
Abstract
A harmonized balance between positive and negative regulation of pattern recognition receptor (PRR)-initiated immune responses is required to achieve the most favorable outcome for the host. This balance is crucial because it must not only ensure activation of the first line of defense against viral infection but also prevent inappropriate immune activation, which results in autoimmune diseases. Recent studies have shown how signal transduction pathways initiated by PRRs are positively and negatively regulated by diverse modulators to maintain host immune homeostasis. However, viruses have developed strategies to subvert the host antiviral response and establish infection. Viruses have evolved numerous genes encoding immunomodulatory proteins that antagonize the host immune system. This review focuses on the current state of knowledge regarding key host factors that regulate innate immune signaling molecules upon viral infection and discusses evidence showing how specific viral proteins counteract antiviral responses via immunomodulatory strategies.
Similar content being viewed by others
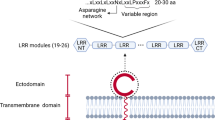
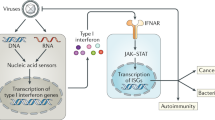

Introduction
Viruses need to hijack the host cell machinery to replicate effectively; however, they must first overcome the host’s defenses. The efficacy of a viral infection depends on the comparative potency of the effector molecules used by the virus and the host. A critical determinant of whether a host succumbs to or can subvert a viral infection is the speed at which the host’s defenses are activated1. Almost all innate immune responses require an extended sequence of actions: pathogen sensing, signal transduction, transcription, translation, protein folding, and transport to the site of action. To initiate signaling upon viral infection, host cells detect viral DNA or RNA using a set of PRRs; these include retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (RNA sensors), cyclic GMP-AMP (cGAMP) synthase (cGAS), interferon gamma inducible protein 16 (IFI16), absent in melanoma 2 (AIM2), and dead-box helicase 41 (DDX41) (DNA sensors)2,3.
Recognition of viral nucleic acids by PRRs triggers transduction of downstream signals mainly via adaptor proteins such as mitochondrial antiviral signaling protein (MAVS) or stimulator of interferon genes (STING), which induce expression of interferon (IFN)-stimulated genes via autocrine or paracrine mechanisms; the products of genes (proinflammatory cytokines, chemokines, and IFNs) inhibit viral replication and spread and induce activation of adaptive immune responses4,5. These antiviral signaling pathways play a crucial role in achieving an optimal outcome for the host; therefore, much attention has been devoted to identifying and understanding the signaling pathways and regulatory factors involved in antiviral innate immunity6 (Figs. 1, 2).
Conventional posttranslational modifications such as polyubiquitination and phosphorylation, unconventional posttranslational modifications such as acetylation and methylation, and other regulatory mechanisms such as physical interactions and translocations affect the production of IFN-β and inflammatory cytokines by targeting innate immune sensors and downstream signaling molecules (e.g., receptors, adaptors, enzymes, and transcription factors)7,8. These aforementioned modifications play a critical role in regulating the production of IFNs and inflammatory cytokines, which can, if production is unchecked, have deleterious effects on the host by promoting the development of autoimmune disorders, allergies, and other immunopathologies, as well as by activating and regulating the cellular status to exacerbate the severity of viral disease9.
It is not surprising that viruses exploit numerous strategies to enhance their replication. To establish efficient, lifelong infection and to initiate viral pathogenesis, a large portion of the viral genome encode numerous immunomodulatory proteins; the function of these proteins is to evade/disrupt the host immune system and ensure viral persistence10. From the perspective of the virus, these actions are critically important because viruses depend on living cells for replication. This review focuses on current knowledge regarding two factors. First, we summarize the posttranslational modifications (PTMs) and other regulatory mechanisms of signaling molecules downstream of the RNA/DNA sensing cascade that regulate efficient IFN responses and/or maintenance of host immune homeostasis. Second, we summarize how RNA/DNA viruses evade transduced host innate immune signals, which are initiated by PRRs, to establish a permissive state in host cells.
Role of PTMs in regulating signal transduction
PTMs play an important role in regulating the stability, activity, subcellular localization, and folding of proteins. Advances in experimental techniques used to map and quantify PTMs have led to marked progress in these areas. Such techniques have identified a number of PTMs that alter the innate immune response by regulating protein function, abundance, catalysis, interactions, or subcellular localization without necessarily requiring induction of a new transcriptional program8,11. Additionally, some of these PTMs are highly dynamic and fully reversible, allowing both initiation and resolution of responses. Phosphorylation, a process by which a phosphoryl group is attached to a serine, threonine, tyrosine, histidine, or aspartate residue, is a well-studied PTM regulated by the opposing actions of protein kinases and phosphatases; this PTM plays an important role in innate immunity11,12. The introduction of a phosphoryl group imparts a negative (–2) charge at physiological pH, resulting in a major biophysical perturbation of protein structure. This is manifested by conformational changes that alter enzymatic activity and/or protein–protein interactions13. Ubiquitination is another important PTM. During ubiquitination, proteins are modified via covalent attachment of a small 76-amino acid protein called ubiquitin, which (as the name implies) is expressed ubiquitously and is highly conserved in all eukaryotes14. Ubiquitination is inversely regulated by ubiquitin activating (E1), ubiquitin-conjugating (E2), and ubiquitin protein ligase (E3) enzymes and by deubiquitinating enzymes (DUBs); thus, it plays a critical role in regulating innate immune signal transduction. In contrast to phosphorylation, a single target site can be modified by a single ubiquitin molecule (monoubiquitination) or by chains of linked ubiquitin molecules (polyubiquitination)15. Ubiquitin chains can be classified topologically into one of four types according to architecture: homogeneous chains, multiple chains (in which one substrate is separately modified by distinct chains), mixed chains (in which a tandem chain contains two linkage types), and branched chains16,17. Lysine 48 (K48)-linked polyubiquitination induces proteasomal degradation of the target protein, whereas K63-linked polyubiquitination mediates signal transduction16,17. Monoubiquitination, linear polyubiquitination, and K6-, K11-, K27-, K29-, and K33-linked ubiquitination are being investigated intensely to determine their divergent roles in innate immunity15. Similar to conventional PTMs, unconventional PTMs also play a role in innate immune signal transduction8. The transfer of acetyl groups from acetyl coenzyme A (acetyl-CoA) to the ε amino acid groups of lysine residues (a process termed acetylation) results in charge neutralization, which alters the biological properties of proteins; in addition, lysine and arginine residues are inversely regulated by methyltransferases (a process termed methylation) and demethylases, and both acetylation and methylation play important roles in innate immune signaling18. Below we summarize the PTMs and other regulatory mechanisms of signaling molecules downstream of the RNA/DNA sensing cascade (also see Tables 1, 2, and 3).

Schematic representation of positive and negative regulatory host factors of Mitochondrial antiviral signaling protein (MAVS), TNF receptor-associated factor (TRAF3), TANK-binding kinase 1 (TBK1), NF-kappa-B essential modulator (NEMO), and IĸB kinase-ε (IKKε) through posttranslational modifications (PTMs) or other regulatory mechanisms and viral proteins interacting with MAVS, TRAF3, TBK1, NEMO, or IKKε for viral evasion of the host immune response. The RLR-MAVS pathway consists of RIG-I and MDA5 as the main viral RNA sensors and the downstream signaling molecules MAVS and TRAF3, which activate IRF3/IRF7 via the kinases IKK and TBK1/IKKε. (Note: Host factors and viral proteins involved in TBK1 regulation upon infection with both RNA and DNA viruses are indicated as being common regulators in the figure.).

Schematic representation of positive and negative regulatory host factors of 2’,3’-cyclic GMP-AMP (2’,3’-cGAMP), stimulator of interferon gene (STING), Interferon regulatory factor 3 (IRF3), and IRF7 through posttranslational modifications (PTMs) or other modifications and viral proteins interacting with cGAMP or STING for viral evasion of the host immune response. The STING-mediated signaling pathway includes cGAS as the key sensor molecule that is mainly involved in the recognition of viral DNA. This recognition triggers cGAMP production and binding of cGAMP with STING, which leads to activation of IRF3/IRF7 and induction of type 1 IFNs. TBK1, IRF3, and IRF7 are involved in the IFN signaling cascade initiated upon sensing of RNA and DNA viruses. (Note: Host factors and viral proteins involved in IRF3/IRF7 regulation upon infection with both RNA and DNA viruses are indicated as being common regulators in the figure.).
Innate immune evasion strategies used by RNA and DNA viruses
Viruses that have evolved with their host develop strategies to evade the innate immune system and ensure their replication and survival. Individual viruses or virus families use different strategies. This review explores the different mechanisms used by RNA and DNA viruses to subvert the functions of individual signaling molecules in the type 1 interferon (IFN) pathway. Many viruses use proteases to cleave target proteins19, while some viral proteins promote the degradation of target innate immune signaling molecules20,21. Furthermore, viral deubiquitinase enzymes remove K63-linked polyubiquitin chains from signaling molecules to prevent their activation22,23, and viral E3 ubiquitin ligases transfer K48-linked polyubiquitin moieties to target molecules to trigger their proteasomal degradation24. Some viral proteins recruit host E3 ubiquitin ligases to polyubiquitinate signaling molecules and increase their proteasomal degradation25. The formation of signaling molecule complexes is crucial for downstream transduction of innate immune signals. Direct interactions with viral proteins inhibit the formation of signaling complexes such as the TRAF3, TANK, and TBK1 complexes26,27. Another important mechanism of immune evasion is physical interaction between viral proteins and host signaling molecules, which prevents activation, dimerization, phosphorylation, or nuclear translocation28,29. Below, we summarize the mechanisms underlying innate immune evasion mediated by viral proteins (also see Tables 4 and 5).
RNA-induced signal transduction and mechanisms underlying viral evasion of host immunity
RLR (RIG-I-like receptor) family receptors are the main PRRs that detect intracellular viral RNA30,31. The RLR family comprises RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). RIG-I and MDA5 are typical PRRs, whereas LGP2 is a regulator of RIG-I and MDA5 mediated signal transduction32,33. RIG-I and MDA5 contain two N-terminal caspase-recruitment domains34, a central DExD/H-box helicase domain, and a C-terminal domain (CTD). RIG-I and MDA5 bind to viral RNA in the cytoplasm via an RNA binding motif30,31, after which the signaling domain interacts with the downstream adaptor molecule MAVS via a CARD-CARD-mediated interaction. This interaction causes aggregation of MAVS to form a prion-like protein complex, which relays the signal to kinases such as TANK-binding kinase 1 (TBK-1) and IĸB kinase-ε (IKKε). Activation of this cascade results in phosphorylation of the transcription factors IFN-regulating factor (IRF)-3 and IRF-731,35,36. Finally, nuclear translocation of IRF-3 and IRF-7 induces the expression of type 1 IFN genes and other antiviral genes37. However, RNA viruses employ strategies to evade these RLR-mediated innate immune responses. Below, we describe the activation and regulatory mechanisms of the major innate signaling molecules, along with the immunomodulatory mechanisms by which viruses evade them.
Regulation of MAVS by host factors
MAVS, also called IPS1, VISA, and CARDIF, is a key adaptor protein for RIG-I-like receptor-initiated signal transduction. Upon viral infection, RIG-I and MDA5 bind to MAVS, thereby activating downstream signal transduction. The MAVS protein, which contains 540 amino acids encoded by the nuclear genome38, is localized predominantly on the mitochondrial outer membrane. However, experimental evidence shows that it also localizes to mitochondrial-associated endoplasmic reticulum membranes and peroxisomes39,40,41. MAVS contains three domains: a CARD, a middle proline-rich region, and a C-terminal transmembrane42 domain. The CARD interacts with CARDs in RIG-I and MDA5, activating MAVS, whereas the proline-rich region interacts with the tumor necrosis factor receptor-related factor (TRAF) family members TRAF2, TRAF3, TRAF5, and TRAF6 to activate downstream signaling43. The TM domain plays a crucial role by ensuring the localization of MAVS to the mitochondrial outer membrane44. Upon binding to the CARDs of RIG-I and MDA5, MAVS rapidly forms prion-like aggregates, which convert other MAVS proteins present on the mitochondrial outer membrane into prion-like aggregates45. Activation of MAVS through aggregation recruits TRAF2, TRAF3, TRAF5, and TRAF6 via the PRR to promote the formation of the TBK1 complex (comprising TBK1, IĸB kinase, IKKε, and NEMO)46. It is not surprising that the expression of MAVS is regulated to ensure that RLR-mediated signaling cascades are not activated rapidly upon stimulation; indeed, its function at this stage of viral infection is to prevent rapid viral replication.
Self-association and prion-like aggregate formation are markers of MAVS activation45. The E3 ubiquitin ligase Tripartite motif-containing protein (TRIM) 31 interacts with MAVS and catalyzes K63-linked polyubiquitination of aa residues K10, K311, and K461 in MAVS to promote the formation of aggregates. Interestingly, this phenomenon occurs upon viral infection in the presence of RIG-I; thus, recruitment of RIG-I may be required for TRIM31-mediated MAVS aggregation upon viral infection47. Moreover, K63-linked polyubiquitination is enhanced by O-linked N-acetyl glucosamine (O-GlcNAc) transferase (OGT)-mediated glycosylation of MAVS48. Another recent study suggested that K27-linked polyubiquitination of K325 in MAVS by the E3 ubiquitin ligase TRIM21 promotes downstream signaling activation. The PRY-SPRY domain of TRIM21 interacts with MAVS, while the RING (Really Interesting New Gene) domain transfers the E3 ubiquitin protein complex to MAVS, resulting in recruitment of TBK1 to MAVS49. K48-linked ubiquitination of MAVS leads to its proteasomal degradation50; thus, proteins that inhibit MAVS K48-linked ubiquitination are positive regulators of MAVS-mediated signaling. Ovarian tumor family deubiquitinase 4 (OTUD4) removes K48-linked ubiquitin chains from MAVS to inhibit its degradation51. Moreover, the expression of cyclophilin A is upregulated upon viral infection; cyclophilin A competes with TRIM25 for binding to MAVS. Inhibiting TRIM25 promotes MAVS ubiquitination and degradation52,53.
Phosphorylation is an important PTM that regulates MAVS signaling. Activated MAVS recruits TBK1 and IKKε to the complex. These kinases mediate the phosphorylation of MAVS, enabling the recruitment of IRF3. Recruited IRF3 is phosphorylated by TBK1, which increases its homodimerization and nuclear translocation. Similar to PTMs, non-PTMs play a crucial role in regulating MAVS signaling12. Importantly, TRAF3 interacts with MAVS (aa 450–468), resulting in activation of MAVS signaling54. Mitofusin 1 (MFN1) binds to MAVS to increase MAVS redistribution; MFN1 positively regulates the RLR-mediated innate antiviral response55. Furthermore, nucleus accumbens-associated 1 (NAC1), a member of the BTB/POZ family, acts as a bridge between MAVS and TBK1, thereby activating downstream signaling56. In addition, focal adhesion kinase (FAK) interacts with MAVS at the mitochondrial membrane in a viral infection-dependent manner to potentiate MAVS-mediated signaling via a kinase-independent mechanism57.
Negative regulation of MAVS is mediated mainly by K48-linked ubiquitination of MAVS, signaling blockade, autophagy, and apoptosis. K48-linked polyubiquitination of MAVS triggers its proteasomal degradation and abrogates RLR-mediated signal transduction. Experimental evidence has shown that several E3 ubiquitin ligases are involved in K48-linked ubiquitination of MAVS and its proteasomal degradation; these ligases include Ring finger protein 5 (RNF5), RNF115, TRIM25, Smurf1, Smurf2, von Hippel-Lindau protein (pVHL), and membrane-associated RING finger protein 5 (MARCH5)52,58,59,60,61,62,63. Importantly, the ubiquitin thioesterase OTU1 (YOD1) cleaves the K63-linked ubiquitin moiety and abrogates the formation of prion-like aggregates by MAVS, thereby attenuating IRF3-mediated production of IFN-β64. Moreover, interactions between several proteins mediate MAVS ubiquitination and degradation via recruitment of E3 ubiquitin ligases. For example, poly(RC) binding protein 1/2 (PCBP1/PCBP2)- and tax1-binding protein 1 (TAX1PB1)-mediated K48-linked ubiquitination of MAVS via AIP4/ITCH triggers proteasomal degradation of MAVS50,65,66. Similarly, Smurf1-mediated K48-linked ubiquitination is upregulated by OTUD167. The E3 ubiquitin ligase RNF125 conjugates ubiquitin to MAVS, thereby suppressing its function, and K27-linked ubiquitination of MAVS mediated by the E3 ubiquitin ligase MARCH8 recruits the autophagy protein NDP52, resulting in lysosomal degradation of MAVS68,69. Additional mechanisms that negatively regulate MAVS-mediated RLR signaling are phosphorylation and degradation of MAVS via Nemo-like kinase (NLK)70. Protein phosphatase magnesium-dependent 1A (PPM1A; also called PP2Cα) is an inherent component of the TBK1/IKKε complex, which targets both MAVS and TBK1/IKKε for dephosphorylation, thereby disrupting MAVS-driven formation of the signaling complex71.
Direct protein–protein interactions and signal blockade are other mechanisms that downregulate MAVS-mediated RLR signaling. Recent studies have shown that lactate, the end product of anaerobic glycolysis, acts as a negative regulator of RLR signal transduction by interacting with the TM domain of MAVS and preventing its mitochondrial localization and aggregation72. Tubulin tyrosine ligase-like protein 12 (TTLL12) interacts with MAVS, TBK1, and IKKε to prevent interactions between MAVS and other molecules. However, upon viral infection, TTLL12 expression decreases, thereby activating downstream MAVS signaling via the release MAVS blockade73. During the late stage of viral infection, MAVS function is negatively regulated by UBX-domain-containing protein 1 (UBXN1). The expression of UBXN1 increases at the late stage of infection, and it then competes with TRAF3/TRAF6 for binding to MAVS74. Similar to UBXN1, LGP2 binds to MAVS and prevents the interaction between MAVS and IKKε75. Additionally, gpatch domain-containing protein 3 (GPATCH3) binds to MAVS to prevent MAVS/TRAF6/TBK1 complex formation76, whereas binding of polo-like kinase 1 (PLK-1) to MAVS disrupts its interaction with TRAF377. The Rho family small guanosine triphosphatase Ras-related C3 botulinum toxin substrate 1 (Rac1) limits the interaction between MAVS and the E3 ligase TRIM31, thereby inhibiting MAVS ubiquitination, aggregation, and activation78. Moreover, physical interactions between the gC1qR79, mitofusin80, ASC81, and PSMA782 proteins and MAVS subvert MAVS function during viral infection.
Regulation of MAVS by viral proteins
From the perspective of the virus, it is important to avoid the host innate immune response during the early stage of infection. Since MAVS plays a critical role as a central adaptor molecule in the RLR-mediated signaling cascade, the genomes of many viruses encode proteins that interfere with MAVS. For example, enterovirus 71 (EV71) cysteine protease 2Apro cleaves MAVS at Gly209, Gly251, and Gly26583. This was the first viral protein found to cleave MAVS at multiple aa residues. The small RNA viruses human rhinovirus C, coxsackievirus B3 (CVB3), and Seneca Valley virus (SVV) encode a cysteine protease, 3Cpro, which cleaves MAVS at Gln148 to prevent signal transduction84,85,86. In addition, CVB3 encodes another MAVS-cleaving protease named 2Apro; however, its cleavage site is unclear87. Porcine reproductive and respiratory syndrome virus (PRRSV) produces a 3C-like serine protease (3CLSP) that cleaves MAVS at Glu26888. Additionally, NS3-4A of hepatitis C virus (HCV)38,89 and the 3ABC precursor of 3C90 of hepatitis A virus91 cleave MAVS to disrupt activation of its downstream signaling92. The E3 ubiquitin ligase-like activity of rotavirus NSP1 means that its interaction with the MAVS CARD or TM domain leads to ubiquitin-dependent proteasomal degradation of MAVS93. Additionally, the structural protein VP3 of RV upregulates the phosphorylation of MAVS, leading to its K48-linked ubiquitination-mediated proteasomal degradation94. Hepatitis B virus (HBV) protein X (HBX) binds to MAVS and promotes its ubiquitination and proteasomal degradation via an unknown E3 ubiquitin ligase95. Additionally, HBV-induced Parkin recruits the linear ubiquitin assembly complex to mitochondria and abrogates IFN-β synthesis96. Severe acute respiratory syndrome coronavirus (SARS-CoV-2) open reading frame 9b (ORF-9b) catalyzes K48-linked ubiquitination of MAVS via the PCBP2-AIP4 axis97. Moreover, HCV infection induces the expression of Golgi protein 73 (GP73), which mediates the proteasomal degradation of MAVS98. HCV infection upregulates NLRX1 and recruits PCBP2 to MAVS, thereby triggering K48-linked ubiquitination and degradation of MAVS with the help of AIP425. In addition, the interaction between the HCV NS5A protein and MAVS prevents the binding of the latter to TRAF3 and TRAF699. The Nipah virus (NiV) V protein interacts directly with UBXN1 to enhance the interaction between MAVS and UBXN1 via protein stabilization100. A recent study showed that the wild-type VP1 (83E) but not the mutant VP1 (83K) protein of foot and mouth disease (FMDV) subverts MAVS signaling by disrupting the interaction between MAVS and TRAF3101. Moreover, the NS1 and N proteins of respiratory syncytial virus attenuate the production of type I IFNs during infection by inhibiting the MAVS/RIG-I interaction and by localizing MAVS in inclusion bodies, respectively102,103. The human metapneumovirus (hMPV) M2-2 protein prevents recruitment of the MAVS downstream adaptors TRAF3, TRAF5, and TRAF6104. Interestingly, a recent study showed that the M protein of SARS-CoV-2 impairs MAVS aggregation and the recruitment of downstream TRAF3, TBK1, and IRF3105, while another study reported that SARS-CoV-2 M2 inhibits RIG-I/MAVS/TRAF3 and TBK-1 complex formation and subsequent nuclear translocation of IRF326. Viral proteins known to interact with or affect MAVS are listed in Table 4.
Regulation of TRAF3 by host factors and viral proteins
TRAF3 (also called Amn, CAP-1, CD40bp, CRAF1, LAP1, or T-BAM) is one of the most enigmatic, ubiquitously expressed members of the TRAF family. The protein contains 568 amino acids (64.295 kDa) and a typical C3HC4 RING finger domain upstream of five zinc fingers, an isoleucine zipper, and a TRAF3 domain in the C-terminal region. The TRAF domain is critical for binding to the cytoplasmic domain of tumor necrosis factor receptor (TNFR) family members and intracellular signaling mediators and for the formation of homo- or heterodimers106,107,108,109. TRAF3 forms a stable complex with MAVS, which recruits kinases and IRF3 to itself, ultimately leading to IRF3 activation and nuclear translocation110.
The E3 ubiquitin ligases DEAD-box helicase 3 (DDX3)110, cIAP1, cIAP2111, galectin 3 binding protein (LGALS3BP)112, TRIM24113, and TRIM35114 trigger K63-linked polyubiquitination of TRAF3. This modification of TRAF3 enables its association with MAVS and TBK1, which activates downstream antiviral signaling. Moreover, the E3 ubiquitin ligase RING finger protein 166 transfers ubiquitin to TRAF3 upon RNA virus infection, thereby activating IFN-β production115. The serine-threonine kinase CK1ɛ interacts with TRAF3 and phosphorylates it on Ser349, which promotes Lys63 (K63)-linked ubiquitination of TRAF3 and subsequent recruitment of the kinase TBK1 to TRAF3116. Osteopontin (OPN) interacts with TRAF3 to inhibit Triad3A-mediated K48-linked polyubiquitination and degradation of TRAF3117. Downstream of kinase 3 (DOK3) interacts with TRAF3 through its tyrosine-rich CTD to induce TRAF3/TBK1 complex formation118, whereas the interaction between TRAF3 and the GTPase-trafficking protein RAB1B facilitates the formation of the TRAF3/MAVS complex119. As mentioned above, K63-linked polyubiquitination plays a critical role in activating TRAF3. Therefore, the deubiquitinases MYSM1120, DUBA121, USP19122, OTUB1, OTUB2123, UCHL1124, and FOSL1125 remove ubiquitin chains from TRAF3 to negatively regulate its function. In addition, scavenger receptor A (SRA) and HSCARG126 negatively regulate the stability of the TRAF3 protein by promoting recruitment of OTUB1 to TRAF3127. K48-linked polyubiquitination and degradation of TRAF3 mediated by estrogen receptor-alpha (ERα)128, WD repeat domain (WDR) 82129, Parkin130, and Triad3A131 is another mechanism that downregulates IFN production via targeting of TRAF3. Linear-ubiquitinated NEMO associates with TRAF3 and disrupts the MAVS-TRAF3 complex, thereby inhibiting IFN activation132.
Since K63-linked polyubiquitination plays an important role in TRAF3-mediated signaling, it comes as no surprise to see that viruses encode proteins that inhibit TRAF3 ubiquitination to overcome host innate responses. The leader proteinase (Lpro) of FMDV133 and the ubiquitin-specific protease (UL36) of herpes simplex virus 1 (HSV-1)134 act as viral deubiquitinases that mediate TRAF3 deubiquitination, leading to downregulation of TRAF3 signaling. The nonstructural protein 2A protease (2Apro) of human enterovirus D68 (EV-D68) cleaves TRAF3 at G462135. The M protein of SARS-CoV forms a complex with TRAF3, TANK, and the TBK1/IKKε complex to inhibit TBK1/IKKε-dependent activation of the IRF3/IRF7 transcription factors27.
Regulation of NEMO by host factors and viral proteins
NF-κB essential modulator (NEMO or IKKγ), which contains 419 aa, is the integral regulatory scaffolding protein of the canonical IKK complex located at the center of both the NF-κB and type I IFN signaling cascades136. The IKK complex comprises two kinases, IKKα and IKKβ, and a regulatory subunit, NEMO137. For appropriate assembly of the IKK complex, NEMO contains two coiled-coil domains (CC1 and CC2) at its N-terminus upstream of a leucine zipper and a C-terminal zinc finger (ZF) domain. In response to RLR signaling, ubiquitinated TBK1 recruits the adaptor protein NEMO via the ubiquitin binding domain. Assembly of the NEMO/TBK1 complex on MAVS activates the TBK1 kinase and phosphorylation of IRF3138. As NEMO plays a critical role in regulating RLR-mediated IFN signaling, several positive and negative host regulatory factors (as well as viral proteins) play roles in regulating NEMO protein function. TRIM23-mediated K27-linked polyubiquitination of NEMO is crucial for virus-induced IRF3-mediated activation of RLR signaling. TRIM23-mediated ubiquitin conjugation occurs when NEMO K165, K309, K325, K326, and K344 are ectopically expressed139. Moreover, K48-linked polyubiquitination of NEMO mediated by the E3 ubiquitin ligases MARCH2 and TRIM29 leads to its proteasomal-dependent degradation140,141. RUN domain Beclin-1-interacting cysteine-rich-containing (Rubicon) interacts with NEMO and removes conjugated ubiquitin moieties from NEMO, thereby inhibiting its activation and subsequent signal transduction upon viral infection142. Additionally, progranulin (PGRN) is expressed during influenza virus infection; PGRN interacts directly with NEMO and recruits A20 (also called TNFAIP3), which removes K63-linked polyubiquitin chains from K264 of NEMO, resulting in impaired activation of downstream signaling143.
Viruses can escape antiviral immune responses by promoting cleavage or degradation of NEMO. Many viruses encode proteases that cleave NEMO independent of proteasomal degradation or apoptosis to inhibit RLR signaling. For example, 3C90 of FMDV specifically targets NEMO at Gln383, cleaving the C-terminal ZF domain from the protein and impairing the ability of NEMO to activate downstream IFN production144. Additionally, the HAV 3C protease (3Cpro) cleaves NEMO at Q304, thereby abolishing its signaling adaptor function and abrogating the induction of IFN-β synthesis145. NSP4, a viral 3C-like serine protease of PRRSV, cleaves NEMO at E166, E171, and E349–S350, while NSP4 of equine arteritis virus, which is similar to NSP4 of PRRSV, cleaves NEMO at E166, E171, Q205, and E349 to inhibit downstream signaling and maintain viral infection146,147. NSP5 of feline infectious peritonitis virus and NSP5 encoded by porcine epidemic diarrhea virus (PEDV) cleave NEMO at Q132, Q205, Q231, and Q231, resulting in downregulation of immune signaling148,149. Similarly, NSP5 of porcine deltacoronavirus (PDCoV) cleaves NEMO at Q231 to impair the ability of NEMO to activate the IFN response and downstream signaling150. Furthermore, ORF9b of SARS-CoV-2 disrupts K63-linked polyubiquitination of NEMO151, thereby downregulating IFN production during SARS-CoV-2 infection.
Regulation of TBK1 by host factors
TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1, also called NAK or T2K) is one of two noncanonical IKKs implicated in regulating the activation of IRF3/IRF7 and the NF-κB signaling pathway. TBK1 is a 729 aa protein (84 kDa) containing an N-terminal kinase domain (KD), a ubiquitin-like domain (ULD), and two C-terminal coiled-coil domains152. The ULD acts as a regulatory domain by binding to the functional domains of TBK1 as well as to substrates such as IRF3/IRF7, thereby enabling the KD to phosphorylate target substrate proteins. Furthermore, the structure of TBK1 is similar to that of the noncanonical kinase IKKε; indeed, both kinases always work together. Cellular expression of TBK1 is ubiquitous; thus, it plays an indispensable role in antiviral innate immunity. Upon infection with RNA viruses, TBK1 is activated by the upstream protein MAVS, and activated TBK1 recruits IRF3 and IRF7; these proteins undergo TBK1-mediated C-terminal phosphorylation to trigger their dimerization and nuclear translocation, an event followed by induction of IFN secretion153.
As a vital kinase that regulates the activation of IRF3/IRF7 and the subsequent expression of IFN, the function of TBK1 must be regulated to maintain immune homeostasis and suppress viral replication. Therefore, several regulatory factors target TBK1 to control its function, while viruses have evolved mechanisms to disable it. Moreover, TRAF family E3 ubiquitin ligase-mediated K63-linked polyubiquitination of intact dimerized TBK1 at Lys30 and Lys401154 results in transautophosphorylation on Ser172, which marks TBK1 for phosphorylation-mediated activation155. Glycogen synthase kinase 3β (GSK3β) facilitates the aforementioned autophosphorylation of TBK1 at Ser172156. TRIM9 short isoform (TRIM9s) facilitates the recruitment of GSK3β to TBK1 upon viral infection157, and Raf kinase inhibitory protein serves as a positive regulator158; both of these proteins promote autophosphorylation of TBK1. Moreover, ubiquilin 2 (UBQLN2) promotes the stability and facilitates the phosphorylation of TBK1159, and Tyr179 (Y179) phosphorylation (catalyzed by the tyrosine kinase Src) is essential for the initiation of TBK1 autophosphorylation160. Ubiquitination also plays a critical role in the activation of TBK1. Mindbomb E3 ubiquitin-protein ligase 1 (MIB1) and MIB2161, ring finger protein 128 (RNF128)162, and neuregulin receptor degradation protein 1 (Nrdp1/RNF41)163 activate TBK1 by promoting its K63-linked ubiquitination. The deubiquitinase complex comprising ubiquitin-specific peptidase 1 (USP1) and USP1-associated factor 1 (UAF1), binds to TBK1 to remove K48-linked polyubiquitination and reverse the degradation process164. The DNA methyltransferase Dnmt3a maintains high expression of the histone deacetylase HDAC9, which maintains deacetylation of TBK1 and increases its kinase activity165, whereas HDAC3 positively regulates TBK1 in the same manner as HDAC9166. Moreover, butyrophilin 3A1 (BTN3A1) interacts with TBK1 to facilitate its dynein-dependent transport to the perinuclear region to promote its association with IRF3 after viral infection167. IFN-induced protein with tetratricopeptide repeats 3 (IFIT3) mediates the bridging of TBK1 to MAVS on mitochondria168. Additionally, the E3 ubiquitin ligase TRIM26 bridges the interaction between NEMO and TBK1, which facilitates immune activation upon viral infection169. Moreover, the homeobox protein MSX1 and docking protein 3 (DOK3) positively regulate TBK1 function to facilitate complex formation, and PLA1A upregulates TBK1 recruitment to mitochondria via modulation of mitochondrial morphology118,170,171.
In contrast, several TBK1-regulating proteins negatively impact TBK1. K48-linked polyubiquitination of TBK1 induced by E3 ubiquitin ligases such as SOCS box-containing 8 (ASB8)172, TRAF-interacting protein173, dual-specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2)174, and THO complex subunit 7 homolog (THOC7)175 triggers proteasomal degradation of TBK1 and ultimately terminates immune activation. Interestingly, USP38 permits K48-linked ubiquitination and subsequent degradation of TBK1 by specifically removing K33-linked ubiquitin chains from the same lysine site on TBK1176. Additionally, Siglec1 recruits TRIM27 and NLRP4 recruits DTX4 to trigger K48-linked polyubiquitination of TBK1177,178. As noted above, K63-linked polyubiquitination plays a crucial role in activating TBK1. Therefore, any protein that disrupts the ubiquitin chain can be considered a negative regulator. For example, the deubiquitinating enzyme cylindromatosis (CYLD) removes K63-linked polyubiquitin moieties from TBK134, and the A20 regulatory complex (comprising the ubiquitin-editing enzyme A20, Tax1-binding protein 1 (TAX1BP1, also called T6BP or TXBP151)179, and ubiquitin-specific protease (USP) 2b (USP2b)180 antagonize K63-linked polyubiquitination of TBK1. Moreover, UBE2S recruits USP15 to TBK1, thereby removing K63-linked polyubiquitin chains181. The Src family kinases Lck, Hck, and Fgr phosphorylate TBK1 directly at Tyr354/394 to prevent its dimerization and activation182. The ADP-ribosylase TIPARP interacts with TBK1 to suppress its activity via ADP-ribosylation183. The phosphatase Cdc25A dephosphorylates TBK1 at its activation site (S172) upon viral infection184. Moreover, upon infection with RNA viruses, protein phosphatase 1B (PPM1B)185, Cdc25A184, and protein phosphatase 4 (PP4)186 dephosphorylate Ser172 of TBK1 to prevent continuous activation of TBK1. Preventing protein–protein interactions is another method of inhibiting TBK1-driven immune activation. NOD-like receptors (e.g., NLRP2)187 and estrogen-related receptor α (ERRα)188 inhibit the interaction between TBK1 and IRF3, while MIP-T3189 prevents the formation of the TRAF3/TBK1 complex. Additionally, ISG56 disrupts the interaction between MITA and VISA or TBK1, while INKIT interacts with TBK1 to impair the recruitment and phosphorylation of IRF3190,191.
Regulation of TBK1 by RNA viral proteins
TBK1 is targeted by viruses to modulate innate immune activation and ensure viral survival and persistent replication. SARS-CoV-2 virus NSP13192, Heartland virus (HRTV) NS193, and dengue virus (DENV) serotype 4 (DENV4) NS194 proteins interact directly with TBK1 to prevent its autophosphorylation. Papain-like protease domain 2 (PLP2) of mouse hepatitis virus A59 (MHV-A59)195 and the short form leader proteinase (Lpro) Lbpro of FMDV133 cleave ubiquitin chains from TBK1 and inactivate its kinase activity. The Us11 protein of HSV-1 interacts with Hsp90, which competes with TBK1 to disrupt the formation of the TBK1/Hsp90 complex. Us11 subsequently mediates TBK1 destabilization via a proteasome-dependent pathway196. Severe fever with thrombocytopenia syndrome bunyavirus (SFTSV) escapes the host immune system by inducing the formation of cytoplasmic inclusion bodies with the help of NS proteins197,198, whereas the NS protein of SFTSV impairs the autophosphorylation of TBK via a direct interaction199. Moreover, the N protein of PEDV200 and the NS protein of HRTV201 inhibit the TBK1/IRF3 interaction by targeting TBK1 directly, while the NS5 protein of Zika virus antagonizes IFN production by blocking TBK1 activation202. A recent study demonstrated that NSP13 of SARS-CoV-2 interacts directly with the MAVS binding domain of TBK1 and disrupts the TBK1-MAVS interaction203. Membrane-anchored PLpro domain (PLpro-TM) of SARS-CoV inhibits STING/TBK1/IKKε-mediated activation of type I IFNs by disrupting the phosphorylation and dimerization of IRF3204. FLIPs (MC159 and MC160) encoded by molluscum contagiosum virus inhibit TBK1 phosphorylation and activation; however, MC159 interacts directly with TBK1, whereas MC160 does not205. Grass carp reovirus (GCRV) inhibits TBK1 activation by removing K63-linked ubiquitination from TBK1 and promoting its K48-linked ubiquitination24.
Regulation of IKKε by host factors and viral proteins
IKKε (originally called IKKi) is a noncanonical member of the IκB kinase family that has been studied extensively due to its ability to promote type I IFN responses. IKKε is a 716 aa protein comprising a KD, a ULD, and a scaffold dimerization domain. The KD of IKKε shares 49% identity and 65% similarity with that of TBK1206. Activation of TBK1 and IKKε promotes phosphorylation and nuclear translocation of IRF3 and 7, leading to transcriptional upregulation of type I IFNs during the induction of the innate immune response207. During the innate immune response, TBK1 and IKKε exhibit functional redundancy, although TBK1 appears to be more important than IKKε. The IKK subunit NEMO promotes activation of TBK1 and IKKε downstream of cytoplasmic DNA signaling, whereby ubiquitinated NEMO recruits IKKβ to facilitate activation of TBK1 or IKKε.
Biochemical analysis has revealed that the interaction between sphingosine 1-phosphate (S1P) lyase and IKKε leads to IKKε-driven activation of IFN signaling208. Viral infection triggers an interaction between DDX3 and IKKε. Expression of DDX3 amplifies TBK1/IKKε-mediated induction of the IFN-β promoter209. DExD/H-box RNA helicase 19 (DDX19) recruits Lamtor2 to form the TBK1/IKKε/Lamtor2/DDX19/IRF3 complex, which suppresses IFN production by promoting degradation of TBK1 and IKKε210. Fascin1, an actin-bundling protein, interacts with IKKε to suppress the RIG-I-mediated signaling cascade in colon cancer cells211.
To date, few studies have been conducted on viral proteins that interfere with the signaling mechanisms of IKKε. NS2B/3 of DENV interacts directly with IKKε; computational analysis revealed that via this interaction, NS2B/3 masks the KD of IKKε and potentially affects its functionality, thereby impairing the phosphorylation and nuclear translocation of IRF3212. Interestingly, NS2 of HCV interacts physically with the IKKε/TBK1 kinase complex, thereby inhibiting IRF3 phosphorylation213. Moreover, the VP35 protein of Ebola virus (EBOV) interacts with IKKε and TBK1 during the early phase of viral infection; this physical interaction with IKKε further prevents the interaction of IKKε with IRF3, IRF7, and MAVS214. Similarly, lymphocytic choriomeningitis virus (LCMV) NP binds to the KD of IKKε to block its autocatalytic activity and its ability to phosphorylate IRF3215. Additionally, ORF8b of Middle East respiratory syndrome coronavirus (MERS-CoV) inhibits HSP-70-dependent IKKε activation, while NS2 of HCV inhibits IKKε-dependent phosphorylation of IRF3213,216.
Regulation of IRF3 by host factors
IRF3 (also called IIAE7) is a master transcription factor responsible for the induction of innate antiviral immunity. It is a 427 aa (47.219 kDa) protein that is expressed ubiquitously in tissues. IRF3 contains an N-terminal DNA binding domain (DBD) and a C-terminal transactivation domain. After considerable research, TBK1 and IKKε were identified as the kinases responsible for IRF3 phosphorylation at its C-terminus, which facilitates the formation of dimers that are then transported to the nucleus136,217 to form a complex with coactivators of the p300/CBP family and initiate the transcription of target genes, including the gene encoding IFN-β218,219. IRF3 contains an active nuclear localization signal that is recognized by importin-α receptors and results in its transport into the nucleus219,220.
Because IRF3 is crucial for RLR-mediated antiviral immune activation, it is not surprising that IRF3 function is both positively and negatively regulated by host proteins or that viruses have evolved mechanisms to abolish protein expression. The long noncoding RNA (lncRNA) lncLrrc55-AS recruits methylesterase 1 (PME-1) to promote the interaction between PME-1 and the phosphatase PP2A, an inhibitor of IRF3 phosphorylation221. Similarly, IRF1 interacts with IRF3 to augment the activation of IRF3 by blocking the interaction between IRF3 and PP2A222. Heat shock protein family D (Hsp60) member 1 facilitates the phosphorylation and dimerization of IRF3 and increases IFN-β induction induced by SeV infection61. The lysine methyltransferase nuclear receptor-binding SET domain 3 (NSD3) binds directly to the IRF3 C-terminal region through its PWWP1 domain and methylates IRF3 at K366. Monomethylation maintains IRF3 phosphorylation by promoting the dissociation of IRF3 from the protein phosphatase PP1cc, thereby promoting the production of type I IFNs223. The deubiquitinating enzyme USP22 deubiquitinates and stabilizes KPNA2 after viral infection, thereby facilitating efficient nuclear translocation of IRF3224.
Regarding the negative regulation of IRF3-mediated signaling, the E3 ubiquitin ligase interacting protein peptidyl-prolyl cis/trans isomerase, NIMA-interacting 1225, and RBCC protein interacting with PKC1 (RBCK1)226, Ro52/TRIM21227, the HECT domain ubiquitin228 E3 ligase RAUL229, and TRIM26230 catalyze the K48-linked polyubiquitination and subsequent proteasomal degradation of IRF3. Moreover, OTUD1 removes viral infection-induced K6-linked ubiquitin moieties from IRF3, resulting in dissociation of IRF3 from the promoter region of its target genes without affecting its protein stability, dimerization, or nuclear translocation231. IFN-induced transmembrane protein 3 (IFITM3) associates with IRF3 and regulates the homeostasis of IRF3 by mediating its autophagic degradation232. Phosphorylation of IRF3 is the key modification that leads to its activation. Therefore, dephosphorylation of IRF3 via phosphatases such as MAPK phosphatase 5 (MKP5)233 and the serine/threonine phosphatase PP2A234 inactivates IRF3. However, Mst1 associates with IRF3 and phosphorylates IRF3 directly at Thr75 and Thr253, which prevents IRF3 homodimerization, reduces its ability to occupy chromatin, and dampens IRF3-mediated transcriptional responses235. Interestingly, the F-box protein FBXO17 decreases IRF3 dimerization and nuclear translocation by recruiting protein phosphatase 2A (PP2A), resulting in dephosphorylation of IRF3236; research suggests that the DDX5 protein facilitates this process during viral infection237. HDAC4 inhibits TBK1- and IKKε-mediated phosphorylation of IRF3 at Ser386 and Ser396238. Sentrin/SUMO-specific protease 2 (SENP2) causes IRF3 deSUMOylation, K48-linked ubiquitination, and degradation239. DEAD-box polypeptide 56 (DDX56) suppresses the nuclear translocation of IRF3 by disrupting the interaction between IRF3 and the nuclear translocation supporter IOP5240. Rubicon specifically interacts with the IRF association domain (IAD) of IRF3, which prevents dimerization of IRF3241. Human argonaute 2 (AGO2) blocks the association of IRF3 with CBP; however, this interaction does not affect the phosphorylation, nuclear translocation, or DNA binding of IRF3242.
Regulation of IRF3 by RNA viral proteins
Due to genomic constraints, the immunomodulatory efforts of most viruses focus on host targets that are key players in the antiviral response. It is not surprising, therefore, that IRF3 is one of these targets. The NS1 proteins of influenza A virus (IAV)243 and porcine hemagglutinating encephalomyelitis virus (PHEV)244, the phosphoprotein (P) of rabies virus (RABV)245, the PLpro protein (with deubiquitination activity) of SARS-CoV-2, the NSP1β protein of PRRSV228, the N protein of Peste des petits ruminants virus (PPRV)247, and the NSP15 protein of PEDV248 inhibit activation of IRF3 to downregulate nuclear translocation. A recent study reported that open reading frame 6 (ORF6) of SARS-CoV-2 binds to the importin karyopherin α 2 (KPNA2), thereby inhibiting the nuclear translocation of IRF3192; in addition, the ORF6, NSP12, and NSP5 proteins inhibit the nuclear translocation of IRF3 to prevent IFN production249,250,251, while the NSP3/papain-like protease cleaves IRF3 to subvert IFN production252. Moreover, NS5 of Japanese encephalitis virus (JEV) interacts with the nuclear transport proteins KPNA2, KPNA3, and KPNA4, which competitively block the interactions between KPNA3 and KPNA4 and one of their cargo molecules, IRF3253. JEV downregulates IRF3 phosphorylation and nuclear translocation, an effect that became more pronounced when the molar ratio of sfRNA to genomic RNA was increased254. The V protein of Sendai virus (SeV) inhibits IRF3 translocation to the nucleus255, and the 3Cpro protein of SVV degrades IRF3 via its protease activity256. The Npro protein of classical swine fever virus (CSFV)257 and the NSP1 protein of RV258 trigger proteasomal degradation of IRF3. FMDV 3A interacts with DDX56 to inhibit type I IFN production by reducing the phosphorylation of IRF3259. Hantavirus260 oncoprotein Tax of human T-cell leukemia virus type 1 (HTLV-1)261, the NS protein of DENV194, and the M protein of MERS-CoV262 downregulate IRF3 phosphorylation. Moreover, two reports revealed that the ML protein of Thogoto virus (THOV) and the NSP1 protein of RV block the dimerization and subsequent nuclear translocation of IRF3263,264.
Regulation of IRF7 by host factors and RNA viral proteins
IRF7 is a 503 aa (55 kDa) protein containing an N-terminal DBD, an IAD, a nuclear export sequence, an autoinhibitory domain, and a signal response domain composed of key serine residues217,265. Unlike IRF3, IRF7 is not expressed ubiquitously in cells; instead, its expression is induced upon pathogen infection or stimulation. However, it is a master regulator of type I IFN gene expression and IFN-dependent innate immune responses266. IKKε and TBK1 are the major kinases responsible for IRF7 phosphorylation and activation267. Nuclear translocation and accumulation of IRF7 trigger the induction of IFN-β and IFN-α expression268. K63-linked polyubiquitination of IRF7 on lysines 444, 446, and 452, a process that is important for its activation prior to its phosphorylation and nuclear translocation, is triggered by TRAF6269. Research has shown that the regulation of IRF7 activity by several negative regulators maintains immune homeostasis. N-Myc and STAT interactor (Nmi) promote K48-linked ubiquitination of IRF7 and its subsequent proteasome-dependent degradation270, whereas Ro52/TRIM21 mediates its ubiquitination-promoted degradation upon upstream signaling activation271. TRIM28 interacts with the SUMO E2 enzymes to increase the SUMOylation of IRF7. TRIM28-mediated SUMOylation of IRF7 increases during viral infection, resulting in transcriptional repression272. The N-terminal deubiquitinase273 domain of the enzyme A20 interacts physically with IRF7 to reduce its K63-linked ubiquitination and negatively regulate transcriptional function274. Moreover, physical interactions between IRF7 and the IFN-inducible p200 family protein IFI204275, activating transcription factor 4 (ATF4), and HSP70276,277 downregulate IRF7 activity, leading to downregulation of innate immune activation. Different RNA viral proteins inhibit IRF7. VP35 of EBOV increases PIAS1-mediated SUMOylation of IRF7, thereby repressing IFN transcription278. In addition, HCV infection impairs the nuclear translocation of IRF-7279. The Zn-binding domain of the CSFV Npro protein interacts directly with IRF7 to subvert its function280. In particular, 3Cpro of SVV was found to reduce IRF7 protein expression and phosphorylation in PK-15 cells256.
DNA virus-induced signal transduction and immune evasion mechanisms
Upon infection with DNA viruses, viral DNA is released into the host cell cytoplasm prior to viral protein synthesis. Cytosolic viral DNA is recognized mainly by cyclic GMP-AMP (cGAMP) synthase (cGAS), which contains a nucleotidyltransferase (NTase) domain. After DNA binding, cGAS synthesizes a second messenger molecule, cyclic GMP-AMP (cGAMP). This cGAMP isomer, called 2’,3’-cGAMP, functions as a second messenger that binds to the ER membrane adaptor STING281,282,283 to induce a conformational change that presumably results in activation of STING. STING then traffics from the ER to the ER-Golgi intermediate compartment and then to the Golgi apparatus284,285. During this process, the carboxyl terminus of STING recruits and activates the kinase TBK1, which in turn phosphorylates the transcription factor IRF3. Phosphorylated IRF3 dimerizes and then enters the nucleus, ultimately leading to the induction of type 1 IFN genes and other antiviral genes286. Although other proteins, such as IFI16, DDX41, and MRE11, also mediate DNA-induced IFN-β production in a STING-dependent manner, only cGAS, which enzymatically generates cGAMP as a second messenger that activates STING, provides a clear molecular mechanism for DNA-stimulated IFN-β production287. However, DNA viruses exploit strategies to evade innate immune responses. Below, we describe the activation and regulation of these mechanisms, along with the immunomodulatory mechanisms by which viruses evade them.
Regulation of 2′,3′-cGAMP by host factors and viral proteins
Upon DNA recognition, cGAS generates the second messenger 2′,3′-cyclic GMP-AMP (2′,3′-cGAMP) by using ATP and GTP284,288. Unlike the secondary messengers in classical bacterial signaling (c-di-GMP and c-di-AMP), 2′,3′-cGAMP contains mixed phosphodiester bonds (G(2′,5′)pA and A(3′,5′)pG)282,289. The intermediate product, called 5′-pppG(2′,5′)pA, is generated by cGAS prior to synthesis of cyclic 2′,3′-cGAMP42. Next, 2′,3′-cGAMP interacts with STING to activate downstream signaling, resulting in strong induction of IFNs, which confer antiviral efficacy288. To date, few studies have examined host factors and viral proteins that regulate 2′,3′-cGAMP function during innate immune activation. A recent study of HSV-1 infection showed that Leucine-rich repeat-containing protein (LRRC) LRRC8A/LRRC8E-containing volume-regulated anion channels transport cGAMP across the plasma membrane to initiate effective antiviral innate immunity290. In contrast, 2′,3′-cGAMP is hydrolyzed predominantly by ectonucleotide pyrophosphatase/phosphodiesterase (ENPP1), thereby preventing STING activation. In general, viruses have evolved mechanisms to antagonize host innate immune activation291,292. However, the antiviral second messenger 2′,3′-cGAMP can be packaged into viral particles, including those of poxviruses, herpesviruses, and retroviruses, thereby enabling its transfer to newly infected cells, where it activates the immune response. Once 2′,3′-cGAMP-carrying virions infect neighboring cells, they activate a STING-dependent antiviral program293,294. Moreover, the poxvirus immune nuclease (poxin) family, a family of 2′,3′-cGAMP-degrading enzymes, has been identified. Vaccinia virus poxin degrades 2′,3′-cGAMP through metal-independent cleavage of the 3′-5′ bond, thereby converting 2′,3′-cGAMP into linear Gp[2′-5′]Ap[3′]. Furthermore, the same study revealed that deletion of the poxin gene (B2R) attenuates vaccinia virus replication in vivo, thereby restricting STING-dependent signaling295.
Regulation of STING by host factors
STING, also called MITA, ERIS, TMEM173, or MPYS, is an ER membrane signaling283 protein of 379 aa; it harbors a predicted TM portion (aa residues 1–173) at the N-terminus, which regulates its cellular localization and homodimerization, since the TM domains cross the ER membrane. It also harbors an intracellular soluble portion (aa residues 174–379) in the CTD, which functions to dock downstream molecules such as TBK1/IKKε and IRF3/IRF7296,297. To initiate signaling, the native ligand cGAMP binds to the V-shaped hydrophilic pocket in the STING dimer. The resulting conformational change exposes the hidden CTT of STING to TBK1 and IRF3298,299. Due to this conformational change, STING is transported from the ER to the ER-Golgi intermediate compartment and then to the Golgi apparatus and perinuclear region300.
Since STING is essential for innate immune responses to cytosolic nucleic acids, its activity is tightly regulated to maintain immune homeostasis while enabling timely activation of downstream signaling to fight against viral infections. Several PTMs are involved in regulating STING function. Among them, K63-linked polyubiquitination plays a critical activating role. Mitochondrial E3 ubiquitin protein ligase 1 (MUL1) catalyzes K63-linked polyubiquitination of STING at K224 to transport TBK1 to IRF3. The ubiquitination-deficient STING mutant K224R fails to translocate to perinuclear puncta in response to a stimulus, suggesting that K63-linked polyubiquitination of STING at K224 is essential for STING trafficking301. The E3 ubiquitin ligases TRAF6302, ubiquitin regulatory X domain-containing protein-3B (UBXN3B)303, and RNF11559 also conjugate K63-linked polyubiquitin chains to STING, thereby strengthening its interaction with IRF3 and TBK1. The E3 ubiquitin ligase complex AMFR and insulin-induced gene 1 (INSIG1) catalyze K27-linked polyubiquitination of STING. This modification acts as an anchoring platform for recruitment of TBK1, thereby facilitating its translocation to perinuclear microsomes304. K48-linked polyubiquitination is one of the main negative regulatory mechanisms of cellular STING protein expression. Therefore, any factor that disrupts the K48-linked polyubiquitin chain may activate signal transduction. The DUBs USP20/USP18305,306, USP44307, CYLD308, OTUD5309, and iRhom2310 remove K48-linked polyubiquitin chains from STING and ultimately boost innate antiviral responses. Palmitoylation plays an important role in regulating protein transport, stability, and cellular localization in host cells. Palmitoylation of STING occurs after its trafficking to the Golgi apparatus; this PTM is essential for activation of STING. Moreover, the palmitoylation inhibitor 2-bromopalmitate (2-BP) impairs STING-mediated IFN induction311,312. Phosphorylation of STING by TBK1 at S366 promotes the recruitment and activation of IRF3313. Moreover, S358 of STING is also phosphorylated, although the kinase responsible is not known314. Interestingly, upon DNA virus infection, the tyrosine kinase CSK phosphorylates STING at Y240 and Y245, which is important for its activation315. The ER-associated proteins ZDHHC1 and transmembrane emp24 protein transport domain-containing 2 (TMED2) associate with STING and mediate its dimerization/aggregation; they also facilitate its trafficking315,316. SNX8 recruits the class III phosphatidylinositol 3-kinase protein VPS34 to STING, thereby facilitating the trafficking of STING from the ER to perinuclear microsomes317.
With respect to the negative regulation of STING, RNF5 impairs STING signaling by modifying it at K150 through K48-linked polyubiquitination, which promotes its degradation318. RNF90 and TRIM29 also promote K48-linked ubiquitination of STING and impair STING signaling319,320; however, the specific aa residue1 that is ubiquitinated is not defined. Moreover, TRIM30α negatively regulates the STING pathway via K48-linked ubiquitination of STING on K275321. In contrast, the DUB USP21 hydrolyzes K27/63-linked polyubiquitin chains322, USP49323 removes K63-linked polyubiquitin chains, and USP13324 removes K33-linked polyubiquitin chains on STING to negatively regulate STING-mediated signaling. PPM1A dephosphorylates STING at S358 and suppresses the formation of perinuclear puncta, thereby suppressing immune responses314. Phosphorylation of Y245 on STING is critical for STING activation. PTPN1 and PTPN2 dephosphorylate STING at Y245 and then promote its degradation via the 20 S proteasome325. Additionally, MRP326 and NLRX1327 interact with STING to downregulate its function, while RIG-I and IL-6 trigger proteasomal degradation of STING in human diploid cells upon dsDNA stimulation328. Autophagy-related gene 9a (Atg9a) colocalizes with STING to disrupt the binding of STING to TBK1329.
Regulation of STING by viral proteins
STING plays a critical role in the host defense against infections with DNA viruses such as HSV-1, vaccinia virus (VVΔE3L), cytomegalovirus (CMV), and baculoviruses330. Therefore, viruses have evolved certain strategies to defeat host innate immunity by antagonizing STING signaling. For example, the ICP27331 protein of HSV is translocated to the cytoplasm during infection, where it interacts with STING and inhibits IRF3 activation. The HSV-1 γ34.5 protein downregulates STING trafficking from the ER to the Golgi by interacting with the N-terminus of STING332, while UL46 of HSV-1, one of the most abundant HSV tegument proteins, interacts with STING to prevent its activation333. The HSV-1 VP1-2 protein deubiquitinates STING and inhibits its downstream signaling334. The human T lymphotropic virus type 1 (HTLV-1) Tax protein also deubiquitinates STING to inhibit its downstream signaling22, while NS4B of HCV cleaves STING directly335, and vIRF1 of KSHV impairs the STING/TBK1 interaction336. Murine CMV (MCMV) encodes a product referred to as M152, which interacts with STING to suppress its activation337. The viral polymerase of HBV interferes with K63-linked polyubiquitination of STING via its reverse transcriptase domain338. The HCMV tegument protein UL82 negatively regulates STING signaling by interacting directly with STING. It then inhibits STING trafficking from the ER to perinuclear punctate structures339. The IE86 protein of HCMV facilitates proteasome-dependent degradation of STING to suppress the secretion of IFN-β1 and CXCL10340,341, and UL42 of HCMV impairs the translocation of STING from the ER to perinuclear punctate structures, which is required for STING activation342. Duck Tembusu virus (DTMUV) NS2B3 cleaves STING by interacting with aa residues 221–225; this method of STING cleavage is not strictly species-specific343.
Regulation of TBK1 by DNA viral proteins
To complete their life cycles in the host, DNA viruses use numerous strategies to evade host immune signaling initiated by RLRs; they do this by targeting TBK1. The Us11 protein of HSV-1 associates with endogenous Hsp90 to disrupt the Hsp90/TBK1 complex, which blocks TBK1 activation. Furthermore, Us11 induces destabilization of TBK1 through a proteasome-dependent pathway that ultimately blocks phosphorylation of IRF 3196. In addition, the UL46 protein of HSV-1 interacts with the C-terminal region of TBK1 to inhibit the interaction of TBK1 and STING333, whereas the gamma(1)34.5 protein forms a complex with TBK1 and disrupts the TBK1/IRF3 interaction, thereby preventing downstream signaling344. ORF11 of murine gammaherpesvirus 68 (MHV-68) interacts directly with TBK1; in particular, it inhibits the TBK1/IRF3 interaction345. The C-terminus and the coiled-coil domain of feline panleukopenia virus (FPV) NS2 interact physically with TBK1, thereby preventing it from being recruited by STING; ultimately, this disrupts the phosphorylation of the downstream protein IRF3346.
Regulation of IRF3 by DNA viral proteins
A number of DNA viral proteins inhibit IRF3 to suppress innate immune signaling. The VP24 protein of HSV-1 and the LANA2 (also called vIRF3) protein of Kaposi’s sarcoma-associated herpesvirus (KHSV) limit the induction of IFN-β by interacting with IRF3 to inhibit its dimerization and phosphorylation29,347. The ICP0 protein (bICP0) encoded by bovine herpesvirus 1 (BoHV-1) induces proteasomal degradation of IRF3 but not IRF7348. Varicella-zoster virus (VZV) is an important alpha herpesvirus that infects only humans. Several VZV viral proteins interfere with IRF3 activity. VZV viral immediate-early protein 62 (IE62) inhibits IRF3 phosphorylation at key serine residues but does not interfere with the IRF3/TBK1 interaction349. ORF47 interacts directly with IRF3, thereby inhibiting subsequent signal transduction, while ORF61 interacts directly with IRF3 and induces its ubiquitination and proteasomal degradation350,351. The nuclear early protein N2 of vaccinia virus inhibits the phosphorylation and nuclear translocation of IRF3351.
Regulation of IRF7 by DNA viral proteins
Different viral proteins inhibit and activate IRF7. The interaction of the Epstein-Barr virus oncoprotein LMP1 with IRF7 catalyzes RIP-dependent K63-linked polyubiquitination and subsequent activation of IRF7352. The VP23 protein of Marek’s disease virus interacts with IRF7 and blocks its binding to TBK1, thereby inhibiting IRF7 phosphorylation and nuclear translocation, resulting in reduced IFN-β production353. The immediate-early nuclear transcription factor RTA encoded by KSHV and HHV8 acts as an ubiquitin E3 ligase to catalyze the polyubiquitination and proteasomal degradation of IRF7354. KSHV vIRF3 interacts specifically with either the DBD or the central IAD of IRF7, which inhibits the DNA binding activity of IRF7355. KSHV vIRF4 interacts specifically with IRF7, thereby inhibiting IRF7 dimerization and ultimately suppressing IRF7-mediated activation of type I IFNs356. LANA2 (also called vIRF3) of KSHV limits the induction of IFN-β by interacting with IRF7 and inhibiting its phosphorylation29.
Conclusions
Over the past few decades, tremendous research progress has been made in identifying and characterizing two antiviral innate immunity pathways: the RLR-MAVS pathway for cytoplasmic RNA sensing and the cGAS-cGAMP-STING pathway for cytosolic DNA recognition. In this review, we summarize the current knowledge of the mechanisms that positively and negatively regulate PRR-mediated immune responses. We also discuss the molecules involved in the two abovementioned signaling pathways, which maintain immune homeostasis to achieve the most favorable outcome for the host. Finally, we explain how viral proteins adapt to escape host antiviral mechanisms to maintain active infection.
Due to advanced biomedical techniques such as fluorescence imaging, mass spectrometry, and nuclear magnetic resonance imaging, we now know much more about the molecular mechanisms and the host and viral factors that regulate signaling. Moreover, each new regulatory and molecular mechanism identified brings the inspiring possibility that we may identify and develop novel immunostimulatory agents, anti-inflammatory agents, vaccines, and antiviral agents that tilt the host-pathogen interaction in favor of the host. Despite tremendous advances in our knowledge regarding the functions and mechanisms of positive and negative regulatory molecules and of escape mechanisms used by viruses to evade innate immune signaling, several intriguing and important aspects regarding the regulation of RNA- and (especially) DNA-initiated signaling pathways and viral escape mechanisms remain elusive. These will be interesting topics for future investigations.
References
Dawson, A. R., Wilson, G. M., Coon, J. J. & Mehle, A. Post-translation regulation of influenza virus replication. Front. Microbiol. 11, 517461 (2020).
Lee, H. C., Chathuranga, K. & Lee, J. S. Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 51, 1–13 (2019).
Barrat, F. J., Elkon, K. B. & Fitzgerald, K. A. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 67, 323–336 (2016).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Sin, W. X., Li, P., Yeong, J. P. & Chin, K. C. Activation and regulation of interferon-β in immune responses. Immunol. Res. 53, 25–40 (2012).
Zhou, Y., He, C., Wang, L. & Ge, B. Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 47, 1414–1426 (2017).
Deribe, Y. L., Pawson, T. & Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010).
Mowen, K. A. & David, M. Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 15, 512–520 (2014).
Mogensen, T. H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273 (2009). Table of Contents.
Chan, Y. K. & Gack, M. U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373 (2016).
Liu, J., Qian, C. & Cao, X. Post-translational modification control of innate immunity. Immunity 45, 15–30 (2016).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Pawson, T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 (2004).
Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 3, 20–26 (2002).
Jiang, X. & Chen, Z. J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 12, 35–48 (2011).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012).
Yau, R. & Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 (2016).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Agbowuro, A. A., Huston, W. M., Gamble, A. B. & Tyndall, J. D. A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 38, 1295–1331 (2018).
Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67 (2010).
Ding, B. et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe 21, 538–547.e534 (2017).
Wang, J., Yang, S., Liu, L., Wang, H. & Yang, B. HTLV-1 Tax impairs K63-linked ubiquitination of STING to evade host innate immunity. Virus Res 232, 13–21 (2017).
Frias-Staheli, N. et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2, 404–416 (2007).
Rao, Y., Ji, J., Liao, Z., Su, H. & Su, J. GCRV hijacks TBK1 to evade IRF7-mediated antiviral immune responses in grass carp Ctenopharyngodon idella. Fish. Shellfish Immunol. 93, 492–499 (2019).
Qin, Y. et al. NLRX1 mediates MAVS degradation to attenuate the hepatitis C virus-induced innate immune response through PCBP2. J. Virol. 91, e01264–17 (2017).
Zheng, Y. et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 5, 299 (2020).
Siu, K. L. et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 284, 16202–16209 (2009).
García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 22, 176–184 (2017).
Lubyova, B., Kellum, M. J., Frisancho, A. J. & Pitha, P. M. Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 279, 7643–7654 (2004).
Yoneyama, M. & Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227, 54–65 (2009).
Loo, Y. M. & Gale, M. Jr. Immune signaling by RIG-I-like receptors. Immunity 34, 680–692 (2011).
Bruns, A. M., Leser, G. P., Lamb, R. A. & Horvath, C. M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55, 771–781 (2014).
Yoneyama, M. et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858 (2005).
Friedman, C. S. et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 (2008).
Xu, L. G. et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19, 727–740 (2005).
Kawai, T. et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005).
Negishi, H., Taniguchi, T. & Yanai, H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb. Perspect. Biol. 10, a028423 (2018).
Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 (2005).
Horner, S. M., Liu, H. M., Park, H. S., Briley, J. & Gale, M. Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl Acad. Sci. USA 108, 14590–14595 (2011).
Bender, S. et al. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog. 11, e1005264 (2015).
Dixit, E. et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681 (2010).
Gao, P. et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Liu, S. et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2, e00785 (2013).
Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682 (2005).
Hou, F. et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461 (2011).
Oshiumi, H. et al. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8, 496–509 (2010).
Liu, B. et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224 (2017).
Li, T. et al. O-GlcNAc transferase links glucose metabolism to MAVS-mediated antiviral innate immunity. Cell Host Microbe 24, 791–803.e796 (2018).
Xue, B. et al. TRIM21 promotes innate immune response to RNA viral infection through Lys27-linked polyubiquitination of MAVS. J. Virol. 92, e00321–18 (2018).
You, F. et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 10, 1300–1308 (2009).
Liuyu, T. et al. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 29, 67–79 (2019).
Castanier, C. et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10, 44 (2012).
Liu, W. et al. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. Elife 6, e24425 (2017).
Paz, S. et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res 21, 895–910 (2011).
Onoguchi, K. et al. Virus-infection or 5’ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 6, e1001012 (2010).
Xia, Z. et al. NAC1 potentiates cellular antiviral signaling by bridging MAVS and TBK1. J. Immunol. 203, 1001–1011 (2019).
Bozym, R. A. et al. Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell Host Microbe 11, 153–166 (2012).
Song, T. et al. c-Abl tyrosine kinase interacts with MAVS and regulates innate immune response. FEBS Lett. 584, 33–38 (2010).
Zhang, Z. D. et al. RNF115 plays dual roles in innate antiviral responses by catalyzing distinct ubiquitination of MAVS and MITA. Nat. Commun. 11, 5536 (2020).
Wang, Y., Tong, X. & Ye, X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 189, 5304–5313 (2012).
Lin, L. et al. HSPD1 interacts with IRF3 to facilitate interferon-beta induction. PLoS ONE 9, e114874 (2014).
Du, J. et al. pVHL Negatively Regulates Antiviral Signaling by Targeting MAVS for Proteasomal Degradation. J. Immunol. 195, 1782–1790 (2015).
Yoo, Y. S. et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 6, 7910 (2015).
Liu, C. et al. The otubain YOD1 suppresses aggregation and activation of the signaling adaptor MAVS through Lys63-linked deubiquitination. J. Immunol. 202, 2957–2970 (2019).
Zhou, X., You, F., Chen, H. & Jiang, Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res 22, 717–727 (2012).
Choi, Y. B., Shembade, N., Parvatiyar, K., Balachandran, S. & Harhaj, E. W. TAX1BP1 restrains virus-induced apoptosis by facilitating itch-mediated degradation of the mitochondrial adaptor MAVS. Mol. Cell. Biol. 37, e00422 (2017).
Zhang, L. et al. Induction of OTUD1 by RNA viruses potently inhibits innate immune responses by promoting degradation of the MAVS/TRAF3/TRAF6 signalosome. PLoS Pathog. 14, e1007067 (2018).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl Acad. Sci. USA 104, 7500–7505 (2007).
Jin, S. et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 68, 308–322.e304 (2017).
Li, S. Z. et al. Phosphorylation of MAVS/VISA by nemo-like kinase (NLK) for degradation regulates the antiviral innate immune response. Nat. Commun. 10, 3233 (2019).
Xiang, W. et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci. Adv. 2, e1501889 (2016).
Zhang, W. et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 178, 176–189.e15 (2019).
Ju, L. G. et al. TTLL12 inhibits the activation of cellular antiviral signaling through interaction with VISA/MAVS. J. Immunol. 198, 1274–1284 (2017).
Wang, P. et al. UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Rep. 3, 1057–1070 (2013).
Komuro, A. & Horvath, C. M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 80, 12332–12342 (2006).
Nie, Y. et al. GPATCH3 negatively regulates RLR-mediated innate antiviral responses by disrupting the assembly of VISA signalosome. PLoS Pathog. 13, e1006328 (2017).
Vitour, D. et al. Polo-like kinase 1 (PLK1) regulates interferon (IFN) induction by MAVS. J. Biol. Chem. 284, 21797–21809 (2009).
Yang, S. et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 5, eaav7999 (2019).
Xu, L., Xiao, N., Liu, F., Ren, H. & Gu, J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl Acad. Sci. USA 106, 1530–1535 (2009).
Yasukawa, K. et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2, ra47 (2009).
Han, Y. et al. Negative regulation of MAVS-mediated innate immune response by ASC. Mol. Cell. Biochem. 445, 35–43 (2018).
Jia, Y. et al. Negative regulation of MAVS-mediated innate immune response by PSMA7. J. Immunol. 183, 4241–4248 (2009).
Wang, B. et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9, e1003231 (2013).
Pang, L. L. et al. The suppression of innate immune response by human rhinovirus C. Biochem. Biophys. Res. Commun. 490, 22–28 (2017).
Mukherjee, A. et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7, e1001311 (2011).
Qian, S. et al. Seneca valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J. Virol. 91, e00823–17 (2017).
Feng, Q. et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 88, 3369–3378 (2014).
Dong, J. et al. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signalling complex to antagonize IFN-β expression. J. Gen. Virol. 96, 3049–3058 (2015).
Li, X. D., Sun, L., Seth, R. B., Pineda, G. & Chen, Z. J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl Acad. Sci. USA 102, 17717–17722 (2005).
Di Sabatino, A. et al. Functional modulation of Crohn’s disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology 133, 137–149 (2007).
Uma, B. & Parvathavarthini, R. Antibacterial effect of hexane extract of sea urchin, Temnopleurus alexandri (Bell, 1884). Int. J. PharmTech Res. 2, 1677–1680 (2010).
Yang, Y. et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl Acad. Sci. USA 104, 7253–7258 (2007).
Nandi, S. et al. MAVS protein is attenuated by rotavirus nonstructural protein 1. PLoS ONE 9, e92126 (2014).
Ding, S. et al. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. Elife 7, e39494 (2018).
Wei, C. et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 185, 1158–1168 (2010).
Khan, M., Syed, G. H., Kim, S. J. & Siddiqui, A. Hepatitis B virus-induced parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 12, e1005693 (2016).
Shi, C. S. et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089 (2014).
Zhang, X. et al. GP73 represses host innate immune response to promote virus replication by facilitating MAVS and TRAF6 degradation. PLoS Pathog. 13, e1006321 (2017).
Refolo, G. et al. Negative regulation of mitochondrial antiviral signaling protein-mediated antiviral signaling by the mitochondrial protein LRPPRC during hepatitis C virus infection. Hepatology 69, 34–50 (2019).
Uchida, S., Horie, R., Sato, H., Kai, C. & Yoneda, M. Possible role of the Nipah virus V protein in the regulation of the interferon beta induction by interacting with UBX domain-containing protein1. Sci. Rep. 8, 7682 (2018).
Ekanayaka, P. et al. Foot-and-mouth disease virus VP1 target the MAVS to inhibit type-I interferon signaling and VP1 E83K mutation results in virus attenuation. PLoS Pathog. 16, e1009057 (2020).
Boyapalle, S. et al. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 7, e29386 (2012).
Lifland, A. W. et al. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 86, 8245–8258 (2012).
Chen, Y. et al. Functional motifs responsible for human metapneumovirus M2-2-mediated innate immune evasion. Virology 499, 361–368 (2016).
Fu, Y. Z. et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell. Mol. Immunol. 18, 613–620 (2020).
Arch, R. H., Gedrich, R. W. & Thompson, C. B. Tumor necrosis factor receptor-associated factors (TRAFs)-a family of adapter proteins that regulates life and death. Genes Dev. 12, 2821–2830 (1998).
Chung, J. Y., Park, Y. C., Ye, H. & Wu, H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci. 115, 679–688 (2002).
Dempsey, P. W., Doyle, S. E., He, J. Q. & Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 14, 193–209 (2003).
Inoue, J. et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp. Cell Res. 254, 14–24 (2000).
Gu, L., Fullam, A., McCormack, N., Höhn, Y. & Schröder, M. DDX3 directly regulates TRAF3 ubiquitination and acts as a scaffold to co-ordinate assembly of signalling complexes downstream from MAVS. Biochem. J. 474, 571–587 (2017).
Mao, A. P. et al. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 285, 9470–9476 (2010).
Xu, G. et al. Inducible LGALS3BP/90K activates antiviral innate immune responses by targeting TRAF6 and TRAF3 complex. PLoS Pathog. 15, e1008002 (2019).
Zhu, Q. et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med 217, e20192083 (2020).
Sun, N. et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 11, 894–914 (2020).
Chen, H. W. et al. Ring finger protein 166 potentiates RNA virus-induced interferon-β production via enhancing the ubiquitination of TRAF3 and TRAF6. Sci. Rep. 5, 14770 (2015).
Zhou, Y. et al. The kinase CK1ɛ controls the antiviral immune response by phosphorylating the signaling adaptor TRAF3. Nat. Immunol. 17, 397–405 (2016).
Zhao, K. et al. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci. Rep. 6, 23771 (2016).
Kim, S. S. et al. DOK3 is required for IFN-β production by enabling TRAF3/TBK1 complex formation and IRF3 activation. J. Immunol. 193, 840–848 (2014).
Beachboard, D. C. et al. The small GTPase RAB1B promotes antiviral innate immunity by interacting with TNF receptor-associated factor 3 (TRAF3). J. Biol. Chem. 294, 14231–14240 (2019).
Panda, S., Nilsson, J. A. & Gekara, N. O. Deubiquitinase MYSM1 regulates innate immunity through inactivation of TRAF3 and TRAF6 complexes. Immunity. 43, 647–659 (2015).
Kayagaki, N. et al. DUBA: a deubiquitinase that regulates type I interferon production. Science 318, 1628–1632 (2007).
Gu, Z., Shi, W., Zhang, L., Hu, Z. & Xu, C. USP19 suppresses cellular type I interferon signaling by targeting TRAF3 for deubiquitination. Future Microbiol 12, 767–779 (2017).
Li, S. et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 285, 4291–4297 (2010).
Karim, R. et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 9, e1003384 (2013).
Cai, B., Wu, J., Yu, X., Su, X. Z. & Wang, R. F. FOSL1 inhibits Type I interferon responses to malaria and viral infections by blocking TBK1 and TRAF3/TRIF interactions. MBio 8, e02161–16 (2017).
Peng, Y., Xu, R. & Zheng, X. HSCARG negatively regulates the cellular antiviral RIG-I like receptor signaling pathway by inhibiting TRAF3 ubiquitination via recruiting OTUB1. PLoS Pathog. 10, e1004041 (2014).
Xie, M. et al. Scavenger receptor A impairs interferon response to HBV infection by limiting TRAF3 ubiquitination through recruiting OTUB1. FEBS J. 287, 310–324 (2020).
Wang, C., Huang, Y., Sheng, J., Huang, H. & Zhou, J. Estrogen receptor alpha inhibits RLR-mediated immune response via ubiquitinating TRAF3. Cell. Signal. 27, 1977–1983 (2015).
Zhu, K. et al. WDR82 negatively regulates cellular antiviral response by mediating TRAF3 polyubiquitination in multiple cell lines. J. Immunol. 195, 5358–5366 (2015).
Xin, D., Gu, H., Liu, E. & Sun, Q. Parkin negatively regulates the antiviral signaling pathway by targeting TRAF3 for degradation. J. Biol. Chem. 293, 11996–12010 (2018).
Nakhaei, P. et al. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 5, e1000650 (2009).
Belgnaoui, S. M. et al. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 12, 211–222 (2012).
Wang, D. et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85, 3758–3766 (2011).
Wang, S., Wang, K., Li, J. & Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 87, 11851–11860 (2013).
Kang, J. et al. Enterovirus D68 protease 2A(pro) targets TRAF3 to subvert host innate immune responses. J. Virol. 95, e01856–20 (2021).
Zhao, T. et al. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 (2007).
Häcker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13 (2006).
Wang, L., Li, S. & Dorf, M. E. NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS ONE 7, e43756 (2012).
Arimoto, K. et al. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl Acad. Sci. USA 107, 15856–15861 (2010).
Chathuranga, K. et al. Negative regulation of NEMO signaling by the ubiquitin E3 ligase MARCH2. EMBO J. 39, e105139 (2020).
Xing, J. et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 17, 1373–1380 (2016).
Wan, Y. et al. Inducible Rubicon facilitates viral replication by antagonizing interferon production. Cell. Mol. Immunol. 14, 607–620 (2017).
Wei, F. et al. Induction of PGRN by influenza virus inhibits the antiviral immune responses through downregulation of type I interferons signaling. PLoS Pathog. 15, e1008062 (2019).
Wang, D. et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86, 9311–9322 (2012).
Wang, D. et al. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J. Virol. 88, 10252–10258 (2014).
Huang, C. et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J. Virol. 88, 10934–10945 (2014).
Chen, J. et al. Arterivirus nsp4 antagonizes interferon beta production by proteolytically cleaving NEMO at multiple sites. J. Virol. 93, e00385–19 (2019).
Chen, S. et al. Feline infectious peritonitis virus Nsp5 inhibits type I interferon production by cleaving NEMO at multiple sites. Viruses 12, 43 (2019).
Wang, D. et al. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J. Virol. 90, 2090–2101 (2016).
Zhu, X. et al. Porcine deltacoronavirus nsp5 inhibits interferon-β production through the cleavage of NEMO. Virology 502, 33–38 (2017).
Wu, J. et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 34, 108761 (2021).
Pomerantz, J. L. & Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18, 6694–6704 (1999).
Takeuchi, O. & Akira, S. Innate immunity to virus infection. Immunol. Rev. 227, 75–86 (2009).
Tu, D. et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3, 747–758 (2013).
Ma, X. et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl Acad. Sci. USA 109, 9378–9383 (2012).
Lei, C. Q. et al. Glycogen synthase kinase 3β regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity 33, 878–889 (2010).
Qin, Y. et al. TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3β to TBK1. Cell Res. 26, 613–628 (2016).
Gu, M. et al. RKIP and TBK1 form a positive feedback loop to promote type I interferon production in innate immunity. EMBO J. 35, 2553–2565 (2016).
Chen, T., Zhang, W., Huang, B., Chen, X. & Huang, C. UBQLN2 Promotes the production of type I interferon via the TBK1-IRF3 PAthway. Cells 9, 1205 (2020).
Li, X. et al. The tyrosine kinase Src promotes phosphorylation of the kinase TBK1 to facilitate type I interferon production after viral infection. Sci. Signal. 10, aae0435 (2017).
Li, S., Wang, L., Berman, M., Kong, Y. Y. & Dorf, M. E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 (2011).
Song, G. et al. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat. Immunol. 17, 1342–1351 (2016).
Wang, C. et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 (2009).
Yu, Z. et al. USP1-UAF1 deubiquitinase complex stabilizes TBK1 and enhances antiviral responses. J. Exp. Med. 214, 3553–3563 (2017).
Li, X. et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 17, 806–815 (2016).
Tang, J. L. et al. Histone deacetylase 3 promotes innate antiviral immunity through deacetylation of TBK1. Protein Cell 12, 261–278 (2020).
Seo, M. et al. MAP4-regulated dynein-dependent trafficking of BTN3A1 controls the TBK1-IRF3 signaling axis. Proc. Natl Acad. Sci. USA 113, 14390–14395 (2016).
Liu, X. Y., Chen, W., Wei, B., Shan, Y. F. & Wang, C. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol. 187, 2559–2568 (2011).
Ran, Y. et al. Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol. 8, 31–43 (2016).
Chen, L. T., Hu, M. M., Xu, Z. S., Liu, Y. & Shu, H. B. MSX1 modulates RLR-mediated innate antiviral signaling by facilitating assembly of TBK1-associated complexes. J. Immunol. 197, 199–207 (2016).
Gao, X. et al. PLA1A participates in the antiviral innate immune response by facilitating the recruitment of TANK-binding kinase 1 to mitochondria. J. Innate Immun. 10, 315–327 (2018).
Guo, Y. et al. E3 ubiquitin ligase ASB8 negatively regulates interferon via regulating TBK1/IKKi homeostasis. Mol. Immunol. 121, 195–203 (2020).
Zhang, M. et al. TRAF-interacting protein (TRIP) negatively regulates IFN-β production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J. Exp. Med 209, 1703–1711 (2012).
An, T. et al. DYRK2 negatively regulates type I interferon induction by promoting TBK1 degradation via Ser527 phosphorylation. PLoS Pathog. 11, e1005179 (2015).
He, T. S. et al. THO complex subunit 7 homolog negatively regulates cellular antiviral response against RNA. Viruses Target. TBK1. Viruses 11, 158 (2019).
Lin, M. et al. USP38 inhibits Type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol. Cell 64, 267–281 (2016).
Zheng, Q. et al. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res 25, 1121–1136 (2015).
Cui, J. et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 13, 387–395 (2012).
Parvatiyar, K., Barber, G. N. & Harhaj, E. W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J. Biol. Chem. 285, 14999–15009 (2010).
Zhang, L., Zhao, X., Zhang, M., Zhao, W. & Gao, C. Ubiquitin-specific protease 2b negatively regulates IFN-β production and antiviral activity by targeting TANK-binding kinase 1. J. Immunol. 193, 2230–2237 (2014).
Huang, L. et al. Ubiquitin-conjugating enzyme 2S enhances viral replication by inhibiting type I IFN production through recruiting USP15 to deubiquitinate TBK1. Cell Rep. 32, 108044 (2020).
Liu, S. et al. Lck/Hck/Fgr-mediated tyrosine phosphorylation negatively regulates TBK1 to restrain innate antiviral responses. Cell Host Microbe 21, 754–768.e755 (2017).
Yamada, T. et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat. Immunol. 17, 687–694 (2016).
Qi, D. et al. Phosphatase Cdc25A negatively regulates the antiviral immune response by inhibiting TBK1 activity. J. Virol. 92, e01118–18 (2018).
Zhao, Y. et al. PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell. Signal. 24, 2197–2204 (2012).
Zhan, Z. et al. Phosphatase PP4 negatively regulates type I IFN production and antiviral innate immunity by dephosphorylating and deactivating TBK1. J. Immunol. 195, 3849–3857 (2015).
Yang, Y. et al. NLRP2 negatively regulates antiviral immunity by interacting with TBK1. Eur. J. Immunol. 48, 1817–1825 (2018).
He, X. et al. ERRα negatively regulates type I interferon induction by inhibiting TBK1-IRF3 interaction. PLoS Pathog. 13, e1006347 (2017).
Ng, M. H. et al. MIP-T3 is a negative regulator of innate type I IFN response. J. Immunol. 187, 6473–6482 (2011).
Li, Y. et al. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl Acad. Sci. USA 106, 7945–7950 (2009).
Lu, B. et al. Induction of INKIT by viral infection negatively regulates antiviral responses through inhibiting phosphorylation of p65 and IRF3. Cell Host Microbe 22, 86–98.e4 (2017).
Xia, H. et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 33, 108234 (2020).
Rezelj, V. V. et al. Differential antagonism of human innate immune responses by tick-borne phlebovirus nonstructural proteins. mSphere 2, e00234–17 (2017).
Dalrymple, N. A., Cimica, V. & Mackow, E. R. Dengue virus NS proteins inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation: dengue virus serotype 1 NS4A is a unique interferon-regulating virulence determinant. mBio 6, e00553–00515 (2015).
Wang, G., Chen, G., Zheng, D., Cheng, G. & Tang, H. PLP2 of mouse hepatitis virus A59 (MHV-A59) targets TBK1 to negatively regulate cellular type I interferon signaling pathway. PLoS ONE 6, e17192 (2011).
Liu, X., Main, D., Ma, Y. & He, B. Herpes simplex virus 1 Inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J. Virol. 92, e00402–18 (2018).
Wu, X. et al. Evasion of antiviral immunity through sequestering of TBK1/IKKε/IRF3 into viral inclusion bodies. J. Virol. 88, 3067–3076 (2014).
Ning, Y. J. et al. Viral suppression of innate immunity via spatial isolation of TBK1/IKKε from mitochondrial antiviral platform. J. Mol. Cell Biol. 6, 324–337 (2014).
Moriyama, M. et al. Two conserved amino acids within the NSs of severe fever with thrombocytopenia syndrome phlebovirus are essential for anti-interferon activity. J. Virol. 92, e00706–18 (2018).
Ding, Z. et al. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J. Virol. 88, 8936–8945 (2014).
Ning, Y. J. et al. Heartland virus NSs protein disrupts host defenses by blocking the TBK1 kinase-IRF3 transcription factor interaction and signaling required for interferon induction. J. Biol. Chem. 292, 16722–16733 (2017).
Lin, S. et al. Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virology 527, 180–187 (2019).
Guo, G. et al. SARS-CoV-2 non-structural protein 13 (nsp13) hijacks host deubiquitinase USP13 and counteracts host antiviral immune response. Signal Transduct. Target. Ther. 6, 119 (2021).
Chen, X. et al. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5, 369–381 (2014).
Randall, C. M., Biswas, S., Selen, C. V. & Shisler, J. L. Inhibition of interferon gene activation by death-effector domain-containing proteins from the molluscum contagiosum virus. Proc. Natl Acad. Sci. USA 111, E265–E272 (2014).
Durand, J. K., Zhang, Q. & Baldwin, A. S. Roles for the IKK-related kinases TBK1 and IKKε in cancer. Cells 7, 139 (2018).
Cai, X., Chiu, Y. H. & Chen, Z. J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 (2014).
Vijayan, M. et al. Sphingosine 1-phosphate lyase enhances the activation of IKKε to promote type I IFN-mediated innate immune responses to influenza A virus infection. J. Immunol. 199, 677–687 (2017).
Schröder, M., Baran, M. & Bowie, A. G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 27, 2147–2157 (2008).
Zhang, K. et al. DDX19 inhibits type I interferon production by disrupting TBK1-IKKε-IRF3 interactions and promoting TBK1 and IKKε degradation. Cell Rep. 26, 1258–1272.e4 (2019).
Matsumura, T. et al. Fascin1 suppresses RIG-I-like receptor signaling and interferon-β production by associating with IκB kinase ϵ (IKKϵ) in colon cancer. J. Biol. Chem. 293, 6326–6336 (2018).
Angleró-Rodríguez, Y. I., Pantoja, P. & Sariol, C. A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IκB kinase epsilon interaction. Clin. Vaccin. Immunol. 21, 29–38 (2014).
Kaukinen, P., Sillanpää, M., Nousiainen, L., Melén, K. & Julkunen, I. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKε and TBK1 functions. J. Med. Virol. 85, 71–82 (2013).
Prins, K. C., Cárdenas, W. B. & Basler, C. F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 83, 3069–3077 (2009).
Pythoud, C. et al. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 86, 7728–7738 (2012).
Wong, L. R. et al. Middle east respiratory syndrome coronavirus ORF8b accessory protein suppresses type I IFN expression by impeding HSP70-dependent activation of IRF3 kinase IKKε. J. Immunol. 205, 1564–1579 (2020).
Honda, K. & Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 (2006).
Servant, M. J. et al. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 278, 9441–9447 (2003).
Takahasi, K. et al. Ser386 phosphorylation of transcription factor IRF-3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells 15, 901–910 (2010).
Lin, R., Heylbroeck, C., Pitha, P. M. & Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996 (1998).
Zhou, Y. et al. Interferon-inducible cytoplasmic lncLrrc55-AS promotes antiviral innate responses by strengthening IRF3 phosphorylation. Cell Res 29, 641–654 (2019).
Wang, J. et al. IRF1 promotes the innate immune response to viral infection by enhancing the activation of IRF3. J. Virol. 94, e01231–20 (2020).
Wang, C. et al. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med 214, 3597–3610 (2017).
Cai, Z. et al. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J. Exp. Med 217, e20191174 (2020).
Saitoh, T. et al. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 7, 598–605 (2006).
Zhang, M. et al. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res 18, 1096–1104 (2008).
Higgs, R. et al. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 181, 1780–1786 (2008).
Beura, L. K. et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 84, 1574–1584 (2010).
Yu, Y. & Hayward, G. S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 (2010).
Wang, P., Zhao, W., Zhao, K., Zhang, L. & Gao, C. TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 11, e1004726 (2015).
Zhang, Z., Wang, D., Wang, P., Zhao, Y. & You, F. OTUD1 negatively regulates type I IFN induction by disrupting noncanonical ubiquitination of IRF3. J. Immunol. 204, 1904–1918 (2020).
Jiang, L. Q. et al. IFITM3 inhibits virus-triggered induction of type I interferon by mediating autophagosome-dependent degradation of IRF3. Cell. Mol. Immunol. 15, 858–867 (2018).
James, S. J. et al. MAPK phosphatase 5 expression induced by influenza and other RNA virus infection negatively regulates IRF3 activation and type I interferon response. Cell Rep. 10, 1722–1734 (2015).
Long, L. et al. Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity 40, 515–529 (2014).
Meng, F. et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev. 30, 1086–1100 (2016).
Peng, D., Wang, Z., Huang, A., Zhao, Y. & Qin, F. X. A novel function of F-Box protein FBXO17 in negative regulation of type I IFN signaling by recruiting PP2A for IFN regulatory factor 3 deactivation. J. Immunol. 198, 808–819 (2017).
Zan, J. et al. RNA helicase DDX5 suppresses IFN-I antiviral innate immune response by interacting with PP2A-Cβ to deactivate IRF3. Exp. Cell Res. 396, 112332 (2020).
Yang, Q. et al. Host HDAC4 regulates the antiviral response by inhibiting the phosphorylation of IRF3. J. Mol. Cell Biol. 11, 158–169 (2019).
Ran, Y. et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 3, 283–292 (2011).
Li, D. et al. DDX56 inhibits type I interferon by disrupting assembly of IRF3-IPO5 to inhibit IRF3 nucleus import. J. Cell Sci. 133, jcs230409 (2019).
Kim, J. H. et al. Rubicon modulates antiviral type I interferon (IFN) signaling by targeting IFN regulatory factor 3 dimerization. J. Virol. 91, e00248–17 (2017).
Wang, S. et al. AGO2 negatively regulates type I interferon signaling pathway by competition binding IRF3 with CBP/p300. Front. Cell. Infect. Microbiol. 7, 195 (2017).
Kuo, R. L. et al. Role of N terminus-truncated NS1 proteins of influenza A virus in inhibiting IRF3 activation. J. Virol. 90, 4696–4705 (2016).
Li, Z. et al. Porcine haemagglutinating encephalomyelitis virus deactivates transcription factor IRF3 and limits type I interferon production. Vet. Microbiol. 252, 108918 (2021).
Rieder, M. et al. Genetic dissection of interferon-antagonistic functions of rabies virus phosphoprotein: inhibition of interferon regulatory factor 3 activation is important for pathogenicity. J. Virol. 85, 842–852 (2011).
Matthews, K., Schäfer, A., Pham, A. & Frieman, M. The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol. J. 11, 209 (2014).
Zhu, Z. et al. Peste des petits ruminants virus nucleocapsid protein inhibits beta interferon production by interacting with IRF3 to block its activation. J. Virol. 93, e00362–19 (2019).
Wu, Y. et al. Porcine epidemic diarrhea virus nsp15 antagonizes interferon signaling by RNA degradation of TBK1 and IRF3. Viruses 12, 599 (2020).
Lei, X. et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11, 3810 (2020).
Wang, W. et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Immunol. 18, 945–953 (2021).
Fung, S. Y., Siu, K. L., Lin, H., Yeung, M. L. & Jin, D. Y. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 17, 1547–1554 (2021).
Moustaqil, M. et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg. Microbes Infect. 10, 178–195 (2021).
Ye, J. et al. Japanese encephalitis virus NS5 inhibits type I interferon (IFN) production by blocking the nuclear translocation of IFN regulatory factor 3 and NF-κB. J. Virol. 91, e00039–17 (2017).
Chang, R. Y. et al. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 166, 11–21 (2013).
Irie, T., Kiyotani, K., Igarashi, T., Yoshida, A. & Sakaguchi, T. Inhibition of interferon regulatory factor 3 activation by paramyxovirus V protein. J. Virol. 86, 7136–7145 (2012).
Xue, Q. et al. Seneca Valley Virus 3C(pro) abrogates the IRF3- and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology 518, 1–7 (2018).
Bauhofer, O. et al. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 81, 3087–3096 (2007).
Barro, M. & Patton, J. T. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl Acad. Sci. USA 102, 4114–4119 (2005).
Fu, S. Z. et al. DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3. Cell. Signal. 64, 109393 (2019).
Matthys, V. S. et al. Hantavirus GnT elements mediate TRAF3 binding and inhibit RIG-I/TBK1-directed beta interferon transcription by blocking IRF3 phosphorylation. J. Virol. 88, 2246–2259 (2014).
Yuen, C. K. et al. Suppression of type I interferon production by human T-cell leukemia virus type 1 oncoprotein tax through inhibition of IRF3 phosphorylation. J. Virol. 90, 3902–3912 (2016).
Lui, P. Y. et al. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg. Microbes Infect. 5, e39 (2016).
Jennings, S., Martínez-Sobrido, L., García-Sastre, A., Weber, F. & Kochs, G. Thogoto virus ML protein suppresses IRF3 function. Virology 331, 63–72 (2005).
Arnold, M. M., Barro, M. & Patton, J. T. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J. Virol. 87, 9813–9821 (2013).
Chen, W. & Royer, W. E. Jr Structural insights into interferon regulatory factor activation. Cell. Signal. 22, 883–887 (2010).
Honda, K. et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 (2005).
Hemmi, H. et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 (2004).
Sato, M. et al. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441, 106–110 (1998).
Ning, S., Campos, A. D., Darnay, B. G., Bentz, G. L. & Pagano, J. S. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28, 6536–6546 (2008).
Wang, J. et al. Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J. Immunol. 191, 3393–3399 (2013).
Higgs, R. et al. Self protection from anti-viral responses-Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS ONE 5, e11776 (2010).
Liang, Q. et al. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187, 4754–4763 (2011).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Ning, S. & Pagano, J. S. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 84, 6130–6138 (2010).
Cao, L. et al. P200 family protein IFI204 negatively regulates type I interferon responses by targeting IRF7 in nucleus. PLoS Pathog. 15, e1008079 (2019).
Liang, Q., Deng, H., Sun, C. W., Townes, T. M. & Zhu, F. Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J. Immunol. 186, 1001–1010 (2011).
Lee, K. J., Lee, H. & Joo, C. H. Negative regulation of IKKε-mediated IRF7 phosphorylation by HSP70. J. Immunol. 204, 2562–2574 (2020).
Chang, T. H. et al. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 5, e1000493 (2009).
Raychoudhuri, A. et al. Hepatitis C virus infection impairs IRF-7 translocation and Alpha interferon synthesis in immortalized human hepatocytes. J. Virol. 84, 10991–10998 (2010).
Fiebach, A. R., Guzylack-Piriou, L., Python, S., Summerfield, A. & Ruggli, N. Classical swine fever virus N(pro) limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J. Virol. 85, 8002–8011 (2011).
Kato, K., Omura, H., Ishitani, R. & Nureki, O. Cyclic GMP-AMP as an endogenous second messenger in innate immune signaling by cytosolic DNA. Annu. Rev. Biochem. 86, 541–566 (2017).
Zhang, X. et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 (2013).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Ablasser, A. et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Diner, E. J. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361 (2013).
Fitzgerald, K. A. et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Gray, E. E. et al. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45, 255–266 (2016).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Gao, J., Tao, J., Liang, W. & Jiang, Z. Cyclic (di)nucleotides: the common language shared by microbe and host. Curr. Opin. Microbiol. 30, 79–87 (2016).
Zhou, C. et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52, 767–781.e6 (2020).
Li, L. et al. Hydrolysis of 2'3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048 (2014).
Kato, K. et al. Structural insights into cGAMP degradation by ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 9, 4424 (2018).
Bridgeman, A. et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science 349, 1228–1232 (2015).
Gentili, M. et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 349, 1232–1236 (2015).
Eaglesham, J. B., Pan, Y., Kupper, T. S. & Kranzusch, P. J. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 566, 259–263 (2019).
Shang, G., Zhang, C., Chen, Z. J., Bai, X. C. & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393 (2019).
Burdette, D. L. & Vance, R. E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 14, 19–26 (2013).
Zhang, C. et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019).
Ergun, S. L. & Li, L. Structural insights into STING signaling. Trends Cell Biol. 30, 399–407 (2020).
Hiller, B. & Hornung, V. STING signaling the enERGIC way. Cell Host Microbe 18, 137–139 (2015).
Ni, G., Konno, H. & Barber, G. N. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2, eaah7119 (2017).
Dunphy, G. et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol. Cell 71, 745–760.e5 (2018).
Yang, L. et al. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat. Commun. 9, 2329 (2018).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Zhang, M. et al. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res 26, 1302–1319 (2016).
Zhang, M. X. et al. USP20 promotes cellular antiviral responses via deconjugating K48-linked ubiquitination of MITA. J. Immunol. 202, 2397–2406 (2019).
Zhang, H. Y. et al. USP44 positively regulates innate immune response to DNA viruses through deubiquitinating MITA. PLoS Pathog. 16, e1008178 (2020).
Zhang, L. et al. The deubiquitinase CYLD is a specific checkpoint of the STING antiviral signaling pathway. PLoS Pathog. 14, e1007435 (2018).
Guo, Y. et al. OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell. Mol. Immunol. 1, 11 (2020).
Luo, W. W. et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066 (2016).
Hansen, A. L., Mukai, K., Schopfer, F. J., Taguchi, T. & Holm, C. K. STING palmitoylation as a therapeutic target. Cell. Mol. Immunol. 16, 236–241 (2019).
Mukai, K. et al. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 (2016).
Konno, H., Konno, K. & Barber, G. N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698 (2013).
Li, Z. et al. PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. 11, e1004783 (2015).
Gao, P., Hu, M. M. & Shu, H. B. CSK promotes innate immune response to DNA virus by phosphorylating MITA. Biochem. Biophys. Res. Commun. 526, 199–205 (2020).
Sun, M. S. et al. TMED2 potentiates cellular IFN responses to DNA viruses by reinforcing MITA dimerization and facilitating Its trafficking. Cell Rep. 25, 3086–3098.e3 (2018).
Wei, J. et al. SNX8 modulates innate immune response to DNA virus by mediating trafficking and activation of MITA. PLoS Pathog. 14, e1007336 (2018).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Yang, B. et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog. 16, e1008387 (2020).
Li, Q. et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov. 4, 13 (2018).
Wang, Y. et al. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. 11, e1005012 (2015).
Chen, Y. et al. p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med. 214, 991–1010 (2017).
Ye, L. et al. USP49 negatively regulates cellular antiviral responses via deconjugating K63-linked ubiquitination of MITA. PLoS Pathog. 15, e1007680 (2019).
Sun, H. et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 8, 15534 (2017).
Xia, T., Yi, X. M., Wu, X., Shang, J. & Shu, H. B. PTPN1/2-mediated dephosphorylation of MITA/STING promotes its 20S proteasomal degradation and attenuates innate antiviral response. Proc. Natl Acad. Sci. USA 116, 20063–20069 (2019).
Chen, H. et al. An alternative splicing isoform of MITA antagonizes MITA-mediated induction of type I IFNs. J. Immunol. 192, 1162–1170 (2014).
Guo, H. et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe 19, 515–528 (2016).
Wu, X. et al. RIG-I and IL-6 are negative-feedback regulators of STING induced by double-stranded DNA. PLoS ONE 12, e0182961 (2017).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl Acad. Sci. U SA 106, 20842–20846 (2009).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Christensen, M. H. et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35, 1385–1399 (2016).
Pan, S., Liu, X., Ma, Y., Cao, Y. & He, B. Herpes simplex virus 1 γ(1)34.5 protein inhibits STING activation that restricts viral replication. J. Virol. 92, e01015–18 (2018).
Deschamps, T. & Kalamvoki, M. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J. Virol. 91, e00535–17 (2017).
Bodda, C. et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 217, e20191422 (2020).
Yi, G. et al. Hepatitis C virus NS4B can suppress STING accumulation to evade innate immune responses. J. Virol. 90, 254–265 (2016).
Ma, Z. et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl Acad. Sci. USA 112, E4306–E4315 (2015).
Stempel, M. et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING’s dual role during MCMV infection. EMBO J. 38, e100983 (2019).
Liu, Y. et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 89, 2287–2300 (2015).
Fu, Y. Z. et al. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21, 231–243 (2017).
Kim, J. E., Kim, Y. E., Stinski, M. F., Ahn, J. H. & Song, Y. J. Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 8, 1854 (2017).
Lee, J. K., Kim, J. E., Park, B. J. & Song, Y. J. Human cytomegalovirus IE86 protein aa 136-289 mediates STING degradation and blocks the cGAS-STING pathway. J. Microbiol. 58, 54–60 (2020).
Fu, Y. Z. et al. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 15, e1007691 (2019).
Wu, Z. et al. Binding of the duck tembusu virus protease to STING is mediated by NS2B and is crucial for STING cleavage and for impaired induction of IFN-β. J. Immunol. 203, 3374–3385 (2019).
Verpooten, D., Ma, Y., Hou, S., Yan, Z. & He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284, 1097–1105 (2009).
Kang, H. R. et al. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J. Virol. 88, 6832–6846 (2014).
Kang, H. et al. Feline panleucopenia virus NS2 suppresses the host IFN-β induction by disrupting the interaction between TBK1 and STING. Viruses 9, 23 (2017).
Zhang, D., Su, C. & Zheng, C. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J. Virol. 90, 5824–5829 (2016).
Saira, K., Zhou, Y. & Jones, C. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J. Virol. 81, 3077–3086 (2007).
Sen, N. et al. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J. Virol. 84, 9240–9253 (2010).
Vandevenne, P. et al. The varicella-zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS ONE 6, e16870 (2011).
Zhu, H. et al. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 85, 11079–11089 (2011).
Huye, L. E., Ning, S., Kelliher, M. & Pagano, J. S. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell. Biol. 27, 2910–2918 (2007).
Gao, L. et al. Inhibition of DNA-sensing pathway by Marek’s disease virus VP23 protein through suppression of interferon regulatory factor 7 activation. J. Virol. 93, e01934–18 (2019).
Yu, Y., Wang, S. E. & Hayward, G. S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70 (2005).
Joo, C. H. et al. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81, 8282–8292 (2007).
Hwang, S. W., Kim, D., Jung, J. U. & Lee, H. R. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochem. Biophys. Res. Commun. 486, 700–705 (2017).
Acknowledgements
This work was supported by the National Research Foundation (Grant Nos. 2018M3A9H4079660, 2019R1A2C2008283, 2021R1A6A1A03045495) and the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM9942011), Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chathuranga, K., Weerawardhana, A., Dodantenna, N. et al. Regulation of antiviral innate immune signaling and viral evasion following viral genome sensing. Exp Mol Med 53, 1647–1668 (2021). https://doi.org/10.1038/s12276-021-00691-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00691-y
This article is cited by
-
The peroxisome: an update on mysteries 3.0
Histochemistry and Cell Biology (2024)
-
Target-dependent RNA polymerase as universal platform for gene expression control in response to intracellular molecules
Nature Communications (2023)
-
Intranasal influenza-vectored COVID-19 vaccine restrains the SARS-CoV-2 inflammatory response in hamsters
Nature Communications (2023)
-
SARS-CoV-2 virus NSP14 Impairs NRF2/HMOX1 activation by targeting Sirtuin 1
Cellular & Molecular Immunology (2022)
