, F. et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl Acad. Sci. USA 103, 3764–3769 (2006).
Sacher, M. Membrane traffic fuses with cartilage development. FEBS Lett. 550, 1–4 (2003).
Mandato, C. et al. Cryptogenic liver disease in four children: a novel congenital disorder of glycosylation. Pediatr. Res. 59, 293–298 (2006).
Wang, Y. et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology 11, 1051–1070 (2001).
Becker, D. J. & Lowe, J. B. Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003).
Lühn, K., Wild, M. K., Eckhardt, M., Gerardy-Schahn, R. & Vestweber, D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nature Genet. 28, 69–72. (2001).
Freeze, H. H. & Aebi, M. Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr. Opin. Struct. Biol. 15, 490–498 (2005).
Ghosh, P., Dahms, N. M. & Kornfeld, S. Mannose 6-phosphate receptors: new twists in the tale. Nature Rev. Mol. Cell Biol. 4, 202–212 (2003).
Kudo, M., Brem, M. S. & Canfield, W. M. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-Hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase α/β-subunits precursor gene. Am. J. Hum. Genet. 78, 451–463 (2006).
Tiede, S. et al. Mucolipidosis II is caused by mutations in GNPTA encoding the α/β GlcNAc-1-phosphotransferase. Nature Med. 11, 1109–1112 (2005).
Fukuda, M. N. HEMPAS. Hereditary erythroblastic multinuclearity with positive acidified serum lysis test. Biochim. Biophys. Acta 1455, 231–239 (1999).
Chui, D. et al. α-Mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell 90, 157–167 (1997).
Leslie, N. D. Insights into the pathogenesis of galactosemia. Annu. Rev. Nutr. 23, 59–80 (2003).
Lai, K., Langley, S. D., Khwaja, F. W., Schmitt, E. W. & Elsas, L. J. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 13, 285–294 (2003).
Sturiale, L. et al. Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 15, 1268–1276 (2005). Patients with uncontrolled galactosaemia show severe underglycosylation of serum proteins, which provides insights into the potential cause of pathogenesis in this disorder.
Prestoz, L. L., Couto, A. S., Shin, Y. S. & Petry, K. G. Altered follicle stimulating hormone isoforms in female galactosaemia patients. Eur. J. Pediatr. 156, 116–120 (1997).
Ridel, K. R., Leslie, N. D. & Gilbert, D. L. An updated review of the long-term neurological effects of galactosemia. Pediatr. Neurol. 33, 153–161 (2005).
Senger, R. S. & Karim, M. N. Variable site-occupancy classification of N-linked glycosylation using artificial neural networks. Biotechnol. Prog. 21, 1653–1662 (2005).
Jones, J., Krag, S. S. & Betenbaugh, M. J. Controlling N-linked glycan site occupancy. Biochim. Biophys. Acta 1726, 121–137 (2005).
Nishikawa, A. & Mizuno, S. The efficiency of N-linked glycosylation of bovine DNase I depends on the Asn-Xaa-Ser/Thr sequence and the tissue of origin. Biochem. J. 355, 245–248 (2001).
Welch, W. J. Role of quality control pathways in human diseases involving protein misfolding. Semin. Cell Dev. Biol. 15, 31–38 (2004).
Lonnqvist, L. et al. A point mutation creating an extra N-glycosylation site in fibrillin-1 results in neonatal Marfan syndrome. Genomics 36, 468–475 (1996).
Raghunath, M., Kielty, C. M. & Steinmann, B. Truncated profibrillin of a Marfan patient is of apparent similar size as fibrillin: intracellular retention leads to over-N-glycosylation. J. Mol. Biol. 248, 901–909 (1995).
Vogt, G. et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nature Genet. 37, 692–700 (2005). A surprising discovery: mutations that create occupied N -glycosylation sites are pathological. Random insertion of glycans might disrupt assembly of signalling complexes.
Morgan, R. et al. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J. Immunol. 173, 7200–7208 (2004).
Martin, P. T. & Freeze, H. H. Glycobiology of neuromuscular disorders. Glycobiology 13, 67R–75R (2003).
Muntoni, F. & Voit, T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul. Disord. 14, 635–649 (2004).
van Reeuwijk, J., Brunner, H. G. & van Bokhoven, H. Glyc-O-genetics of Walker–Warburg syndrome. Clin. Genet. 67, 281–289 (2004).
Muntoni, F., Brockington, M., Torelli, S. & Brown, S. C. Defective glycosylation in congenital muscular dystrophies. Curr. Opin. Neurol. 17, 205–209 (2004).
Willer, T. et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl Acad. Sci. USA 101, 14126–14131 (2004).
Manya, H. et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl Acad. Sci. USA 101, 500–505 (2004).
van Reeuwijk, J. et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker–Warburg syndrome. J. Med. Genet. 42, 907–912 (2005).
Yoshida, A. et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 1, 717–724 (2001).
Taniguchi, K. et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 12, 527–534 (2003).
Liu, J. et al. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech. Dev. 123, 228–240 (2006).
Mercuri, E. et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch. Neurol. 63, 251–257 (2006).
Kanagawa, M. et al. Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell 117, 953–964 (2004).
Barresi, R. et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat. Med. 10, 696–703 (2004). References 92 and 93 stress the importance of LARGE and provide insights into how it might function.
Nguyen, H. H., Jayasinha, V., Xia, B., Hoyte, K. & Martin, P. T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl Acad. Sci. USA 99, 5616–5621 (2002).
Rando, T. A. Artificial sweeteners — enhancing glycosylation to treat muscular dystrophies. N. Engl. J. Med. 351, 1254–1256 (2004).
Engvall, E. & Wewer, U. M. The new frontier in muscular dystrophy research: booster genes. FASEB J. 17, 1579–1584 (2003).
Gotte, M. & Kresse, H. Defective glycosaminoglycan substitution of decorin in a patient with progeroid syndrome is a direct consequence of two point mutations in the galactosyltransferase I (β4GalT-7) gene. Biochem. Genet. 43, 65–77 (2005).
Zak, B. M., Crawford, B. E. & Esko, J. D. Hereditary multiple exostoses and heparan sulfate polymerization. Biochim. Biophys. Acta 1573, 346–355 (2002).
Jaeken, J. & Carchon, H. Congenital disorders of glycosylation: a booming chapter of pediatrics. Curr. Opin. Pediatr. 16, 434–439 (2004).
Lin, X. et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol. 224, 299–311 (2000).
Stickens, D., Zak, B. M., Rougier, N., Esko, J. D. & Werb, Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 132, 5055–5068 (2005).
Dawson, P. A. & Markovich, D. Pathogenetics of the human SLC26 transporters. Curr. Med. Chem. 12, 385–396 (2005).
Venkatachalam, K. V. Human 3′-phosphoadenosine 5′-phosphosulfate (PAPS) synthase: biochemistry, molecular biology and genetic deficiency. IUBMB Life 55, 1–11 (2003).
Ten Hagen, K. G., Fritz, T. A. & Tabak, L. A. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 13, 1R–16R (2003).
Fukumoto, S. Post-translational modification of Fibroblast Growth Factor 23. Ther. Apher. Dial. 9, 319–322 (2005).
Ju, T. & Cummings, R. D. Protein glycosylation: chaperone mutation in Tn syndrome. Nature 437, 1252 (2005). This paper indicates that somatic mutations that affect a glycosyltransferase-specific chaperone might be involved in autoimmune disease.
Simpson, M. A. et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nature Genet. 36, 1225–1229 (2004).
Yamashita, T. et al. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc. Natl Acad. Sci. USA 102, 2725–2730 (2005).
Proia, R. L. Gangliosides help stabilize the brain. Nature Genet. 36, 1147–1148 (2004).
Smith, L. J. Paroxysmal nocturnal hemoglobinuria. Clin. Lab. Sci. 17, 172–177 (2004).
Hanaoka, N. et al. Immunoselection by natural killer cells of PIGA mutant cells missing stress-inducible ULBP. Blood 107, 1184–1191 (2006).
Taniguchi, N., Nakamura, K., Narimatsu, H., von der Lieth, C. W. & Paulson, J. Human Disease Glycomics/Proteome Initiative workshop and the 4th HUPO annual congress. Proteomics 6, 12–13 (2006).
Sweet collaborations [Editorial]. Nature Methods 2, 799 (2005).
Young, W. W. Jr. Organization of Golgi glycosyltransferases in membranes: complexity via complexes. J. Membr. Biol. 198, 1–13 (2004).
Sprong, H. et al. Association of the Golgi UDP-galactose transporter with UDP-galactose:ceramide galactosyltransferase allows UDP-galactose import in the endoplasmic reticulum. Mol. Biol. Cell 14, 3482–3493 (2003).
Demetriou, M., Granovsky, M., Quaggin, S. & Dennis, J. W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409, 733–739. (2001). This paper reveals that modest changes in glycan structure can have dramatic, clinically important effects on signalling.
Ohtsubo, K. et al. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123, 1307–1321 (2005).
Reumers, J. et al. SNPeffect: a database mapping molecular phenotypic effects of human non-synonymous coding SNPs. Nucleic Acids Res. 33, D527–D532 (2005).
Westphal, V. et al. Reduced heparan sulfate accumulation in enterocytes contributes to protein-losing enteropathy in a congenital disorder of glycosylation. Am. J. Pathol. 157, 1917–1925 (2000).
Bode, L. & Freeze, H. H. Applied glycoproteomics — approaches to study genetic–environmental collisions causing protein-losing enteropathy. Biochim. Biophys. Acta 1760, 547–559 (2005).
Lenz, D. et al. Protein-losing enteropathy in patients with Fontan circulation: is it triggered by infection? Crit. Care 7, 185–190 (2003).
Eklund, E. A. & Freeze, H. H. The congenital disorders of glycosylation — a multifaceted group of syndromes. NeuroRx 3, 254–263 (2006).
Acknowledgements
I am indebted to G. Srikrishna, L. Bode, and C. Kranz for critical reading of this manuscript, E. Eklund for figure designs and the National Institutes of Health for support.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Related links
Glossary
- Hypomorphic allele
-
A mutation that causes a partial decrease in the activity of the gene product.
- Heterosis
-
Describes situations in which individuals who are heterozygous for a specific pair of alleles have a fitness advantage over those who carry either homozygous genotype.
- Isoelectric focusing
-
A method that is used to separate proteins within an electric field on the basis of their isoelectric points.
- Electrospray ionization
-
An ionization technique that is used in the mass-spectrometric analysis of large biomolecules. Charged droplets of analyte solution are used to produce gas-phase ions for analysis.
- Thrombocytopaenia
-
Decreased number of blood platelets.
- Neutropaenia
-
A type of leukopaenia that mainly affects neutrophils.
- Complex-type glycans
-
N-glycans that contain multiple GlcNAc-based branches, usually terminated with sialic acid or galactose.
- Dysmorphology
-
A morphological defect that results from an abnormal developmental process.
- Interferon
-
A pro-inflammatory cytokine that is produce by T cells in response to antigenic or mitogenic stimuli.
- Reichert's membrane
-
The first basement membrane that is formed during mouse embryonic development; it is not present in humans.
- Variable expressivity
-
The variability in severity of a disease-causing genetic trait.
- Haploinsufficiency
-
The inability of the remaining wild-type allele to compensate for a heterozygous loss-of-function mutation.
- Sialyl LewisX antigen
-
A branched O-linked glycan, Sia-Gal-GlcNAc, in which fucose is bound to GlcNAc. It functions as a ligand for selectin, which is important for leukocyte trafficking.
- Leukopaenia
-
Reduction in the number of circulating leukocytes.
- Protein-losing enteropathy
-
Increased enteric loss of serum protein, especially albumin, that causes hypoproteinaemia.
Rights and permissions
About this article
Cite this article
Freeze, H. Genetic defects in the human glycome. Nat Rev Genet 7, 537–551 (2006). https://doi.org/10.1038/nrg1894
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrg1894
This article is cited by
-
Glycosylation and behavioral symptoms in neurological disorders
Translational Psychiatry (2023)
-
Severe phenotypes of B3GAT3-related disorder caused by two heterozygous variants: a case report and literature review
BMC Medical Genomics (2022)
-
A comprehensive review of global alignment of multiple biological networks: background, applications and open issues
Network Modeling Analysis in Health Informatics and Bioinformatics (2022)
-
N- and O-glycan analysis for the detection of glycosylation disorders
Egyptian Journal of Medical Human Genetics (2021)
-
HILIC-UPLC-MS for high throughput and isomeric N-glycan separation and characterization in Congenital Disorders Glycosylation and human diseases
Glycoconjugate Journal (2021)
