
Abstract
Multilocus sequence typing (MLST) was proposed in 1998 as a portable sequence-based method for identifying clonal relationships among bacteria. Today, in the whole-genome era of microbiology, the need for systematic, standardized descriptions of bacterial genotypic variation remains a priority. Here, to meet this need, we draw on the successes of MLST and 16S rRNA gene sequencing to propose a hierarchical gene-by-gene approach that reflects functional and evolutionary relationships and catalogues bacteria 'from domain to strain'. Our gene-based typing approach using online platforms such as the Bacterial Isolate Genome Sequence Database (BIGSdb) allows the scalable organization and analysis of whole-genome sequence data.
Similar content being viewed by others

Main
Advances in nucleotide-sequencing technology have provided unparalleled access to the enormous genetic diversity that has accumulated in the bacterial domain during 3.5–4 billion years of evolution1. Numerous sets of whole-genome sequencing (WGS) data for bacterial isolates (Box 1) are available2, and metagenomic studies using these technologies continue to reveal further, seemingly boundless, diversity in bacterial communities3. Faced with this plethora of information, microbiologists must develop structured means of describing this diversity and of linking phenotype and genotype, thereby facilitating an improved understanding of the microbiological world. Given that we have precise information on the function of only a very small proportion of bacterial genes, and no knowledge at all about most, this is a formidable, if extremely exciting, challenge.
Here, we focus primarily on pathogenic bacteria, although the concepts discussed are applicable more widely to all bacteria and archaea. Bacterial pathogens played a crucial part in the development of experimental microbiology and remain the most intensively studied prokaryotes more than 100 years later4. Pathogens have emerged across the diversity of the bacterial — but, interestingly, not the archaeal — domain on many occasions and are both polyphyletic and highly diverse. Thus, although pathogens represent only a tiny subset of the bacterial world, the challenges faced by the clinical microbiology laboratory are representative of those faced by microbiology as a whole.
Taxonomic and functional analyses are based on the observations that diversity among bacteria is not continuous and that distinct, stable types with particular properties exist5. These founding principles of microbiology6 have been upheld by much subsequent research, but the study of such clusters remains largely descriptive, and the evolutionary mechanisms that led to cluster emergence and persistence remain incompletely understood7,8. Structuring is also evident within bacterial genomes, as diversity is unevenly distributed among genes and genomic regions9. Further, many bacterial genomes are flexible, containing both 'core' and 'accessory' components10 (Box 1). Sequence variation in the core genome represents an important starting point if different organisms are to be compared, but all types of variation, including that in genes, intergenic regions and episomes, must be catalogued effectively to make comprehensive associations between phenotype and genotype.
Pre-WGS cataloguing of diversity
A major advance in defining bacterial diversity was the proposal, by the late Carl Woese and colleagues, of a universal and 'natural' — that is, genealogical — classification system based on small-subunit 16S rRNA gene sequences11. The variation in these core genes, which are present in all bacteria, has been extensively exploited for the investigation of taxonomic relationships12, including defining and identifying species13,14, and forms the basis of many metagenomic community studies15. For these applications, 16S rRNA gene sequences provide an inordinate quantity of information in relation to their small size, and they were seminal in establishing that 'prokaryotes' are not a monophyletic group, but contain two of the three domains of life: Bacteria and Archaea12. Despite these successes, 16S rRNA sequences provide only limited resolution among closely related bacteria: many bacterial species exhibit identical 16S rRNA sequences among diverse isolates, and even species groups within genera are often poorly resolved, if at all.
The need for higher-resolution characterization of isolates has led to the development of a wide range of strain-typing methods16, including multilocus sequence typing (MLST)17, which has become the method of choice for typing many organisms18. MLST was designed to accommodate the conflicting signals of vertical and horizontal genetic transfer (Box 1) that are present in bacterial populations19 by examining the genome at multiple 'housekeeping' gene loci17,20. This concept had been used previously by multilocus enzyme electrophoresis (MLEE)21, which examines metabolic enzyme variation, but the indexing of gene sequence variation by MLST dramatically improved resolution, reproducibility and portability. The development of curated online MLST reference data sets (such as those found in the PubMLST database collection) provided both portable nomenclature schemes and the possibility of analysing the sequences to infer evolutionary relationships17,18.
Like MLEE, MLST uses alleles as the unit of comparison, rather than nucleotide sequences. In allele-based comparisons among isolates, each allelic change is counted as a single genetic event, regardless of the number of nucleotide polymorphisms involved. This provides a simple and effective correction for the fact that in many bacteria, common horizontal genetic transfer events account for many more polymorphisms among specimens than rarer point mutations22. The MLST approach retains information at all loci and avoids the need to categorize which changes are recent point mutations and which are due to recombination. As MLST schemes record the sequences of allelic variants, MLST data can also be used for sequence-based analyses when this is appropriate18,20,23.
In most MLST schemes, seven 'MLST loci' are indexed, for which each unique sequence for each locus is assigned an arbitrary and unique allele number. The designations for each of the loci are incorporated into an allelic profile (for example, 2-3-4-3-8-4-6), or a sequence type (ST), which is also assigned a numerical designation (for example, ST11). The ST and allelic designations are related to their respective allelic profiles and sequences in the MLST databases20, and each ST thus summarizes thousands of base pairs of information. In those bacteria that were most extensively examined by MLST, many hundreds of alleles at each locus and thousands of STs (see the PubMLST database collection) have been identified. Although an ST represents only a tiny percentage of the 'conserved' parts of the genome in question, the large number of STs in many bacterial populations demonstrates the importance of an expandable means of summarizing and comparing data. The allele and ST designations can be used in definitions of strains or grouped into clonal complexes or lineages (Box 1) as an improved understanding of the biological population structure emerges18,20.
Unfortunately, the variability of housekeeping genes among different bacteria makes it impossible to develop MLST schemes for anything but closely related bacteria. Consequently, even within genera (for example, in the genus Streptococcus24,25,26,27), it is necessary to have more than one MLST scheme, targeting different loci. In addition, MLST does not provide sufficient discrimination for all typing purposes, including resolving differences among variants of single-clone, low diversity, asexual pathogens such as Bacillus anthracis28 and Yersinia pestis29 or isolates of more diverse pathogens that belong to the same lineage. For example, at least two distinct sublineages within the ST11 clonal complex of Neisseria meningitidis are indistinguishable by MLST30. Therefore, MLST alone is not always sufficient for applications such as contact tracing in epidemics or for characterizing single-clone pathogens31, and in these cases MLST can be supplemented with additional typing schemes that index more variable loci, such as antigen genes32,33 or variable-number tandem repeats (VNTRs)34. To conclude, before the advent of WGS data, multiple approaches were needed to address the range of isolate identification and typing requirements, many of which were specific for particular organisms.
Post-WGS cataloguing of diversity
The complete closed genome sequence of a bacterium, be it from an isolate or a sequence reconstructed from a metagenomic study, is the ultimate 'molecular bar-code' for typing and taxonomic purposes. Rapid, very high-throughput sequencing methods35 are removing the practical constraints that framed the design of previously used approaches, as it is now possible to generate accurate WGS data for bacterial isolates in a single experiment36. Current WGS technologies do not generate complete genome sequences; rather, they produce data that can be used to reconstruct most of the genome sequence37. The relative performances of different sequencing platforms have been recently reviewed38,39 and are not further discussed here, but it is reasonable to suppose that the already very high data quality will continue to improve and that large numbers of very high-quality complete genomes are not too distant a prospect.
Although WGS data are easily and efficiently stored as strings of several million As, Cs, Gs and Ts, systems are required that can summarize and organize the variation present in large numbers of genome sequences within a practical framework. Faced with the wealth of data becoming available, novel systems for the description of genetic diversity can be designed from first principles, and a number of criteria required to guide this enterprise are evident from the successes of rRNA sequencing and MLST. Such systems should be:
-
Universal, in that they are applicable to all bacteria
-
Natural, reflecting genealogical relationships while retaining the capacity to describe closely related organisms with distinct properties
-
Understandable, so that the output and the process by which the system has been arrived at are transparent, easily interpreted and reproducible, and where possible the system should be backwards compatible with previous approaches
-
Expandable, to account for the incompleteness of our knowledge of diversity, and flexible enough to accommodate changes in this knowledge
-
Portable, because methods need to be easily carried out in any laboratory and the data need to be freely exchanged by the use of generic methodologies, reagents and bioinformatics pipelines
-
Technology independent, so that the data used are independent of the means of their collection (this means that schemes adopted now need to retain their validity as data improve)
-
Readily available to the entire community
-
Scalable, so that methods are sufficiently fast and inexpensive to be useable in real time for large or small numbers of isolates (this scalability is especially important for clinical applications and large-scale bacterial population analyses)
-
Able to accommodate a wide range of variation so that they can encompass both close and distant genealogical relationships
-
Broadly accepted by those who use them and open to contributions from members of the community.
These apparently straightforward criteria are yet to be met by a broadly adopted approach, and the successes and failures of currently implemented approaches to cataloguing bacterial diversity can be measured against them.
A variety of means have been used to identify WGS variation among bacterial specimens. A popular method has been to construct phylogenies on the basis of SNPs (single-nucleotide differences among samples) that have been identified either by the mapping of short-read sequence data to a reference genome, or by aligning de novo-assembled sequences to a reference genome37,40. This approach has been used successfully to investigate the epidemiology and evolution of a number of single-clone pathogens or members of the same lineage41,42,43,44,45,46,47,48,49,50,51,52, but it is limited by its requirement for a reference sequence or whole-genome alignment40. The analysis of diverse or highly recombining organisms in this way will prove challenging because the number of total polymorphisms increases as the number of polymorphisms conforming to a clonal model of descent decreases (Box 1). Alternatively, methods based on sequence similarity — for example, the use of 'coloured' de Bruijn graphs to detect genetic variation — eliminate the need for a reference genome or alignment53; however, such approaches are yet to be widely used.
Arguably the most versatile means of examining sequence variation among sets of WGS data, and the most natural to microbiologists, is the gene-by-gene (or 'MLST-like') approach to de novo-assembled genomes, which can be applied to diverse specimens without the need for high-quality reference genomes54,55,56. Most bacterial genomes are dominated by single-copy functional genes, the majority of which are protein coding57, whereas pseudogenes, paralogues and intergenic regions represent only a small percentage of the genome9. Functional protein-coding genes have numerous advantages as a basis for isolate characterization, as illustrated by the success of MLST, and gene-based typing methods are both easily understood and compatible with previous schemes. Although chromosomal protein-coding genes are a useful unit of analysis, other genomic regions such as pseudogenes and intergenic regions can also be considered as typing loci and indexed in the same way. The basis of the gene-by-gene approach is a de novo assembly followed by annotation, and various assembly algorithms are available for this process. The continuing improvements in sequencing and assembly technologies58, in combination with the efforts of initiatives such as the Genomics Standards Consortium59, mean that de novo assemblies which contain most, if not all, of the genome of a given organism are likely to become the norm in the very near future60.
An important consideration in the design of typing schemes is the level of resolution required among specimens. This, in turn, depends on the particular question being addressed, as some questions require more discrimination among specimens than others. To use the clinical setting as an example: very high resolution is necessary for the detection of outbreaks and the investigation of within-patient variation43; lower resolution is required to determine the membership of a particular clonal complex or lineage61; and even lower resolution is sufficient for determining the species causing an infection37,62,63. The gene-by-gene approach is inherently hierarchical and scalable, as fewer genes can be used for lower-resolution typing, whereas higher levels of resolution can be attained by increasing the number of genes included in the analysis. However, to establish such a system, it is necessary to be able to store, organize and access genome sequence data, and for this enterprise, databases are essential.
Gene-by-gene typing infrastructure
The need for effective data repositories is well recognized, as is evident from the interest generated by initiatives such as the Global Microbial Identifier63,64,65, and the power of such infrastructure is illustrated by the success of the 16S rRNA sequence and MLST databases20,62. For example, there are many MLST databases available on a number of websites (see PubMLST, the MLST homepage, the MLST databases at the ERI, UCC (Environmental Research Institute, University College Cork, Ireland) and the Institut Pasteur MLST databases), and these databases enable data generated in different laboratories to be efficiently compared. The first MLST database software to be developed66,67,68 stored only isolate information, and allele and ST definitions. Newer platforms such as the Bacterial Isolate Genome Sequence Database (BIGSdb)69 extend the MLST, or gene-by-gene, approach to WGS data. BIGSdb encompasses all the functionality of the software formerly used for the MLST databases hosted on PubMLST67,70. It is open source and runs on the Linux operating system using standard PC hardware and, to our knowledge, is currently the only internet server platform to offer the gene-by-gene approach to genome analysis.
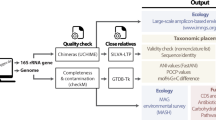
BIGSdb links any type of contiguous sequence data, from single genes to complete closed genomes, with provenance and phenotype data (metadata) for the isolates from which the sequences were derived; it also stores allele and locus definitions. There is no inherent limit on the number of isolate, allele or locus records, and loci can be grouped into an unlimited number of schemes (groups of loci that are analysed together, such as the MLST loci). As all this information is text based, very large data sets can be stored on modest computer equipment. BIGSdb has three main components: an isolate provenance and genome database, a sequence definition database, and typing and analysis schemes (Fig. 1). The isolate database stores assembled contiguous sequences (contigs) for each isolate, along with the designations of each locus that have been defined in the database for that isolate. The sequence definition database contains all known alleles for each locus that has been defined, and these alleles are usually identified by integers. As new variants are identified, this expands to index the diversity of new alleles for both known loci and newly identified genes. Although loci are usually genes (especially protein-coding genes), any sequence string, nucleotide or peptide-coding region can be defined as a locus, so that intergenic regions, for example, can be included in this approach.

The gene-by-gene approach can be used to integrate whole-genome sequencing (WGS) data with isolate data, facilitating easy storage and retrieval for downstream analyses. WGS data can be obtained in several ways: DNA can be isolated from a bacterial isolate or community sample and sequenced on an appropriate platform, short-read data can be obtained from the Sequence Read Archive (SRA), and assembled genomes can be downloaded from public databases (for example, GenBank). In the cases of sequencing and SRA data, short-read data are assembled with an appropriate algorithm, directed by the sequencing platform used. Assembled contigs are uploaded to a Bacterial Isolate Genome Sequence Database (BIGSdb), and they can then be compared against either sets of reference genes or a reference genome using algorithms such as BLAST. Reference gene sets can be tailored to meet particular requirements and thus range from collections of loci that are useful for epidemiological investigations to subsets of genes with functional relevance, for example in a metabolic pathway. For comparisons based on both reference gene sets and reference genomes, the nucleotide sequences remain accessible, but loci are assigned allele designations to generate an allelic profile as for multilocus sequence typing (MLST). The Genome Comparator module in BIGSdb can be used to produce a distance matrix based on allelic profiles, and this matrix can in turn be used to visualize relationships between isolates using an appropriate algorithm, such as NeighbourNet in SplitsTree. Alternatively, downstream analyses of the aligned sequence data can be carried out by exporting sequence data to external packages. In addition to a distance matrix and sequence alignments, the Genome Comparator outputs also include a table showing which loci are identical and which are different among the isolates examined. rMLST, ribosomal MLST.
The sequence data stored in BIGSdb can be generated by any means and are uploaded as contigs. Sequence quality is controlled at the assembly stage before incorporation into the database, but not in the database itself, although experiment tags can be used to indicate the nature and type of the data included. Different types of data can be stored side-by-side in the sequence bin and can then be retrieved and analysed as required. Data can be uploaded from public archives, such as GenBank or the Integrated Microbial Genomes (IMG) database71, and the accession numbers for these repositories provide links between these databases and the BIGSdb record (Fig. 1).
When sequence data are deposited in BIGSdb, they are scanned against the definitions databases using an appropriate algorithm (currently BLAST72, but other algorithms could be used). When a known allele is identified, this is recorded as a designation for that locus in the isolate record, and the position of the allele in the contig is marked (tagged) (Fig. 1). This process is rapid, and any new alleles identified can be assigned in the definition database; genomic data are periodically rescanned in an iterative process, resulting in an expanding and increasingly comprehensive catalogue of genomic diversity. Thus, as well as linking sequence data to phenotype, BIGSdb is a population-based annotation tool that enables the identification of particular genes and allelic variants within WGS data, maintaining a record of the known variation of that gene across the samples stored in the database. The assignment of alleles to loci can be defined using sequence similarity criteria, and this definition is stored in the locus record.
The grouping of defined loci into schemes is particularly useful for typing and taxonomy applications but has many other possible uses, including functional annotation. Combinations of alleles within schemes can be linked with other data — for example, to associate the sequences of genes with notable phenotypes such as antimicrobial resistance or susceptibility54. These schemes are not limited to given groups of organisms, but can be applied to any isolate in the database, which is particularly useful for the annotation of accessory genes that might be widely distributed in diverse organisms, or core genes that are shared among them. When a scheme has been defined, it can be readily applied to other isolates in the database.
The Genome Comparator module of BIGSdb facilitates gene-based comparative genomics by examining groups of shared genes in any number of isolates across any number of genes. For example, it can use an annotated reference genome as the source of comparison sequences to BLAST against other genomes in the database. This generates whole-genome profiles that can be analysed by standard distance methods such as NeighbourNet73 implemented within the SplitsTree package74, simultaneously providing lists of loci that are identical, variable, missing or truncated (incomplete genes located at the ends of contigs) among data sets. BIGSdb allows queries through web-based interfaces, making annotated genome content accessible to external databases and analysis software as well as to users querying the data directly (Fig. 1).
Sequence data and nomenclature
With a tool such as BIGSdb, the gene-by-gene approach can be used to catalogue genomic sequence variation and to map this onto existing or novel nomenclature schemes by associating particular alleles or combinations of alleles with names or designations. Nomenclature schemes are important means of communication which have to be agreed on among those who use them. They should be stable and preferably backwards compatible; however, they do require occasional modification as new insights are obtained, because stability should not be allowed to override accuracy in the description of biological diversity. By contrast, if determined correctly, sequence data and data summaries such as STs are absolute and require modification only to correct errors. The relationship between sequence data and nomenclature schemes therefore has to be transparent, and these two things should not be confused (Fig. 2).

Hierarchical nomenclature schemes are artificial constructs that are developed to facilitate communication and are subject to change as new information becomes available. Nomenclature schemes are dependent on various factors, including sequence relationships, and are ideally genealogically based. The challenge is to map whole-genome sequencing (WGS) data to nomenclature schemes transparently but flexibly at a range of resolutions. The highest discrimination is required for studies of bacterial isolates from one patient or from very closely related transmission chains; these isolates can be thought of as having undergone microevolution. Such studies will require comparisons of whole genomes using whole-genome multilocus sequence typing (MLST)30. Progressively lower resolution is required for studies of isolates with more distant common ancestors and, therefore, with more genetic differences. These relationships are best studied using the core genome common to the set of isolates of interest. Genes encoding ribosomal proteins are a particularly useful subset of core genes, and ribosomal MLST75 accommodates many levels of genealogical relationships, from clonal complexes and lineages to species78, genera and beyond75. In a database such as the Bacterial Isolate Genome Sequence Database (BIGSdb), multiple gene-by-gene schemes can be implemented alongside other, more conventional sequence-based schemes69. Particular genotype summaries of genes or collections of genes can be associated with particular nomenclature schemes, enabling the database to deliver a plain-language report to the user.
Different levels of sequence information can be associated with different taxonomic levels. Analysis of a single locus, for example, is often sufficient to distinguish many groups (phylum, class, order, family or genus), whereas determining speciation and subspeciation requires higher resolution, which can be attained by increasing the number of loci analysed. Researchers can apply a set of MLST approaches, each using different numbers of loci and each suitable for addressing different levels of isolate discrimination (Fig. 2). The highest level of resolution using a gene-by-gene approach can be termed whole-genome MLST (wgMLST), in which all the loci of a given isolate are compared to equivalent loci in other isolates. The wgMLST approach is applicable to single-clone pathogens with closed genomes or to very closely related variants of more diverse organisms. This analysis can involve the entire genome if loci corresponding to intergenic regions are also defined. Few bacteria share all loci, so comparisons of the core genome of a given group (core-genome MLST (cgMLST)) provides high-resolution data across a group of related but not identical isolates. Other subsets of loci might have particular applications, such as conventional seven-locus MLST. It has been found that a scheme based on the 53 ribosomal protein loci present in most bacteria — ribosomal MLST (rMLST) — is both highly flexible and informative75.
rMLST for taxonomy and typing
For the purposes of bacterial typing and taxonomy, the ribosomal protein subunit (rps) genes have the advantages of being universally present but differentially variable76; indeed, although the variability of some rps genes has prevented their inclusion in sets of core genes77, this variation permits high levels of discrimination among closely related isolates30,75. The rps genes have the further advantage of being distributed across the genome, offering some stability in the face of horizontal genetic transfer. An rMLST allelic profile, or ribosomal sequence type (rST), can accurately summarize the relationships between bacterial genomes, providing a manageable basis for universal bacterial systematics and typing, as exemplified by the use of rMLST to define species groupings and strain types within the genus Neisseria30,75,78.
A database that catalogues variation in the 53 rps genes across the bacterial domain has been established (rMLST on PubMLST) using the BIGSdb platform75. This database currently includes more than 30,000 sets of assembled WGS data obtained from publicly available sources. All of these data can be indexed with rMLST, providing the basis for an efficient and rapid identification and characterization scheme76. The Genome Comparator tool on PubMLST can explore the rMLST loci within very large collections of WGS data from diverse bacterial isolates to identify clusters of isolates rapidly, and these clusters can then be investigated at higher resolution with approaches such as cgMLST and wgMLST as required30,79 (Fig. 2). As this database contains WGS data, it has applications beyond indexing just the rMLST loci, and schemes for any set of genes present in the de novo assemblies can be established within the database, as illustrated below for staphylococci.
Applying gene-by-gene analysis
Several members of the genus Staphylococcus are human and animal pathogens, and methicillin-resistant Staphylococcus aureus (MRSA) strains prove particularly problematic, largely because of their role in nosocomial outbreaks80,81. Consequently, staphylococci have been subjected to many molecular epidemiology studies80, including WGS analyses41,42,43,44,45,46,82. Many areas of the biology and pathology of these organisms have been investigated, including their evolution (particularly the origins of MRSA) in hospitals and communities83,84, the spread of clones among animals and humans85, the dynamics of colonization and infection86, and outbreak detection and control87. Although WGS is increasingly used in these investigations, it is difficult to compare the data generated in different studies, as they are typically available only as short reads. This problem is not confined to staphylococci. Further, the various SNP-based approaches used in different studies have not been standardized. Here, we illustrate how the gene-by-gene approach, implemented with the BIGSdb platform, can be used for combined analyses of data emanating from different studies of this important group of organisms.
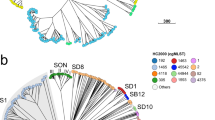
WGS data for 926 staphylococci (889 S. aureus isolates and 37 genomes from 20 other species) were obtained from the Sequence Read Archive (SRA), GenBank and the IMG database (Box 2; Supplementary information S1,S2,S3 (tables)). For data from the SRA, draft genomes were assembled de novo, and contigs were uploaded to the PubMLST website. Using the BIGSdb web interface and SplitsTree, species groups were resolved among 52 staphylococci. Nucleotide sequence-based analysis of the rMLST loci defined groups within the genus and provided insights into the species assignments of two recently described staphylococcal isolates, Staphylococcus sp. OJ82 (Ref. 88) and Staphylococcus sp. AL1 (Ref. 89) (Fig. 3a). Furthermore, an allele-based rMLST analysis of 669 S. aureus isolates identified 229 unique rSTs based on 51 rps loci (two paralogous loci, rpsN and rpmG, were excluded for the purposes of this analysis) and resolved the isolates into lineages that corresponded to previously described clonal complexes83,90, also indicating genealogical relationships among the clonal complexes (Fig. 3b).

These analyses were carried out using both the Genome Comparator module within the Bacterial Isolate Genome Sequence Database (BIGSdb) platform and data publicly available within the PubMLST website. The phylogenetic networks were generated using the NeighbourNet algorithm in SplitsTree (v4.12.3). a | Resolution of 52 staphylococcal isolates on the basis of nucleotide sequence diversity at 51 ribosomal multilocus sequence typing (rMLST) loci, permitting the determination of the species assignment of two recently described isolates, Staphylococcus sp. OJ82 and Staphylococcus sp. AL1. Staphylococcus sp. OJ82 probably corresponds to Stapylococcus equorum, whereas Staphylococcus sp. AL1 is related to, but distinct from, S. equorum and Staphylococcus saprophyticus. All species shown are in the genus Staphylococcus, except for Macrococcus caseolyticus. b | The diversity of 669 Staphylococcus aureus isolates on the basis of allelic diversity at 51 rMLST loci. The extensive diversity of S. aureus is illustrated here; the rMLST clustering is congruent with MLST clonal complexes (CCs; indicated) and indicates relationships among the isolates. c | Resolution of multidrug-resistant S. aureus (MRSA) isolates from an outbreak in a special-care baby unit41, using a gene-by-gene comparison to a reference genome (S. aureus subsp. aureus HO 5096 0412). Twenty isolates obtained from a health care worker are indicated with the letter H and shown in red, whereas patient isolates are indicated with a letter P. Groups of isolates from patients who were members of the same family are shown in the same colour. Reticulations in the diagrams indicate departures from a strictly tree-like phylogeny; this can have a number of causes, including homoplasy as a result of recombination, mutation or lack of resolution. Such graphs are rapidly produced and represent the relationships among sets of genome data; these relationships can then be readily used to resolve isolate relationships in clinical and other settings. Scale bars represent distances calculated from the nucleotide sequence alignment (part a) and number of loci (parts b,c).
The detection and control of S. aureus outbreaks remains a high priority in health care systems41,42,43. The gene-by-gene approach described above can rapidly establish very high-resolution relationships among isolates, as demonstrated by an analysis of isolate data from a study that described an MRSA outbreak in a special-care baby unit43. Genome Comparator was used to compare all the isolates to a reference genome (from isolate S. aureus subsp. aureus HO 5096 0412) and provided a web-based visualization of the relationships among isolates from patients and from a health care worker (Fig. 3c); such a visualization would be easily and rapidly generated and interpreted in a health care setting. This analysis demonstrated that isolates from babies, mothers and their partners were closely related to each other and to those from a health-care worker, suggesting that there was an ongoing outbreak. Along with other available data, these results could have been interpreted in such a way that prompted interventions to prevent further transmission. Similar results were presented in the original report, but the gene-by-gene approach is independent of a specialized bioinformatics pipeline. This approach is appropriate for the very high-resolution analysis of isolates from a clinical setting, achieving a resolution equivalent to that obtained by SNP-based approaches.
The analyses presented here are not exhaustive; rather, they are a demonstration of the potential of the gene-by-gene approach as implemented in BIGSdb. This approach can be extended to any set of isolates and any combination of genes using web-based tools. BIGSdb can be used to store, organize and interpret WGS data from many different investigations in a single analysis; however, the database is sufficiently compact that WGS data from tens of thousands of isolates can be stored on a modestly sized personal computer for use in settings where Internet connectivity is poor or sporadic, or where clinically sensitive data need to be stored separately. The analyses relied on straightforward assumptions and did not require processor- or memory-intensive algorithms, nor did they involve selective removal of data that do not conform to a particular evolutionary model. An additional advantage of using such an approach is that Genome Comparator automatically provides lists of those loci that are identical and those that are variable, enabling genetic differences to be rapidly identified.
Conclusions and future directions
WGS studies of bacteria are currently in a golden age, with many resources and much interest being dedicated to the area as a whole and especially to WGS applications for clinical problems36,37,38,63. However, when this initial excitement has passed, it will be necessary to move into a more prosaic exploitation phase, which will require the construction of sustainable infrastructures — sustainable both intellectually (that is, the concept of relating WGS data to typing and taxonomy) and physically (that is, the use of inter-operable data sets that are archived in accessible databases). MLST offers mixed precedents for this process. On the positive side, the engagement of communities of scientists working on particular areas has enabled the assembly of many high-quality data sets, and this 'bottom up' approach is essential to ensure that appropriate typing schemes are developed and maintained by those people who need to use them. These efforts have largely been 'amateur', however, in the sense that they are not directly funded, and most MLST databases rely on enthusiastic community members. The careful curation of sequences and maintenance of typing schemes are essential 'public goods' that all researchers in the field wish to use, but to which few are willing, or able, to contribute.
A number of initiatives are currently underway to build such infrastructures, and these initiatives will have to balance community involvement and sustainability (Box 2). The development of multiple approaches, as long as they are open access, inter-operable and sustainable, is important at this stage, when it is unclear exactly what the most effective and broadly acceptable means of exploiting WGS data will be. However, maintaining databases is challenging in the face of contracting resources as this is not a high-impact activity, although it is essential. The PubMLST model discussed here is one approach to these problems; this approach attempts to balance centralization (in the form of a curated set of reference genes) with affordability, open access and community involvement. The task of relating WGS data to particular nomenclature schemes is of sufficient magnitude and complexity that a whole community of scientists and others will need to engage with the project.
As with all paradigm shifts and 'disruptive technologies', the exploitation of WGS is simultaneously exciting and confusing, and presents great opportunities along with major challenges. Ultimately, however, the availability of large volumes of WGS data promises the development of a new type of microbiology research in which population and evolutionary approaches are integrated with functional and structural studies by the organized presentation and interpretation of biodiversity data.
References
Ciccarelli, F. D. et al. Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287 (2006).
Medini, D. et al. Microbiology in the post-genomic era. Nature Rev. Microbiol. 6, 419–430 (2008).
Rusch, D. B. et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 5, e77 (2007).
Fournier, P. E. & Raoult, D. Prospects for the future using genomics and proteomics in clinical microbiology. Annu. Rev. Microbiol. 65, 169–188 (2011).
Stackebrandt, E. et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 52, 1043–1047 (2002).
Koch, R. An address on bacteriological research. Br. Med. J. 2, 380–383 (1890).
Fraser, C., Alm, E. J., Polz, M. F., Spratt, B. G. & Hanage, W. P. The bacterial species challenge: making sense of genetic and ecological diversity. Science 323, 741–746 (2009).
Buckee, C. O. et al. Role of selection in the emergence of lineages and the evolution of virulence in Neisseria meningitidis. Proc. Natl Acad. Sci. USA 105, 15082–15087 (2008).
Bentley, S. D. & Parkhill, J. Comparative genomic structure of prokaryotes. Annu. Rev. Genet. 38, 771–792 (2004).
Tettelin, H., Riley, D., Cattuto, C. & Medini, D. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477 (2008).
Woese, C. R., Kandler, O. & Wheelis, M. L. Towards a natural system of organisms — proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl Acad. Sci. USA 87, 4576–4579 (1990).
Fox, G. E. et al. The phylogeny of prokaryotes. Science 209, 457–463 (1980).
Clarridge, J. E. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 17, 840–862 (2004).
Vamosi, S. M., Heard, S. B., Vamosi, J. C. & Webb, C. O. Emerging patterns in the comparative analysis of phylogenetic community structure. Mol. Ecol. 18, 572–592 (2009).
Hugenholtz, P., Goebel, B. M. & Pace, N. R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180, 4765–4774 (1998).
Sabat, A. J. et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 18, 17–30 (2013).
Maiden, M. C. J. et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl Acad. Sci. USA 95, 3140–3145 (1998).
Perez-Losada, M., Cabezas, P., Castro-Nallar, E. & Crandall, K. A. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infection Genet. Evol. 16, 38–53 (2013).
Maynard Smith, J., Smith, N. H., O'Rourke, M. & Spratt, B. G. How clonal are bacteria? Proc. Natl Acad. Sci. USA 90, 4384–4388 (1993).
Maiden, M. C. J. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60, 561–588 (2006).
Selander, R. K. et al. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl. Environ. Microbiol. 51, 837–884 (1986).
Didelot, X. & Maiden, M. C. Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322 (2010).
Turner, K. M. & Feil, E. J. The secret life of the multilocus sequence type. Int. J. Antimicrob. Agents 29, 129–135 (2007).
Do, T. et al. Population structure of Streptococcus oralis. Microbiology 155, 2593–2602 (2009).
Webb, K. et al. Development of an unambiguous and discriminatory multilocus sequence typing scheme for the Streptococcus zooepidemicus group. Microbiology 154, 3016–3024 (2008).
Coffey, T. J. et al. First insights into the evolution of Streptococcus uberis: a multilocus sequence typing scheme that enables investigation of its population biology. Appl. Environ. Microbiol. 72, 1420–1428 (2006).
Enright, M. C. & Spratt, B. G. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144, 3049–3060 (1998).
Priest, F. G., Barker, M., Baillie, L. W., Holmes, E. C. & Maiden, M. C. Population structure and evolution of the Bacillus cereus group. J. Bacteriol. 186, 7959–7970 (2004).
Achtman, M. et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl Acad. Sci. USA 101, 17837–17842 (2004).
Jolley, K. A. et al. Resolution of a meningococcal disease outbreak from whole genome sequence data with rapid web-based analysis methods. J. Clin. Microbiol. 50, 3046–3053 (2012).
Holt, K. E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nature Genet. 40, 987–993 (2008).
Jolley, K. A., Brehony, C. & Maiden, M. C. Molecular typing of meningococci: recommendations for target choice and nomenclature. FEMS Microbiol. Rev. 31, 89–96 (2007).
Dingle, K. E., McCarthy, N. D., Cody, A. J., Peto, T. E. & Maiden, M. C. Extended sequence typing of Campylobacter spp., United Kingdom. Emerg. Infect. Dis. 14, 1620–1622 (2008).
Adair, D. M. et al. Diversity in a variable-number tandem repeat from Yersinia pestis. J. Clin. Microbiol. 38, 1516–1519 (2000).
Parkhill, J. & Wren, B. W. Bacterial epidemiology and biology — lessons from genome sequencing. Genome Biol. 12, 230 (2011).
Pallen, M. J., Loman, N. J. & Penn, C. W. High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Curr. Opin. Microbiol. 13, 625–631 (2010).
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E. A. & Crook, D. W. Transforming clinical microbiology with bacterial genome sequencing. Nature Rev. Genet. 13, 601–612 (2012).
Loman, N. J. et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nature Rev. Microbiol. 10, 599–606 (2012).
Junemann, S. et al. Updating benchtop sequencing performance comparison. Nature Biotech. 31, 294–296 (2013).
Croucher, N. J., Harris, S. R., Grad, Y. H. & Hanage, W. P. Bacterial genomes in epidemiology-present and future. Philos. Trans. R. Soc. B 368, 20120202 (2013).
Köser, C. U. et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. New Engl. J. Med. 366, 2267–2275 (2012).
Eyre, D. W. et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2, e001124 (2012).
Harris, S. R. et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 31, 130–136 (2012).
McAdam, P. R. et al. Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc. Natl Acad. Sci. USA 109, 9107–9112 (2012).
Holden, M. T. et al. A genomic portrait of the emergence, evolution and global spread of a methicillin resistant Staphylococcus aureus pandemic. Genome Res. 23, 653–664 (2013).
Young, B. C. et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc. Natl Acad. Sci. USA 109, 4550–4555 (2012).
Mutreja, A. et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477, 462–465 (2011).
Bryant, J. M. et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381, 1551–1560 (2013).
He, M. et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl Acad. Sci. USA 107, 7527–7532 (2010).
Cui, Y. J. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl Acad. Sci. USA 110, 577–582 (2013).
Walker, T. M. et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13, 137–146 (2013).
Grad, Y. H. et al. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc. Natl Acad. Sci. USA 109, 3065–3070 (2012).
Iqbal, Z., Turner, I. & McVean, G. High-throughput microbial population genomics using the Cortex variation assembler. Bioinformatics 29, 275–276 (2013).
Jolley, K. A. & Maiden, M. C. Automated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplar. Euro Surveill. 18, 20379 (2013).
Sheppard, S. K., Jolley, K. A. & Maiden, M. C. J. A gene-by-gene approach to bacterial population genomics: whole genome MLST of Campylobacter. Genes 3, 261–277 (2012).
Bratcher, H. B., Bennett, J. S. & Maiden, M. C. J. Evolutionary and genomic insights into meningococcal biology. Future Microbiol. 7, 873–885 (2012).
Konstantinidis, K. T. & Tiedje, J. M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl Acad. Sci. USA 101, 3160–3165 (2004).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Chain, P. S. et al. Genomics. Genome project standards in a new era of sequencing. Science 326, 236–237 (2009).
Hunt, M. et al. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 14, R47 (2013).
Enright, M. C. et al. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl Acad. Sci. USA 99, 7687–7692 (2002).
Harmsen, D., Rothganger, J., Frosch, M. & Albert, J. RIDOM: ribosomal differentiation of medical micro-organisms database. Nucleic Acids Res. 30, 416–417 (2002).
Koser, C. U. et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 8, e1002824 (2012).
Aarestrup, F. M. et al. Integrating genome-based informatics to modernize global disease monitoring, information sharing, and response. Emerg. Infect. Dis. 18, e1 (2012).
Carrico, J. A., Sabat, A. J. & Friedrich, A. W., Ramirez, M. & ESCMID Study Group for Epidemiological Markers (ESGEM). Bioinformatics in bacterial molecular epidemiology and public health: databases, tools and the next-generation sequencing revolution. Euro Surveill. 18, 32–40 (2013).
Chan, M. S., Maiden, M. C. & Spratt, B. G. Database-driven multi locus sequence typing (MLST) of bacterial pathogens. Bioinformatics 17, 1077–1083 (2001).
Jolley, K. A., Chan, M. S. & Maiden, M. C. mlstdbNet — distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5, 86 (2004).
Aanensen, D. M. & Spratt, B. G. The multilocus sequence typing network: mlst.net. Nucleic Acids Res. 33, W728–W733 (2005).
Jolley, K. A. & Maiden, M. C. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595 (2010).
Jolley, K. A. & Maiden, M. C. AgdbNet — antigen sequence database software for bacterial typing. BMC Bioinformatics 7, 314 (2006).
Markowitz, V. M. et al. The integrated microbial genomes system: an expanding comparative analysis resource. Nucleic Acids Res. 38, D382–D390 (2010).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Bryant, D. & Moulton, V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 21, 255–265 (2004).
Huson, D. H. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73 (1998).
Jolley, K. A. et al. Ribosomal multi-locus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015 (2012).
Yutin, N., Puigbo, P., Koonin, E. V. & Wolf, Y. I. Phylogenomics of prokaryotic ribosomal proteins. PLoS ONE 7, e36972 (2012).
Wu, M. & Eisen, J. A. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9, R151 (2008).
Bennett, J. S. et al. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158, 1570–1580 (2012).
Cody, A. J. et al. Real-time genomic epidemiology of human Campylobacter isolates using whole genome multilocus sequence typing. J. Clin. Microbiol. 51, 2526–2534 (2013).
Stefani, S. et al. Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 39, 273–282 (2012).
Widerstrom, M., Wistrom, J., Sjostedt, A. & Monsen, T. Coagulase-negative staphylococci: update on the molecular epidemiology and clinical presentation, with a focus on Staphylococcus epidermidis and Staphylococcus saprophyticus. Eur. J. Clin. Microbiol. Infect. Dis. 31, 7–20 (2012).
Harris, S. R. et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474 (2010).
Lindsay, J. A. Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 98–103 (2010).
Uhlemann, A. C., Otto, M., Lowy, F. D. & Deleo, F. R. Evolution of community- and healthcare-associated methicillin-resistant Staphylococcus aureus. Infect. Genet. Evol. http://dx.doi.org/10.1016/j.meegid.2013.04.030 (2013).
Fitzgerald, J. R. Livestock-associated Staphylococcus aureus: origin, evolution and public health threat. Trends Microbiol. 20, 192–198 (2012).
Fitzgerald, J. R. Evolution of Staphylococcus aureus during human colonization and infection. Infect. Genet. Evol. http://dx.doi.org/10.1016/j.meegid.2013.04.020 (2013).
Lindsay, J. A. Evolution of Staphylococcus aureus and MRSA during outbreaks. Infect. Genet. Evol. http://dx.doi.org/10.1016/j.meegid.2013.04.017 (2013).
Sung, J. S., Chun, J., Choi, S. & Park, W. Genome sequence of the halotolerant Staphylococcus sp. strain OJ82, isolated from Korean traditional salt-fermented seafood. J. Bacteriol. 194, 6353–6354 (2012).
Chong, T. M., Tung, H. J., Yin, W. F. & Chan, K. G. Insights from the genome sequence of quorum-quenching Staphylococcus sp. strain AL1, isolated from traditional Chinese soy sauce brine fermentation. J. Bacteriol. 194, 6611–6612 (2012).
Holt, D. C. et al. A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol. Evol. 3, 881–895 (2011).
Milkman, R. & McKane, M. in Population Genetics of Bacteria (eds Baumberg, S., Young, J. P. W., Wellington, E. M. H. & Saunders, J. R.) 127–142 (Cambridge Univ. Press, 1995).
Lapage, S. P. et al. International Code of Nomenclature of Bacteria: Bacteriological Code, 1990 Revision (American Society for Microbiology, 1992).
Maynard Smith, J., Dowson, C. G. & Spratt, B. G. Localized sex in bacteria. Nature 349, 29–31 (1991).
Spratt, B. G. Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature 332, 173–176 (1988).
Fernandez-Suarez, X. M. & Galperin, M. Y. The 2013 nucleic acids research database issue and the online molecular biology database collection. Nucleic Acids Res. 41, D1–D7 (2013).
Acknowledgements
The authors acknowledge the IMG, GenBank and SRA databases, isolates from which were included in the analyses presented in this article. All isolate genomes can be accessed at the rMLST database on PubMLST and are identified under the project names Maiden 2013 Nat Rev SCBU for the outbreak analysis, Maiden 2013 Nat Rev S aureus for the other S. aureus analyses and Maiden 2013 Nat Rev rMLST for the genus Staphylococcus rMLST analysis, with original database accession numbers provided as available. The authors are grateful to J. S. Bennett, H. B. Bratcher, C. Brehony, A. J. Cody, F. Colles, O. B. Harrison, D. M. Hill, S. K. Sheppard, E. R. Watkins and H. Wimalarathna, as well as many other collaborators, for their contributions to the development of the context of this work and for comments on the manuscript. M.C.J.M. is a Wellcome Trust Senior Fellow in Basic Biomedical Sciences. M.J.J.v.R. is funded by the Clarendon Fund and Merton College, Oxford University, UK, and J.E.B. is funded by the Patho-NGen-Trace consortium. The research from the Patho-NGen-Trace project leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013; grant 278864).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
FURTHER INFORMATION
Supplementary information
Supplementary information S1 (table)
staphylococcal rMLST analysis (PDF 391 kb)
Supplementary information S2 (table)
238 isolates with unique rSTs derived from 699 S. aureus isolates included in species level rMLST analysis (PDF 1830 kb)
Supplementary information S3 (table)
S. aureus isolates from an outbreak in a UK Special Care Baby Unit (PDF 475 kb)
Rights and permissions
About this article
Cite this article
Maiden, M., van Rensburg, M., Bray, J. et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11, 728–736 (2013). https://doi.org/10.1038/nrmicro3093
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro3093
This article is cited by
-
Investigating environmental transmission to resolve a Bacillus cereus group outbreak in a neonatal intensive care unit using core genome multilocus sequence typing
Antimicrobial Resistance & Infection Control (2024)
-
Performance evaluation of core genome multilocus sequence typing for genotyping of Mycobacterium tuberculosis strains in China: based on multicenter, population-based collection
European Journal of Clinical Microbiology & Infectious Diseases (2024)
-
Deciphering the functional diversity of the gut microbiota of the black soldier fly (Hermetia illucens): recent advances and future challenges
Animal Microbiome (2023)
-
cgMSI: pathogen detection within species from nanopore metagenomic sequencing data
BMC Bioinformatics (2023)
-
Whole genome sequencing of Salmonella enterica serovars isolated from humans, animals, and the environment in Lagos, Nigeria
BMC Microbiology (2023)
