
Abstract
The global number of patients with depression increases in correlation to exposure to social stress. Chronic stress does not trigger depression in all individuals, as some remain resilient. The underlying molecular mechanisms that contribute to stress sensitivity have been poorly understood, although revealing the regulation of stress sensitivity could help develop treatments for depression. We previously found that striatal Shati/Nat8l, an N-acetyltransferase, was increased in a depression mouse model. We investigated the roles of Shati/Nat8l in stress sensitivity in mice and found that Shati/Nat8l and brain-derived neurotrophic factor (BDNF) levels in the dorsal striatum were increased in stress-susceptible mice but not in resilient mice exposed to repeated social defeat stress (RSDS). Knockdown of Shati/Nat8l in the dorsal striatum induced resilience to RSDS. In addition, blockade of BDNF signaling in the dorsal striatum by ANA-12, a BDNF-specific receptor tropomyosin-receptor-kinase B (TrkB) inhibitor, also induced resilience to stress. Shati/Nat8l is correlated with BDNF expression after RSDS, and BDNF is downstream of Shati/Nat8l pathways in the dorsal striatum; Shati/Nat8l is epigenetically regulated by BDNF via histone acetylation. Our results demonstrate that striatal Shati/Nat8l–BDNF pathways determine stress sensitivity through epigenetic regulation. The striatal Shati/Nat8l–BDNF pathway could be a novel target for treatments of depression and could establish a novel therapeutic strategy for depression patients.
Similar content being viewed by others


Introduction
In recent years, the number of patients with mood disorders, including major depressive disorder (depression), has increased. Depression is one of the most serious disorders among all mental illnesses, and symptoms include loss of motivation and energy and the presence of suicidal thoughts. Depression is characteristically resistant to treatment. Generally, antidepressant drugs require long periods before the effects are seen and do not have high response rates. Approximately 30% of depression patients have resistance to treatment with antidepressants [1, 2]. For these reasons, discovering novel targets of antidepressants and establishing therapeutic strategies that work for all patients is desired.
Stress is closely related to depression, and several studies have suggested that depression onset is triggered by stressful environments [3, 4]. Vulnerability to depression onset is caused by depressive episodes such as stress, thereby leading to depression onset in response to even mild stress [5, 6]. However, not all individuals are vulnerable to depression onset, as some remain resilient [7]. In behavioral experiments, mice exposed to the same stressful conditions can also be classified as either stress-susceptible (those that show depressive behaviors) or resilient (those that do not) [8]. Revealing the underlying regulatory mechanisms of stress sensitivity could offer insight in understanding and developing treatments for depression.
Shati/Nat8l was previously identified in the brains of psychosis model mice [9]. Shati/Nat8l, which we focused on here, is expressed in the whole brain and is involved in the reward system that, in the medial prefrontal cortex (mPFC), attenuates methamphetamine addiction [10]. Shati/Nat8l has N-acetyl transfer activity and predominantly regulates the amount of N-acetyl aspartate (NAA) [11], which is converted acetyl-CoA [12]. NAA is biosynthesized to N-acetyl aspartylglutamate (NAAG) by condensation with glutamic acid by NAAG synthetase [13]. We found that Shati/Nat8l in the dorsal striatum is increased in depression model mice [14]. In a previous human study, it was shown that Shati/Nat8l could be a diagnostic biomarker for depression [15], and NAA levels are altered in the brains of patients with depression [16]. These reports indicate that Shati/Nat8l could be associated with the pathogenesis of depression. However, the detailed contribution of Shati/Nat8l to depression remains elusive.
Stress-induced alterations in brain-derived neurotrophic factor (BDNF) levels in the brain have been proposed to be involved in the mechanism of depression onset [17, 18]. BDNF expression is altered in various brain regions in human patients with depression and depression model mice. Interestingly, the role of BDNF in the central nervous system is region- and circuit-specific [19,20,21]. BDNF in the hippocampus and the mPFC has an antidepressant effect, whereas BDNF in the ventral tegmental area (VTA)–nuclear accumbent (NAc) circuits has a pro-depressant efficacy [22, 23]. On the other hand, there are no reports on the roles that BDNF in the dorsal striatum has in depression. It has been reported that BDNF is controlled by epigenetic mechanisms, including histone methylation and acetylation and DNA methylation [24,25,26].
In this study, we examined the knockdown effects of Shati/Nat8l in the dorsal striatum to stress sensitivity in mice to clarify the role of striatal Shati/Nat8l in depression and further analyzed the mechanisms with a focus on BDNF.
Materials and methods
Animals and environments
Male 8-week-old C57Bl/6J mice (Nihon SLC, Hamamatsu, Japan) and Shati/Nat8lflox/flox mice (described below) were used. All mice were housed under controlled temperature (25 °C ± 1 °C), humidity (50% ± 5%), and a 12-h light/dark conditions (light, 7:00–19:00; dark, 19:00–7:00) with ad libitum access to food and water. All experiments were performed with permission from the Committee for Animal Experiments of the University of Toyama (A2018PHA-5, 13) and according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Production of Shati/Nat8lflox/flox mice
We entrusted Unitech Co., Ltd. (Kashiwa, Japan) to produce Shati/Nat8lflox/flox mice. The Shati/Nat8l targeting construct was built using conventional cloning. Exons of the Shati/Nat8l coding gene were flanked by loxP sites, followed by an FRT-flanked neomycin cassette, in the construction process. Next, the targeting vector was linearized and inserted into embryonic stem cells. Cloned embryonic stem cells were inserted into blastocysts of C57BL/6J mice to produce chimeric mice. The chimeric mice were crossed with C57BL/6J mice to generate heterozygous mice carrying one loxP-flanked allele (Shati/Nat8lflox/+). Shati/Nat8lflox/+ mice were crossed with CAG-Flp mice to remove the neomycin cassette. Shati/Nat8lflox/+ mice without the neomycin cassette and Flp cassette were then produced by crossing Shati/Nat8lflox/+ mice without the neomycin cassette with C57BL/6J mice. Shati/Nat8lflox/+ mice without the neomycin cassette and Flp cassette were then crossed with Shati/Nat8lflox/+ to generate Shati/Nat8lflox/flox. This study was performed with permission from the Board of Safety Committee for Recombination DNA Experiments of the University of Toyama (G2015PHA-15).
Production and microinjection of adeno‐associated virus (AAV) vectors
The production and microinjection of AAV vectors were performed as previously reported [27, 28]. Briefly, an expression cassette including the CMV promoter and cDNA encoding either Cre recombinase or the enhanced green fluorescent protein sequence was inserted in a plasmid for the AAV-Cre or a mock vector, respectively. The AAV plasmid, AAV3 rep/AAV9 vp expression plasmid, and helper plasmid (Agilent Technologies, Santa Clara, CA) were co-transfected into HEK293 cells to produce AAV-Cre or mock vectors. AAV-Cre induced neuron-specific Cre-recombinase activity. Shati/Nat8lflox/flox mice were injected with AAV-Cre vectors to generate the conditional knockdown of Shati/Nat8l. The AAV-Cre or mock vectors (1 × 1010–1012 units) were injected into the bilateral dorsal striatum (AP, 0.5 mm; ML, ±2.0 mm; DV, 3.5 mm), mPFC (AP, 1.7 mm; ML, ±0.3 mm; DV, 1.5 mm), or hippocampus (AP, −1.6 mm; ML, ±1.0 mm; DV, 1.35 mm) [29] after mice were anesthetized with a combination of anesthetics (medetomidine [0.3 mg/kg], midazolam [4.0 mg/kg], and butorphanol [5.0 mg/kg]). Four weeks after the microinjection, the mice were used in experiments. Shati/Nat8lflox/flox mice that received the mock vector injection were used as a control group.
Repeated social defeat stress (RSDS)
RSDS was performed as previously described [8, 30]. Retired male ICR mice (4–5 months old) were used as stressors. Briefly, ICR mice were screened for aggressive behaviors before the experiments. ICR mice that attacked for more than 60 s out of 180 s during the screening were used. C57BL/6J mice were exposed to daily physical attack by ICR mice for 10 min. After direct contact with ICR mice, C57BL/6J mice and ICR mice were placed in cages separated by a transparent board, allowing indirect contact for 24 h until the next exposure to physical stress. Naive mice as controls were also single-housed and were in contact with other mice for 10 min daily.
Strong-RSDS
ICR mice that attacked for more than 90 s out of 180 s during the screening were used. The experimental schedule and methods were the same as those of the normal RSDS.
Behavioral tests
Twenty-four hours after the final defeat session, the defeated mice and naive mice were used for behavioral tests. All behavioral tests were performed in the light condition of mice.
Social interaction test
The social interaction test was performed as previously reported [8, 31]. Mice were placed in an open-field area (40 × 40 × 30 cm) with a small cage for the ICR mice. Their activity was monitored for 150 s under the condition in which no ICR mouse was present in the small cage as a pretest. The mice were then returned to their home cage for 30 s. Next, the mice were placed again in an open-field area, and their activity was monitored for 150 s under the condition in which an ICR mouse was placed in the small cage as a post-test. The time spent in the interaction and corner avoidance zones in the post-test was measured, and the social interaction ratio was calculated to assess social interaction behavior. The social interaction ratio was defined as follows: (the time in the interaction zone with the ICR mouse)/(the time in the interaction zone without the ICR mouse). Susceptible mice or resilient mice were classified based on the social interaction ratio (susceptible: social interaction ratio <1.0; resilient: social interaction ratio ≥1.0) [32].
Sucrose preference test
The sucrose preference test was performed as previously reported [31]. The consumption of water and 1% sucrose for 12 h were measured. Sucrose preference was defined as follows: (the amount of sucrose consumed)/(the total amount of water and sucrose consumed).
Locomotor activity test
The locomotor activity test was performed as previously reported [14]. In brief, mice were placed in an open-field area (40 × 40 × 30 cm), and their locomotor activity was measured with Scanet MV-40AQ (Melquest) for 60 min.
Tail suspension test
The tail suspension test was performed as previously reported [14, 33]. Mice were suspended with adhesive tape from a suspension bar (height, 12 cm) for 6 min. The immobility time from 1 to 6 min was measured.
Forced swimming test
The forced swimming test was performed as previously reported [14]. In brief, mice were placed individually in a blinded cylinder (diameter, 21 cm; height, 22.5 cm) containing 22 °C water (depth, 18 cm). The mice were forced to swim for 6 min. The immobility time from 1 to 6 min was measured with Scanet MV-40AQ (Melquest, Toyama, Japan).
Immunostaining
Immunostaining was performed as previously described [34]. Briefly, 20-μm frozen sections were prepared using a cryostat (CM3050S, Leica Wetzlar, Germany). The fixing, permeabilizing, washing, and blocking procedures were performed, after which the sections were incubated with primary antibodies against rabbit anti-Cre recombinase (#18771, 1:40, IBL, Fujioka, Japan) and mouse anti-NeuN (MAB377, 1:500, St. Louis, MO, USA) at 4 °C overnight. Next, the sections were washed with Tris-buffered solution with Tween-20 and then incubated with CFTM 488 goat anti-rabbit IgG (H + L) (#20111, 1:1000, Biotium, Hayward, CA) and CFTM 594 goat anti-mouse IgG (H + L) (#20113, 1:1000, Biotium) as the secondary antibodies for 2 h at room temperature. The sections were washed and incubated with DAPI staining solution. After the sections were washed and mounted, immunofluorescence images were obtained using an AxioCam icC1 (Carl Zeiss, Jena, Germany).
Real-time RT-PCR analysis
RT-PCR was performed as previously described [31]. The sequences of the primers used are as follows: Shati/Nat8l: forward, 5′-GTGATTCTGGCCTACCTGGA-3′; reverse, 5′-CCACTGTGTTGTCCTCCTCA-3′; BDNF IV: forward, 5′-CAGAGCAGCAGCTGCCTTGATGTT-3′; reverse, 5′-GCCTTGTCCGTGGACGTTTA-3′; 36B4: forward, 5′- ACCCTGAAGTGCTCGACATC-3′; reverse, 5′- AGGAAGGCCTTGACCTTTTC-3′.
Western blotting
Western blotting was performed as previously described [35]. Primary antibodies against rabbit anti-BDNF (ab108319, 1:1000, Abcam, Cambridge, UK) and mouse anti-GAPDH (M171-3, 1:1000, MBL) were used. Secondary antibodies (RPN2124, anti-rabbit IgG; 1:5000, anti-mouse IgG; 1:5000, GE Healthcare, Chicago, IL, USA) were applied. The proteins were detected using an ECL Prime kit (GE Healthcare) and quantified using Image J software.
Microinfusion
Microinfusion was performed as previously reported [31]. After the mice were anesthetized, guide cannulas (AG-4, Eicom, Kyoto, Japan) were implanted into the bilateral dorsal striatum (AP, 0.5 mm; ML, ±2.0 mm; DV, 3.0 mm). ANA-12 (tropomyosin-receptor-kinase B (TrkB) inhibitor, 1 µg/0.5 μL per side) (S7745, Selleck Biotech, Tokyo, Japan) was infused using an injector EPS-64 microsyringe pump (Eicom) through the guide cannulas. This dose has been reported not to affect behaviors in mice [36], and ANA-12 was demonstrated to be stable without degradation and inhibit BDNF signaling for 60 min [37]. Ringer solution (147-mM NaCl, 4-mM KCl, and 2.3-mM CaCl2) was infused as a control. The ANA-12 or ringer solution was infused 15 min before each social defeat stress session for 10 days as previously reported [38].
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed as previously described [39]. The acetylation and methylation levels of histone H3 and DNA methylation levels at the promoter sites of BDNF IV were detected using the ChIP assay. A Dynabeads™ Protein G Immunoprecipitation Kit was used (Thermo Fisher Scientific K.K., Tokyo, Japan) to precipitate the crosslinked chromatin. Antibodies (2 µg for 25 µg of chromatin) against anti-acetylated H3K9 (ab10812, Abcam), anti-H3K4 (ab176799, Abcam), anti-total acetylated H3 (ab47915, Abcam), anti-methylated H3K9 (ab8898, Abcam), or anti-5-methylcytosine (5-mC) (C15200081, Diagenode, Liege, Belgium) were applied. Normal rabbit IgG (#2729, Cell Signaling Technology) was used as a negative control. The DNA samples were purified used for RT-PCR. The primers for the BDNF IV promoter have been previously reported [24]. Quantification was performed using Image J software, and the levels of epigenetic modification were normalized with input controls for each sample. The sequences of the primers used were as follows: BDNF IV: forward, 5′-GCGCGGAATTCTGATTCTGGTAAT-3′; reverse, 5′-GAGAGGGCTCCACGCTGCCTTGACG-3′.36B4: forward, 5′-ACCCTGAAGTGCTCGACATC-3′; reverse, 5′- AGGAAGGCCTTGACCTTTTC-3′.
Statistical analysis
Data were analyzed with Prism version 5. The Student’s t test or the chi-squared test was used to analyze the data between the two groups. One-way ANOVA followed by the Bonferroni’s post hoc test was used for the analysis of the data between multiple groups. Two-way ANOVA followed by the Bonferroni’s or Tukey–Kramer’s post hoc test was applied to analyze the data between two factors. Correlations were analyzed using Pearson’s r. Power analysis was applied to analyze the validity of sample size in the experiments using the G*Power 3.1 software [40, 41]. All data are presented as the mean ± standard error.
Results
Shati/Nat8l expression in the dorsal striatum, but not mPFC or hippocampus, correlates explicitly with sensitivity to RSDS
First, we assessed depressive behaviors after RSDS in a social interaction test. The time in the interaction zone was significantly reduced in susceptible mice compared to stress-naive mice and resilient mice (Fig. 1a) (F2,53 = 40.15, p < 0.0001; one-way ANOVA). The time in the avoidance zone significantly increased in susceptible mice compared to stress-naive mice and resilient mice (Fig. 1b) (F2,35 = 17.18, p < 0.0001; one-way ANOVA). The interaction ratio was significantly decreased in susceptible mice compared to stress-naive mice and resilient mice (Fig. 1c) (F2,53 = 56.04, p < 0.0001; one-way ANOVA). The methods for classification into the resilient or susceptible groups in this study were validated by these results. Shati/Nat8l mRNA levels were quantified by real-time PCR immediately after the social interaction tests. Shati/Nat8l mRNA levels were significantly increased in the dorsal striatum of susceptible mice compared with stress-naive mice (Fig. 1d) (F2,34 = 31.44, p < 0.0001; one-way ANOVA). We then confirmed the correlation between Shati/Nat8l expression in the dorsal striatum and social behavior (Fig. 1e, f) [(vs. time in interaction zone: r = −0.567, p = 0.0054) (vs. interaction ratio: r = −0.708, p = 0.0002; Pearson’s r)]. We also performed correlation analyses exclusively in mice exhibiting depressive-like behavior (stress-susceptible mice). we confirmed the correlation between striatal Shati/Nat8l mRNA expression in stress-susceptible mice and interaction ratio (Fig. S1) (r = −0.726, p = 0.0001). However, such an elevation of Shati/Nat8l mRNA observed in the dorsal striatum was not detected in the mPFC or hippocampus (Fig. 1d). In addition, there were no correlation between Shati/Nat8l mRNA levels in the mPFC or hippocampus and social behavior (Fig. 1g, h) (mPFC: r = −0.224, p = 0.634, hippocampus r = 0.136, p = 0.545; Pearson’s r). These results are in agreement with previous studies showing that chronic stress induces region-specific increases of Shati/Nat8l mRNA in the dorsal striatum [14, 31].

a Social interaction test was performed after repeated social defeat stress (RSDS). There was a significant difference in time spent in the interaction zone in susceptible mice compared with naive mice and resilient mice under the condition in which ICR mice were present (F2,53 = 40.15, p < 0.0001). Naive, n = 22; resilient, n = 15; susceptible, n = 19; ***p < 0.001 vs. susceptible mice (one-way ANOVA with Bonferroni post hoc tests). b There was a significant difference in time spent in the avoidance zone in susceptible mice compared with naive mice and resilient mice under the condition in which ICR mice were present (F2,53 = 17.18, p < 0.0001;). Naive, n = 22; resilient, n = 15; susceptible, n = 19; ***p < 0.005 vs. susceptible mice, **p < 0.01 vs. susceptible mice (one-way ANOVA with Bonferroni post hoc tests). c There was a significant difference in the interaction ratio in susceptible mice compared with naive mice and resilient mice under the condition in which ICR mice were present (F2,53 = 56.04, p < 0.0001). Naive, n = 22; resilient, n = 15; susceptible, n = 19; ***p < 0.005 vs. susceptible mice (one-way ANOVA with Bonferroni post hoc tests). d Shati/Nat8l mRNA levels in the dorsal striatum, but not mPFC or hippocampus, of susceptible mice were changed by RSDS compared with naive mice and resilient mice (F2,34 = 31.44, p < 0.0001). Striatum: naive, n = 16; resilient, n = 10; susceptible, n = 12; ***p < 0.005 vs. susceptible mice, mPFC: naive, n = 8; resilient, n = 8; susceptible, n = 8, hippocampus: naive, n = 8; resilient, n = 8; susceptible, n = 8 (one-way ANOVA with Bonferroni post hoc tests). Shati/Nat8l mRNA expression in the dorsal striatum was strongly correlated with social interaction behavior including time in interaction zone (e) (r = −0.567, p = 0.0054) and interaction ratio (f) (r = −0.708, p = 0.0002) Resilient, n = 10; susceptible, n = 12 (Pearson’s r). Social interaction behavior was not correlated with Shati/Nat8l mRNA expression in the mPFC (g) or hippocampus (h), after RSDS. mPFC: resilient, n = 8; susceptible, n = 8, hippocampus: resilient, n = 8; susceptible, n = 8 (Pearson’s r).
Knockdown of Shati/Nat8l in the dorsal striatum induces resilience to RSDS
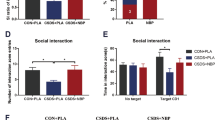
We generated Shati/Nat8l conditional knockdown in the dorsal striatum of mice (Shati cKD mice) using the Cre/loxP system for investigation of whether Shati/Nat8l in the striatum is involved in determining stress resilience. We performed microinjection of AAV-Cre into the dorsal striatum of Shati/Nat8lflox/flox mice and of AAV-Mock into the dorsal striatum as a control (mock mice). The specific Cre-positive cells (green signals) in the dorsal striatal neurons were observed in Shati cKD mice (Fig. 2a). The significant decrease of Shati/Nat8l mRNA was confirmed in the dorsal striatum of Shati cKD mice compared with that of mock mice (Fig. 2b) (t5 = 10.59, p = 0.0001; Student’s t test). In this series of experiments, mock mice and Shati cKD mice were exposed to strong-RSDS (S-RSDS) to evaluate resilience, followed by the behavioral tests to assess depression (Fig. 2c). There was no difference in the social interaction time between mock mice and Shati cKD mice without social defeat stress. While mock mice with S-RSDS showed strong suppression of social interaction, the Shati cKD mice with S-RSDS did not show a lower social interaction time (Fig. 2d) (main effect of virus: F1,30 = 25.30, p < 0.0001; main effect of stress: F1,30 = 25.71, p < 0.0001; interaction effect: F1,30 = 25.20, p < 0.0001; two-way ANOVA). Furthermore, the ratio of stress resilience was increased fourfold by knockdown of Shati/Nat8l in the dorsal striatum (Fig. 2e) (p = 0.0071; significance was analyzed using the chi-squared test). Mock mice showed decreased sucrose preference by S-RSDS, while Shati cKD mice with S-RSDS did not (Fig. 2f) (main effect of virus: F1,30 = 9.214, p = 0.3799; main effect of stress: F1,30 = 1.434, p = 0.2405; interaction effect: F1,30 = 9.214, p = 0.0049; two-way ANOVA). After S-RSDS, Shati cKD mice showed more resilience to stress and a decrease in immobility time than mock mice in the tail suspension and forced swimming test (Fig. 2g, h) [(tail suspension test: main effect of virus: F1,30 = 1.028, p = 0.3187; main effect of stress: F1,30 = 1.849, p = 0.184; interaction effect: F1,30 = 6.437, p = 0.0166) (forced swimming test: main effect of virus: F1,30 = 0.6166, p = 0.4385; main effect of stress: F1,30 = 10.99, p = 0.0024; interaction effect: F1,30 = 2.988, p = 0.0942; two-way ANOVA)]. It is possible that the experimental procedures and methods could have affected the basic activity observed in the behavioral test. Thus, we measured the locomotor activity of these mice subjected to S-RSDS and/or microinjection into the dorsal striatum and confirmed that experimental protocols had no influence on motor function (Fig. 2i). These results suggest that the stress-responsive factor Shati/Nat8l in the dorsal striatum regulates social stress resilience.

a Cre recombinase was expressed in the injection site (the dorsal striatum) of Shati cKD mice using immunohistochemistry (Cre-positive cells: green, NeuN-positive cells: red). Entire portion (left figure); scale bars: 500 μm. Magnified portion (right figure); scale bars: 100 μm. b Shati/Nat8l mRNA expression in the dorsal striatum decreased in Shati cKD mice compared with mock mice (t5 = 10.59, p = 0.0001). Mock, n = 4; Shati cKD, n = 3; ***p < 0.005 vs. mock mice (student t test). c The timeline of experiments. Mice were exposed to S-RSDS 4 weeks after microinjection. Twenty-four hours after the last session of S-RSDS, the behavioral tests were performed. d Mock mice showed decreased social interaction after S-RSDS. Shati cKD mice did not show decreased social interaction after S-RSDS (main effect of virus: F1,30 = 25.30, p < 0.0001; main effect of stress: F1,30 = 25.71, p < 0.0001; interaction effect: F1,30 = 25.20, p < 0.0001). Naive-mock, n = 9; naive-Shati cKD, n = 8; S-RSDS-mock, n = 9; S-RSDS-Shati cKD, n = 8; ***p < 0.005 vs. naive-mock mice; ###p < 0.005 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests). e The proportion of susceptible mice after S-RSDS was significantly decreased by knockdown of Shati/Nat8l in the dorsal striatum (p = 0.0071). Mock: resillient, n = 2, susceptible, n = 7; Shati cKD: resillient, n = 7, susceptible, n = 1. (Chi-squared test). f Mock mice showed decreased sucrose preference after S-RSDS. Shati cKD mice did not show decreased sucrose preference after S-RSDS (main effect of virus: F1,30 = 9.214, p = 0.3799; main effect of stress: F1,30 = 1.434, p = 0.2405; interaction effect: F1,30 = 9.214, p = 0.0049). Naive-mock, n = 9, Naive-Shati cKD, n = 8; S-RSDS-Mock, n = 9; S-RSDS-Shati cKD, n = 8. *p < 0.05 vs. naive-mock mice; #p < 0.05 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests). g Shati cKD mice did not show extended immobility time in the tail suspension test compared with mock mice after S-RSDS (main effect of virus: F1,30 = 1.028, p = 0.3187; main effect of stress: F1,30 = 1.849, p = 0.184; interaction effect: F1,30 = 6.437, p = 0.0166). Naive-mock, n = 9; naive-Shati cKD, n = 8; S-RSDS-Mock, n = 9; S-RSDS-Shati cKD, n = 8. ##p < 0.01 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests). h Shati cKD mice did not show extended immobility time in the forced swimming test compared with mock mice after S-RSDS (main effect of virus: F1,30 = 0.6166, p = 0.4385; main effect of stress: F1,30 = 10.99, p = 0.0024; interaction effect: F1,30 = 2.988, p = 0.0942;). Naive-mock, n = 9; Naive-Shati cKD, n = 8; S-RSDS-Mock, n = 9; S-RSDS-Shati cKD, n = 8; ##p < 0.05 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests). i There were no differences in locomotor activity. Naive-mock, n = 9; naive-Shati cKD, n = 8; S-RSDS-Mock, n = 9; S-RSDS-Shati cKD, n = 8 (two-way ANOVA with Bonferroni post hoc tests).
We investigated the contribution of Shati/Nat8l in these regions by generating Shati/Nat8l conditional knockdown mice of the mPFC (PFC-Shati cKD mice) and hippocampus (HIP-Shati cKD mice), although we are confident that the expression of Shati/Nat8l in the mPFC and hippocampus does not correlate with stress sensitivity. Shati/Nat8l mRNA levels were decreased in the PFC- and HIP-Shati ckD mice (Fig. S2a, f) (PFC-Shati cKD: t14 = 10.33, p < 0.0001; HIP-Shati cKD: t14 = 7.362, p < 0.0001; Student’s t test). PFC- and HIP-Shati cKD mice did not show stress resilience or susceptibility in any behavioral experiments (Fig. S2b–e, g–j). These findings strengthen the evidence of involvement of Shati/Nat8l in stress sensitivity in the dorsal striatum rather than in the hippocampus and mPFC.
Antagonism of striatal BDNF signaling elevated by RSDS suppresses stress sensitivity
Previous reports have shown that BDNF levels in the mPFC and hippocampus are decreased in depression model mice [42, 43], and we confirmed the similar expression patterns of BDNF mRNA in these regions after RSDS (Fig. S3a, b) (mPFC: F2,21 = 26.19, p < 0.0001; hippocampus: F2,21 = 15.71, p < 0.0001; one-way ANOVA). Thus, we investigated BDNF expression levels in the dorsal striatum, where Shati/Nat8l determines stress sensitivity. Surprisingly, BDNF mRNA levels in the dorsal striatum increased after social defeat stress in susceptible mice in contrast to those in the mPFC and hippocampus (Fig. 3a) (F2,35 = 16.55, p < 0.0001; one-way ANOVA); such elevation was not observed in resilient mice. In addition, increased mRNA levels exhibit a clear correlation with decreased social behavior (Fig. 3b) (r = −0.643, p = 0.0011; Pearson’s r). Protein levels of BDNF were also increased in the dorsal striatum in the susceptible mice, but not the resilient mice, compared with the stress-naive controls (Figs. 3c and S4) (F2,21 = 8.11, p = 0.0025; one-way ANOVA).

a BDNF mRNA in the dorsal striatum of susceptible mice was changed by RSDS compared with naive mice and resilient mice (F2,35 = 16.55, p < 0.0001). Naive, n = 16; resilient, n = 10; susceptible, n = 12; ***p < 0.005 vs. Susceptible mice (one-way ANOVA with Bonferroni post hoc tests). b Social interaction behavior was strongly correlated with BDNF mRNA expression in the dorsal striatum after RSDS (r = −0.643, p = 0.0011). Resilient, n = 10; susceptible, n = 12 (Pearson’s r). c Representative images of BDNF protein. BDNF protein levels in the dorsal striatum of susceptible mice were changed by RSDS compared with naive mice and resilient mice (F2,21 = 8.11, p = 0.0025). Naive, n = 8; resilient, n = 8; susceptible, n = 8; **p < 0.01 vs. susceptible mice (one-way ANOVA with Bonferroni post hoc tests). d Timeline of experiments. ANA-12 (1 µg/0.5 μL per side) or ringer solution was infused 15 min before each social defeat stress session for 10 days. The behavioral tests were performed 24 h after the last session of RSDS. e Ringer-treated mice showed decreased social interaction after RSDS in the social interaction test. ANA-12-treated mice did not show decreased social interaction after RSDS (main effect of virus: F1,25 = 7.924, p = 0.0094; main effect of stress: F1,25 = 12.32, p = 0.0017; interaction effect: F1,25 = 9.785, p = 0.0044). Naive-ringer, n = 8; Naive-ANA-12, n = 7; RSDS-Ringer, n = 7; RSDS-ANA-12, n = 7; ***p < 0.005 vs. Naive-Ringer mice; ###p < 0.005 vs. RSDS-ANA-12 mice (two-way ANOVA with Bonferroni post hoc tests). f Ringer-treated mice showed decreased sucrose preference after RSDS in the sucrose preference test. ANA-12-treated mice did not show decreased sucrose preference after RSDS (main effect of virus: F1,25 = 16.86, p = 0.0004; main effect of stress: F1,25 = 13.98, p = 0.0010; interaction effect: F1,25 = 9.540, p = 0.0049). Naive-Ringer, n = 8; naive-ANA-12, n = 7; RSDS-Ringer, n = 7; RSDS-ANA-12, n = 7. ***p < 0.005 vs. Naive-Ringer mice; ###p < 0.005 vs. RSDS-ANA-12 mice (two-way ANOVA with Bonferroni post hoc tests). g Ringer-treated mice showed extended immobility time after RSDS in the tail suspension test. ANA-12-treated mice did not show extended immobility time after RSDS (main effect of virus: F1,24 = 1.959, p = 0.1744; main effect of stress: F1,24 = 4.771, p = 0.0389; interaction effect: F1,24 = 8.684, p = 0.0070). Naive-Ringer, n = 8; Naive-ANA-12, n = 7; RSDS-Ringer, n = 6; RSDS-ANA-12, n = 7; *p < 0.05 vs. naive-Ringer mice; #p < 0.05 vs. RSDS-ANA-12 mice (two-way ANOVA with the Tukey–Kramer’s post hoc test). h Ringer-treated mice showed extended immobility time after RSDS in the forced swimming test. ANA-12-treated mice did not show extended immobility time after RSDS (main effect of virus: F1,25 = 8.339, p = 0.0079; main effect of stress: F1,25 = 1.936, p = 0.1763; interaction effect: F1,25 = 12.60, p = 0.0016). Naive-Ringer, n = 8; naive-ANA-12, n = 7; RSDS-Ringer, n = 7; RSDS-ANA-12, n = 7; ***p < 0.005 vs. naive-Ringer mice; #p < 0.05 vs. RSDS-ANA-12 mice (two-way ANOVA with Bonferroni post hoc tests). i There were no differences in locomotor activity; naive-Ringer, n = 8; naive-ANA-12, n = 7; RSDS-Ringer, n = 7; RSDS-ANA-12, n = 7 (two-way ANOVA with Bonferroni post hoc tests).
We assessed the effect of ANA-12, a noncompetitive antagonist of the BDNF-specific receptor TrkB to investigate the involvement of striatal BDNF elevated by RSDS in stress resilience. ANA-12 binds selectively and directly to TrkB and blocks the downstream effects of TrkB signaling without affecting TrkA and TrkC [37]. ANA-12 was infused daily into the dorsal striatum 15 min before each social defeat stress exposure for a total of 10 days, followed by behavioral tests (Fig. 3d). ANA-12 was confirmed to confer no effect on social interaction without RSDS (Fig. 3e). While ringer-treated mice with RSDS showed significantly decreased social interaction, ANA-12-treated mice with RSDS did not (Fig. 3e) (main effect of virus: F1,25 = 7.924, p = 0.0094; main effect of stress: F1,25 = 12.32, p = 0.0017; interaction effect: F1,25 = 9.785, p = 0.0044; two-way ANOVA). ANA-12-treated mice with RSDS did not have the lower sucrose preference that was observed in ringer-treated mice (Fig. 3f) (main effect of virus: F1,25 = 16.86, p = 0.0004; main effect of stress: F1,25 = 13.98, p = 0.0010; interaction effect: F1,25 = 9.540, p = 0.0049; two-way ANOVA). The increases of immobility time by RSDS in the tail suspension and forced swimming tests were both suppressed by microinfusion of ANA-12 into the dorsal striatum (Fig. 3g, h) [(tail suspension test: main effect of virus: F1,24 = 1.959, p = 0.1744; main effect of stress: F1,24 = 4.771, p = 0.0389; interaction effect: F1,24 = 8.684, p = 0.0070) (forced swimming test: main effect of virus: F1,25 = 8.339, p = 0.0079; main effect of stress: F1,25 = 1.936, p = 0.1763; interaction effect: F1,25 = 12.60, p = 0.0016; two-way ANOVA)]. Microinfusion of ANA-12 and/or social defeat stress did not affect locomotor activity (Fig. 3i). These results suggest that BDNF-TrkB signaling in the dorsal striatum upregulated by social stress induces stress vulnerability.
Striatal BDNF is a downstream protein of Shati/Nat8l
We investigated the functional relationship of stress sensitivity between BDNF and Shati/Nat8l in the dorsal striatum since both striatal Shati/Nat8l and BDNF regulate stress sensitivity. Notably, BDNF mRNA levels displayed a positive correlation to Shati/Nat8l mRNA levels after social defeat stress (Fig. 4a) (r = 0.753, p < 0.0001; Pearson’s r). In the mPFC and hippocampus, however, no correlations were observed between BDNF mRNA levels and Shati/Nat8l mRNA levels (mPFC: r = 0.175, p = 0.434, hippocampus r = 0.210, p = 0.346; Pearson’s r) (Fig. 4b, c). These results indicate that BDNF regulation by RSDS-induced elevation of Shati/Nat8l occurs specifically in the dorsal striatum.

a BDNF mRNA in the dorsal striatum of susceptible mice was correlated with Shati/Nat8l mRNA expression after RSDS (r = 0.753, p < 0.0001). Resilient n = 10, susceptible n = 12. (Pearson’s r). BDNF mRNA was not correlated with Shati/Nat8l mRNA expression after RSDS in mPFC (b) and hippocampus (c), mPFC: resilient, n = 8; susceptible, n = 8, Hippocampus: resilient, n = 8; susceptible, n = 8 (Pearson’s r). d BDNF mRNA in the dorsal striatum of mock mice increased after S-RSDS compared with naive-mock mice. Shati cKD mice did not show increased BDNF mRNA levels in the dorsal striatum after S-RSDS (main effect of virus: F1,15 = 3.871, p = 0.0679; main effect of stress: F1,15 = 10.86, p = 0.0049; interaction effect: F1,15 = 6.267, p = 0.0243). Naive-mock, n = 6; naive-Shati cKD, n = 4; S-RSDS-Mock, n = 5; S-RSDS-Shati cKD, n = 4; *p < 0.05 vs. naive-mock mice; ##p < 0.01 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests). e Representative images of BDNF protein. BDNF protein in the dorsal striatum of mock mice increased after S-RSDS compared with naive-mock mice. Shati cKD mice after S-RSDS did not show increased BDNF protein levels in the dorsal striatum compared with S-RSDS-mock mice. BDNF protein levels in naive-Shati cKD mice were suppressed compared with naive-mock mice (main effect of virus: F1,20 = 35.48, p < 0.0001; main effect of stress: F1,20 = 137.6, p < 0.0001; interaction effect: F1,20 = 29.41, p < 0.0001). Naive-mock, n = 6; naive-Shati cKD, n = 6; S-RSDS-Mock, n = 6; S-RSDS-Shati cKD, n = 6; ***p < 0.005, **p < 0.01 vs. naive-mock mice; ###p < 0.005 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests).
BDNF levels in Shati cKD mice were examined to determine whether BDNF is a downstream protein of Shati/Nat8l. The increase of BDNF mRNA by RSDS was suppressed in Shati cKD mice (Fig. 4d) (main effect of virus: F1,15 = 3.871, p = 0.0679; main effect of stress: F1,15 = 10.86, p = 0.0049; interaction effect: F1,15 = 6.267, p = 0.0243; two-way ANOVA). The protein levels of BDNF in the dorsal striatum also did not increase after RSDS in the Shati cKD mice (Fig. 4e). BDNF protein levels were decreased in Shati cKD mice, even without RSDS (Figs. 4e and S5) (main effect of virus: F1,20 = 35.48, p < 0.0001; main effect of stress: F1,20 = 137.6, p < 0.0001; interaction effect: F1,20 = 29.41, p < 0.0001; two-way ANOVA). Therefore, BDNF in the dorsal striatum is suggested to be regulated by Shati/Nat8l under exposure to social stress.
Shati/Nat8l epigenetically regulates striatal BDNF expression via histone H3K9 acetylation
Possible epigenetic mechanisms, including acetylation and methylation of histones and DNA methylation, were assessed by ChIP analysis to investigate the mechanisms of elevation of BDNF mRNA by RSDS in the dorsal striatum of susceptible mice. The levels of modifications in the BDNF IV promoter region were caused by altered expression of BDNF IV, but not other variants, under social defeat stress [24]. Five markers of epigenetic modifications were selected: acetylated H3K4 (H3K4ac), acetylated H3K9 (H3K9ac), total acetylated H3 (total H3ac), methylated H3K9 (H3K9me), and methylated DNA (5-mC) in BDNF IV promoter regions. H3K4ac levels were too low to be detected in the mouse brains. H3K9ac levels in the susceptible mice were significantly increased compared with stress-naive mice and resilient mice (Figs. 5a, b and S6a, b) (F2,20 = 22.78, p < 0.0001; one-way ANOVA). This finding is consistent with the increased BDNF levels in the dorsal striatum of susceptible mice after RSDS because histone acetylation generally enhances gene transcription. On the other hand, total H3 acetylation, H3K9me, and 5-mC levels were not changed in any of the three groups (Figs. 5c–e and S6c–e). There were no changes in the mPFC and hippocampus in H3K9ac levels after social defeat stress, unlike in the dorsal striatum (Fig. S7a, b).

a Epigenetic modification levels, including H3K9 acetylation, total histone acetylation, H3K9 methylation and DNA methylation in the BDNF promoter regions in the dorsal striatum were detected by electrophoresis. Quantification of histone H3K9 acetylation (b), total histone H3 acetylation (c), histone H3K9 methylation (d) or DNA methylation (e). Histone H3K9 acetylation in the BDNF IV promoter region in the dorsal striatum of susceptible mice was changed compared with naive mice and resilient mice (F2,20 = 22.78, p < 0.0001). Total histone acetylation, H3K9 methylation, and DNA methylation in the BDNF IV promoter regions in the dorsal striatum were not changed compared with naive mice and resilient mice. H3K9 acetylation: naive, n = 7; resilient, n = 8; susceptible, n = 8, total histone acetylation: naive, n = 8; resilient, n = 8; susceptible, n = 8, H3K9 methylation: naive, n = 8; resilient, n = 8; susceptible, n = 8, DNA methylation: naive, n = 8; resilient, n = 8; susceptible, n = 8; ***p < 0.005 vs. susceptible mice (one-way ANOVA with Bonferroni post hoc tests). f Epigenetic modification levels, including H3K9 acetylation and total histone acetylation in the BDNF IV promoter regions in the dorsal striatum were detected by electrophoresis. Quantification of histone H3K9 acetylation (g) or total histone H3 acetylation (h). Histone H3K9 acetylation in the BDNF IV promoter region in the dorsal striatum of mock mice increased after S-RSDS compared with naive-mock mice. Shati cKD mice after S-RSDS showed a decrease in histone H3K9 acetylation in the BDNF promoter region in the dorsal striatum compared with S-RSDS-mock mice (main effect of virus: F1,12 = 9.036, p = 0.0109; main effect of stress: F1,12 = 56.73, p < 0.0001; interaction effect: F1,12 = 10.58, p = 0.0069). Total histone H3 acetylation were not affected by knockdown of Shati/Nat8l in the dorsal striatum or S-RSDS. H3K9 acetylation: naive-mock, n = 8; naive-Shati cKD, n = 8; S-RSDS-Mock, n = 8; S-RSDS-Shati cKD, n = 8, total histone acetylation: naive-mock, n = 8; naive-Shati cKD, n = 8; S-RSDS-Mock, n = 8; S-RSDS-Shati cKD, n = 8; **p < 0.01 vs. naive-mock mice; ###p < 0.005 vs. S-RSDS-Shati cKD mice (two-way ANOVA with Bonferroni post hoc tests).
We next measured H3K9ac levels in Shati cKD mice to investigate whether Shati/Nat8l epigenetically modulates BDNF mRNA through acetylation of histone H3K9. Elevation of H3K9ac levels after RSDS was abolished by striatal knockdown of Shati/Nat8l (Figs. 5f, g and S8a, b) (main effect of virus: F1,12 = 9.036, p = 0.0109; main effect of stress: F1,12 = 56.73, p < 0.0001; interaction effect: F1,12 = 10.58, p = 0.0069; two-way ANOVA). Total H3 acetylation levels were not changed (Figs. 5h and S8c). Our results indicate that BDNF levels after social defeat stress in the dorsal striatum were controlled by Shati/Nat8l via histone H3K9 acetylation.
Discussion
The elucidation of the mechanisms regulating stress sensitivity is expected to facilitate the development of therapeutic strategies for depression. However, the underlying molecular mechanisms that contribute to resilience against chronic stress have remained poorly understood. In this study, we identified the elevation of striatal Shati/Nat8l and BDNF expression in mice susceptible to social stress and that knockdown of Shati/Nat8l or blockade of BDNF signaling in the dorsal striatum strongly induces stress resilience. BDNF expression and related histone acetylation were decreased in Shati cKD mice. These results demonstrate that the Shati/Nat8l–BDNF pathway in the dorsal striatum contributes to social stress sensitivity.
Although the protocol of social defeat stress in female mice has been established [44, 45], male mice were used in this study because this protocol is complex and the aggressivity of ICR mice against female mice was not sufficient to expose S-RSDS. Furthermore, we aimed to investigate the Shati/Nat8l–BDNF pathway in stress sensitivity without the possible effects of the estrous cycle. Therefore, male mice were used to expose the validated social stress by ICR mice and prevent the estrous cycle effects in depression pathogenesis. However, our study does not deny the necessity of an experiment in female mice. A future study in female mice with appropriate procedures to target striatal Shati/Nat8l–BDNF pathway is warranted.
RSDS is a well-established method for generating a mouse model for depression in terms of expression, construction, and predictive validity. It has been reported that depression model mice subjected to social defeat stress show reduction in social interaction ability, anhedonia [46], and helplessness [47] under the stressor environment, which are reflective of the symptoms of human depression and were rescued by antidepressant treatment using like selective serotonin reuptake inhibitors (SSRI) [48, 49]. Herein, we demonstrated that Shati/Nat8l mRNA levels were increased by RSDS only in the dorsal striatum of susceptible mice. Importantly, the dispersion in the expression of striatal Shati/Nat8l in the stress-naive group was not confirmed, suggesting that the increase of Shati/Nat8l was induced in response to social defeat stress. In addition, we showed that striatal knockdown of Shati/Nat8l, which was increased by RSDS only in the dorsal striatum of susceptible mice, induced resilience to social stress in behavioral tests. These results are consistent with the findings of our previous study in which overexpression of Shati/Nat8l in the dorsal striatum resulted in vulnerability to RSDS and even subthreshold social stress [31]. Our findings indicate that the strong involvement of striatal Shati/Nat8l in stress sensitivity. Shati/Nat8l has been investigated as the molecule related to addiction [50, 51] and cognitive function [52, 53]. The evidence from this study affords a deep understanding of the crucial role of Shati/Nat8l in sensitivity to social stress.
BDNF is involved in the pathogenesis of many psychiatric diseases such as schizophrenia, autism, and depression [54,55,56]. BDNF is a well-known neurotrophic factor that plays important roles in learning and memory as well as neuronal survival [57,58,59]. BDNF in the hippocampus and mPFC have significant functions in antidepressant effects [18], and its expression is reduced in those regions in depressive patients and mice [60, 61]. In contrast, BDNF is upregulated in NAc of depressive subjects and exerts pro-depressant effects [62, 63]. These results suggest that BDNF has brain region-specific functions related to stress response and disease development. In this study, enhanced BDNF production in the dorsal striatum by RSDS was observed in susceptible mice but not resilient mice. In addition, blocking of BDNF signaling in the dorsal striatum induced a strong resilience to RSDS. This is the first report on the action of BDNF in the dorsal striatum in an animal model of depression.
There had been no reports of the dorsal striatal function in stress sensitivity prior to our previous study [31]. The present study provides proof that the dorsal striatum plays an important role in stress sensitivity. Recently, the number of reports that demonstrated the involvement of the dorsal striatum in emotional behaviors, motivation, and negative-decision making have been increasing [64, 65]. Networks regulate such functions with other brain regions [66, 67]. The involvement of mPFC as a modulator of stress sensitivity among networks within the dorsal striatum is possible. Dendritic atrophy in the mPFC by chronic stress is reportedly correlated with dorsolateral striatum hypertrophy, thus affecting decision making [68]. mPFC atrophy was confirmed in depressive patients exposed to stress [69], whereas the dendritic shortening and reduced spine density in mPFC neurons by RSDS was observed only in the stress-susceptible group [70]. Furthermore, the hypertrophy of the dorsolateral striatum by chronic stress is consistent with our results showing striatal BDNF upregulation by RSDS because BDNF is known to be essential for neurogenesis. We also focused on the dorsal raphe nucleus. Neurons in the dorsal raphe nucleus are rich in serotonin, whose shortage is strongly related to depression, and SSRI supplementation is the first-line treatment. Indeed, the hereditary reduction of serotonin in the brain induces social defeat stress in vulnerable mice [71]. In our previous study, overexpression of Shati/Nat8l in the dorsal striatum caused stress susceptibility accompanied by a decrease in serotonin [14]. Such impairment of stress sensitivity was recovered by activation of the dorsal raphe nucleus or systemic administration of SSRI [31]. These reports strongly suggest that overexpression of Shati/Nat8l in the dorsal striatum inactivates serotonergic systems from the dorsal raphe nucleus, followed by suppression of serotonin release. In this context, resilience to RSDS in Shati cKD mice is probably caused by activation of the serotonergic system from the dorsal raphe nucleus. We showed that Shati/Nat8l–BDNF pathways regulate stress sensitivity. This result indicates that the serotonergic system from the dorsal raphe nucleus could be downstream of the Shati/Nat8l–BDNF pathways in the dorsal striatum, and the controlled serotonin release in the brain by striatal Shati/Nat8l–BDNF pathways determines sensitivity to stress.
However, note that our results do not exclude the possibility that other brain regions are also related to depression pathogenesis. In fact, we reported decreased BDNF in the mPFC and hippocampus of susceptible mice after social defeat stress. The involvement of BDNF in the mPFC and hippocampus in depression were established [42, 72], and consistent results regarding these regions were obtained here. In addition, the mesolimbic system, including VTA-NAc circuits, might be involved in depression pathogenesis. Our results showed pro-depressant function of striatal BDNF similar to that of BDNF/TrkB signaling in the VTA-NAc dopaminergic circuit in socially defeated mice [73]. Although our previous study could not confirm Shati/Nat8l alteration in the NAc in certain depression model mice, including social defeated mice [14, 31], considering the interaction between the dorsal striatum and NAc, striatal BDNF signaling might interact with neuronal circuits in the NAc. The detailed mechanisms underlying this negative regulation of stress sensitivity by striatal BDNF must be clarified in the future.
Previous studies reported that epigenetic regulation is greatly involved in altering BDNF expression [74, 75]. Our study showed that histone acetylation was enhanced in stress-susceptible mice, whereas histone and DNA methylation were not affected. We strongly suggest that histone acetylation regulates the elevation of striatal BDNF expression by RSDS. We selected the BDNF IV promoter because it is the most well-characterized BDNF promoter; epigenetic modifications at this site are altered in patients with depression and stress-induced depression model mice, and these alterations were reversed by antidepressant treatments [25, 76]. The increase of H3K9ac levels in the BDNF IV promoter region in the dorsal striatum in susceptible mice is consistent with the increase of BDNF in those mice, as histone acetylation loosens chromatin structures to facilitate the gene transcription. In addition, Shati/Nat8l exists upstream of BDNF. Shati/Nat8l dominantly produces NAA [11], which is finally converted to acetyl-CoA [12]. One possibility of enhanced acetylation of H3K9 in susceptible mice is that NAA production elevated by upregulated Shati/Nat8l, followed by an increase of acetyl-CoA, a substrate of histone acetylation. In fact, we confirmed that the NAA levels in the dorsal striatum are increased in Shati/Nat8l-overexpression mice, which exhibit vulnerability to social stress [31]. Another possibility is that Shati/Nat8l affects histone deacetylase (HDAC). It is reported that social defeat stress decreases HDACs levels [77]. Decreased HDACs could be mediated by Shati/Nat8l, which might be an underlying mechanism of enhanced histone acetylation. All of these possibilities should be considered when elucidating the pathogenesis of depression.
In this study, we showed that BDNF mRNA and H3K9 acetylation levels are regulated by Shati/Nat8l in the dorsal striatum, and striatal BDNF-TrkB signaling plays an important role in stress sensitivity. However, whether BDNF regulated by histone acetylation and synthesized in the dorsal striatum is directly involved in stress sensitivity remains unclear. VTA-dorsal medial striatal dopaminergic neuronal projections could release BDNF [73]. Therefore, TrkB activation in VTA-dorsal medial striatal dopaminergic neuron projections, followed by upregulation of BDNF release, might also be involved in stress sensitivity.
Finally, we discussed the reinforcement of resilience for stress suppresses the progression of depression from a clinical standpoint. Reduction of damage from stress would synergize therapeutics for depression and accelerate the recovery of its pathology. In this study, we clarified the contribution of BDNF in the striatum to stress sensitivity. Thus, the decrease of striatal BDNF is an appealing therapeutic strategy for depression. However, it is difficult to directly target BDNF itself for treatment in depression because BDNF also has antidepressant effects in other brain regions, including the mPFC and hippocampus. Considering that the Shati/Nat8l–BDNF pathway specifically works to the dorsal striatum in response to stress, modulation of Shati/Nat8l would allow BDNF to be striatum specifically reduced. Our series of studies provide evidence to suggest that the striatal Shati/Nat8l–BDNF pathway regulates the dorsal raphe nucleus, which controls the serotonergic system in the brain. Recently, R-ketamine has received focus as an effective approach to treating depression, and a single treatment of ketamine showed rapid and sustained antidepressant effects even in patients with treatment resistance [78]. This effect of R-ketamine is reported to be mediated by the activation of serotoninergic systems in the dorsal raphe nucleus through the projection from mPFC via AMPA receptor stimulation in the mPFC [79], although other circuits have been reported. However, safety in long-term treatment must be taken into consideration because ketamine has psychological side effects such as dissociation and as a psychotomimetic and has abuse potential [80]. Targeting the dorsal raphe nucleus, which regulates serotonin in the whole brain, by Shati/Nat8l indicates a potent antidepressant efficacy similar to ketamine. Therefore, Shati/Nat8l is a promising candidate as a novel target of treatment for depression by modulating stress sensitivity.
Our results demonstrate that the Shati/Nat8l–BDNF pathways in the dorsal striatum determine sensitivity to stress in the pathogenesis of depression via epigenetic regulation. We have provided the first evidence supporting the role of striatal BDNF in depression pathology. The present results further indicate that Shati/Nat8l–BDNF signaling in the dorsal striatum should be considered a potential new target for treating depression and might establish therapeutic strategies for all patients, including those with treatment resistance.
Funding and disclosure
This research was supported by the grant-in-aid for Scientific Research (KAKENHI) (B) [JSPS KAKENHI Grant Number, 26293213, JP21H02632] (SM, AN), Kobayashi Foundation (AN), and Smoking Research Foundation Grant for Biomedical Research and Foundation (AN). HM has been supported by Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan. The funders had no role in study design, data collection or analysis, decision to publish, or preparation of the manuscript. The authors declare no competing interests.
References
Al-Harbi KS. Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer Adherence. 2012;6:369–88.
Keller MB, Gelenberg AJ, Hirschfeld RM, Rush AJ, Thase ME, Kocsis JH, et al. The treatment of chronic depression, part 2: a double-blind, randomized trial of sertraline and imipramine. J Clin Psychiatry. 1998;59:598–607.
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 2008;455:894–902.
Yang L, Zhao Y, Wang Y, Liu L, Zhang X, Li B, et al. The effects of psychological stress on depression. Curr Neuropharmacol. 2015;13:494–504.
Monroe SM, Harkness KL. Life stress, the “kindling” hypothesis, and the recurrence of depression: considerations from a life stress perspective. Psychol Rev. 2005;112:417–45.
Technow JR, Hazel NA, Abela JR, Hankin BL. Stress sensitivity interacts with depression history to predict depressive symptoms among youth: prospective changes following first depression onset. J Abnorm Child Psychol. 2015;43:489–501.
Pearson-Leary J, Zhao C, Bittinger K, Eacret D, Luz S, Vigderman AS, et al. The gut microbiome regulates the increases in depressive-type behaviors and in inflammatory processes in the ventral hippocampus of stress vulnerable rats. Mol Psychiatry. 2019;25:1068–79.
Golden SA, Covington HE 3rd, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat Protoc. 2011;6:1183–91.
Niwa M, Nitta A, Mizoguchi H, Ito Y, Noda Y, Nagai T, et al. A novel molecule “shati” is involved in methamphetamine-induced hyperlocomotion, sensitization, and conditioned place preference. J Neurosci. 2007;27:7604–15.
Haddar M, Uno K, Azuma K, Muramatsu SI, Nitta A. Inhibitory effects of Shati/Nat8l overexpression in the medial prefrontal cortex on methamphetamine‐induced conditioned place preference in mice. Addict Biol. 2020;25:e12749.
Toriumi K, Mamiya T, Song Z, Honjo T, Watanabe H, Tanaka J, et al. Deletion of SHATI/NAT8L decreases the N-acetylaspartate content in the brain and induces behavioral deficits, which can be ameliorated by administering N-acetylaspartate. Eur Neuropsychopharmacol. 2015;25:2108–17.
Singhal NK, Huang H, Li S, Clements R, Gadd J, Daniels A, et al. The neuronal metabolite NAA regulates histone H3 methylation in oligodendrocytes and myelin lipid composition. Exp Brain Res. 2017;235:279–92.
Becker I, Lodder J, Gieselmann V, Eckhardt M. Molecular characterization of N-acetylaspartylglutamate synthetase. J Biol Chem. 2010;285:29156–64.
Miyamoto Y, Iegaki N, Fu K, Ishikawa Y, Sumi K, Azuma S, et al. Striatal N-acetylaspartate synthetase Shati/Nat8l regulates depressive behaviors via mGluR3-mediated serotonergic suppression in mice. Int J Neuropsychopharmacol. 2017;20:1027–35.
Miyanishi H, Uno K, Iwata M, Kikuchi Y, Yamamori H, Yasuda Y, et al. Investigating DNA methylation of SHATI/NAT8L promoter sites in blood of unmedicated patients with major depressive disorder. Biol Pharm Bull. 2020;43:1067–72.
Reynolds LM, Reynolds GP. Differential regional N-acetylaspartate deficits in postmortem brain in schizophrenia, bipolar disorder and major depressive disorder. J Psychiatr Res. 2011;45:54–9.
Caviedes A, Lafourcade C, Soto C, Wyneken U. BDNF/NF-κB signaling in the neurobiology of depression. Curr Pharm Des. 2017;23:3154–63.
Zhang JC, Yao W, Hashimoto K. Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr Neuropharmacol. 2016;14:721–31.
Boulle F, Kenis G, Cazorla M, Hamon M, Steinbusch HW, Lanfumey L, et al. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog Neurobiol. 2012;98:197–206.
Covington HE 3rd, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 2011;71:656–70.
Tadic A, Muller-Engling L, Schlicht KF, Kotsiari A, Dreimüller N, Kleimann A, et al. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol Psychiatry. 2014;19:281–3.
Jiang C, Lin WJ, Sadahiro M, Labonté B, Menard C, Pfau ML, et al. VGF function in depression and antidepressant efficacy. Mol Psychiatry. 2017;23:1632–42.
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–61.
Xu H, Wang J, Zhang K, Zhao M, Ellenbroek B, Shao F, et al. Effects of adolescent social stress and antidepressant treatment on cognitive inflexibility and Bdnf epigenetic modifications in the mPFC of adult mice. Psychoneuroendocrinology. 2018;88:92–101.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–25.
Dong E, Tueting P, Matrisciano F, Grayson DR, Guidotti A. Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl Psychiatry. 2016;6:e711.
Iida A, Takino N, Miyauchi H, Shimazaki K, Muramatsu S. Systemic delivery of tyrosine-mutant AAV vectors results in robust transduction of neurons in adult mice. Biomed Res Int. 2013:2013;974819.
Krzyzosiak A, Szyszka-Niagolov M, Wietrzych M, Gobaille S, Muramatsu S, Krezel W. Retinoid x receptor gamma control of affective behaviors involves dopaminergic signaling in mice. Neuron. 2010;66:908–20.
Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Compact. 3rd ed. Amsterdam: Elsevier; 2008.
Fox ME, Lobo MK. The molecular and cellular mechanisms of depression: a focus on reward circuitry. Mol Psychiatry. 2019;24:1798–815.
Uno K, Miyanishi H, Sodeyama K, Fujiwara T, Miyazaki T, Muramatsu SI, et al. Vulnerability to depressive behavior induced by overexpression of striatal Shati/Nat8l via the serotonergic neuronal pathway in mice. Behav Brain Res. 2019;376:112227.
Heshmati M, Christoffel DJ, LeClair K, Cathomas F, Golden SA, Aleyasin H, et al. Depression and social defeat stress are associated with inhibitory synaptic changes in the nucleus accumbens. J Neurosci. 2020;40:6228–33.
Furukawa-Hibi Y, Nitta A, Fukumitsu H, Somiya H, Furukawa S, Nabeshima T, et al. Overexpression of piccolo C2A domain induces depressive behaviors in mice. Neuroreport. 2010;21:1177–81.
Sumi K, Uno K, Matsumura S, Miyamoto Y, Furukawa-Hibi Y, Muramatsu SI, et al. Induction of neuronal axon outgrowth by Shati/Nat8l by energy metabolism in mice cultured neurons. Neuroreport. 2015;26:740–6.
Sumi K, Uno K, Noike H, Tomohiro T, Hatanaka Y, Furukawa-Hibi Y, et al. Behavioral impairment in SHATI/NAT8L knockout mice via dysfunction of myelination development. Sci Rep. 2017;7:16872.
Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014;17:27–9.
Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–57.
Koo JW, Labonté B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–78.
Uno K, Miyazaki T, Sodeyama K, Miyamoto Y, Nitta A. Methamphetamine induces Shati/Nat8L expression in the mouse nucleus accumbens via CREB- and dopamine D1 receptor-dependent mechanism. PLoS ONE. 2017;12:e0174196.
Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91.
Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149–60.
Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, et al. Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology. 2016;233:3647–57.
Qiao H, An SC, Xu C, Ma XM. Role of proBDNF and BDNF in dendritic spine plasticity and depressive-like behaviors induced by an animal model of depression. Brain Res. 2017;1663:29–37.
Harris AZ, Atsak P, Bretton ZH, Holt ES, Alam R, Morton MP, et al. A novel method for chronic social defeat stress in female mice. Neuropsychopharmacology. 2018;43:1276–83.
van Doeselaar L, Yang H, Bordes J, Brix L, Engelhardt C, Tang F, et al. Chronic social defeat stress in female mice leads to sex-specific behavioral and neuroendocrine effects. Stress. 2021;24:168–80.
Li X, Wang H, Chen Q, Li Z, Liu C, Yin S, et al. Felbamate produces antidepressant-like actions in the chronic unpredictable mild stress and chronic social defeat stress models of depression. Fundam Clin Pharm. 2019;33:621–33.
Yang B, Ren Q, Ma M, Chen QX. Hashimoto K antidepressant effects of (+)-MK-801 and (−)-MK-801 in the social defeat stress model. Int J Neuropsychopharmacol. 2016;19:pyw080.
Kudryashov NV, Kalinina TS, Shimshirt AA, Korolev AO, Volkova AV, Voronina TA. Antidepressant-like effect of fluoxetine may depend on translocator protein activity and pretest session duration in forced swimming test in mice. 2018;29:375–8.
Ripoll N, David DJ, Dailly E, Hascoët M, Bourin M. Antidepressant-like effects in various mice strains in the tail suspension test. Behav Brain Res. 2003;143:193–200.
Haddar M, Uno K, Hamatani K, Muramatsu SI, Nitta A. Regulatory system of mGluR group II in the nucleus accumbens for methamphetamine‐induced dopamine increase by the medial prefrontal cortex. Neuropsychopharmacol Rep. 2019;39:209–16.
Toriumi K, Kondo M, Nagai T, Hashimoto R, Ohi K, Song Z, et al. Deletion of SHATI/NAT8L increases dopamine D1 receptor on the cell surface in the nucleus accumbens, accelerating methamphetamine dependence. Int J Neuropsychopharmacol. 2014;17:443–53.
Haddar M, Azuma K, Izuo N, Uno K, Asano T, Muramatsu SI, et al. Impairment of cognitive function induced by Shati/Nat8l overexpression in the prefrontal cortex of mice. Behav Brain Res. 2021;397:112938.
Toriumi K, Tanaka J, Mamiya T, Alkam T, Kim HC, Nitta A. Shati/Nat8l knockout mice show behavioral deficits ameliorated by atomoxetine and methylphenidate. Behav Brain Res. 2018;339:207–14.
Armeanu R, Mokkonen M, Crespi B. Meta-analysis of BDNF levels in autism. Cell Mol Neurobiol. 2017;37:949–54.
Molendijk ML, Spinhoven P, Polak M, Bus BA, Penninx BW, Elzinga BM. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol Psychiatry. 2014;19:791–800.
Zhang Y, Fang X, Fan W, Tang W, Cai J, Song L, et al. Interaction between BDNF and TNF-α genes in schizophrenia. Psychoneuroendocrinology. 2018;89:1–6.
Numakawa T, Odaka H, Adachi N. Actions of brain-derived neurotrophin factor in the neurogenesis and neuronal function, and its involvement in the pathophysiology of brain diseases. Int J Mol Sci. 2018;19:E3650.
Radiske A, Rossato JI, Gonzalez MC, Köhler CA, Bevilaqua LR, Cammarota M. BDNF controls object recognition memory reconsolidation. Neurobiol Learn Mem. 2017;142:79–84.
Vignoli B, Battistini G, Melani R, Blum R, Santi S, Berardi N, et al. Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. 2016;92:873–87.
Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47.
Pandey GN, Ren X, Rizavi HS, Conley RR, Roberts RC, Dwivedi Y. Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int J Neuropsychopharmacol. 2008;11:1047–61.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8.
Amemori KI, Amemori S, Gibson DJ, Graybiel AM. Striatal microstimulation induces persistent and repetitive negative decision-making predicted by striatal beta-band oscillation. Neuron. 2018;99:829–41.e6.
Lamers A, Toepper M, Fernando SC, Schlosser N, Bauer E, Woermann F, et al. Nonacceptance of negative emotions in women with borderline personality disorder: association with neuroactivity of the dorsal striatum. J Psychiatry Neurosci. 2019;44:303–12.
Costa G, Serra M, Marongiu J, Morelli M, Simola N. Influence of dopamine transmission in the medial prefrontal cortex and dorsal striatum on the emission of 50-kHz ultrasonic vocalizations in rats treated with amphetamine: effects on drug-stimulated and conditioned calls. Prog Neuropsychopharmacol Biol Psychiatry. 2020;97:109797.
Lago T, Davis A, Grillon C, Ernst M. Striatum on the anxiety map: small detours into adolescence. Brain Res. 2017;1654:177–84.
Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ, et al. Chronic stress causes frontostriatal reorganization and affects decision-making. Science. 2009;325:625–5.
Belleau EL, Treadway MT, Pizzagalli DA. The impact of stress and major depressive disorder on hippocampal and medial prefrontal cortex morphology. Biol Psychiatry. 2019;85:443–53.
Shinohara R, Taniguchi M, Ehrlich AT, Yokogawa K, Deguchi Y, Cherasse Y, et al. Dopamine D1 receptor subtype mediates acute stress-induced dendritic growth in excitatory neurons of the medial prefrontal cortex and contributes to suppression of stress susceptibility in mice. Mol Psychiatry. 2018;23:1717–30.
Sachs BD, Ni JR, Caron MG. Brain 5-HT deficiency increases stress vulnerability and impairs antidepressant responses following psychosocial stress. Proc Natl Acad Sci U S A. 2015;112:2557–62.
Fang W, Zhang J, Hong L, Huang W, Dai X, Ye Q, et al. Metformin ameliorates stress-induced depression-like behaviors via enhancing the expression of BDNF by activating AMPK/CREB-mediated histone acetylation. J Affect Disord. 2020;260:302–13.
Koo JW, Chaudhury D, Han MH, Nestler EJ. Role of mesolimbic brain-derived neurotrophic factor in depression. Biol Psychiatry. 2019;86:738–48.
Li J, Zhang DD, Wang CQ, Shi M, Wang LL. Protective effects of low-intensity pulsed ultrasound on aluminum overload-induced cerebral damage through epigenetic regulation of brain-derived neurotrophic factor expression. Biosci Rep. 2019;39:BSR20181185.
Viana Borges J, Souza de Freitas B, Antoniazzi V, de Souza Dos Santos C, Vedovelli K, Naziaseno Pires V, et al. Social isolation and social support at adulthood affect epigenetic mechanisms, brain-derived neurotrophic factor levels and behavior of chronically stressed rats. Behav Brain Res. 2019;366:36–44.
Seo MK, Ly NN, Lee CH, Cho HY, Choi CM, Nhu LH, et al. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology. 2016;105:388–97.
Karnib N, El-Ghandour R, El Hayek L, Nasrallah P, Khalifeh M, Barmo N, et al. Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology. 2019;44:1152–62.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64.
Fukumoto K, Iijima M, Chaki S. The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5-HT neurons in the DRN. Neuropsychopharmacology. 2016;41:1046–56.
Shin C, Kim YK. Ketamine in major depressive disorder: mechanisms and future perspectives. Psychiatry Investig. 2020;17:181–92.
Acknowledgements
We thank Naomi Takino and Mika Ito for technical assistance in producing the Shati/Nat8l AAV vectors. We also thank Dr Naotaka Izuo for the technical assistance in writing the manuscript.
Author information
Authors and Affiliations
Contributions
HM and AN designed the project, HM conducted all experiments, SM provided the AAV-Cre vectors, HM analyzed the data. HM wrote the draft of manuscript. AN revised and did form the final manuscript.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Miyanishi, H., Muramatsu, Si. & Nitta, A. Striatal Shati/Nat8l–BDNF pathways determine the sensitivity to social defeat stress in mice through epigenetic regulation. Neuropsychopharmacol. 46, 1594–1605 (2021). https://doi.org/10.1038/s41386-021-01033-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01033-2
This article is cited by
-
Cannabinoid Type 1 Receptors in the Basolateral Amygdala Regulate ACPA-Induced Place Preference and Anxiolytic-Like Behaviors
Neurochemical Research (2022)
-
Knockdown of Piccolo in the Nucleus Accumbens Suppresses Methamphetamine-Induced Hyperlocomotion and Conditioned Place Preference in Mice
Neurochemical Research (2022)
-
Shati/Nat8l Overexpression Improves Cognitive Decline by Upregulating Neuronal Trophic Factor in Alzheimer’s Disease Model Mice
Neurochemical Research (2022)
-
N-Acetyl Transferase, Shati/Nat8l, in the Dorsal Hippocampus Suppresses Aging-induced Impairment of Cognitive Function in Mice
Neurochemical Research (2022)
