
Abstract
To flourish, cancers greatly depend on their surrounding tumor microenvironment (TME), and cancer-associated fibroblasts (CAFs) in TME are critical for cancer occurrence and progression because of their versatile roles in extracellular matrix remodeling, maintenance of stemness, blood vessel formation, modulation of tumor metabolism, immune response, and promotion of cancer cell proliferation, migration, invasion, and therapeutic resistance. CAFs are highly heterogeneous stromal cells and their crosstalk with cancer cells is mediated by a complex and intricate signaling network consisting of transforming growth factor-beta, phosphoinositide 3-kinase/AKT/mammalian target of rapamycin, mitogen-activated protein kinase, Wnt, Janus kinase/signal transducers and activators of transcription, epidermal growth factor receptor, Hippo, and nuclear factor kappa-light-chain-enhancer of activated B cells, etc., signaling pathways. These signals in CAFs exhibit their own special characteristics during the cancer progression and have the potential to be targeted for anticancer therapy. Therefore, a comprehensive understanding of these signaling cascades in interactions between cancer cells and CAFs is necessary to fully realize the pivotal roles of CAFs in cancers. Herein, in this review, we will summarize the enormous amounts of findings on the signals mediating crosstalk of CAFs with cancer cells and its related targets or trials. Further, we hypothesize three potential targeting strategies, including, namely, epithelial–mesenchymal common targets, sequential target perturbation, and crosstalk-directed signaling targets, paving the way for CAF-directed or host cell-directed antitumor therapy.
Similar content being viewed by others


Introduction
Cancer, as a major public health problem worldwide, is the second leading cause of death with an estimated 10.0 million globally in 2020.1,2 Majority of cancer deaths from cancers are caused by local recurrence and/or distant organ/tissue metastasis.3,4 If the cancers are identified in the early stage and occur in the original lesion site, the total 5-year relative survival rate of the ten most common cancers is ~34.2–100%, with a local recurrence rate of <16.1% after surgery, radiation, and/or chemotherapy, while the total 5-year relative survival rate drops to 2.5–30.2% for advanced cancers with frequent recurrences and/or metastasis,5,6 thereby requiring more aggressive treatments, including immunotherapy, biological therapy, or targeted therapy, etc. However, the recurrent or metastatic cancers can exhibit quick progression and/or become resistant to therapeutic strategies, which are exclusively or mainly aimed at cancer cells. One of the main reasons for the failure of cancer treatment is that the tumor microenvironment (TME) is fully or partially ignored in the development of antitumor therapy.7 Since cancer progression is highly associated with the physiological state of TME, targeting nonneoplastic stromal components that substantially contribute to tumor progression are considered for cancer treatment.8
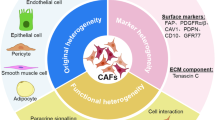
Cancer-associated fibroblasts (CAFs), as the major components of TME, have been extensively explored and are known to be involved in diverse cellular processes, including cell differentiation, proliferation, and stemness; extracellular matrix (ECM) remodeling; and cell migration and apoptosis, all of which can exert critical roles in tumor biological behaviors, including tumorigenesis, tumor growth, energy metabolism, tumor immunity, angiogenesis, tumor progression, recurrence, and metastasis.9,10,11 The biological activities of CAFs are mediated by various intracellular and extracellular factors, especially those in signaling pathways closely related to cancer progression, which might be targeted for anticancer therapy. Since CAFs exert molecular and functional heterogeneity in different cancers and even in different stages of the same type of tumor and because of the specific crosstalk between CAFs and cancer cells,12 any therapeutic strategies developed should exploit the specificity and diversity of CAFs to optimize treatment efficacy for targeted therapy. To better understand the nature of CAFs, herein, we summarize historic milestones of the basic research and clinical studies on CAFs, especially those focused on precursors of CAFs and CAFs isolation, heterogeneity, signaling pathways, and involvement in cancer therapy and therapy resistance, and suggest new potential therapeutic strategies.
Definition of fibroblasts
Fibroblasts were firstly identified in the 1850s as spindle-shaped cells in connective tissue that can synthesize collagen13 and originated mostly from the primitive mesenchyme of the mesoderm,14 and, in some cases, partially from the neural crest of the ectoderm.15,16 Indeed, the definition of fibroblasts is constantly changing. It is widely accepted that fibroblasts in normal tissues are generally embedded within fibrillar ECM as single resting mesenchymal cells.17 They are also defined by their cell morphology, tissue location, and lack of the lineage markers that are expressed by other cells, such as epithelial cells and leukocytes.15 Currently, vimentin is widely used to compare fibroblasts with cells expressing epithelial markers, and fibroblast‐specific protein 1 (FSP1, also known as S100A4) is considered as a reliable marker for quiescent fibroblasts.18 However, vimentin and FSP1 are also expressed by cells in mesenchymal lineages in addition to fibroblasts; thus, cellular shape and location are frequently combined for the identification of fibroblasts,19,20,21,22,23 demonstrating that details on the lineage of fibroblasts remain to be determined.
Quiescent fibroblasts in the interstitial space are the major producers of ECM under normal physiological conditions and can be reversibly activated to facilitate repair and regeneration in response to tissue damage.24 Preceding their functioning in the regeneration stage, quiescent fibroblasts are activated into myofibroblasts and then accumulate at the sites of repair for wound healing.25 In these cases, activated fibroblasts secrete transforming growth factor-beta (TGF-β) and acquire a contractile phenotype via the expression of α-smooth muscle actin (α-SMA), thereby effectively closing wounds.26 In addition, fibroblasts are also critical for the homeostasis of adjacent epithelial cells, acting in an indirect paracrine manner, similar to that of growth factors27,28 or via direct mesenchymal–epithelial cell interactions.29,30 In angiogenesis with increased production of vascular endothelial growth factor A (VEGFA),31 the immune response, and keratinocyte proliferation, fibroblasts play roles by secreting cytokines and chemokines.27,32 Further, ECM development mediated by fibroblasts in lymph nodes acts as a “highway” to transport potential antigens and contributes to the migration of leukocytes,33 indicating that the structural roles of fibroblasts allow effective immune responses. Interestingly, when wounds heal, activated fibroblasts are restored to the quiescent phenotype owing to apoptosis,34 indicating that reversibility is a hallmark feature of fibroblasts associated with tissue repair.
From fibroblasts to CAFs in TME
Cellular sources and heterogeneity of CAFs
Cancers, as ongoing and unabated injurious stimuli, initiate fibroblasts irreversibility transition, driving acquisition of cancer-associated phenotypes (Fig. 1). The irreversibility transitions could be driven in a variety of ways. First, TME as a reservoir of growth factors, cytokines, and other factors signals to resident fibroblasts contributing to the transformation of normal fibroblasts (NFs) to CAFs. A diverse set of factors, including TGF-β1, osteopontin (OPN), and interleukin-1β (IL-1β), etc., which are released from cancer cells and/or immune cells,35 induce the transition of stromal fibroblasts to CAFs by regulating the TGF-β and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways.36,37,38,39 Then, exosomes also play essential roles in cellular communications, promoting fibroblasts to acquire new receptors or even genetic material from the cancer cells. Cancer-derived exosomes shuttling cargos such as microRNAs (miRNAs), long noncoding RNA (lncRNA) Gm26809, or TGF-β1 to reprogram NFs into CAFs via the downstream mitogen-activated protein kinase (MAPK), NF-κB, signal transducers and activators of transcription 3 (STAT3), or TGF-β signaling cascades.38,40,41,42 Also, a shift in energy metabolism such as aerobic glycolysis is potentially considered as a priming event in the conversion of NFs into CAFs. Lysophosphatidic acid (LPA), TGF-β1, or platelet-derived growth factor (PDGF) from cancer cells are able to induce aerobic glycolysis of fibroblasts via hypoxia-inducible factor-1α (HIF-1α) pathway; fibroblasts can also be metabolically reprogrammed via caveolin-1 (CAV-1) downregulation or cancer cell-derived mitochondrial transfer.43,44,45,46 In addition, various evidence has proven that the conversion of NFs into CAFs is accompanied by changes in the self-expression of certain components. For instance, Yes-associated protein 1 (YAP1) in NFs, as a transcriptional coactivator, modulates the transcription of SRC by forming a protein compound with TEA domain transcription factor-1 (TEAD1), resulting in cytoskeletal protein activation and ultimately transformation into CAFs.47 Overall, induced by cancer cells and TME, etc., NFs are activated to CAFs. Compared to resting NFs, CAFs acquire enhanced proliferative and secretory capabilities, which contribute to ECM remodeling, autocrine activation, and immunomodulatory function. Activated CAFs are characterized by different markers that are expressed at low levels or not expressed in NFs (Table 1). Among them, all or part of a combination with α-SMA, fibroblast-activated protein (FAP), and PDGF receptor α/β (PDGFRα/β) could be used to distinguish CAFs from NFs in cancers. Some markers, since CAFs are suggested to represent a heterogeneous population of cells,48 are required to characterize this heterogeneity.

Conversion from normal fibroblasts (NFs) to cancer-associated fibroblasts (CAFs). a Grow factors and cytokines such as transforming growth factor-beta 1 (TGF-β1), osteopontin (OPN), and IL-1β combined with their reporters in NFs, then activated the downstream effector including miRNAs and CD44, etc. to regulate the targeted gene expression of CAFs through TGF-β/Smads and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways. b Cancer-derived exosomes shuttling cargos such as miRNAs and lncRNAs transformed NFs to CAFs via the downstream signals including TGF-β/Smads, Janus kinase/signal transducers and activators of transcription (JAK/STAT), NF-κB and mitogen-activated protein kinase (MAPK) cascades. c NF-CAF conversion was driven by glucose metabolism reprogramming and hypoxia-inducible factor-1α (HIF-1α) signaling pathway was implicated in this glycolysis. d Changes in cellular homeostasis triggered the self-propelled conversion by regulating the cytoskeletal proteins activation and secreted phenotype through the JAK/STAT and p53 signaling pathways. Fli-1 leukemia integration 1, TCF12 transcription factor 12, SOCS1 suppressor of cytokine signaling 1, B4GALT3 β-1,4-galactosyltransferases III, IGF2R insulin-like growth factor 2 receptor, LPA lysophosphatidic acid, VDAC voltage-dependent anion channel, YAP1 Yes-associated protein 1, TEAD1 TEA domain transcription factor-1, SDF-1 stromal cell-derived factor-1, also known as CXCL12, EREG epiregulin, ROS reactive oxygen species
It is becoming clear that there are subpopulations of CAFs for distinct functional states, raising the question of what determines the CAFs’ heterogeneity. Overwhelming evidence suggests that CAFs’ heterogeneity includes different organs/tissues, sources, functions, secretion types, and others.49,50 The alterations in CAFs show a remarkable spectrum of organs/tissues specificity. For example, CAV-1 was found to induce glycometabolic reprogramming in breast CAFs,51 while CAV-1-induced aerobic glycolysis was not completely verifiable in oral CAFs.52 Therefore, some alterations in CAFs appear only in cancers from one or a few tissue types, instead of a pan-cancer genome and transcriptome commonalities. The heterogeneity of CAFs in the same organ or tissue is likely held to depend on their precursor fibroblasts.53 Generally, CAFs are derived from the activated local tissue-resident fibroblasts, fibrocytes recruited from bone marrow, mesenchymal stem cells (MSCs) and stellate cells, or are the products of the mesenchymal transition of epithelial and endothelial cells, and the transdifferentiation of pericytes, smooth muscle cells, and adipocytes.54,55 Depending on their origin, the functions, and markers of CAF subtypes are diverse and unique. The CAF subtypes from local tissue-resident fibroblasts are similar to myofibroblasts with high expression of cytoskeletal proteins like α-SMA for cell contraction, while the CAF subtypes are derived from perivascular cells might be associated with metastasis. However, drawing definitive conclusions on the cellular origins of CAFs is difficult because currently there is no available means to track the conversion between cell states directly or to collect longitudinal samples from the same lesion in human tissue. Mouse models with well-characterized disease progression have been created to shed light on the origin of CAFs.56 In a mouse model of breast cancer, three transcriptionally diverse subpopulations of CAFs were defined via various lineage sources.57 In addition, the cues emanating from molecular phenotypes or secretion phenotypes might also determine the CAFs’ heterogeneity. Recently, single-cell RNA-sequencing and conventional RNA-sequencing of human tissues have allowed better unbiased assessment of heterogeneous CAFs.20,58,59 By analyzing a combination of classical markers, such as FAP and PDGFRβ, CAFs in breast cancer were distinguished by levels of marker expression.58 Another classic way to identify CAFs involves analyzing the different secretory phenotypes exhibited in different subtypes. For instance, elastin and collagen levels are distinctively expressed in CAFs of the lung TME.60
Accordingly, the high heterogeneity in CAFs raises an interesting question: If CAFs would switch in distinct functional states or subtypes? As an answer to this question, it has been suggested that IL-1 signaling induces the generation of inflammatory CAFs, and TGF-β antagonizes CAF switching from an inflammatory phenotype to a myofibroblast phenotype.61,62 Taken together, all these evidences show that the discovery of the heterogeneity of CAFs revealed a remarkably complex and diverse portrait.
Methods for isolation and culture of CAFs
Progression in heterogeneity studies requires more accurate methods for isolation and culture of CAFs. Without question, fibroblasts are easily isolated and cultured on plastic, e.g., human skin, mouse ears, and tail tips can be used as sources to isolate fibroblasts that can be digested and cultured in a medium.63 Using the typical curettage method combined with trypsinization or enzyme digestion methods for CAF primary cell culture, this model is unnecessary to purify cells prior to culture because of their rapid initial proliferation of fibroblasts. Antibiotics and additional washing steps are usually included in the culture process to prevent infections with bacterial and/or mycoplasma. Epithelial cells growing either in groups or scattered among the CAFs can be easily removed because of differences in adhesion ability and tolerance to trypsin of these two cell types, greatly contributing to further research on CAFs.64 In 2006, our group separated the CAFs from human oral cancer tissues using this curettage method.22 However, in these studies involving CAFs, caveats were included to suggest subtle variations in various subtypes requiring the need for new markers. CAF subtypes can be identified through multicolor flow cytometry (fluorescence-activated cell sorting). After tissue digestion, lineage markers are used to exclude hematopoietic, epithelial, and endothelial cells, and various combinations of CAF markers are used for CAF subtype identification.58 CAF subtypes can also be identified through single-cell transcriptomics and mass cytometry methods.57,65,66,67,68 Although the α-SMA and FAP staining for distinction CAFs from NFs are available, putative CAF subtype identification methods still require more reproducibility, validation, and repeated optimization.
In primary cell cultures, early passaged and immortalized CAFs have functions that can be directly investigated in vitro, and it is crucial to replicate the TME considering the intricate interactions among tumor cells, CAFs, and other stromal cells. The crosstalk of CAFs with cancer cells has been evaluated through various culture patterns. Cells can be directly cocultured and indirectly cocultured in Transwell chambers or conditioned medium (CM).69,70 Indeed, we extended the two-dimensional (2D) culture of fibroblasts from oral precancerous lesions with the addition of Candida.71 Furthermore, techniques differ for cell culture in 2D and 3D, with the latter allowing patterns of growth in vitro to better mimic that of the tissue architecture in vivo.72 One type of 3D coculture is implemented through the use of reconstituted matrices. The solid porous scaffold is based on a range of natural and synthetic materials and serves as a membrane providing a platform that can be added to a mixture of different cells, including CAFs. For instance, this scaffold‐based technology can be used to replicate tissue architecture, which is composed of alternate layers of cells, and especially for tumors of the epithelium with CAFs.73,74 Aggregate culture platforms of particular interest are scaffold‐free systems, also referred to as spheroids or organoids, in which heterogeneous populations can be evaluated for drug resistance and sensitivity or can be used to establish hypoxic cancer models.72,75,76 However, organoids commonly contained only epithelial cells and lack fibroblasts and types of other cells, such as immune and endothelial cells.77 To overcome these limitations of organoids, a multilayer bladder called an “assembloids” has been created by reconstituting tissue stem cells with stromal components representative of an organized architecture.78 In sum, either classical 2D/3D cultures or assembloids will benefit functional studies of CAFs in the context of the gradually accepted importance of TME.
Major signaling pathways and targeted therapies in CAFs
Many signaling pathways have been explored extensively in CAF-mediated cancer progression for their roles in carcinogenesis, tumor growth, cell migration and invasion, energy metabolism, and cancer recurrence and metastasis. Various endogenous and exogenous factors in CAFs, including biomarkers, cytokines, chemokines, miRNAs, and lncRNAs, are involved in the regulation of these signaling pathways. Several major signal cascades affect not only the biological behaviors of CAFs themselves but also the crosstalk between CAFs and cancer cells. Therefore, in this section, we will discuss how signaling pathways regulate the CAFs, the crosstalk of CAFs with cancer cells, and the targeted therapies.
TGF-β signaling pathway
TGF-β signaling pathway in CAFs and its targeted therapy
Were it not for the fibroblasts, TGF-β would more likely be discovered many years later, because TGF-β was initially identified by its ability to stimulate the growth of rat fibroblasts.79 In the canonical TGF-β signaling pathway, one group of TGF-β superfamily ligands, including the TGF-β/Activin/Nodal, bind to TGF-β type II receptor (TGF-βRII), which phosphorylates TGF-βRI. The binding of TGF-βRII and TGF-βRI propagates signaling by phosphorylating Smad2/3, while Smad1/5/8/9 are mediated by another group of TGF-β ligands, such as bone morphogenetic protein (BMP), through binding of BMP-RII and BMP-RI. Phosphorylated Samd2/3 heterotrimerize with Smad4 and translocate into the nucleus as a transcription factor complex, subsequently regulating the transcription of TGF-β target genes. Inhibitory Samd6/7 binds to activated type I receptors and then inhibit signal transduction of the TGF-β family.80,81 In the noncanonical TGF-β signaling pathway, TGF-β superfamily ligands can activate Rho, extracellular signal-regulated kinase (ERK), Janus kinase/STAT3, and phosphoinositide 3-kinase (PI3K)/AKT pathways in CAFs.51,82,83
Over the past nearly four decades, TGF-β has been further explored and found to be widely produced by nearly all cell types including CAFs, and the TGF-β signaling pathway has been found to have pleiotropic effects on CAF behaviors through autocrine and paracrine mechanisms.15,38,84 Resident NFs can be induced to transition into CAFs by TGF-β1 in various tumors, including bladder, breast, colorectal, and pancreatic cancer,38,85,86 indicating that TGF-β1-driven CAF generation is a common event during cancer development. Mechanistically, TGF-β1 alters the target gene expression of stromal fibroblasts through the canonical TGF-β signaling pathway, leading to differential gene expressions such as α-SMA and FAP in CAFs.37,87 After treatment with TGF-β1, MSCs were induced to differentiate into CAFs through the activation of the JAK/STAT3 signaling cascade, and inhibition of TGF-β/Smads signaling pathway reduced the transformation.84,88,89 These data suggest that both canonical and noncanonical TGF-β pathways exhibit roles in promoting CAFs generation. In addition, CAF proliferation was attenuated by a TGF-β receptor inhibitor (LY2109761) in hepatocellular carcinoma (HCC),90 and CAF migration was enhanced by TGF-β1 through overexpression of the tight junction protein occludin in colon cancer.83 Paracrine TGF-β caused the activation of noncanonical TGF-β/RhoA/ROCK axis signaling, as well as the TGF-β canonical pathway that induced transcriptional regulation of Snail1 and Twist1 target genes to increase CAFs contractility and ECM remodeling.82 Of note, since the cellular biological behaviors are driven by energy, as a hallmark of cancer, metabolic reprogramming of CAFs is defined as reverse Warburg effect (RWE), characterized by increased lactate, glutamine, nucleotides, fatty acids, and pyruvate derived from aerobic glycolysis.51,91 Recently, studies have supported the supposition that TGF-β signaling pathway plays a critical role in RWE mainly through metabolic reprogramming-related proteins, including CAV-1 and isocitrate dehydrogenase-3α (IDH3α).92,93 Mechanistically, CAV-1 interacted with the TGF-βRI, and induced its degradation, and then suppressed TGF-β-dependent Smad2 phosphorylation and nuclear translocation.94 TGF-β overexpression in CAFs decreased mitochondrial activity and increased glycolysis via CAV-1 downregulation in breast cancer, and TGF-β1-induced CAFs switched metabolic programming from oxidative phosphorylation to aerobic glycolysis by downregulating IDH3α in colon cancer.92,93 Importantly, CAF-specific endoglin (TGF-β family coreceptor) targeted by a neutralizing antibody (TRC105) decreased the metastatic spread of colorectal cancer cells to the liver in vivo.37 Summary, not only the components of canonical and noncanonical TGF-β signaling pathways in CAFs could be targeted for antitumor therapy but also the biomarkers such as CAF-derived CAV-1 and endoglin, etc., have great potential to be targeted in cancer treatment.
TGF-β signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
The CAF-mediated TGF-β pathway contributes to cancer progression by regulating many physiological processes, including cancer cell proliferation, migration, invasion, and metastasis.95 Previous studies showed that TGF-β-activated CAFs secreted growth factors, including TGF-β, fibroblast growth factor 2/7 (FGF2/7), VEGF, PDGF, and hepatocyte growth factor (HGF), to promote cancer cell proliferation.96 CAFs stimulated gastric cancer cell migration and invasion, which were attenuated by Smad2 small interfering RNA (siRNA) and anti-TGF-β-neutralizing antibody.97 TGF-β-activated ECM remodeling in CAFs created biochemical and mechanical stimuli for the invasion of cancer cells.92,98,99,100 Intriguingly, HCC cells were found to have high levels of connective tissue growth factor (CTGF) as a consequence of elevated TGF-β1 expression, and LY2109761 (a TGF-β receptor inhibitor) not only suppressed CAF proliferation but also alleviated CTGF expression, thereby reducing tumor growth and dissemination,90 indicating that TGF-βR-targeted therapy seems to have good efficacy in terms of antitumor metastasis. Consistently, CAFs contributed to the “education” of cancer cells, changing their behavior from indolent or nonaggressive into that of an invasive and metastatic phenotype.101,102 However, in our preliminary studies and the data from Wang lab, TGF-βRI mutation was detected in ~19% of head and neck squamous cell carcinoma (HNSCC) with metastasis and decreased or abrogated TGF-βRII/TGF-βRIII expression was evident in 35.3% of human oral squamous cell carcinoma (OSCC) on the protein level and in >70% of human HNSCC at the messenger RNA levels.103,104,105 These data have illustrated that TGF-βR-targeted therapy exerts strict indications, and its mutation needs to be detected before targeting TGF-βR. Notably, the nutrients recycled through the RWE via CAF-derived CAV-1 could be transferred into adjacent tumor cells to promote cancer progression in a paracrine fashion,94 while loss of CAV-1 in the tumor–stroma led to activated TGF-β signaling to trigger the epithelial–mesenchymal transition (EMT) of cancer cells,92,106,107 demonstrating that CAF-derived CAV-1 plays a paradoxical role in tumor progression, and any targeted strategies used to exploit the dual roles of CAV-1 in TGF-β signaling pathway should be developed with consideration of the ability of CAV-1 to transition from acting as a tumor promotor to acting as a suppressor to optimize treatment efficacy.
Indeed, the effect of autocrine TGF-β signaling on CAFs remained unclear until a study demonstrated that the establishment of TGF-β autocrine signaling pathways induced CAF formation during breast cancer progression,108 indicating that tumor-derived TGF-β, in a positive-feedback loop, could affect the biological characteristics of CAFs and that the crosstalk between CAFs and cancer cells is not unidirectional but bidirectional (Fig. 2). For instance, tumor-derived TGF-β was likely to recruit CAFs affiliated with the invasive front and at the bone metastatic disease to promote tumor development.102 Cancer cell-induced reactive oxygen species (ROS) promoted the loss of CAV-1 in CAFs via autophagy and then activated HIF-1α under ROS-induced pseudohypoxic conditions.45,109 In ovarian cancer through TGF-β signaling pathway, tumor-derived lysophosphatidic acid and exosomes promoted the differentiation of MSCs to CAFs,17,102,110,111,112,113 and cancer-derived TGF-β stimulated the expression of IL-6, C–X-C motif chemokine 12 (CXCL12), and VEGFA in CAFs to induce metastasis.114 Accumulating evidence suggests that abundant miRNAs in CAFs have regulatory roles in tumor progression (Table 2); for instance, targeting miR-101 attenuated TGF-β signal transduction by downregulating TGF-βR1 and Smad2 in HCC cells to suppress vascular mimicry (VM) formation.115 In fact, since the bidirectional crosstalk between the CAFs and cancer cells, any therapeutic strategy targeted CAFs or cancer cells might not obtain optimize efficacy. Thus, using a systems biology strategy, we combined experimental and computational analyses for the prediction of epithelial targets in an interactive network of proteins and found that TGF-βRIII would be targeted as an epithelial–mesenchymal common target (EMCT) in OSCC.116 In summary, the paracrine and autocrine TGF-β signaling pathway contributes extensively to the crosstalk of CAFs and cancer cells, and EMCTs show great potential for use in targeted therapy, while more studies are still needed to determine which TGF-β signaling component can serve as a common target in antitumor therapy.

TGF-β signaling pathway-mediated crosstalk between CAFs and cancer cells. The canonical TGF-β signaling pathways consist of TGF-β/Activin/Nodal-Smads pathway and bone morphogenetic protein/growth differentiation factor/Müllerian-inhibiting substance (BMP/GDF/MIS)-Smads pathway. Noncanonical pathways represent those that activate TGF-βR, but induce no-Smads pathway. Within tumor microenvironment (TME), a large number of TGF-β protein secreted by cancer cells mediated the transformation of NFs into CAFs supporting cancer progression by activation TGF-β signaling pathway, particularly canonical pathway. Activated CAFs can be orchestrated by TME to maintain their status and promote their proliferation and migration. In turn, these adaptations would also be contributed to the formation of tumor-promoting TME. CAF-secreted factors regulated extracellular matrix (ECM) remodeling to accelerate cancer invasion and metastasis indirectly. On the other hand, most factors derived from CAFs can directly mediate intricate regulation on the cancer cells. Most proteins, such as ECM proteins, GDF, TGF-β, IL-11, and CXCL12, could activate the pathway of cancer cells to exert biological functions, including promoting cancer growth, migration, invasion, and metastasis, through receptor–ligand binding. Genetic information would also transfer from CAFs into cancer cells by extracellular vehicles. CAF-derived exosomal miR-93 and miR-423 would be endocytosed by cancer cells and then promoted cancer chemoresistance and radioresistance. FOXA1 forkhead box protein A1, MMP2 matrix metalloproteinase 2, GP130 glycoprotein 130, CTGF connective tissue growth factor, IDH3α isocitrate dehydrogenase-3α, CAV-1 caveolin-1
PI3K/AKT/mTOR signaling pathway
PI3K/AKT/mTOR signaling pathway in CAFs and its targeted therapy
PI3K, as an intracellular phosphatidylinositol kinase, encompasses p85 and p110.117 Transmembrane growth factor receptors include epidermal growth factor receptor (EGFR), G-protein-coupled estrogen receptor (GPER), VEGF, and insulin growth factor receptor 1, etc. and can activate PI3K and then phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3).118,119 Then, PIP3 binds to the phosphoinositide-dependent kinase 1 (PDK1) and PDK2, and subsequently recruits AKT to the plasma membrane and phosphorylates the threonine/serine (Thr308/Ser473) phosphorylation site to activate AKT.117,118,119 Activated AKT can phosphorylate and activate its substrate mammalian target of rapamycin (mTOR) via direct and indirect pathways.120,121,122 Phosphatase and tensin homolog (PTEN), as a tumor suppressor that dephosphorylates PIP3 into PIP2 for inactivation of AKT and PDK1, negatively regulating the PI3K/AKT/mTOR signaling pathway.117,123
The PI3K/AKT/mTOR signaling pathway is crucial to many aspects of cell differentiation, growth, apoptosis, and mobility.124,125,126 Ample evidence has concluded that PI3K/AKT pathway mainly promotes the differentiation of diverse cells into CAFs (Fig. 3). For instance, tumor-derived exosomal miRNA-21, which directly targets PTEN, drove hepatic stellate cell differentiation into CAFs by downregulating PTEN and activating PDK1/AKT signaling pathway.55 BMP2 activated the PI3K/AKT and MEK/ERK signaling pathways and induced the transition from pericytes to CAFs, and Noggin (BMP signaling pathway inhibitor) inhibited PI3K/AKT and MAPK signaling pathways and reversed the pericyte–CAFs transition.127 The Notch signaling pathway also promoted CAF differentiation from human bone MSCs via AKT pathway.128 In addition, there could be a potential correlation between CAF survival and AKT signaling pathway. B7-H3 has been recognized as a co-stimulatory molecule in immune responses.129 In renal cell carcinoma, B7-H3 silencing increased apoptosis and prevented the cell cycle process and simultaneously inhibited AKT phosphorylation,130 suggesting that AKT pathways might play a role in promoting CAF proliferation and in inhibiting the apoptosis induced by B7-H3. In another study, overexpression of Noggin in CAFs decreased CAF proliferation.131 Further, the PI3K/AKT signaling pathway affected the CAF motility. GW4064, as an activator of farnesoid X receptor (FXR), significantly reduced cell migration, and this inhibition was also found in cells expressing wild-type AKT.132,133,134,135 Interestingly, the PI3K/AKT inhibitor LY294002 significantly potentiated the inhibitory effects mediated by GW4064,132 illustrating that PI3K/AKT signaling pathway was involved in the CAF motility mediated by FXR.

PI3K/AKT/mTOR signaling pathway in CAFs and the crosstalk of CAFs with cancer cells. In the CAFs, by the receptor–ligand binding, activated phosphatidylinositol-3-kinase (PI3K) can phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), while miRNA-21 could attenuate the inhibition of phosphatase and tensin homolog (PTEN) on PIP3. As a result, PIP3 activated phosphoinositide-dependent kinase 1 (PDK1)/AKT signaling cascade to transfer the rapamycin target protein (mTOR) into the nuclei, subsequently regulating the expression of targeted genes associated with differentiation into CAFs and motility, etc. Notch signaling pathway was also involved in CAF differentiation via AKT signaling pathway. B7-H3 promoted AKT phosphorylation for proliferation in CAFs, while AKT phosphorylation was involved in the inhibitory effects on the migration mediated by GW4064. Similarly, CAF-derived HGF, IGF-2, IL-22, and CXCL5 can activate PI3K/AKT/mTOR signaling axis, while CXCL12 can inhibit PTEN. Nodal-induced activation of Smad2/3 could activate AKT phosphorylation and lncRNA UCA1 collaborated with mTOR. Consequently, CAF-mediated PI3K/AKT signaling pathway regulated the cell proliferation, migration, and stemness in cancer cells. FXR farnesoid X receptor, HGF hepatocyte growth factor, IGR insulin growth factor receptor, GPER G-protein-coupled estrogen receptor, EGFR epidermal growth factor receptor, VEGFR vascular endothelial growth factor, DAPT, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester, VCAM-1 vascular cell adhesion molecule-1, SPARC secreted protein acidic and rich in cysteine
PI3K/AKT/mTOR signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
Many studies on the PI3K/AKT/mTOR pathway in CAFs have shown that activation of this cascade promoted various cancer behaviors, especially cell proliferation (Fig. 3). The fact that PI3K/AKT signaling pathways regulated CAF-mediated cancer cell proliferation in oral,136 lung,137,138 gastric,139 colon,140 endometrial,141 and anal142 cancers. Mechanistically, in gastric cancer, a neutralizing antibody against Nodal attenuated CAF-induced cancer cell proliferation through Nodal-induced activation of the Smad2/3/AKT signal axis.139 In another study, blockade of PTEN phosphorylation by siRNA led to the promotion of colon cancer cell proliferation upon stimulation with CXCL12 through the activation of PI3K/AKT signaling pathway.140 In contrast, Subramaniam et al. found that a specific PI3K inhibitor (LY294002) reversed the CAF-mediated cell proliferation in endometrial cancer.141 These findings suggest that the role of AKT signaling axis in various cancers seems to be tumor/tissue-specific and/or that different inhibitors affect different signal transduction pathways to promote cell proliferation. These observations raise the question: Do different inhibitors attenuate CAF-mediated proliferation via PI3K/AKT signaling pathway in the same type of tumor? In general, blocking vascular cell adhesion molecule-1 (VCAM-1) by siRNA138 and inhibiting IL-22 with an anti-IL-22 antibody,137 CAF-CM-promoted proliferation was attenuated via factors downstream of PI3K/AKT signaling cascade in the same type of lung cancer.
In addition, in colorectal cancer, CAFs increased the adhesion of cancer cells to endothelial cells and the migration of cancer cells in liver or lung metastasis by upregulating CD44 through HGF/MET/AKT signal pathway.143 VM was reported to be facilitated by the cancer cells with sufficient plasticity to form vascular networks for the perfusion of rapidly growing tumors and metastases.144,145 Kim et al. provided data showing that CAF-CM-induced VM was closely associated with a high level of erythropoietin-producing human hepatocellular receptor A2 (EphA2).146 Interestingly, both an EphA2 inhibitor (siRNA) and a PI3K inhibitor (LY294002) decreased VM induced by CAF-CM and suggested that the EphA2/PI3K or HGF/PI3K signaling pathway was involved in CAF-CM-mediated VM,146,147 implying that both EphA2 and HGF might be potential therapeutic targets for cancer anti-vascular treatment in gastric cancer. Of note, downregulation of CAF-derived secreted protein acidic and rich in cysteine (SPARC) can lead to dedifferentiation of gastric cancer cells to CD44+/CD24− cancer stem cell (CSC)-like cells, and the AKT/mTOR and MEK/ERK signaling pathways might be involved in these processes,148 indicating that CAF-derived SPARC maintained tumor stemness through the AKT/mTOR signaling pathway. Further, both miRNAs and lncRNAs, the two most studied classes of noncoding RNAs (ncRNAs), are crucial regulators of gene expression and interact closely with the PI3K/AKT/mTOR pathway during oncogenesis.136,149,150,151 For instance, in colorectal cancer, CAFs upregulated lncRNA UCA1 in cancer cells and collaborated with mTOR to suppress the miR-143, thereof leading to an increase in KRAS protein and resulting in regulation of the EMT and cell invasion and migration.152 Similar to the EMCT discussed above, Ogier et al. provided data showing that 7E3 blocked neuregulin 1 (NRG1)-mediated HER3 and AKT/MAPK signals to inhibit tumor growth in pancreatic cancer,153 demonstrating that NRG1 expressed by CAFs and cancer cells is an EMCT candidate. Overall, the CAF-mediated PI3K/AKT signaling pathway regulated cell proliferation, migration, VM, and stemness, and both miRNAs and lncRNAs were involved in this signal cascade. Although various PI3K/AKT inhibitors have been used in many studies, maximizing their utility in CAF-targeted therapy remains challenging. Optimization of the tumor-type selection strategies, the EMCT, and combinatory approaches will help to improve the efficacy of these agents.
MAPK signaling pathway
MAPK signaling pathway in CAFs and its targeted therapy
MAPK signaling pathways comprises signaling cascades involving three major kinases: ERK, c-Jun-N-terminal kinase (JNK), and p38 (MAPK14).154,155 Components of the MAPK pathways respond to various input signals, including cytokines, chemokines, growth factors, and stress, etc., signals. Therefore, the MAPK pathway is divided into mitogen- and stress-activated MAPK pathways, with classical representatives being ERK as the mitogen-responsive MAPKs and JNK and p38 as the stress-responsive MAPKs.156,157 Once the phosphorylation of ERK1/2, JNK1/2/3, or p38 is induced by an upstream cascade, these kinases are translocated into the nucleus where they activate transcription factors, subsequently leading to the regulation of gene expression.157,158
First, it was reported that miR-211 directly targeted the insulin-like growth factor 2 receptor to activate the MAPK signaling pathway, resulting in CAF generation.40 Gastric cancer cell-derived exosomes induced pericytes to form CAFs by activating PI3K/AKT and MEK/ERK pathways; however, BMP pathway inhibition reversed the cancer exosome-induced CAF transition.127 Surprisingly, CAFs could utilize lipids endogenously synthesized by a gold nanoparticle to induce the expression of lipogenesis genes such as fatty acid synthase (FASN), sterol response element-binding protein 2, and fatty acid-binding protein 3, and thus maintain a quiescent phenotype.159 In addition, Ando et al. showed that eicosapentaenoic acid, a polyunsaturated fatty acid, decreased the expression of IL-6 and VEGF in CAFs by inhibiting the ERK pathway, thereby reducing the cancer angiogenesis in vitro.160,161,162 Indeed, fatty acids are necessary for the basic functions of nearly all cell types including CAFs,163 and FASN is a key lipogenic enzyme in the biogenesis of fatty acids that generates palmitate from malonyl-CoA and acetyl-CoA in the presence of nicotinamide adenine dinucleotide phosphate.164,165 Intriguingly, 17β-estradiol (E2) and G1 upregulated FASN involved in the metabolism of fatty acids in CAFs via EGFR/ERK signaling cascade.166 In fact, MAPK signal was found to be involved not only in the metabolism of fatty acids but also in glycolysis in CAFs. We have found that CAF-lncRNA H19 regulated the levels of phosphorylated ERK, JNK, and p38 and further promoted glycolysis reprogramming in OSCC,52 demonstrating that the activated MAPK signaling may contribute to glucose metabolism in CAFs. In human lung cancer, CAFs displayed significantly higher migration activity in response to PDGF-BB than fibroblasts derived from noncancerous tissues and were presumed to be more dependent on ERK1/2 signaling for enhanced migration activity.167 Similarly, in another study, Eck et al. found that compared to the NFs in the mammary tissue, CAFs expressed increased CXCR4 and that AMD3100 (a CXCR4 inhibitor) suppressed the phosphorylation of ERK1/2 caused by CXCL12, subsequently leading to less invasive and migratory CAF phenotypes.168 In addition, it was reported that tissue inhibitor of metalloproteinase-1 (TIMP-1) could enhance prostate CAFs proliferation and migration in vitro and activate the ERK1/2 signaling pathway in CAFs.169 However, TIMP-1 significantly promoted CAF proliferation and motility but not the proliferation of tumor cells in prostate cancer,169 suggesting that the TIMP-1-mediated accumulation of prostate CAFs likely resulted from both enhanced infiltration and expansion of prostate CAFs within the tumors. With regard to ECM, upregulated Snail1 was found in CAFs, and it was required for the fibrogenic response of CAFs exposed to a stiff matrix.170 Mechanistically, increased ERK2 activity augmented the nuclear accumulation of Snail1 to decrease cytosolic proteasome degradation, and Snail1 affected the expression and activity of YAP1 in CAFs exposed to a stiff matrix.170
MAPK signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
MAPK signaling pathways, as ubiquitous signal transduction, regulate almost all aspects of cellular function in cancers.157,171 For cell proliferation mediated by CAFs in MAPK signal, endometrial cancer cell proliferation was prompted significantly by CAF-CM compared to NF-CM through the phosphorylated ERK, and it could be reversed by U0126 (an ERK selective inhibitor).141 Similarly, CAF-derived epiregulin significantly enhanced cancer cell proliferation through a downstream effector of ERK, and ERK inhibitors U0126 and PD98059 counteracted epiregulin-induced promotion of tumor growth in colitis-associated cancer.172 These findings suggest that MAPK/ERK signaling pathway is evolutionarily conserved and that U0126 is a highly effective depressant of this cascade. Notably, it was shown that Twist1 exhibited a dual role in CAFs and cancer cells in the EMT process: on the one hand, Twist1 promoted the expression and secretion of CXCL12 from CAFs, and its knockdown in CAFs inhibited tumor growth; on the other hand, activated CXCL12/CXCR4 signaling promoted EMT process through ERK/AKT-Twist1-MMP1/E-cadherin pathway in esophageal cancer cells.173
Of note, one of the most important characteristics of the MAPK pathway in the crosstalk between CAFs and cancer cells (Fig. 4), we propose, is the extensive cross-signaling between MAPK pathways and other cascades, such as PI3K/AKT signal, JAK/STAT cascade, and TGF-β pathway, in various cancers.127,147,174 For instance, blocking VCAM-1 suppressed proliferation and invasion of CAF-CM-treated cancer cells by activating the MAPK/AKT signaling pathway.138 Consistently, CAFs secreted urokinase plasminogen activator (uPA) to promote cancer cell proliferation, migration, and invasion through PI3K/AKT and MAPK/ERK signaling pathways in esophageal squamous cell carcinoma (ESCC).175 In another study, CAFs promoted the viability of neuroblastoma cells by increasing their proliferation and inhibiting their apoptosis through co-activation of the JAK2/STAT3 and MEK/ERK1/2 signaling pathways.176 In a mouse model of neuroblastoma, inhibition of JAK2/STAT3 and MEK/ERK/1/2 by ruxolitinib and trametinib treatment, respectively, potentiated the tumor response to etoposide and suppressed tumor progression.176 In summary, MAPK signaling pathways have great potential as targets in cancer therapy, and currently, the most extensively studied MAPK signal is the ERK pathway. An alternative approach, unlike the EMCT, we suspect, which is supported by the observation with CAF-derived epiregulin,172 CAF-derived periostin177 or CAF-secreted uPA,175 is that the dual targeting of the key biomarker in CAFs and its vital downstream effector of MAPK signaling axis in cancer cells may optimize the efficacy of blocking the crosstalk between CAFs and cancer cells in targeted therapy.

MAPK signaling pathway in CAFs and the crosstalk of CAFs with cancer cells. In CAFs, by E2 and G1, the EGFR/ERK signaling upregulated FASN expression for the metabolism of fatty acids. The PDGF-BB and SDF-1 could stimulate the higher invasive and migratory capability of CAFs via ERK1/2 phosphorylation. FA activated RAS upregulating SNAIL1 via ERK2 signaling, which mediated the fibrogenic response of CAFs. TIMP-1 enhanced CAF proliferation and migration and activated ERK1/2 signaling pathway in CAFs by the production of soluble factors. In the crosstalk between CAFs and cancer cells, CAF-derived EREG and POSTN could enhance the cancer cell proliferation and tumor growth by the downstream effector of ERK1/2. VCAM-1 and CAF-derived IL-32 increased the proliferation, invasion, metastasis, and EMT in cancer cells by activating p38/MAPK signaling pathway. Activated CXCL12/CXCR4 signal promoted EMT process through ERK/AKT-Twist1-MMP1 pathway. EPA decreased the expression of IL‑6 and VEGF secretion in CAFs by inhibition of ERK phosphorylation, thereof affecting angiogenesis. E2 17β-estradiol, G1 1-(4-(6-bromobenzo[1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)-ethanone, 1-[(3aS,4R,9bR-rel)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone, PDGF-BB platelet-derived growth factor-BB, FA focal adhesions, TIMP-1 tissue inhibitor of metalloproteinase-1, AP1 activating protein 1, FASN fatty acid synthase, SRF serum response factor, TCF ternary complex factor, POSTN periostin, EPA eicosapentaenoic acid, ECM extracellular matrix, EMT epithelial–mesenchymal transition
In addition, MAPK/p38 or MAPK/JNK signaling pathway also plays important roles in the crosstalk of CAFs with cancer cells (Fig. 4). Blockage of MAPK/p38 pathway diminished IL-32-induced EMT markers, cell invasion, and metastasis in breast cancer.178 Li et al. found that DSF/Cu increased cellular ROS levels and activated the apoptosis-related MAPK pathway without inducing a significant change in JNK or p38 expression.179 However, stress-activated MAPK pathways, including JNK cascade and p38 pathway, continued to exert the complementary functions in CAF-targeted MAPK signaling pathways in cancer treatment.
Wnt signaling pathway
Wnt signaling pathway in CAFs and its targeted therapy
Wnt signaling pathway includes 19 Wnt ligands and more than 15 receptors, which can be classified into canonical and noncanonical signaling pathways.116,180 In the canonical cascade, in the absence of Wnt ligands, cytoplasmic β-catenin combines with Axin complex and phosphorylates by glycogen synthase kinase 3β (GSK3β), leading to β-catenin degradation in the cytoplasm via β-TrCP200 ubiquitination.181,182 Conversely, in the presence of Wnt ligands, including Wnt1, Wnt2, and Wnt3a, the ligands combine with Fzd/LRP (LDL-receptor-related protein) receptors, and then, LRP receptors are phosphorylated by GSK3β, thereby causing the release of β-catenin from the Axin complex and translocation from the cytoplasm into the nucleus for targeted gene expression, including CD44, c-Myc, and cyclin D1.183 β-Catenin is not involved in the noncanonical Wnt signaling process. Through the binding of FZD receptors or ROR1/ROR2/RYK coreceptors, Wnt/RCP and Wnt/Ca2+ signaling cascades are activated for transcriptional responses and/or cytoskeletal rearrangement.184,185
In Wnt/β-catenin signaling, CAF-derived β-catenin became a major concern, as it is seemed to be a relatively early-stage event in carcinogenesis. For instance, many CAFs infiltrated into and/or around invasive tissue in the presence of high β-catenin levels in human melanoma.186 Through a new conditional gene knockout system (Col1α2-CreER mouse), β-catenin was depleted in dermal fibroblasts, causing cell cycle arrest and suppressing cell proliferation and chemical factor and ECM protein production.186 Similarly, in colorectal cancer, Mosa et al. generated a Wnt3HA/HAAPCmin/+ mouse model and demonstrated a direct role of Wnt signaling in fibroblast activation, contractility, and CAF phenotypic plasticity.187 Importantly, β-catenin ablation reduced the expression of PDGFRα and FSP1 with no obvious cytoskeletal rearrangement in stromal fibroblasts,188 noting that other signaling pathways might be implicated in the process of cytoskeletal rearrangement mediated by noncanonical Wnt signaling cascade. It has been shown that β-catenin also forms a β-catenin/E-cadherin complex that contributes to the motility and migration of fibroblasts.189 In HNSCC, periostin is highly produced and secreted by CAFs.190 CAF-derived periostin was found to be a potential ligand for protein tyrosine kinase 7 (PTK7) and was correlated with Wnt/β-catenin signal activation.190 In addition, unlike DKK1/2/4, which suppressed the Wnt/β-catenin signaling cascade, DKK3 neither interacted with LRP5/6 nor fulfilled the antagonistic role of a bona fide member of the DKK family in the canonical Wnt signaling pathway.191,192 However, DKK3 decreased the stability of Kremen to increase LRP6 membrane localization and stabilization of β-catenin.191,193 Interestingly, in CAFs, heat-shock factor-1 interacted with the DKK3 locus and upregulated the expression of DKK3,194 indicating that DKK3 might be a target for blocking of the Wnt/β-catenin signaling.
Wnt signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
Wnt signaling pathway is aberrantly activated in various cancers, including melanoma,186 esophageal,195 head and neck,190,196 breast,193 gastric,197 liver,198 ovarian,193 and colorectal cancers,193,199 and its genetic alterations are frequent, at ~66.55%, in cancers.200 What distinguishes the Wnt signaling pathway in CAFs from other pathways? Notably, in contrast to the studies on mutations in APC, RNF43, ZNRF3, AXIN1/2, and CTNNB1 detected in human colorectal adenocarcinoma,201 endometrial cancer,202 HCC,203 and gastric cancer,204 few studies have been published related to their alterations of these genes in CAFs. To further address the role of cancer cell mutations in CAFs, using a 3D coculture model, Zhou et al. found that melanoma growth was suppressed by CAF deactivation induced by β-catenin ablation, which led to the reduced production of paracrine factors and ECM proteins.188 Similarly, CAF-derived periostin promoted the CSC phenotype, tumor progression, and metastasis via canonical Wnt/β-catenin signaling pathway in HNSCC.190 Mechanistically, CAF-derived periostin bound to PTK7 on the cancer cell membrane and transferred the signals to disheveled proteins by LRP6, thereby inducing the phosphorylation of GSK3β and the hypophosphorylation of β-catenin, leading to the translocation of β-catenin from the cytoplasm to the nuclei.190 However, it was reported that β-catenin-mediated Wnt signaling was dispensable for the function of CAFs in ECM remodeling and promoting cell proliferation and invasion in breast cancer.193 This suggests that Wnt/β-catenin signaling pathway affects the crosstalk of CAFs and cancer cells in highly specific tumor types. Interestingly, by generating Wnt-independent tumor organoids, which secreted the Wnt antagonist Sfrp1, Mosa et al. found that Sfrp1 or genetic depletion of β-catenin strongly decreased the number of cancer-associated myofibroblasts (myCAFs: α-SMA+/Acta2+).187 Coculture of this tumor organoid with inflammatory CAFs (iCAFs: IL-6+/Tnfa+/IL-1a+) resulted in the upregulation of Vim and Zeb1, while myCAFs reverse this upregulation,187 indicating that the EMT process could be induced by Sfrp1 and that tumor behaviors were differentially regulated via Wnt signaling pathway in specific CAF subtypes.
In addition, CAF-derived Wnts can lead to cell growth and other biological functions of cancer cells (Fig. 5). For example, Wnt2 protein secreted by CAFs enhanced cell invasion and migration in colorectal cancer205 and angiogenesis by shifting the balance towards proangiogenic signaling in colon cancer.206 It is likely that treatment with CAF-CM and an elevated autophagy rate augmented the levels of β-catenin and P-GSK3β, which are the key proteins in the Wnt/β-catenin pathway, thereby promoting tumor progression.207 Notably, the upregulation of Wnt proteins in CAFs was explainable with both intrinsic and extrinsic aspects. On the one hand, Wnt5a was enriched by the loss of H3K27me3 in CAFs, and inhibition of secreted Wnt5a from CAFs suppressed cancer cell growth and migration in gastric cancer.208 On the other hand, Taxotere treatment enhanced Wnt16 expression in CAFs and this in turn might have contributed to the proliferation, invasion, and chemoresistance of breast cancer cells.209 Taken together, these findings show that the attenuated Wnt signaling cascade in CAFs could contribute as a suppressor of tumor progression, in a manner similar to that described for reduction in tumor cell-intrinsic Wnt signaling activity; however, more studies are required to dissect the underlying potential mechanisms and genetic factors related to CAF-mediated Wnt signaling in cancer progression.

Crosstalk of different signaling pathways among CAFs, cancer cells, and immune cells. A reservoir of biological behaviors of CAFs, including CAFs generation, proliferation, ECM remodeling, and energy metabolism, etc. were regulated by several major signals like TGF-β and PI3K/AKT/mTOR signaling pathways. Importantly, CAF-mediated signaling pathways like JAK/STAT, Wnt, Hippo, MAPK, EGFR, and NF-κB signal were widely involved in cancer cells proliferation, stemness, invasion, migration, metastasis, angiogenesis, epithelial–mesenchymal transition (EMT) process, and therapeutic resistance. CAF-mediated signaling pathways did not always display with individual effects, but commonly crossed to each other to form a signaling network in cancer progression by the cross-connections such as STAT3, AKT, and YAP1. As the great source of cytokines, chemokines, and growth factors, CAF-secreted factors, including TGF-β1, IL-6, IL-8, CXCL1, CXCL12, and PGE2, etc., affect proliferation and activation of T cell, recruitment and activation of myeloid-derived suppressor cells (MDSCs), differentiation, and polarization of monocytes/macrophages, etc. PGE2 prostaglandin E2
JAK/STAT signaling pathway
JAK/STAT signaling pathway in CAFs and its targeted therapy
JAK/STAT signaling pathway is a signal cascade stimulated by many kinds of cytokines and consists of a host of ligands and several tyrosine kinase-related receptors with four tyrosine kinase JAK and seven transcription factor STAT family members, suppressors of cytokine signaling proteins, and multiple STAT-dependent operons.210,211 JAK enzymes share a common domain structure consisting of seven JAK-homology domains.212 Typically, cytokines, chemokines, and/or growth factors integrate with tyrosine kinase-related receptors, and the latter recruits JAK, activating receptor and JAK. The phosphorylated tyrosine on the receptor molecule binds with the SH2 site of STAT.213 STAT binding to the receptor triggers the tyrosine phosphorylation of STAT, leading to STAT dimer formation and its translocation into the nuclei where it targets gene expression.214
JAK/STAT signaling pathway is constitutively activated in CAFs. In TME, CAF-derived cytokines, including IL-6, IL-10, IL-11, and IL-22, act as ligands for JAK/STAT signal cascade (Table 3). Intriguingly, IL-6, as a pro-inflammatory cytokine, partnered with GP130 to activate STAT3, while IL-10, as an anti-inflammatory cytokine, did not interact GP130 but promoted the phosphorylation of STAT3,215,216,217 demonstrating that different cytokines are likely to activate the same STAT protein. Actomyosin contractility plays a key role in ECM remodeling by CAFs to permit cell migration. GP130-IL6ST signaling influenced JAK1-derived actomyosin-mediated contractility through the phosphorylation of MLC2 in CAFs and promoted ECM remodeling.218,219 Consistently, cytokine oncostatin M not only promoted actomyosin-mediated contractility and ECM remodeling by CAFs through GP130-IL6ST, JAK1, and ROCK signal axes but also induced CAF generation through the JAK/STAT signaling pathway.220 In addition, aberrant DNA methylation contributed to the maintenance of the phenotype of CAFs via the JAK/STAT cascade.221 Since STAT3 acetylation caused the epigenetic modification-dependent loss of SRC homology phosphatase-1 (SHP-1) and dephosphorylates JAK1, SHP-1 knockout led to the sustained constitutive phosphorylation of JAK1 and STAT3, which maintained the contractility- and invasion-promoting properties of CAFs.221,222 To attenuate the effect of specific cytokines on JAK/STAT signal, therapeutic approaches, including blocking cytokine antibodies or inhibitors, are warranted to identify the tumor-promoting roles of CAFs. Targeted inhibition, such as that induced by 5-azacytidine and ruxolitinib treatment, resulted in the sustained abrogation of JAK1/STAT3 phosphorylation and rescued SHP-1 expression, thereby inhibiting the tumor-promoting invasive phenotypes of CAFs.223,224 In an analysis of miRNAs in CAFs through JAK/STAT signaling pathway, miR-210 increased the expression of matrix metalloproteinase 9 (MMP9), FGF2, and VEGFA by activating the JAK2/STAT3 signaling pathway for proangiogenesis and ten-eleven translocation 2 was identified as the target of miR-210 in CAFs, which was implicated in proangiogenic switching.225 In addition, p53 was reported to regulate the CAF properties through STAT3 signaling, and CAF activation, migration, and invasion could be clearly inhibited by Stattic (Y705), an inhibitor of STAT3.226
JAK/STAT signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
Constitutive activation of JAKs and STATs was first identified as being associated with malignancy,227 and accumulating evidence has shown that CAF-mediated JAK/STAT signaling pathway is widely involved in human cancers, including prostate,228 lung,229 breast,230 colorectal,231 gastric,232 endometrial,233 liver,234 and oral235 cancers, through various tumor biological processes, including in increased cell plasticity, proliferation, migration, invasion, EMT, angiogenesis, and metastasis. Notably, IL-6 represents the most investigated cytokine regulating the crosstalk between CAFs and cancer cells (Table 3). In HCC, CAF-derived IL-6 facilitated HCC cell EMT, which in turn activated the IL-6/IL-6R/STAT3 axis in a positive-feedback loop to promote the expression of TG2 for the acquisition of EMT phenotypes.234 IL-6 binding with GP130 could trigger STAT3 activation, and this response could be suppressed by the inhibition of netrin-1.236 Netrin-1, as a multifunctional secreted glycoprotein, is highly expressed in colon CAFs, and its blocking antibody (Net1-mAb) inhibited cancer stemness, plasticity, and intercellular signaling between CAFs and cancer cells by suppressing the IL-6/JAK2/STAT3 signaling pathway.236
As the upstream of IL-6, epiregulin-induced CAF activation promote EMT by activating IL-6/JAK2/STAT3 signaling axis, which could be inhibited by a JAK2 inhibitor (AG490).235 Intriguingly, the migration of melanoma cells was dependent on GP130-IL6ST/JAK1-ROCK signaling; however, although it was not necessary in cancer cells, this signaling pathway was required for CAF-induced ECM remodeling to promote the invasion of squamous cell carcinoma (SCC),237 indicating that the targeted therapy of JAK/STAT signaling pathway for SCC invasion might not be the epithelium but CAFs. Further, cytokine signaling of GP130-IL6ST/JAK1 cascade mediated actomyosin contractility in cancer cells and CAFs to promote SCC invasion.237 Estrogen in CAF-CM promoted gastric cancer cell proliferation and invasion via IL-6/STAT3 signal axis, and these two processes could be inhibited by an IL-6-neutralizing antibody and STAT3 siRNA, respectively.236
In addition, CAF-derived IL-17a significantly promoted the migration and invasion of gastric cancer cells by activating the JAK2/STAT3 signaling pathway, and the effects of CAF-mediated cancer progression were inhibited significantly by treatment with an IL-17a-neutralizing antibody or JAK2 inhibitor (AG490).232 In addition, Heichler et al. found that IL-11 was frequently overexpressed in colorectal cancer and acted as a signal transducer and activator of STAT3, which was inversely correlated with patient prognosis.231 Taken together, therapeutic agents targeting the JAK/STAT signaling pathway, including blocking antibodies against Netrin-1, cytokines including IL-6, IL-11, and IL-17a, or inhibitors of JAK kinase such as AG490 or STAT activity such as STAT3 siRNA, could be useful agents in antitumor treatment.
EGFR signaling pathway
EGFR signaling pathway in CAFs and its targeted therapy
EGFR belongs to the ErbB family of receptors, which includes ErbB1/EGFR/HER1, ErbB2/HER2/Neu, ErbB3/HER3, and ErbB4/HER4.238 ErbB family members can be activated by the following ligands: amphiregulin, betacellulin, EGF, heparin-binding EGF-like growth factor, TGF-α, epiregulin, epigen, and NRGs.239,240 Structurally, EGFR family members share a common domain arrangement that comprises a cysteine-rich extracellular domain, a hydrophobic transmembrane domain, and an intracellular tyrosine kinase domain. Specifically, the extracellular region of EGFR is subdivided into four domains, and the intracellular tyrosine kinase domain is highly conserved with variable phosphorylation sites.241,242 The EGFR signaling pathway is activated by ligand-induced receptor dimerization, in which the tyrosine residues in the intrinsic kinase domain of one receptor cross-phosphorylate specific residues in the C-terminal tail of the partnering receptor to recruit functional proteins.243,244
EGFR is expressed in almost all nonneoplastic cell types in TME, including CAFs, except mature cells in the lymphohematopoietic system.245,246 GPER was first reported as a GPCR gene in breast cancer,247 and Luo et al. found that GPER expression was abundant in breast CAFs.248 G15 (a selective GPER antagonist), AG (an inhibitor of EGFR), and U0126 (an inhibitor of ERK1/2) significantly inhibited GPER-mediated proliferation and cell cycle changes in breast CAFs induced by E2, G1, and tamoxifen,248 implying that GPER/EGFR/ERK signaling pathway was activated in this process. Of note, zinc chloride (ZnCl2) increased GPER-targeted CTGF in breast CAFs.249 Since CTGF was reported to have a role in the migration of different cell types,250 Pisano et al. found that ZnCl2-stimulated migration of CAFs was abolished by knockdown of GPER or CTGF, and this migratory response could be rescued by the addition of CTGF.249 In mammals, α1,6-fucosyltransferase (FUT8), as the only enzyme that catalyzes core α1,6-fucosylation (CF), was reported to be overexpressed in CAFs in 82% of lung adenocarcinoma cases, and upregulated FUT8 could prompt the CF modification at high levels.251 Downregulation of either EGFR or FUT8 reduced the phosphorylation of EGFR; however, the blockade of EGFR signaling was rescued by an EGFR activator in FUT8 down-regulated CAFs,251 demonstrating that EGFR activity in CAFs was regulated by this FUT8/CF treatment.
EGFR signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
Currently, EGFR tyrosine kinase inhibitors (EGFR-TKIs) are being effectively used for anticancer therapy, while CAF-derived survival signaling to cancer cells can modify EGFR-TKI efficacy.252,253,254,255,256 In HNSCC, it was reported that EGF-containing fibulin-like ECM protein-1 (FBLN3) was secreted by CAFs but not NFs, and CAF-derived FBLN3 could increase anchorage-independent growth and tumor sphere formation and maintain cancer stemness.257 Interestingly, targeting the EGFR signaling pathway with gefitinib effectively inhibited CAF-mediated cancer stemness,257 demonstrating that CAF-derived factors such as FBLN3 in the EGFR signal cascade promoted cancer stemness properties are potential therapeutic targets to effectively block CAF-promoted CSC niche formation. In another study, it was shown that FUT8/CF in CAFs prompted the proliferation of cancer cells through EGFR signal cascade in non-small-cell lung cancer,251 suggesting that EGFR signaling in CAFs exerted a catalytic effect on CAF-mediated cancer progression and could be regulated by the CF modification of EGFR.
In addition, the CAF-mediated EGFR signaling pathway plays promoting roles in tumor invasion and metastasis (Fig. 5). It was demonstrated that the collective invasion of SCC cells could be driven by the matrix-dependent mechano-sensitization to EGFR signaling.258 Because receptor tyrosine kinase (RTK) can interact exclusively with activated integrins, the ECM determines the type of RTK/integrin interaction proceeds on the cellular membrane, and this selectivity may change the intracellular location or conformation of EGFR.99,259 Given that the L-type calcium channel CAV1.1 functions with ECM stiffness and is triggered by EGFR signaling activation, calcium channel blockers may suppress SCC invasion and metastasis, and an EGFR blockade could trigger the EMT process in HNSCC.258 Notably, Gao et al. found that CAFs associated with high-grade serous ovarian cancer contributed to the formation of heterotypic spheroids in malignant ascites and that these CAF-centered spheroids recruited floating ovarian cancer cells, resulting in premetastatic niche formation at an early stage.260 In summary, these evidences support the supposition that the CAF-mediated EGFR signaling pathway is essential for several cellular functions, including maintenance of cancer stemness, cell proliferation and invasion, and metastasis. Importantly, in contrast to EGFR overexpression in tumor cells, which was positively correlated with the overall survival period of patients with several cancers,261 EGFR overexpression in CAFs had no significant relation to the prognosis of patients with colorectal cancer,246 indicating that EGFR in CAFs might not be an independent prognostic factor for survival evaluation in patients with cancers.
Hippo signaling pathway
Hippo signaling pathway in CAFs and its targeted therapy
Hippo signaling pathway was originally discovered to be an important regulator of organ size in Drosophila.262 In mammals, the canonical Hippo signaling cascade consists of mammalian sterile 20-like (MST) kinases, l (LATS) kinases, and adaptor proteins Salvador homolog 1 and Mps one binder kinase activator protein. Central to this cascade signals from MST1/2 to the oncogenic of transcriptional cofactors YAP1 and its paralog transcriptional coactivator with PDZ-binding motif (TAZ).263 The major target transcription factors regulated by YAP/TAZ are the TEAD family.263 Notably, in noncanonical Hippo signaling, for instance, phosphorylated YAP1/TAZ binds directly to an angiomotin family proteins, which is in the Crumbs complex, to α-catenin, β-catenin, PTPN14, and Scribble in adherens junctions and to ZO-1/2 in tight junctions, subsequently leading to the regulation of YAP1/TAZ localization and activity independently of LATS.264
In contrast to other classical signal transduction pathways, such as TGF-β or Wnt signal, the Hippo pathway does not seem to be involved in special extracellular ligands or transmembrane receptors, but is regulated by upstream signals and involved in cell morphology and polarity and cell–cell and cell–ECM adhesion.265,266,267,268 Studying prostate cancer, Shen et al. found that the YAP1/TEAD1 protein complex transformed NFs to CAFs by activating cytoskeletal proteins and actin by regulating SRC.47 In addition, the proliferation of CAFs was significantly inhibited by siYAP1 or the inhibitor verteporfin,47 indicating that YAP1 had multiple effects on CAFs. YAP activation in CAFs269,270 controlled the expression of several cytoskeletal regulators, including ANLN and CTGF, and regulated actomyosin contractility and ECM remodeling in CAFs via MYL9/myosin light chain-2,269 and in agreement, the gain of YAP function in CAFs was associated with reactivation of actomyosin contractility and SRC,193 supporting the supposition that YAP/TAZ activity of CAFs was primarily associated with its effect on ECM remodeling. In another study, Calvo et al. found that YAP depletion by siRNA caused weakening of the ability of CAFs to physically contract collagen-rich matrices, and fewer focal adhesions and fewer essential structures were formed for force transmission between the cytoskeleton and matrix; however, depletion of TAZ had little effect on these processes,269 indicating that TAZ is not the only downstream component of the YAP-mediated signaling pathway. Interestingly, in breast cancer, the activity of the upstream negative regulators MST1/2 was not different between NFs and CAFs, while the activity of LATS kinases and phosphorylated YAP was augmented in CAFs,269 suggesting that YAP was not activated by the attenuated activity of the canonical MST/LATS signaling pathway in CAFs.
Hippo signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
CAF-mediated Hippo signaling pathway tumor-promoting activities are mainly related to cell proliferation and invasion and angiogenesis (Fig. 5). In prostate cancer, CAFs with high YAP1 expression could prompt the proliferation of cancerous epithelial cells and were more likely to cause the distant metastasis.47 Similarly, Shen et al. found that knocking down YAP1 or SRC in CAFs attenuated the promotion of CM on the proliferation and invasion capacity of human prostate cancer cells,47 indicating that CAFs in prostate cancer could promote tumor cell proliferation and invasion, which was highly dependent on the paracrine activity of YAP1 and/or SRC in CAFs. Strikingly, we found that YAP expression in primary HNSCC cells was associated with Nodal-induced metastasis; however, YAP knockdown in HNSCC cells was not associated with changes in EMT.271 Notably, CAF-derived miR-221 could trigger proliferative and migratory effects on MDA-MB 231 and SkBr3 breast cancer cells by interfering with the A20/c-Rel/CTGF signaling pathway.272 MiR-221 was reported to be strongly upregulated and closely related to the invasion and advanced clinical stages of patients with breast cancer.273 In general, targeting miR-221 by specific inhibitors such as LNA-i-miR-221 might cause a suppressive effect on cancer progression, especially breast tumors.274,275 Consistently, the ability of CAFs to promote cancer cell invasion was significantly dependent on YAP function, and loss of YAP function reduced the formation of fibrous collagen networks by CAFs and suppressed angiogenesis in vivo.269 In addition, in a mouse model, treatment with Y27632 (a potent, ATP-competitive inhibitor of ROCK1 and ROCK2) reduced the nuclear localization of YAP in CAFs and inhibited angiogenesis,269 indicating that the metastatic function of YAP in HNSCC may not be a result of EMT.
With respect to the molecular mechanism of CAF-mediated YAP/TAZ signaling pathway in the crosstalk between CAFs and cancer cells, it was reported that DKK3 in CAFs potentiated the Wnt and Hippo signaling pathways.193 DKK3 knockdown in CAFs decreased the levels of unphosphorylated β-catenin and TAZ, reduced nuclear YAP/TAZ translocation, and inhibited β-catenin and YAP/TAZ transcriptional activity in breast, colorectal, and ovarian cancers.193 DKK3 reduced YAP/TAZ degradation through the Wnt/β-catenin signaling pathway, thus acting in parallel to YAP/TAZ regulation mediated through mechanotransduction.269,276 Although DKK3 could activate β-catenin in CAFs, the inhibition of β-catenin by RNA interference did not affect CAF-mediated ECM remodeling or cell growth or invasion in cancer,193 indicating that exogenous inhibitors targeted to Wnt/β-catenin signaling axis might not attenuate aggressive behaviors of CAFs and/or that Wnt/β-catenin signaling pathway might be one of the upstream regulators of the YAP/TAZ signaling cascade. In support of this notion, DKK proteins were reported to negatively affect the surface expression of Kremen, and DKK3 was shown to inhibit Wnt signaling by triggering LRP5/6 internalization through the formation of a ternary complex with Kremen1/2 receptors.191,192,277 In contrast, Ferrari et al. found that Kremen1, but not LRP6, immunoprecipitated with DKK3 in CAFs, while DKK3 silencing led to Kremen1 localization to the cell periphery,193 demonstrating that DKK3-mediated LRP6 regulation could activate β-catenin and YAP/TAZ, with the latter being the main mediator of DKK3 functions in the crosstalk between CAFs and cancer cells.
NF-κB signaling pathway
NF-κB signaling pathway in CAFs and its targeted therapy
As a ubiquitous transcription factor, NF-κB consists of five different proteins: RelA (p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52).278 Generally, core components of NF-κB signaling pathway are inhibitors of NF-κB (IκB) proteins, namely, the IκB kinase (IKK) complex and NF-κB transcription factor.279,280 In the canonical NF-κB pathway, activation is triggered by the binding of ligands (e.g., TNF-α, IL-1β) to their respective receptors (e.g., Toll-like receptors (TLRs), TNFR, and IL-1R), which leads to the phosphorylation and activation of IKK, thereof initiating the phosphorylation of IκB proteins.281,282 In the noncanonical NF-κB pathway, ligands such as CD40 bind to their cognate receptors. Then, this binding leads to activation of NF-κB by NF-κB-inducing kinase, which phosphorylates IKKα and facilitates IKKα phosphorylation of p100 for p52 generation. RelB/p52 heterodimers then translocate into the nucleus, subsequently leading to the activation of noncanonical NF-kB target gene expression.283,284,285
CD146, a cell membrane protein, was knocked down to promote CAFs activation, which might have been caused by the modulation of NF-κB activity.286 In another study, Wu et al. found that gastric cancer-derived HTRA1 promoted CAFs generation from NFs through the activation of the NF-κB/bFGF/FGF2 signaling pathway.287 Furthermore, CXCR2 signaling in CAFs promoted the CAF acquisition of secretory phenotype by activating NF-κB,288 noting that NF-κB signaling pathway was involved in regulating CAF factor secretion. Similarly, in OSCC, CAFs were stimulated with IL-1β and exhibited increased CXCL1 secretion in an NF-κB-dependent manner.289 Cancer-derived IL-1α increased the expression of COX-2, CXCL8, CCL20, and IL-6 in CAFs to form an inflammatory environment in pancreatic cancer.290 In regard to the immune response, it was reported that decreased α-SMA in CAFs was observed after their incubation with the polysaccharide MPSSS and impaired the immunosuppressive effect of CAFs through TLR4/NF-κB signaling, but there was no obvious effect on CAF viability.291 Ligustilide had no effect on CAFs’ viability, but reversed the immunosuppressive function of CAFs through the TLR4/NF-κB signaling pathway.292 In sum, these evidences suggest that NF-κB signaling pathway is mainly implicated in the activation, secretory phenotype, and immunosuppressive functions of CAFs.
NF-κB signaling pathway-mediated crosstalk of CAFs with cancer cells and its targeted therapy
Fundamentally, CAF-driven NF-κB was reported as a pro-inflammatory gene signature critical for tumor progression (Fig. 5). For instance, CAF-derived CXCL1, IL-6, and COX-2, known as targets of the NF-κB transcription factor, were correlated with tumor-promoting inflammation and tumor invasiveness in human breast cancer.293 It was reported that oxidative stress triggered NF-κB activation and STAT3 in CAFs to upregulate CCL2, and inhibition of CCL2 could reduce tumor growth of oral cancer in a mouse model.294 IL-6 secreted by CAFs could trigger the induction of neoplastic OPN to promote the growth, migration, and invasion of cancer cells in HNSCC via the integrin αvβ3/NF-κB pathway.295 Notably, in SCC, the pro-inflammatory signaling driven by CAFs was NF-κB-dependent.36 In sum, these data suggest that CAF-driven NF-κB signaling plays a central role in mediating protumor inflammation. Interestingly, TLR4 expressed by tumor cells was significantly associated with decreased recurrence; however, its overexpression in CAFs was independently associated with increased recurrence in patients with colorectal cancer,296 indicating that TLR4 in CAFs might not be an independent prognostic factor for recurrence of colorectal cancer.
In addition, CAFs could mediate the EMT and tumor stemness through a pro-inflammatory signature strictly dependent on COX-2-, NF-κB-, and HIF-1α-related signaling cascades.297 In pancreatic cancer, phosphorylated NF-κB was positively correlated with CAV-1 expression, and knockdown of CAV-1 in CAFs could reduce the invasiveness and motility of cancer cells, but did not affect cell proliferation.298 Importantly, overexpression of Smad7 in IKKβ-deficient CAFs prevented HGF secretion, and pharmacological inhibition of Met in a CAC model supported that enhanced tumor promotion was dependent on HGF-Met signaling in the mucosa of IKKβ-mutant animals,299 suggesting that a tumor-suppressive function of IKKβ/NF-κB in CAFs might be related to HGF release. In our preliminary study, we found that topically applied Tat-Smad7 penetrated cells in both healthy oral mucosa and oral cancer, attenuating NF-κB signaling-related inflammation.300 In addition, the effect of antisense oligonucleotide-miR-221 transfection in CAFs caused the inhibition of migration/invasion by downregulating NF-κB in pancreatic cancer.301 In addition, ablation of Saa3, a member of the serum amyloid A apolipoprotein family, in pancreatic tumor cells rendered them insensitive to the inhibitory effect of Saa3-null CAFs,302 suggesting that saa3 in CAFs may provide potential therapeutic benefit to pancreatic ductal adenocarcinoma (PDAC) patients. Taken together, these results suggest that CAV-1, Smad7, saa3, and miRNAs might be candidates to target the CAF-mediated NF-κB signaling pathway in cancers.
Other signaling pathways in CAFs, crosstalk of CAFs with cancer cells, and its targeted therapy
CAF-induced signaling pathways not discussed above, including Notch,303,304,305 Hedgehog,306,307,308 p53,309,310,311,312 Rho/Rock,313,314 HIF-1α,44,315,316,317 and peroxisome proliferator-activated receptor (PPAR)318,319,320 signaling cascades, have been widely studied to understand CAFs crosstalk with cancer cells. Indeed, covering all aspects of all signaling pathways of CAFs in cancer progression is beyond the scope of this review. However, similar to the signaling pathways we summarized above, other signaling pathways exhibit unique characteristics. For instance, HIF-1α signal in CAFs promotes tumor progression mainly by regulating glycolytic metabolism. Radhakrishnan et al. demonstrated that LPA-induced glycolytic shift was the earliest, potentially priming event of the NF-CAF transition, and it was mediated through LPA/LPAR/HIF-1α signaling axis.44 In another example, CAF-specific RWE increased the expression of fructose bisphosphatase in cancer cells, leading to the modulation of HIF-1α function in non-small-cell lung cancer cells.321 Thus, all these signaling hubs in CAFs have great potential as targets for blocking CAFs crosstalk with cancer cells, and further investigations are warranted to identify the specific functions of these targets.
Immunotheray driven by CAFs
In addition to CAFs, the TME contains an array of other nonneoplastic cells, including resident and infiltrating immune cells. Particular emphasis has been placed on T lymphocytes, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and others. The immune cells in the TME play a dual role during cancer onset and progression, as they can mediate tumor clearance by killing immunogenic neoplastic cells and contribute to tumor escape by shaping tumor immunogenicity.322,323 Immunotherapies, harnessing the dual roles of the immune system in battling cancer, have emerged as new pillars within oncotherapy.324,325
To date, CAFs have been found mainly to regulate and rewire the TME to favor the malignant biological properties of tumors through the interaction of CAFs and T lymphocytes. In colorectal cancer, CD70 expression in CAFs and the CD70/CD27 axis affected the expansion and differentiation of T lymphocytes by activating the NF-κB signaling pathway.326 Chakravarthy et al. found that pan-cancer ECM dysregulation was linked to CAF-mediated TGF-β signaling, which was closely related to immunological activity and could predict failure of PD-1 blockade.327 Meanwhile, the combination of TGF-β blockers and anti-PD-L1 antibodies promoted T cell penetration into the tumor center, improving tumor responsiveness to anti-PD-L1 therapy.328 Activated CAFs can also produce TGF-β to form a positive-feedback loop for CAFs activation. Therefore, inhibition of TGF-β signaling, such as through treatment with galunisertib (LY2157299) or AVID200, is worthy of further exploration as a method to improve PD-L1 immunotherapy.329,330,331,332 In addition to TGF-β blockers, pirfenidone (PFD)-targeting CAFs possessed inhibitory effects on migration and decreased the expression of PD-L1 by targeting CCL17 and TNF-β. The regulation of CAFs through PFD treatment may reduce the acquisition of CAF-mediated immunosuppressive capacity in breast cancer cells, thereby leading to increased efficacy of chemotherapy.333 In addition, highly expressed presenilin 1 in CAFs also regulated tumor-infiltrating lymphocyte populations in the TME via the Wnt/β-catenin pathway. Inhibition of presenilin 1 by IL-1β promoted the proliferation and penetration of CD8+ T cells in multiple ovarian models and retrieved functional CD8+ T cells in the TME, which may improve the efficacy of current immunotherapies.334 The angiotensin receptor blocker losartan can drive myofibroblast CAFs into a quiescent state, attenuate immunosuppression, and increase T lymphocyte activity, thereby significantly improving the response of breast cancer cells to immune checkpoint blockers.335 Feig et al. found that pancreatic cancer cells coated with the CAF-derived CXCL12 caused the depletion of T cells and contributed to cancer unresponsiveness to α-PD-L1; administering a CXCR4 (a CXCL12 receptor) inhibitor, AMD3100, blocked CAF-directed immune evasion of the tumor to increase T cell infiltration in cancer cells and greatly diminish cancer volume when administered in combination with α-PD-L1.336 These studies demonstrate that the dysregulation of CAFs contributes to tumor-induced immunosuppression and that immunotherapy driven by CAFs sensitizes tumors to T cells, stimulating strong antitumor cellular immunity and tumor regression.
In addition, immunotherapies targeting TAMs have drawn significant attention, as they deplete and/or reprogram TAMs to block their protumor functions or restore their tumoricidal properties.337 Colony-stimulating factor-1 receptor (CSF1R) inhibitors targeting TAMs successfully in diffuse-type giant cell tumors failed to deliver an antitumor effect in other tumor models, as CSF1R inhibition altered CXCL1 and other chemokine secretion by CAFs and triggered a profound increase in protumor polymorphonuclear myeloid-derived suppressor cell (PMN-MDSC) recruitment to tumors; combined inhibition of CSF1R and CXCR2 (a CXCL1 receptor on granulocytes) blocked CAF-mediated MDSC recruitment and reduced tumor growth.338 In OSCC, CAFs educated macrophages to suppress T cell proliferation, which was restored by the neutralization of TGF-β, IL-10, or arginase I.339 The CAF–TAM interaction was also regulated by pleiotropic glycoprotein stanniocalcin-1, which was secreted by CAFs and partially suppressed TAM differentiation.340 In colorectal cancer, CAFs could attract monocytes via IL-8/CXCR2 pathway to induce their polarization.341 Thus, CAFs play crucial roles in shaping the immunosuppressive TME by educating TAMs to induce a protumor phenotype, and methods for reversing CAF-mediated immunosuppression in TAM-targeted therapeutics need to be considered. In addition, CAFs can functionally sculpt other immune cells in the TME through their high secretory ability. CAF-derived cytokines, including IL-6 and CXCL12, induced the generation and activation of MDSCs to favor HCC progression.342 HCC CAFs recruited normal dendritic cells and educated them to acquire a tolerogenic phenotype through IL-6/STAT3 signaling.343 Taking these observations together, it can be concluded that crosstalk between CAFs and T cells, TAMs, MDSCs, etc. is involved in the formation of tumor immunosuppression (Fig. 5), and combination therapy driven by CAFs and immunotherapies might be an effective and promising strategy for treating insensitive tumors.
Meanwhile, the heterogeneity of intratumoral CAFs impels a paradigm shift in CAFs behaviors, especially immune-related function. Although the classification criteria for CAF subtype are diverse and are in a state of flux, some subpopulations of CAFs with an immunomodulating phenotype likely make more distinct contributions to tumor immunity. Based on the cellular source, Bartoschek et al. found that mCAFs from resident fibroblast can also regulate the immune response.57 Unlike this, from the perspective of function, in PDAC, iCAFs revealing a pro-inflammatory gene signature with IL-6 and CXCL12 expression promoted tumor growth, angiogenesis, and macrophage recruitment.344 Similarly, in breast cancer, CAF-S1 fibroblasts attracted or regulated the function and differentiation of T lymphocytes by secreting CXCL12 to format an immunosuppressive environment.58 Further, the CAF subtypes with a high level of IL-6 and FAP, which associated with a large cluster of pathways involved in immune regulation and the worse survival outcomes in ovarian cancer.345 These data suggest that the classification of different subpopulations based on CAFs’ function seems to be much more reliable in tumor immunity.
Therapeutic resistance caused by CAFs
At present, a number of CAF-mediated anticancer therapies have been reported, of which most are in phases of preclinical trials. Overall, CAF-mediated anticancer therapies mainly include the following five application approaches: inhibiting transformation from normal fibroblasts to CAF, promoting transformation from CAF to normal fibroblasts, inhibiting tumor development and progression, activating immunity system, and reversing tumor chemoresistance (Table 4). We found that breast cancer is the most widely studied for targeting CAFs in cancer treatment. Interestingly, no trials of pancreas cancer were found in the inhibition of tumor development and progression; however, a majority of studies in reversing tumor chemoresistance were related to pancreas cancer, suggesting that CAF-mediated chemoresistance might be tumor- or organ-specific. The interaction between tumor cells and CAFs or ECM blunts the response to cancer cell-directed therapies, including chemotherapy, small-molecule drug-based therapy, and radiotherapy (RT).346
Chemotherapy utilizing traditional cytotoxic antitumor drugs plays therapeutic roles in the lung, pancreatic, colorectal cancer, and other malignant tumors.347,348,349,350 First, CAFs generate physical obstacles around malignant elements by secreting a dense desmoplastic matrix into the TME. Laminin A1 secreted by CAFs, as well as tissue transglutaminase produced by PDAC cells, altered the response to gemcitabine,351 and breast CAFs increased MMP1 secretion synergistically with type IV collagen to promote Taxotere resistance via the TGF-β signaling, while GM6001 (an MMP1 inhibitor) recovered cancer cell chemosensitivity,352 perhaps by establishing physical barriers or inducing microvasculature compression through CAFs, and overcoming CAF-driven ECM hindrance of chemotherapeutic drugs delivery. Then, CAFs released regulatory molecules into tumor tissue in a paracrine-353,354,355,356 or exosome-driven357 manner to protect tumor cells from being eliminated. CAFs that were cocultured with cancer cells secreted IL-6 and upregulated the expression of CXCR7 through the STAT3/NF-κB pathway, which enhanced the proliferation and resistance of ESCC to cisplatin.218 In HNSCC, CAFs can upregulate autophagy by increasing the secretion of IL-6 and IL-8, thereby reducing cell sensitivity to cisplatin.358 In addition, CAFs can promote tumor stemness through the secretion of IL-17a and POSTN.190,359,360 Together, these data suggest that CAFs stimulated by agents in chemotherapy activate pro-survival signaling pathways, including those promoting the proliferation, stemness, and autophagy of cancer cells, to enhance treatment resistance (Fig. 6). In addition, activated CAFs can reprogram tumor cell metabolism to maintain tumor cell survival and protect them from apoptosis induced by treatment and/or stress. Augmented pyruvate in CAFs fed to tumor cells increased the activity of the tricarboxylic acid cycle to promote the survival of lymphoma cells.361 EMT mediated by CAFs decreased cellular adhesion, which was beneficial to tumor metastasis and resistance.362

CAF-mediated therapeutic resistance in anticancer treatment. a CAFs secreted dense desmoplastic matrix, including laminin A1, type IV collagen into the TME hindering the delivery of drugs, and increasing the resistance. b CAFs released regulatory molecules such as periostin, Nodal, EGF, IGF-2, and IL-6 or exosomes, which activated pro-survival signaling pathways, including proliferation, stemness, and apoptosis of cancer cells enhancing therapeutic resistance. c CAFs fed increased pyruvate, lactate, or GSSG to tumor cells by increasing energy supply and decreasing ROS. It also activated IGF-2 to increase autophagy via downstream pathways and the changes of metabolism in CAFs and tumor cells maintain the tumor survival in treatment. d CAFs released regulatory factors, including IGF-1, HGF, CXCL12, IL-6, and TGF-β1, to mediate EMT process through downstream ANAX2 and p38 pathways, subsequently increasing metastasis and resistance. TG2 tissue transglutaminase 2, LAMA1 laminin A1, Bcl-2 B cell lymphoma 2, Bax Bcl-2-associated X protein, Sox-2 SRY (sex-determining region Y)-box 2, TCA cycle tricarboxylic acid cycle, PP2A protein phosphatase 2A, CRM1 chromosomal region maintenance 1, GSSG glutathione disulfide, GSH glutathione, ANXA2 annexin A2
Small-molecule drugs targeting signaling pathways for tumor progression and growth have received attention and are widely used in the treatment of lung, breast, head and neck, and liver cancer.363,364,365 In comparing the potential mechanisms of CAF-mediated drug resistance that are substantially similar to those of chemical resistance, certain aspects of CAF-mediated drug resistance are unique (Fig. 6). To respond to drugs, the tumor–stroma or TME targets specific pathways and attenuates the effects of the drugs. For instance, excess CAF-secreted EGF can competitively bind to EGFR with cetuximab and activate MAPK to promote cetuximab resistance in colorectal cancer.253 Similarly, expressed PDGF-C in CAFs promotes angiogenesis and inhibits the effect of VEGF inhibitor.366
Similar to chemotherapy, CAF-induced pro-survival signaling, including proliferation, autophagy, and stemness, is able to induce resistance to RT (Fig. 6).367,368,369 In addition, the formation of a hypoxic microenvironment and the EMT driven by CAFs can also confer radioresistance onto cancer cells.370,371,372,373 In particular, we concluded that CSCs played an important role in the poor efficacy of RT and that CSCs survived more easily due to their potential innate radioresistance, which was partially induced by CAFs.374 CAFs induced a reduction response to antitumor treatment by maintaining activity via an autocrine periostin loop even during RT,371 and caused radiation-induced fibrosis, which was associated with retinoic acid or TGF-β,375 and with a severe adverse response to RT.376 These properties of CAFs make them potential targets for sensitizing tumors exposed to RT.
Challenges and future perspectives
In the present review, we concluded that several CAF-mediated signaling pathways exerted a supportive role in cancer progression. Key signaling pathway components, biomarkers in CAFs and CAF-derived factors, miRNAs, lncRNAs, etc., were predicted or found to have great potential for targeted therapy. Importantly, several clinical trials on CAFs have also been performed and shown that CAFs have a promising future in cancer therapy (Table 5). However, there are also multiple hurdles that need to be overcome before targeting CAFs in cancer treatment.
First, how would decreasing the number of CAFs directly attenuate their tumor-promoting effects? Ample evidence suggests that FAP is an excellent target for CAFs, and antibodies against FAP and other FAP-targeting drugs are in development.377 T cell-mediated CAFs could be depleted by a DNA vaccine targeting FAP.378,379 Although FAP-specific CAR-T cells can selectively kill CAFs, they have been found to cause extensive lethal osteotoxicity and cachexia due to FAP expression in MSCs.338,380,381 In this regard, nanoparticles based on ZnF16pc loading and FAP-specific single-chain variable fragments or αFAP-Z@FRT can target FAP to effectively and safely deplete CAFs.382 In addition to CAF depletion, sibrolizumab (an anti-FAP monoclonal antibody) has been shown to target and prevent the activation of FAP+ CAFs to inhibit tumor growth.383,384 From another perspective, Noggin (a BMP signaling pathway inhibitor) can reverse the pericyte-CAF transition to decrease the number of CAFs.127 Notably, in addition to CAF depletion, the prevention of CAF generation and CAF normalization highly depend on their cellular phenotypes (Fig. 7a). Thus, there is an urgent need to enhance the understanding of the heterogeneity of CAFs and find more specific therapeutic targets.

CAF-driven targeted therapies and alternative targeting avenues, including epithelial–mesenchymal common target (EMCT), sequential target perturbation (STP), and crosstalk-directed signaling target (CDST) for CAF-directed or host cell-directed antitumor therapy. a Besides the inhibition of NFs to CAFs, there are still two major ways to decrease the CAFs number in TME: (i) targeting specific markers of CAFs by monoclonal antibody (mAb), recombinant virus, anti-precursor cell to deplete CAFs; (ii) revert the activated CAFs to quiescent phenotype by retinoic acid (RA), angiotensin receptor blockers (ARBs), gold nanoparticle (GNP), etc. b Both simultaneous overexpression of the same molecular protein in CAFs and cancer cells like TGF-βRIII and NRG1 has the potential to be targeted as EMCT. c In the regard to CAF-mediated signaling pathways in crosstalk with cancer cells, CDST is targeting simultaneously two different components of one signaling cascade in cancer cells and CAFs, respectively, in which the trigger locating in the upstream of CAFs (Target I), while effector locating in the downstream of cancer cells (Target II). d EGFR-TKI targets the mut-EGFR in cancer cells, while EGFR is up-expressed in CAFs. STP on cancer cells and CAFs, targeting the EGFR in cancer cells firstly (Step①), then in CAFs (Step②), may promote NK cell function to enhance antitumor efficacy and also avoid the IGF-2-mediated chemoresistance after long-term treatment. Since there are different functions in cancer cells and CAFs, STP also aims at targeting CAFs to block the protumor effect firstly (Step①: such as targeting ECM remodeling to move the barrier for CD8+ T cell infiltration) and then aiming at cancer cells (Step②: such as performing PD-1/PD-L1 to antitumor immunotherapy) might obtain well therapeutic efficacy
Second, since targeting cancer cells can unexpectedly render a therapeutic failure or even an acceleration of cancer, the option of EMCT targeting both in cancer cells and their adjacent stromal cells have become an attractive alternative. We have previously demonstrated that the common target perturbation of TGF-βRIII in oral cancerous epithelial cells and CAFs in conjunction with multiple antitumor effects that include depression of angiogenesis and metastasis and promotion of CAFs transition to an NF type, attenuating the CAF support of tumors.385 This finding suggests that the candidates for EMCTs are based on their simultaneous overexpression (Fig. 7b). Indeed, differentially expressed proteins in cancer cells and CAFs are common events in tumor progression. For these cases, we propose an alternative approach: sequential target perturbation (STP) in cancer cells and CAFs might lead to therapeutic efficacy, with targeting of CAFs to block the protumor effect first and then treating cancer cells to realize anticancer effects (Fig. 7d).
Third, to date, targeting two components of one signaling pathway related to specific tumor biology in cancer cells has been performed to treat malignancies.386,387 Since the crosstalk between cancer cells and CAFs is mediated by complexed signaling networks, we propose a novel therapeutic approach involving crosstalk-directed signaling targets (CDSTs) that simultaneously target two different components of one signaling cascade in cancer cells and CAFs, in which the trigger is located upstream of the CAFs and the effector is located in the downstream of the cancer cells (Fig. 7c). For instance, as CAF-derived IL-6 is beneficial for ECM remodeling218,219 and CAF generation220 to fuel tumor progression, and because IL-6 activates the IL-6/JAK/STAT3 signaling pathway in cancer cells,236,237 combining an IL-6-neutralizing antibody236 to treat the CAFs and the JAK inhibitor tofacitinib to treat the cancer cells388,389 might be valuable to study the targeted therapy. CAFs and their related signaling and/or downstream effectors in cancer cells are underexplored targets for cancer therapy.
Fourth, immunotherapy is considered an established pillar of cancer treatment; however, the current questions to answer include why does immunotherapy work well in some cancers but not at all in others? One of the major reasons that cancers fail to respond to anti-PD-1/PD-L immune checkpoint therapy is that CD8+ T cells cannot infiltrate into the TME of “Cold Tumor.”390 An important reason for this failure is CAF-mediated ECM forming a physical barrier to prevent CD8+ T cell infiltration and limits the delivery of drugs.391,392 Currently, there are three strategies for targeting CAF-mediated ECM remodeling: targeting the producer of ECM (activated CAFs),393 targeting the signaling pathways for ECM remodeling,394,395 and targeting ECM components.396,397 In this regard, with a method similar to STP, we propose that targeting CAF-mediated ECM to move the barrier first and then performing cancer immunotherapy might optimize therapeutic efficacy. In addition, since CAFs can recruit the PMN-MDSCs for protumor progression and resistance, attenuating this recruitment is a potential future cancer treatment. In sum, promising CAF-driven immunotherapy might focus on ECM normalization, prevention of disturbances from non-therapeutic immune cells, and combining other antitumor therapies with immunotherapy to maximize the efficacy of cancer immunotherapy.
In conclusion, since CAF-mediated signaling pathways have been implicated in the crosstalk with cancer cells that promote tumor progression and can affect the antitumor therapeutic efficacy, considering the evidence presented in this review, our group hypothesizes that EMCTs, STP, and CDSTs are alternative targeting avenues for CAF- or host cell-directed antitumor therapy.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. https://doi.org/10.3322/caac.21660 (2021).
Gupta, G. P. & Massagué, J. Cancer metastasis: building a framework. Cell 127, 679–695 (2006).
Mahvi, D. A., Liu, R., Grinstaff, M. W., Colson, Y. L. & Raut, C. P. Local cancer recurrence: the realities, challenges, and opportunities for new therapies. CA Cancer J. Clin. 68, 488–505 (2018).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. https://seer.cancer.gov/statfacts (2021).
Fang, H. & Declerck, Y. A. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 73, 4965–4977 (2013).
Roma-Rodrigues, C., Mendes, R., Baptista, P. V. & Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 20, 840 (2019).
Zhang, H. et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 19, 43 (2020).
Tommelein, J. et al. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front. Oncol. 5, 63 (2015).
Knops, A. M. et al. Cancer-associated fibroblast density, prognostic characteristics, and recurrence in head and neck squamous cell carcinoma: a meta-analysis. Front. Oncol. 10, 565306 (2020).
Kanzaki, R. & Pietras, K. Heterogeneity of cancer-associated fibroblasts: opportunities for precision medicine. Cancer Sci. 111, 2708–2717 (2020).
Virchow, R. (eds). Die Cellularpathologie in Ihrer Begründung Auf Physiologische Und Pathologische Gewebelehre (A. Hirschwald, 1858).
Hay, E. D. An overview of epithelio-mesenchymal transformation. Acta Anat. 154, 8–20 (1995).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Sharpe, P. T. Neural crest and tooth morphogenesis. Adv. Dent. Res. 15, 4–7 (2001).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016).
Zhang, W. et al. S100a4 is secreted by alternatively activated alveolar macrophages and promotes activation of lung fibroblasts in pulmonary fibrosis. Front. Immunol. 9, 1216 (2018).
Dave, J. M. & Bayless, K. J. Vimentin as an integral regulator of cell adhesion and endothelial sprouting. Microcirculation 21, 333–344 (2014).
Driskell, R. R. et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013).
Kikuchi, N. et al. Nuclear expression of S100A4 is associated with aggressive behavior of epithelial ovarian carcinoma: an important autocrine/paracrine factor in tumor progression. Cancer Sci. 97, 1061–1069 (2006).
Liu, Y. et al. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral Dis. 12, 375–380 (2006).
Österreicher, C. H. et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl Acad. Sci. USA 108, 308–313 (2011).
Rognoni, E. et al. Fibroblast state switching orchestrates dermal maturation and wound healing. Mol. Syst. Biol. 14, e8174 (2018).
Fintha, A., Gasparics, Á., Rosivall, L. & Sebe, A. Therapeutic targeting of fibrotic epithelial-mesenchymal transition-an outstanding challenge. Front. Pharmacol. 10, 388 (2019).
Hinz, B. The myofibroblast: paradigm for a mechanically active cell. J. Biomech. 43, 146–155 (2010).
Shinde, A. V. & Frangogiannis, N. G. Fibroblasts in myocardial infarction: a role in inflammation and repair. J. Mol. Cell. Cardiol. 70, 74–82 (2014).
Wiseman, B. S. & Werb, Z. Stromal effects on mammary gland development and breast cancer. Science 296, 1046–1049 (2002).
Le Guen, L., Marchal, S., Faure, S. & de Santa Barbara, P. Mesenchymal-epithelial interactions during digestive tract development and epithelial stem cell regeneration. Cell. Mol. Life Sci. 72, 3883–3896 (2015).
Werner, S., Krieg, T. & Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Invest. Dermatol. 127, 998–1008 (2007).
Sakai, N. et al. Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci. Rep. 7, 5392 (2017).
Carr, M. J., Li, Y., Rezakhanlou, A. M. & Ghahary, A. Keratinocyte-releasable factors stimulate the expression of granulocyte colony-stimulating factor in human dermal fibroblasts. J. Cell. Biochem. 118, 308–317 (2017).
Brown, F. D. & Turley, S. J. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J. Immunol. 194, 1389–1394 (2015).
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C. & Brown, R. A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 (2002).
Kuzet, S. E. & Gaggioli, C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 365, 607–619 (2016).
Erez, N., Truitt, M., Olson, P., Arron, S. T. & Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 17, 135–147 (2010).
Li, Q. et al. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci. Rep. 3, 2038 (2013).
Ringuette Goulet, C. et al. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Mol. Cancer Res. 16, 1196–1204 (2018).
Sharon, Y. et al. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 75, 963–973 (2015).
Dror, S. et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 18, 1006–1017 (2016).
Hu, T. & Hu, J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle 18, 3085–3094 (2019).
Shelton, M. et al. The role of CAF derived exosomal microRNAs in the tumour microenvironment of melanoma. Biochim. Biophys. Acta 1875, 188456 (2021).
Orang, A. V., Petersen, J., McKinnon, R. A. & Michael, M. Z. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol. Metab. 23, 98–126 (2019).
Radhakrishnan, R. et al. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 442, 464–474 (2019).
Yoshida, G. J. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 34, 111 (2015).
Zhang, Z. et al. Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell. Mol. Life Sci. 77, 1115–1133 (2020).
Shen, T. et al. YAP1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J. Exp. Clin. Cancer Res. 39, 36 (2020).
Mhaidly, R. & Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: role in immunosuppression and new therapies. Semin. Immunol. 48, 101417 (2020).
LeBleu, V. S. & Neilson, E. G. Origin and functional heterogeneity of fibroblasts. FASEB J. 34, 3519–3536 (2020).
Ishii, G., Ochiai, A. & Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 99, 186–196 (2016).
Pavlides, S. et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8, 3984–4001 (2009).
Yang, J. et al. Glycolysis reprogramming in cancer-associated fibroblasts promotes the growth of oral cancer through the lncRNA H19/miR-675-5p/PFKFB3 signaling pathway. Int. J. Oral Sci. 13, 12 (2021).
Chen, X. & Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 18, 99–115 (2019).
Liu, T. et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 12, 86 (2019).
Zhou, Y. et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 37, 324 (2018).
Luo, J. J., Young, C. D., Zhou, H. M. & Wang, X. J. Mouse models for studying oral cancer: impact in the era of cancer immunotherapy. J. Dent. Res. 97, 683–690 (2018).
Bartoschek, M. et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150 (2018).
Costa, A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479 (2018). e410.
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Costea, D. E. et al. Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 73, 3888–3901 (2013).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Ding, N. et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 153, 601–613 (2013).
Bliss, L. A. et al. Use of postmortem human dura mater and scalp for deriving human fibroblast cultures. PLoS ONE 7, e45282 (2012).
Seluanov, A., Vaidya, A. & Gorbunova, V. Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 5, 2033 (2010).
Lambrechts, D. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Li, H. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017).
Puram, S. V. et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624 (2017). e1624.
Sebastian, A. et al. Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers 12, 1307 (2020).
Li, S. et al. The fibroblast TIAM2 promotes lung cancer cell invasion and metastasis. J. Cancer 10, 1879–1889 (2019).
Richards, K. E. et al. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 36, 1770–1778 (2017).
Cheng, R. et al. Reduced CX3CL1 secretion contributes to the susceptibility of oral leukoplakia-associated fibroblasts to Candida albicans. Front. Cell. Infect. Microbiol. 6, 150 (2016).
van Grunsven, L. A. 3D in vitro models of liver fibrosis. Adv. Drug Deliv. Rev. 121, 133–146 (2017).
Balachander, G. M., Balaji, S. A., Rangarajan, A. & Chatterjee, K. Enhanced metastatic potential in a 3D tissue scaffold toward a comprehensive in vitro model for breast cancer metastasis. ACS Appl. Mater. Interfaces 7, 27810–27822 (2015).
Croce, S., Peloso, A., Zoro, T., Avanzini, M. A. & Cobianchi, L. A hepatic scaffold from decellularized liver tissue: food for thought. Biomolecules 9, 813 (2019).
Mehta, G., Hsiao, A. Y., Ingram, M., Luker, G. D. & Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Rel. 164, 192–204 (2012).
Zanoni, M. et al. Modeling neoplastic disease with spheroids and organoids. J. Hematol. Oncol. 13, 97 (2020).
Fan, H., Demirci, U. & Chen, P. Emerging organoid models: leaping forward in cancer research. J. Hematol. Oncol. 12, 142 (2019).
Kim, E. et al. Creation of bladder assembloids mimicking tissue regeneration and cancer. Nature 588, 664–669 (2020).
Frolik, C. A., Dart, L. L., Meyers, C. A., Smith, D. M. & Sporn, M. B. Purification and initial characterization of a type beta transforming growth factor from human placenta. Proc. Natl Acad. Sci. USA 80, 3676–3680 (1983).
Liu, T. & Feng, X. H. Regulation of TGF-beta signalling by protein phosphatases. Biochem. J. 430, 191–198 (2010).
Shi, Y. & Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003).
Erdogan, B. & Webb, D. J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 45, 229–236 (2017).
Karagiannis, G. S. et al. Collective migration of cancer-associated fibroblasts is enhanced by overexpression of tight junction-associated proteins claudin-11 and occludin. Mol. Oncol. 8, 178–195 (2014).
Shangguan, L. et al. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 30, 2810–2819 (2012).
Hawinkels, L. J. et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 33, 97–107 (2014).
Untergasser, G. et al. Profiling molecular targets of TGF-beta1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech. Ageing Dev. 126, 59–69 (2005).
Lamprecht, S. et al. Teaming up for trouble: cancer cells, transforming growth factor-β1 signaling and the epigenetic corruption of stromal naïve fibroblasts. Cancers 10, 61 (2018).
Barcellos-de-Souza, P. et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells 34, 2536–2547 (2016).
Tan, H. X. et al. TGFβ1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. OncoTargets Ther. 12, 5323–5334 (2019).
Mazzocca, A. et al. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 51, 523–534 (2010).
Martinez-Outschoorn, U. E. et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 43, 1045–1051 (2011).
Guido, C. et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 11, 3019–3035 (2012).
Zhang, D. et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 10, 1335–1348 (2015).
Martinez-Outschoorn, U. E., Sotgia, F. & Lisanti, M. P. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 15, 4–5 (2012).
Shi, X., Young, C. D., Zhou, H. & Wang, X. Transforming growth factor-β signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules 10, 1666 (2020).
Hasegawa, T. et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-β signaling. Int. J. Cancer 134, 1785–1795 (2014).
Fuyuhiro, Y. et al. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 103, 797–805 (2012).
Carloni, V., Luong, T. V. & Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 34, 834–843 (2014).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012).
Tao, L., Huang, G., Song, H., Chen, Y. & Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 14, 2611–2620 (2017).
Yoshida, G. J., Azuma, A., Miura, Y. & Orimo, A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 20, 2256 (2019).
Chen, T. et al. Novel inactivating mutations of transforming growth factor-beta type I receptor gene in head-and-neck cancer metastases. Int. J. Cancer 93, 653–661 (2001).
Lu, S. L. et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 20, 1331–1342 (2006).
Meng, W. et al. Downregulation of TGF-beta receptor types II and III in oral squamous cell carcinoma and oral carcinoma-associated fibroblasts. BMC Cancer 11, 88 (2011).
Yoshida, G. J. Emerging role of epithelial-mesenchymal transition in hepatic cancer. J. Exp. Clin. Cancer Res. 35, 141 (2016).
Zeisberg, M. & Neilson, E. G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437 (2009).
Kojima, Y. et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl Acad. Sci. USA 107, 20009–20014 (2010).
Pavlides, S. et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 9, 3485–3505 (2010).
Cho, J. A. et al. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 123, 379–386 (2011).
Jeon, E. S. et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells 26, 789–797 (2008).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model. Mech. 11, dmm029447 (2018).
Motohara, T. et al. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 38, 2885–2898 (2019).
Ko, S. Y. et al. HOXA9 promotes ovarian cancer growth by stimulating cancer-associated fibroblasts. J. Clin. Invest. 122, 3603–3617 (2012).
Yang, J. et al. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 383, 18–27 (2016).
Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341 (2010).
LoPiccolo, J., Blumenthal, G. M., Bernstein, W. B. & Dennis, P. A. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist. Update 11, 32–50 (2008).
Weichhart, T. & Säemann, M. D. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis. 67, iii70–iii74 (2008).
Knowles, M. A., Platt, F. M., Ross, R. L. & Hurst, C. D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 28, 305–316 (2009).
Loewith, R. et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 (2002).
Pópulo, H., Lopes, J. M. & Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 13, 1886–1918 (2012).
Courtney, K. D., Corcoran, R. B. & Engelman, J. A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 28, 1075–1083 (2010).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004).
Lien, E. C., Dibble, C. C. & Toker, A. PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62–71 (2017).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017).
Ning, X., Zhang, H., Wang, C. & Song, X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer-associated fibroblasts. Med. Sci. Monit. 24, 2350–2359 (2018).
Wang, Y. M., Wang, W. & Qiu, E. D. Osteosarcoma cells induce differentiation of mesenchymal stem cells into cancer associated fibroblasts through Notch and Akt signaling pathway. Int. J. Clin. Exp. Pathol. 10, 8479–8486 (2017).
Chapoval, A. I. et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2, 269–274 (2001).
Zhang, S., Zhou, C., Zhang, D., Huang, Z. & Zhang, G. The anti-apoptotic effect on cancer-associated fibroblasts of B7-H3 molecule enhancing the cell invasion and metastasis in renal cancer. OncoTargets Ther. 12, 4119–4127 (2019).
Pham, L. K. et al. Contextual effect of repression of bone morphogenetic protein activity in prostate cancer. Endocr. Relat. Cancer 20, 861–874 (2013).
Barone, I. et al. Activation of farnesoid X receptor impairs the tumor-promoting function of breast cancer-associated fibroblasts. Cancer Lett. 437, 89–99 (2018).
Catalano, S. et al. Inhibition of Leydig tumor growth by farnesoid X receptor activation: the in vitro and in vivo basis for a novel therapeutic strategy. Int. J. Cancer 132, 2237–2247 (2013).
Modica, S., Murzilli, S., Salvatore, L., Schmidt, D. R. & Moschetta, A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res. 68, 9589–9594 (2008).
Yang, F. et al. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 67, 863–867 (2007).
Li, Y. Y. et al. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine 36, 209–220 (2018).
Li, H. et al. Interleukin-22 secreted by cancer-associated fibroblasts regulates the proliferation and metastasis of lung cancer cells via the PI3K-Akt-mTOR signaling pathway. Am. J. Transl. Res. 11, 4077–4088 (2019).
Zhou, Z. et al. VCAM-1 secreted from cancer-associated fibroblasts enhances the growth and invasion of lung cancer cells through AKT and MAPK signaling. Cancer Lett. 473, 62–73 (2020).
Pang, T. et al. Cancer-associated fibroblasts promote malignancy of gastric cancer cells via Nodal signalling. Cell Biochem. Funct. 38, 4–11 (2020).
Ma, J. et al. Fibroblast-derived CXCL12 regulates PTEN expression and is associated with the proliferation and invasion of colon cancer cells via PI3k/Akt signaling. Cell Commun. Signal. 17, 119 (2019).
Subramaniam, K. S. et al. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 8, e68923 (2013).
Cacheux, W. et al. Interaction between IGF2-PI3K axis and cancer-associated-fibroblasts promotes anal squamous carcinogenesis. Int. J. Cancer 145, 1852–1859 (2019).
Zhang, R., Qi, F., Shao, S., Li, G. & Feng, Y. Human colorectal cancer-derived carcinoma associated fibroblasts promote CD44-mediated adhesion of colorectal cancer cells to endothelial cells by secretion of HGF. Cancer Cell Int. 19, 192 (2019).
Williamson, S. C. et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 7, 13322 (2016).
Xiang, T. et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 9, 5009 (2018).
Kim, H. S. et al. Role of EphA2-PI3K signaling in vasculogenic mimicry induced by cancer-associated fibroblasts in gastric cancer cells. Oncol. Lett. 18, 3031–3038 (2019).
Ding, X. et al. HGF derived from cancer‑associated fibroblasts promotes vascularization in gastric cancer via PI3K/AKT and ERK1/2 signaling. Oncol. Rep. 40, 1185–1195 (2018).
Ma, Y. et al. Low expression of SPARC in gastric cancer-associated fibroblasts leads to stemness transformation and 5-fluorouracil resistance in gastric cancer. Cancer Cell Int. 19, 137 (2019).
Benetatos, L., Voulgaris, E. & Vartholomatos, G. The crosstalk between long non-coding RNAs and PI3K in cancer. Med. Oncol. 34, 39 (2017).
Dong, P. et al. The impact of microRNA-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 12, 231 (2014).
Klingenberg, M., Matsuda, A., Diederichs, S. & Patel, T. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J. Hepatol. 67, 603–618 (2017).
Jahangiri, B., Khalaj-Kondori, M., Asadollahi, E. & Sadeghizadeh, M. Cancer-associated fibroblasts enhance cell proliferation and metastasis of colorectal cancer SW480 cells by provoking long noncoding RNA UCA1. J. Cell Commun. Signal. 13, 53–64 (2019).
Ogier, C. et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett. 432, 227–236 (2018).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Imajo, M., Tsuchiya, Y. & Nishida, E. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life 58, 312–317 (2006).
Kim, E. K. & Choi, E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Lee, S., Rauch, J. & Kolch, W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 21, 1102 (2020).
Khokhlatchev, A. V. et al. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93, 605–615 (1998).
Hossen, M. N. et al. Gold nanoparticle transforms activated cancer-associated fibroblasts to quiescence. ACS Appl. Mater. Interfaces 11, 26060–26068 (2019).
Adam, O. Dietary fatty acids and immune reactions in synovial tissue. Eur. J. Med. Res. 8, 381–387 (2003).
Ando, N. et al. Eicosapentaenoic acid suppresses angiogenesis via reducing secretion of IL‑6 and VEGF from colon cancer‑associated fibroblasts. Oncol. Rep. 42, 339–349 (2019).
Pappalardo, G., Almeida, A. & Ravasco, P. Eicosapentaenoic acid in cancer improves body composition and modulates metabolism. Nutrition 31, 549–555 (2015).
de Carvalho, C. & Caramujo, M. J. The various roles of fatty acids. Molecules 23, 2583 (2018).
Menendez, J. A. & Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 (2007).
Menendez, J. A., Oza, B. P., Colomer, R. & Lupu, R. The estrogenic activity of synthetic progestins used in oral contraceptives enhances fatty acid synthase-dependent breast cancer cell proliferation and survival. Int. J. Oncol. 26, 1507–1515 (2005).
Santolla, M. F. et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17β-estradiol in cancer cells and cancer-associated fibroblasts. J. Biol. Chem. 287, 43234–43245 (2012).
Ishii, G. et al. Fibroblasts associated with cancer cells keep enhanced migration activity after separation from cancer cells: a novel character of tumor educated fibroblasts. Int. J. Oncol. 37, 317–325 (2010).
Eck, S. M., Côté, A. L., Winkelman, W. D. & Brinckerhoff, C. E. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 7, 1033–1044 (2009).
Gong, Y. et al. TIMP-1 promotes accumulation of cancer associated fibroblasts and cancer progression. PLoS ONE 8, e77366 (2013).
Zhang, K. et al. Mechanical signals regulate and activate SNAIL1 protein to control the fibrogenic response of cancer-associated fibroblasts. J. Cell Sci. 129, 1989–2002 (2016).
Burotto, M., Chiou, V. L., Lee, J. M. & Kohn, E. C. The MAPK pathway across different malignancies: a new perspective. Cancer 120, 3446–3456 (2014).
Neufert, C. et al. Tumor fibroblast-derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J. Clin. Invest. 123, 1428–1443 (2013).
Zhu, X. et al. Dual role of twist1 in cancer-associated fibroblasts and tumor cells promoted epithelial-mesenchymal transition of esophageal cancer. Exp. Cell Res. 375, 41–50 (2019).
Wang, X. et al. Cancer-associated fibroblasts-stimulated interleukin-11 promotes metastasis of gastric cancer cells mediated by upregulation of MUC1. Exp. Cell Res. 368, 184–193 (2018).
Tian, B. et al. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget 8, 42300–42313 (2017).
Borriello, L. et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 77, 5142–5157 (2017).
Kikuchi, Y. et al. The niche component periostin is produced by cancer-associated fibroblasts, supporting growth of gastric cancer through ERK activation. Am. J. Pathol. 184, 859–870 (2014).
Wen, S. et al. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 442, 320–332 (2019).
Li, Y. et al. Disulfiram/copper induces antitumor activity against both nasopharyngeal cancer cells and cancer-associated fibroblasts through ROS/MAPK and ferroptosis pathways. Cancers 12, 138 (2020).
Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 51, 1357–1369 (2017).
Latres, E., Chiaur, D. S. & Pagano, M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 18, 849–854 (1999).
MacDonald, B. T., Tamai, K. & He, X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 (2009).
Metcalfe, C., Mendoza-Topaz, C., Mieszczanek, J. & Bienz, M. Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J. Cell Sci. 123, 1588–1599 (2010).
Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell. Signal. 22, 717–727 (2010).
Kikuchi, A., Yamamoto, H., Sato, A. & Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 291, 21–71 (2011).
Zhou, L., Yang, K., Randall Wickett, R. & Zhang, Y. Dermal fibroblasts induce cell cycle arrest and block epithelial-mesenchymal transition to inhibit the early stage melanoma development. Cancer Med. 5, 1566–1579 (2016).
Mosa, M. H. et al. A Wnt-induced phenotypic switch in cancer-associated fibroblasts inhibits EMT in colorectal cancer. Cancer Res. 80, 5569–5582 (2020).
Zhou, L., Yang, K., Wickett, R. R., Kadekaro, A. L. & Zhang, Y. Targeted deactivation of cancer-associated fibroblasts by β-catenin ablation suppresses melanoma growth. Tumor Biol. 37, 14235–14248 (2016).
Bielefeld, K. A. et al. Fibronectin and beta-catenin act in a regulatory loop in dermal fibroblasts to modulate cutaneous healing. J. Biol. Chem. 286, 27687–27697 (2011).
Yu, B. et al. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 9, 1082 (2018).
Mao, B. et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 417, 664–667 (2002).
Mao, B. et al. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411, 321–325 (2001).
Ferrari, N. et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 10, 130 (2019).
Scherz-Shouval, R. et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158, 564–578 (2014).
Kudo, J. et al. Aberrant nuclear localization of beta-catenin without genetic alterations in beta-catenin or Axin genes in esophageal cancer. World J. Surg. Oncol. 5, 21 (2007).
Soulieres, D. et al. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J. Clin. Oncol. 22, 77–85 (2004).
Clements, W. M. et al. Beta-catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 62, 3503–3506 (2002).
Laurent-Puig, P. et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 120, 1763–1773 (2001).
Pan, T., Xu, J. & Zhu, Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. Int. J. Mol. Med. 39, 9–20 (2017).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Novellasdemunt, L. et al. USP7 is a tumor-specific WNT activator for APC-mutated colorectal cancer by mediating β-catenin deubiquitination. Cell Rep. 21, 612–627 (2017).
Giannakis, M. et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 46, 1264–1266 (2014).
Zucman-Rossi, J. et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 26, 774–780 (2007).
Nanki, K. et al. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 174, 856–869 (2018). e817.
Aizawa, T. et al. Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. 8, 6370–6382 (2019).
Unterleuthner, D. et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 23, 159–177 (2020).
Wang, M. et al. Cancer-associated fibroblasts autophagy enhances progression of triple-negative breast cancer cells. Med. Sci. Monit. 23, 3904–3912 (2017).
Maeda, M. et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut 69, 243–251 (2020).
Xiang, L., Song, Z. & Rong, G. Taxotere-induced WNT16 expression in carcinoma-associated fibroblasts might associate with progression and chemoresistance of breast cancer. Ann. Clin. Lab. Sci. 50, 205–212 (2020).
Ihle, J. N. & Kerr, I. M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 11, 69–74 (1995).
Levy, D. E. & Darnell, J. E. Jr. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 (2002).
Hammarén, H. M., Virtanen, A. T., Raivola, J. & Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 118, 48–63 (2019).
Quintás-Cardama, A. & Verstovsek, S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin. Cancer Res. 19, 1933–1940 (2013).
Stroud, R. M. & Wells, J. A. Mechanistic diversity of cytokine receptor signaling across cell membranes. Sci. STKE 2004, re7 (2004).
Delgoffe, G. M., Murray, P. J. & Vignali, D. A. Interpreting mixed signals: the cell’s cytokine conundrum. Curr. Opin. Immunol. 23, 632–638 (2011).
Ina, K. et al. Intestinal fibroblast-derived IL-10 increases survival of mucosal T cells by inhibiting growth factor deprivation- and Fas-mediated apoptosis. J. Immunol. 175, 2000–2009 (2005).
Murray, P. J. The JAK-STAT signaling pathway: input and output integration. J. Immunol. 178, 2623–2629 (2007).
Qiao, Y. et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 37, 873–883 (2018).
Shintani, Y. et al. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J. Thorac. Oncol. 11, 1482–1492 (2016).
Nightingale, J. et al. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J. Am. Soc. Nephrol. 15, 21–32 (2004).
Albrengues, J. et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 6, 10204 (2015).
Sengupta, T. K., Schmitt, E. M. & Ivashkiv, L. B. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proc. Natl Acad. Sci. USA 93, 9499–9504 (1996).
Hu, M. et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 37, 899–905 (2005).
Mishra, R., Haldar, S., Suchanti, S. & Bhowmick, N. A. Epigenetic changes in fibroblasts drive cancer metabolism and differentiation. Endocr. Relat. Cancer 26, R673–R688 (2019).
Fan, J., Xu, G., Chang, Z., Zhu, L. & Yao, J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin. Sci. 134, 807–825 (2020).
Teramoto, K. et al. Clinical significance of PD-L1-positive cancer-associated fibroblasts in pN0M0 non-small cell lung cancer. Lung Cancer 137, 56–63 (2019).
Leonard, W. J. & O’Shea, J. J. Jaks and STATs: biological implications. Annu. Rev. Immunol. 16, 293–322 (1998).
Giannoni, E. et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 70, 6945–6956 (2010).
Vicent, S. et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res. 72, 5744–5756 (2012).
Sun, X. et al. Tumor suppressor HIC1 is synergistically compromised by cancer-associated fibroblasts and tumor cells through the IL-6/pSTAT3 axis in breast cancer. BMC Cancer 19, 1180 (2019).
Heichler, C. et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 69, 1269–1282 (2020).
Zhang, J. et al. Cancer-associated fibroblasts promote the migration and invasion of gastric cancer cells via activating IL-17a/JAK2/STAT3 signaling. Ann. Transl. Med. 8, 877 (2020).
Wang, X., Sun, X., Mu, L. & Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition in endometrial cancer cells by regulating pituitary tumor transforming gene. Cancer Invest. 37, 134–143 (2019).
Jia, C. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition via the transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in hepatocellular carcinoma. Int. J. Biol. Sci. 16, 2542–2558 (2020).
Wang, Y. et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via JAK2-STAT3 pathway. J. Exp. Clin. Cancer Res. 38, 274 (2019).
Sung, P. J. et al. Cancer-associated fibroblasts produce Netrin-1 to control cancer cell plasticity. Cancer Res. 79, 3651–3661 (2019).
Sanz-Moreno, V. et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20, 229–245 (2011).
Olayioye, M. A., Neve, R. M., Lane, H. A. & Hynes, N. E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167 (2000).
Citri, A. & Yarden, Y. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516 (2006).
Yarden, Y. & Sliwkowski, M. X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 (2001).
Ferguson, K. M. et al. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 (2003).
Ogiso, H. et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 (2002).
Siwak, D. R. et al. Targeting the epidermal growth factor receptor in epithelial ovarian cancer: current knowledge and future challenges. J. Oncol. 2010, 568938 (2010).
Tzahar, E. et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 16, 5276–5287 (1996).
Normanno, N. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 (2006).
Shin, N. et al. Cancer-associated fibroblasts and desmoplastic reactions related to cancer invasiveness in patients with colorectal cancer. Ann. Coloproctol. 35, 36–46 (2019).
Carmeci, C., Thompson, D. A., Ring, H. Z., Francke, U. & Weigel, R. J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45, 607–617 (1997).
Luo, H. et al. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr. Relat. Cancer 21, 355–369 (2014).
Pisano, A. et al. GPER, IGF-IR, and EGFR transduction signaling are involved in stimulatory effects of zinc in breast cancer cells and cancer-associated fibroblasts. Mol. Carcinogen. 56, 580–593 (2017).
Aguiar, D. P. et al. New strategy to control cell migration and metastasis regulated by CCN2/CTGF. Cancer Cell Int. 14, 61 (2014).
Li, F. et al. α1,6-Fucosyltransferase (FUT8) regulates the cancer-promoting capacity of cancer-associated fibroblasts (CAFs) by modifying EGFR core fucosylation (CF) in non-small cell lung cancer (NSCLC). Am. J. Cancer Res. 10, 816–837 (2020).
Costa, D. et al. Targeting the epidermal growth factor receptor can counteract the inhibition of natural killer cell function exerted by colorectal tumor-associated fibroblasts. Front. Immunol. 9, 1150 (2018).
Garvey, C. M. et al. Anti-EGFR therapy induces EGF secretion by cancer-associated fibroblasts to confer colorectal cancer chemoresistance. Cancers 12, 1393 (2020).
Mink, S. R. et al. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol. Cancer Res. 8, 809–820 (2010).
Suzuki, E. et al. Secretion of high amounts of hepatocyte growth factor is a characteristic feature of cancer-associated fibroblasts with EGFR-TKI resistance-promoting phenotype: a study of 18 cases of cancer-associated fibroblasts. Pathol. Int. 69, 472–480 (2019).
Vaquero, J. et al. The IGF2/IR/IGF1R pathway in tumor cells and myofibroblasts mediates resistance to egfr inhibition in cholangiocarcinoma. Clin. Cancer Res. 24, 4282–4296 (2018).
Álvarez-Teijeiro, S. et al. Factors secreted by cancer-associated fibroblasts that sustain cancer stem properties in head and neck squamous carcinoma cells as potential therapeutic targets. Cancers 10, 334 (2018).
Grasset, E. M. et al. Matrix stiffening and EGFR cooperate to promote the collective invasion of cancer cells. Cancer Res. 78, 5229–5242 (2018).
Sieg, D. J. et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2, 249–256 (2000).
Gao, Q. et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 216, 688–703 (2019).
Nicholson, R. I., Gee, J. M. & Harper, M. E. EGFR and cancer prognosis. Eur. J. Cancer 37, S9–S15 (2001).
Justice, R. W., Zilian, O., Woods, D. F., Noll, M. & Bryant, P. J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546 (1995).
Zheng, Y. & Pan, D. The Hippo signaling pathway in development and disease. Dev. Cell 50, 264–282 (2019).
Nishio, M. et al. In Innovative Medicine: Basic Research and Development (eds Nakao, K., Minato, N. & Uemoto, S.) 79–94 (Springer, 2015).
Yang, C. C. et al. Differential regulation of the Hippo pathway by adherens junctions and apical-basal cell polarity modules. Proc. Natl Acad. Sci. USA 112, 1785–1790 (2015).
Bao, M., Xie, J., Piruska, A. & Huck, W. T. S. 3D microniches reveal the importance of cell size and shape. Nat. Commun. 8, 1962 (2017).
Ma, S., Meng, Z., Chen, R. & Guan, K. L. The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604 (2019).
Rognoni, E. & Walko, G. The roles of YAP/TAZ and the Hippo pathway in healthy and diseased skin. Cells 8, 411 (2019).
Calvo, F. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Zanconato, F., Battilana, G., Cordenonsi, M. & Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 29, 26–33 (2016).
Ge, L. et al. Yes-associated protein expression in head and neck squamous cell carcinoma nodal metastasis. PLoS ONE 6, e27529 (2011).
Santolla, M. F. et al. miR-221 stimulates breast cancer cells and cancer-associated fibroblasts (CAFs) through selective interference with the A20/c-Rel/CTGF signaling. J. Exp. Clin. Cancer Res. 37, 94 (2018).
Eissa, S., Matboli, M., Sharawy, A. & El-Sharkawi, F. Prognostic and biological significance of microRNA-221 in breast cancer. Gene 574, 163–167 (2015).
Gallo Cantafio, M. E. et al. Pharmacokinetics and pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in mice and non-human primates. Mol. Ther. 5, e336 (2016).
Gullà, A. et al. A 13 mer LNA-i-miR-221 inhibitor restores drug sensitivity in melphalan-refractory multiple myeloma cells. Clin. Cancer Res. 22, 1222–1233 (2016).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Nakamura, R. E. & Hackam, A. S. Analysis of Dickkopf3 interactions with Wnt signaling receptors. Growth Factors 28, 232–242 (2010).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Hoesel, B. & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86 (2013).
Huang, D. B., Vu, D. & Ghosh, G. NF-kappaB RelB forms an intertwined homodimer. Structure 13, 1365–1373 (2005).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362 (2008).
Perkins, N. D. & Gilmore, T. D. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 13, 759–772 (2006).
Cildir, G., Low, K. C. & Tergaonkar, V. Noncanonical NF-κB signaling in health and disease. Trends Mol. Med. 22, 414–429 (2016).
Sun, S. C. Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 (2011).
Xiao, G., Harhaj, E. W. & Sun, S. C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409 (2001).
Zheng, B. et al. CD146 attenuation in cancer-associated fibroblasts promotes pancreatic cancer progression. Mol. Carcinogen. 55, 1560–1572 (2016).
Wu, H., Ma, S., Xiang, M. & Tong, S. HTRA1 promotes transdifferentiation of normal fibroblasts to cancer-associated fibroblasts through activation of the NF-κB/bFGF signaling pathway in gastric cancer. Biochem. Biophys. Res. Commun. 514, 933–939 (2019).
Awaji, M. et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 34, 9405–9418 (2020).
Wei, L. Y. et al. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through CXCL1. Oral Oncol. 88, 115–123 (2019).
Tjomsland, V. et al. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 13, 664–675 (2011).
Xu, Y. et al. MPSSS impairs the immunosuppressive function of cancer-associated fibroblasts via the TLR4-NF-κB pathway. Biosci. Rep. 39, BSR20182171 (2019).
Ma, J. et al. Ligustilide inhibits the activation of cancer-associated fibroblasts. Life Sci. 218, 58–64 (2019).
Erez, N., Glanz, S., Raz, Y., Avivi, C. & Barshack, I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 437, 397–402 (2013).
Li, X. et al. A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis 35, 1362–1370 (2014).
Qin, X. et al. Cancer-associated fibroblast-derived IL-6 promotes head and neck cancer progression via the osteopontin-NF-kappa B signaling pathway. Theranostics 8, 921–940 (2018).
Eiró, N. et al. Toll-like receptor-4 expression by stromal fibroblasts is associated with poor prognosis in colorectal cancer. J. Immunother. 36, 342–349 (2013).
Giannoni, E., Bianchini, F., Calorini, L. & Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox Signal. 14, 2361–2371 (2011).
Yamao, T. et al. Cellular senescence, represented by expression of caveolin-1, in cancer-associated fibroblasts promotes tumor invasion in pancreatic cancer. Ann. Surg. Oncol. 26, 1552–1559 (2019).
Pallangyo, C. K., Ziegler, P. K. & Greten, F. R. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 212, 2253–2266 (2015).
Luo, J. et al. Smad7 promotes healing of radiotherapy-induced oral mucositis without compromising oral cancer therapy in a xenograft mouse model. Clin. Cancer Res. 25, 808–818 (2019).
Ali, S. et al. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am. J. Cancer Res. 5, 1251–1264 (2015).
Djurec, M. et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl Acad. Sci. USA 115, E1147–E1156 (2018).
Du, Y. et al. Intracellular Notch1 signaling in cancer-associated fibroblasts dictates the plasticity and stemness of melanoma stem/initiating cells. Stem Cells 37, 865–875 (2019).
Pazolli, E. et al. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 72, 2251–2261 (2012).
Pelon, F. et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 11, 404 (2020).
Chong, Y. et al. Galectin-1 from cancer-associated fibroblasts induces epithelial-mesenchymal transition through β1 integrin-mediated upregulation of Gli1 in gastric cancer. J. Exp. Clin. Cancer Res. 35, 175 (2016).
Valenti, G. et al. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 77, 2134–2147 (2017).
Xu, G. et al. Exosomal miRNA-139 in cancer-associated fibroblasts inhibits gastric cancer progression by repressing MMP11 expression. Int. J. Biol. Sci. 15, 2320–2329 (2019).
Fang, Y. et al. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 383, 111543 (2019).
Mertens, J. C. et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 73, 897–907 (2013).
Procopio, M. G. et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 17, 1193–1204 (2015).
Schmid, J. O. et al. Cancer cells cue the p53 response of cancer-associated fibroblasts to cisplatin. Cancer Res. 72, 5824–5832 (2012).
Erdogan, B. et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 216, 3799–3816 (2017).
Stadler, S. et al. Colon cancer cell-derived 12(S)-HETE induces the retraction of cancer-associated fibroblast via MLC2, RHO/ROCK and Ca(2+) signalling. Cell. Mol. Life Sci. 74, 1907–1921 (2017).
Becker, L. M. et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. 31, 107701 (2020).
De Francesco, E. M. et al. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 15, R64 (2013).
Murata, T. et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 71, 6633–6642 (2011).
Chan, J. S. K. et al. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 37, 160–173 (2018).
Johansson, A. C. et al. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol. Cancer Res. 10, 1158–1168 (2012).
Li, D. et al. SERS analysis of carcinoma-associated fibroblasts in a tumor microenvironment based on targeted 2D nanosheets. Nanoscale 12, 2133–2141 (2020).
Duda, P., Janczara, J., McCubrey, J. A., Gizak, A. & Rakus, D. The reverse Warburg effect is associated with Fbp2-dependent Hif1α regulation in cancer cells stimulated by fibroblasts. Cells 9, 205 (2020).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Lei, X. et al. Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett. 470, 126–133 (2020).
O’Donnell, J. S., Teng, M. W. L. & Smyth, M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167 (2019).
Yang, Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest. 125, 3335–3337 (2015).
Inoue, S. et al. CD70 expression in tumor-associated fibroblasts predicts worse survival in colorectal cancer patients. Virchows Arch. 475, 425–434 (2019).
Chakravarthy, A., Khan, L., Bensler, N. P., Bose, P. & De Carvalho, D. D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 9, 4692 (2018).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Derynck, R., Turley, S. J. & Akhurst, R. J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 18, 9–34 (2021).
Holmgaard, R. B. et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6, 47 (2018).
Joyce, C. E. et al. TGFβ signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond Syndrome. J. Clin. Invest. 129, 3821–3826 (2019).
Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 (2005).
Aboulkheyr, Es,H., Zhand, S., Thiery, J. P. & Warkiani, M. E. Pirfenidone reduces immune-suppressive capacity of cancer-associated fibroblasts through targeting CCL17 and TNF-beta. Integr. Biol. 12, 188–197 (2020).
Zhang, H. et al. CTL attenuation regulated by PS1 in cancer-associated fibroblast. Front. Immunol. 11, 999 (2020).
Chauhan, V. P. et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl Acad. Sci. USA 116, 10674–10680 (2019).
Feig, C. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl Acad. Sci. USA 110, 20212–20217 (2013).
Ngambenjawong, C., Gustafson, H. H. & Pun, S. H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 114, 206–221 (2017).
Kumar, V. et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 32, 654–668.e655 (2017).
Takahashi, H. et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 8, 8633–8647 (2017).
Kamata, T. et al. Fibroblast-derived STC-1 modulates tumor-associated macrophages and lung adenocarcinoma development. Cell Rep. 31, 107802 (2020).
Zhang, R. et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 10, 273 (2019).
Deng, Y. et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 36, 1090–1101 (2017).
Cheng, J. T. et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 5, e198 (2016).
Hosein, A. N., Brekken, R. A. & Maitra, A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17, 487–505 (2020).
Hussain, A. et al. Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J. Exp. Med. 217, e20191094 (2020).
Yan, Y. et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 38, 171 (2019).
Houthuijzen, J. M. & Jonkers, J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 37, 577–597 (2018).
Kobayashi, H. et al. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 282–295 (2019).
Sun, Y. et al. Cancer associated fibroblasts tailored tumor microenvironment of therapy resistance in gastrointestinal cancers. J. Cell. Physiol. 233, 6359–6369 (2018).
von Ahrens, D., Bhagat, T. D., Nagrath, D., Maitra, A. & Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 10, 76 (2017).
Lee, J. et al. Tissue transglutaminase activates cancer-associated fibroblasts and contributes to gemcitabine resistance in pancreatic cancer. Neoplasia 18, 689–698 (2016).
Cui, Q. et al. Upregulating MMP-1 in carcinoma-associated fibroblasts reduces the efficacy of Taxotere on breast cancer synergized by collagen IV. Oncol. Lett. 16, 3537–3544 (2018).
Izar, B. et al. Bidirectional cross talk between patient-derived melanoma and cancer-associated fibroblasts promotes invasion and proliferation. Pigment Cell Melanoma Res. 29, 656–668 (2016).
Liu, Y., Zhang, J., Sun, X., Su, Q. & You, C. Down-regulation of miR-29b in carcinoma associated fibroblasts promotes cell growth and metastasis of breast cancer. Oncotarget 8, 39559–39570 (2017).
Long, X. et al. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 10, 375 (2019).
Xu, Y., Zhang, Z., Zhang, L. & Zhang, C. Novel module and hub genes of distinctive breast cancer associated fibroblasts identified by weighted gene co-expression network analysis. Breast Cancer 27, 1017–1028 (2020).
Wei, J. R., Dong, J. & Li, L. Cancer-associated fibroblasts-derived gamma-glutamyltransferase 5 promotes tumor growth and drug resistance in lung adenocarcinoma. Aging 12, 13220–13233 (2020).
New, J. et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 77, 6679–6691 (2017).
Lotti, F. et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 210, 2851–2872 (2013).
Ren, J. et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 8, 3932–3948 (2018).
Sakamoto, A. et al. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 110, 269–278 (2019).
Rong, G., Kang, H., Wang, Y., Hai, T. & Sun, H. Candidate markers that associate with chemotherapy resistance in breast cancer through the study on Taxotere-induced damage to tumor microenvironment and gene expression profiling of carcinoma-associated fibroblasts (CAFs). PLoS ONE 8, e70960 (2013).
Chen, T. H., Chang, P. M. & Yang, M. H. Novel immune-modulating drugs for advanced head and neck cancer. Head Neck 41, 46–56 (2019).
Schuppan, D., Ashfaq-Khan, M., Yang, A. T. & Kim, Y. O. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 68-69, 435–451 (2018).
Yang, S., Zhang, Z. & Wang, Q. Emerging therapies for small cell lung cancer. J. Hematol. Oncol. 12, 47 (2019).
Francia, G., Emmenegger, U. & Kerbel, R. S. Tumor-associated fibroblasts as “Trojan Horse” mediators of resistance to anti-VEGF therapy. Cancer Cell 15, 3–5 (2009).
Chen, X. et al. Exosome-mediated transfer of miR-93-5p from cancer-associated fibroblasts confer radioresistance in colorectal cancer cells by downregulating FOXA1 and upregulating TGFB3. J. Exp. Clin. Cancer Res. 39, 65 (2020).
Liu, L. et al. Cancer associated fibroblasts-derived exosomes contribute to radioresistance through promoting colorectal cancer stem cells phenotype. Exp. Cell Res. 391, 111956 (2020).
Wang, Y. et al. Cancer-associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine 17, 45–56 (2017).
Al-Assar, O. et al. Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother. Oncol. 111, 243–251 (2014).
Erkan, M. et al. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 132, 1447–1464 (2007).
Horsman, M. R. & Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res 57, i90–i98 (2016).
Zhang, H. et al. Cancer-associated fibroblast-promoted LncRNA DNM3OS confers radioresistance by regulating DNA damage response in esophageal squamous cell carcinoma. Clin. Cancer Res. 25, 1989–2000 (2019).
Liu, Y., Yang, M., Luo, J. & Zhou, H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. Int. J. Oral Sci. 12, 19 (2020).
Forrester, H. B., Li, J., Leong, T., McKay, M. J. & Sprung, C. N. Identification of a radiation sensitivity gene expression profile in primary fibroblasts derived from patients who developed radiotherapy-induced fibrosis. Radiother. Oncol. 111, 186–193 (2014).
Straub, J. M. et al. Radiation-induced fibrosis: mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 141, 1985–1994 (2015).
Fitzgerald, A. A. & Weiner, L. M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 39, 783–803 (2020).
Lappano, R., Rigiracciolo, D. C., Belfiore, A., Maggiolini, M. & De Francesco, E. M. Cancer associated fibroblasts: role in breast cancer and potential as therapeutic targets. Expert Opin. Ther. Targets 24, 559–572 (2020).
Miao, L., Guo, S., Lin, C. M., Liu, Q. & Huang, L. Nanoformulations for combination or cascade anticancer therapy. Adv. Drug Deliv. Rev. 115, 3–22 (2017).
de Sostoa, J. et al. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer 7, 19 (2019).
Freedman, J. D. et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 78, 6852–6865 (2018).
Zhou, S. et al. FAP-targeted photodynamic therapy mediated by ferritin nanoparticles elicits an immune response against cancer cells and cancer associated fibroblasts. Adv. Funct. Mater. 31, 2007017 (2021).
Hofheinz, R. D. et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26, 44–48 (2003).
Siska, P. J., Singer, K., Evert, K., Renner, K. & Kreutz, M. The immunological Warburg effect: Can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol. Rev. 295, 187–202 (2020).
Meng, W. et al. A systems biology approach identifies effective tumor-stroma common targets for oral squamous cell carcinoma. Cancer Res. 74, 2306–2315 (2014).
Faratian, D. et al. Trastuzumab and pertuzumab produce changes in morphology and estrogen receptor signaling in ovarian cancer xenografts revealing new treatment strategies. Clin. Cancer Res. 17, 4451–4461 (2011).
Weiss, A. et al. Identification of a synergistic multi-drug combination active in cancer cells via the prevention of spindle pole clustering. Cancers 11, 1612 (2019).
Ham, I. H. et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 18, 68 (2019).
Hosseini, A. et al. Janus kinase inhibitors: a therapeutic strategy for cancer and autoimmune diseases. J. Cell. Physiol. 235, 5903–5924 (2020).
Noman, M. Z. et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 6, eaax7881 (2020).
Puré, E. & Lo, A. Can targeting stroma pave the way to enhanced antitumor immunity and immunotherapy of solid tumors? Cancer Immunol. Res. 4, 269–278 (2016).
van Elsas, M. J., van Hall, T. & van der Burg, S. H. Future challenges in cancer resistance to immunotherapy. Cancers 12, 935 (2020).
Chronopoulos, A. et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 7, 12630 (2016).
Jiang, H. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 22, 851–860 (2016).
Olive, K. P. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009).
Hauge, A. & Rofstad, E. K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 18, 207 (2020).
Provenzano, P. P. et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429 (2012).
Yin, Z. et al. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J. Hematol. Oncol. 12, 101 (2019).
Li, J., Guan, J., Long, X., Wang, Y. & Xiang, X. mir-1-mediated paracrine effect of cancer-associated fibroblasts on lung cancer cell proliferation and chemoresistance. Oncol. Rep. 35, 3523–3531 (2016).
Zhang, J. et al. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed. Pharmacother. 74, 215–221 (2015).
Yang, F. et al. MiR-210 in exosomes derived from CAFs promotes non-small cell lung cancer migration and invasion through PTEN/PI3K/AKT pathway. Cell Signal. 73, 109675 (2020).
Shen, H. et al. Reprogramming of normal fibroblasts into cancer-associated fibroblasts by miRNAs-mediated CCL2/VEGFA signaling. PLoS Genet. 12, e1006244 (2016).
Tao, S. et al. Elevating microRNA-1-3p shuttled by cancer-associated fibroblasts-derived extracellular vesicles suppresses breast cancer progression and metastasis by inhibiting GLIS1. Cancer Gene Ther. https://doi.org/10.1038/s41417-020-00244-x (2020).
Gao, Y. et al. CD63(+) cancer-associated fibroblasts confer tamoxifen resistance to breast cancer cells through exosomal miR-22. Adv. Sci. 7, 2002518 (2020).
Verghese, E. T. et al. MiR-26b is down-regulated in carcinoma-associated fibroblasts from ER-positive breast cancers leading to enhanced cell migration and invasion. J. Pathol. 231, 388–399 (2013).
Dou, D. et al. Cancer-associated fibroblasts-derived exosomes suppress immune cell function in breast cancer via the miR-92/PD-L1 pathway. Front. Immunol. 11, 2026 (2020).
Sun, Y. et al. Primed atypical ductal hyperplasia-associated fibroblasts promote cell growth and polarity changes of transformed epithelium-like breast cancer MCF-7 cells via miR-200b/c-IKKβ signaling. Cell Death Dis. 9, 122 (2018).
Tang, X. et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 23, 132–145 (2016).
Du, Y. E. et al. MiR-205/YAP1 in activated fibroblasts of breast tumor promotes VEGF-independent angiogenesis through STAT3 signaling. Theranostics 7, 3972–3988 (2017).
Bronisz, A. et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 14, 159–167 (2011).
Wang, H. et al. MicroRNA-181d-5p-containing exosomes derived from CAFs promote EMT by regulating CDX2/HOXA5 in breast cancer. Mol. Ther. 19, 654–667 (2020).
Liu, Y., Yang, Y., Du, J., Lin, D. & Li, F. MiR-3613-3p from carcinoma-associated fibroblasts exosomes promoted breast cancer cell proliferation and metastasis by regulating SOCS2 expression. IUBMB Life 72, 1705–1714 (2020).
Kim, J. E. et al. The stromal loss of miR-4516 promotes the FOSL1-dependent proliferation and malignancy of triple negative breast cancer. Cancer Lett. 469, 256–265 (2020).
Wu, H. J., Hao, M., Yeo, S. K. & Guan, J. L. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 39, 2539–2549 (2020).
Tang, X. et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 452, 79–89 (2019).
Musumeci, M. et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 30, 4231–4242 (2011).
Zhang, Y. et al. Loss of exosomal miR-146a-5p from cancer-associated fibroblasts after androgen deprivation therapy contributes to prostate cancer metastasis. J. Exp. Clin. Cancer Res. 39, 282 (2020).
Josson, S. et al. miR-409-3p/-5p promotes tumorigenesis, epithelial-to-mesenchymal transition, and bone metastasis of human prostate cancer. Clin. Cancer Res. 20, 4636–4646 (2014).
Shan, G. et al. Cancer-associated fibroblast-secreted exosomal miR-423-5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF-β signaling pathway. Exp. Mol. Med. 52, 1809–1822 (2020).
Bullock, M. D. et al. Pleiotropic actions of miR-21 highlight the critical role of deregulated stromal microRNAs during colorectal cancer progression. Cell Death Dis. 4, e684 (2013).
Yang, X. et al. miR-31 affects colorectal cancer cells by inhibiting autophagy in cancer-associated fibroblasts. Oncotarget 7, 79617–79628 (2016).
Hu, J. L. et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 18, 91 (2019).
Ast, V. et al. MiR-192, miR-200c and miR-17 are fibroblast-mediated inhibitors of colorectal cancer invasion. Oncotarget 9, 35559–35580 (2018).
Shi, L., Wang, Z., Geng, X., Zhang, Y. & Xue, Z. Exosomal miRNA-34 from cancer-associated fibroblasts inhibits growth and invasion of gastric cancer cells in vitro and in vivo. Aging 12, 8549–8564 (2020).
Yang, T. S. et al. MicroRNA-106b in cancer-associated fibroblasts from gastric cancer promotes cell migration and invasion by targeting PTEN. FEBS Lett. 588, 2162–2169 (2014).
Li, P. et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 25, 588–603 (2015).
Wang, R. et al. Downregulation of miRNA-214 in cancer-associated fibroblasts contributes to migration and invasion of gastric cancer cells through targeting FGF9 and inducing EMT. J. Exp. Clin. Cancer Res. 38, 20 (2019).
Liu, X. et al. Exosomal miR-29b from cancer-associated fibroblasts inhibits the migration and invasion of hepatocellular carcinoma cells. Transl. Cancer Res. 9, 2576–2587 (2020).
Zhang, Z. et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 397, 33–42 (2017).
Fang, T. et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 9, 191 (2018).
Utaijaratrasmi, P. et al. The microRNA-15a-PAI-2 axis in cholangiocarcinoma-associated fibroblasts promotes migration of cancer cells. Mol. Cancer 17, 10 (2018).
Zhang, X. et al. Extracellular vesicles-encapsulated microRNA-10a-5p shed from cancer-associated fibroblast facilitates cervical squamous cell carcinoma cell angiogenesis and tumorigenicity via Hedgehog signaling pathway. Cancer Gene Ther. https://doi.org/10.1038/s41417-020-00238-9 (2020).
Zhang, L., Yao, J., Li, W. & Zhang, C. Micro-RNA-21 regulates cancer-associated fibroblast-mediated drug resistance in pancreatic cancer. Oncol. Res. 26, 827–835 (2018).
Shen, Z. et al. Cancer-associated fibroblasts promote cancer cell growth through a miR-7-RASSF2-PAR-4 axis in the tumor microenvironment. Oncotarget 8, 1290–1303 (2017).
Qin, X. et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 20, 12 (2019).
Wang, X. et al. Loss of exosomal miR-3188 in cancer-associated fibroblasts contributes to HNC progression. J. Exp. Clin. Cancer Res. 38, 151 (2019).
Zhou, X. et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 37, 242 (2018).
Wang, J. W., Wu, X. F., Gu, X. J. & Jiang, X. H. Exosomal miR-1228 from cancer-associated fibroblasts promotes cell migration and invasion of osteosarcoma by directly targeting SCAI. Oncol. Res. 27, 979–986 (2019).
Au Yeung, C. L. et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 7, 11150 (2016).
Guo, H. et al. Cancer-associated fibroblast-derived exosomal microRNA-98-5p promotes cisplatin resistance in ovarian cancer by targeting CDKN1A. Cancer Cell Int. 19, 347 (2019).
Mitra, A. K. et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2, 1100–1108 (2012).
Aprelikova, O. et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle 9, 4387–4398 (2010).
Aprelikova, O. et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 32, 3246–3253 (2013).
Li, B. L. et al. Loss of exosomal miR-148b from cancer-associated fibroblasts promotes endometrial cancer cell invasion and cancer metastasis. J. Cell. Physiol. 234, 2943–2953 (2019).
Huang, Y. H. et al. Cancer-associated fibroblast-derived interleukin-1β activates protumor C-C motif chemokine ligand 22 signaling in head and neck cancer. Cancer Sci. 110, 2783–2793 (2019).
De Marco, P. et al. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci. Rep. 6, 24354 (2016).
Zhang, X. & Hwang, Y. S. Cancer-associated fibroblast stimulates cancer cell invasion in an interleukin-1 receptor (IL-1R)-dependent manner. Oncol. Lett. 18, 4645–4650 (2019).
Subramaniam, K. S. et al. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 6, 200–213 (2016).
Wang, L. et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 8, 76116–76128 (2017).
Li, Y. et al. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J. Mol. Med. 97, 243–255 (2019).
Cheteh, E. H. et al. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Dis. 6, 42 (2020).
Ding, X. et al. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 9, 867 (2018).
Goulet, C. R. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 19, 137 (2019).
Pan, M. S. et al. Gallbladder cancer-associated fibroblasts promote vasculogenic mimicry formation and tumor growth in gallbladder cancer via upregulating the expression of NOX4, a poor prognosis factor, through IL-6-JAK-STAT3 signal pathway. J. Exp. Clin. Cancer Res. 39, 234 (2020).
Wu, X. et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 8, 20741–20750 (2017).
Zhang, Y., Cong, X., Li, Z. & Xue, Y. Estrogen facilitates gastric cancer cell proliferation and invasion through promoting the secretion of interleukin-6 by cancer-associated fibroblasts. Int. Immunopharmacol. 78, 105937 (2020).
Xiong, S. et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 8, 302–316 (2018).
Cheng, Y. et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 9, 422 (2018).
Zhai, J. et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 454, 37–43 (2019).
Ma, J., Song, X., Xu, X. & Mou, Y. Cancer-associated fibroblasts promote the chemo-resistance in gastric cancer through secreting IL-11 targeting JAK/STAT3/Bcl2 pathway. Cancer Res. Treat. 51, 194–210 (2019).
Tao, L. et al. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 6, 38408 (2016).
Fukui, H. et al. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br. J. Cancer 111, 763–771 (2014).
Yin, S. Y. et al. Induction of IL-25 secretion from tumour-associated fibroblasts suppresses mammary tumour metastasis. Nat. Commun. 7, 11311 (2016).
Chen, S. F. et al. The paracrine effect of cancer-associated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J. Pathol. 231, 180–189 (2013).
Li, Z. et al. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 145, 1946–1957 (2019).
Bian, L., Sun, X., Jin, K. & He, Y. Oral cancer-associated fibroblasts inhibit heat-induced apoptosis in Tca8113 cells through upregulated expression of Bcl-2 through the Mig/CXCR3 axis. Oncol. Rep. 28, 2063–2068 (2012).
Zhang, F. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Fut. Oncol. 16, 2619–2633 (2020).
Izumi, D. et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int. J. Cancer 138, 1207–1219 (2016).
Li, D. et al. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 6, 2192–2206 (2016).
Sugihara, H. et al. Cancer-associated fibroblast-derived CXCL12 causes tumor progression in adenocarcinoma of the esophagogastric junction. Med. Oncol. 32, 618 (2015).
Wald, O. et al. Interaction between neoplastic cells and cancer-associated fibroblasts through the CXCL12/CXCR4 axis: role in non-small cell lung cancer tumor proliferation. J. Thorac. Cardiovasc. Surg. 141, 1503–1512 (2011).
Wang, M. et al. The primary growth of laryngeal squamous cell carcinoma cells in vitro is effectively supported by paired cancer-associated fibroblasts alone. Tumor Biol. 39, 1–12 (2017).
Noh, K. H. et al. Crosstalk between prostate cancer cells and tumor-associated fibroblasts enhances the malignancy by inhibiting the tumor suppressor PLZF. Cancers 12, 1083 (2020).
Yang, T. et al. Down-regulation of KLF5 in cancer-associated fibroblasts inhibit gastric cancer cells progression by CCL5/CCR5 axis. Cancer Biol. Ther. 18, 806–815 (2017).
Inoue, C. et al. PD-L1 induction by cancer-associated fibroblast-derived factors in lung adenocarcinoma cells. Cancers 11, 1257 (2019).
Allaoui, R. et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat. Commun. 7, 13050 (2016).
Peng, C. et al. Integrin αvβ6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. Biosci. Rep. 38, BSR20180243 (2018).
Wei, L. et al. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 9, 1065 (2018).
Teng, F. et al. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 9, 8 (2016).
Sun, Y. et al. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumor Biol. 39, 712592 (2017).
Hegab, A. E. et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 249, 193–205 (2019).
Knuchel, S., Anderle, P., Werfelli, P., Diamantis, E. & Rüegg, C. Fibroblast surface-associated FGF-2 promotes contact-dependent colorectal cancer cell migration and invasion through FGFR-SRC signaling and integrin αvβ5-mediated adhesion. Oncotarget 6, 14300–14317 (2015).
Suh, J., Kim, D. H., Lee, Y. H., Jang, J. H. & Surh, Y. J. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinogen. 59, 1028–1040 (2020).
Sun, C. et al. FGF9 from cancer-associated fibroblasts is a possible mediator of invasion and anti-apoptosis of gastric cancer cells. BMC Cancer 15, 333 (2015).
Bruzzese, F. et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 74, 3408–3417 (2014).
Kumar, D. et al. Mitigation of tumor-associated fibroblast-facilitated head and neck cancer progression with anti-hepatocyte growth factor antibody ficlatuzumab. JAMA Otolaryngol. Head Neck Surg. 141, 1133–1139 (2015).
Peng, H. et al. Cancer-associated fibroblasts enhance the chemoresistance of CD73(+) hepatocellular carcinoma cancer cells via HGF-Met-ERK1/2 pathway. Ann. Transl. Med. 8, 856 (2020).
Lau, E. Y. et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 15, 1175–1189 (2016).
Yi, Y. et al. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta 1864, 793–803 (2018).
Tommelein, J. et al. Radiotherapy-activated cancer-associated fibroblasts promote tumor progression through paracrine IGF1R activation. Cancer Res. 78, 659–670 (2018).
Zhang, Q., Yang, J., Bai, J. & Ren, J. Reverse of non-small cell lung cancer drug resistance induced by cancer-associated fibroblasts via a paracrine pathway. Cancer Sci. 109, 944–955 (2018).
Sun, D. Y., Wu, J. Q., He, Z. H., He, M. F. & Sun, H. B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 235, 116791 (2019).
Yu, Y. et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 110, 724–732 (2014).
Shen, J. et al. Cancer-associated fibroblasts-derived VCAM1 induced by H. pylori infection facilitates tumor invasion in gastric cancer. Oncogene 39, 2961–2974 (2020).
Gao, M. Q. et al. Human breast cancer-associated fibroblasts enhance cancer cell proliferation through increased TGF-α cleavage by ADAM17. Cancer Lett. 336, 240–246 (2013).
Bauer, J. et al. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin A signaling. Sci. Rep. 10, 50 (2020).
Wang, L. et al. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 110, 1609–1620 (2019).
Satoyoshi, R., Kuriyama, S., Aiba, N., Yashiro, M. & Tanaka, M. Asporin activates coordinated invasion of scirrhous gastric cancer and cancer-associated fibroblasts. Oncogene 34, 650–660 (2015).
Bae, C. A. et al. Inhibiting the GAS6/AXL axis suppresses tumor progression by blocking the interaction between cancer-associated fibroblasts and cancer cells in gastric carcinoma. Gastric Cancer 23, 824–836 (2020).
Miki, Y. et al. CD9-positive exosomes from cancer-associated fibroblasts stimulate the migration ability of scirrhous-type gastric cancer cells. Br. J. Cancer 118, 867–877 (2018).
Bonneau, C. et al. A subset of activated fibroblasts is associated with distant relapse in early luminal breast cancer. Breast Cancer Res. 22, 76 (2020).
Zhu, H. F. et al. Cancer-associated fibroblasts promote colorectal cancer progression by secreting CLEC3B. Cancer Biol. Ther. 20, 967–978 (2019).
Li, X., Li, Q., Yu, X., Li, H. & Huang, G. Reverse of microtubule-directed chemotherapeutic drugs resistance induced by cancer-associated fibroblasts in breast cancer. OncoTargets Ther. 12, 7963–7973 (2019).
Begum, A. et al. Direct interactions with cancer-associated fibroblasts lead to enhanced pancreatic cancer stem cell function. Pancreas 48, 329–334 (2019).
Gong, J. et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 11, 267 (2020).
He, X. J. et al. Expression of galectin-1 in carcinoma-associated fibroblasts promotes gastric cancer cell invasion through upregulation of integrin β1. Cancer Sci. 105, 1402–1410 (2014).
Tang, D. et al. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumor Biol. 37, 1889–1899 (2016).
Ren, J. et al. Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 21, 109 (2019).
Kugeratski, F. G. et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci. Signal. 12, eaan8247 (2019).
Du, X. et al. HIC-5 in cancer-associated fibroblasts contributes to esophageal squamous cell carcinoma progression. Cell Death Dis. 10, 873 (2019).
Zhao, X. L. et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J. Pathol. 243, 376–389 (2017).
Sun, K. et al. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine 41, 370–383 (2019).
Li, Q. et al. Lysyl oxidase promotes liver metastasis of gastric cancer via facilitating the reciprocal interactions between tumor cells and cancer associated fibroblasts. EBioMedicine 49, 157–171 (2019).
Xuefeng, X. et al. Epithelial-mesenchymal transition and metastasis of colon cancer cells induced by the FAK pathway in cancer-associated fibroblasts. J. Int. Med. Res. 48, 300060520931242 (2020).
Wang, X. et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin β1-FAK signaling pathway. Int. J. Cancer 141, 998–1010 (2017).
Zhang, A. et al. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 6, 463–470 (2017).
Principe, S. et al. Proteomic analysis of cancer-associated fibroblasts reveals a paracrine role for MFAP5 in human oral tongue squamous cell carcinoma. J. Proteome Res. 17, 2045–2059 (2018).
Zhang, D. et al. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci. Rep. 7, 16231 (2017).
Fullár, A. et al. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer 15, 256 (2015).
Hassona, Y., Cirillo, N., Heesom, K., Parkinson, E. K. & Prime, S. S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 111, 1230–1237 (2014).
Choi, S. Y. et al. NAB 2-expressing cancer-associated fibroblast promotes HNSCC progression. Cancers 11, 388 (2019).
Liu, C. et al. LSD1 stimulates cancer-associated fibroblasts to drive Notch3-dependent self-renewal of liver cancer stem-like cells. Cancer Res. 78, 938–949 (2018).
Che, Y. et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 9, 759 (2018).
Wei, W. F. et al. Periostin(+) cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol. Oncol. 15, 210–227 (2021).
Chu, L. et al. Periostin secreted by carcinoma-associated fibroblasts promotes ovarian cancer cell platinum resistance through the PI3K/Akt signaling pathway. Technol. Cancer Res. Treat. 19, 153303382097753 (2020).
Underwood, T. J. et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 235, 466–477 (2015).
Elmusrati, A. A., Pilborough, A. E., Khurram, S. A. & Lambert, D. W. Cancer-associated fibroblasts promote bone invasion in oral squamous cell carcinoma. Br. J. Cancer 117, 867–875 (2017).
You, J. et al. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM 112, 581–590 (2019).
Li, Y. et al. Stress-induced upregulation of TNFSF4 in cancer-associated fibroblast facilitates chemoresistance of lung adenocarcinoma through inhibiting apoptosis of tumor cells. Cancer Lett. 497, 212–220 (2021).
Hanley, C. J. et al. Targeting the myofibroblastic cancer-associated fibroblast phenotype through inhibition of NOX4. J. Natl Cancer Inst. 110, 109–120 (2018).
Lv, Y. et al. Nanoplatform assembled from a CD44-targeted prodrug and smart liposomes for dual targeting of tumor microenvironment and cancer cells. ACS Nano 12, 1519–1536 (2018).
Zhang, Q. et al. LY2157299 monohydrate, a TGF-βR1 inhibitor, suppresses tumor growth and ascites development in ovarian cancer. Cancers 10, 260 (2018).
Miao, L. et al. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Res. 77, 719–731 (2017).
Yao, Y. et al. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J. Exp. Clin. Cancer Res. 37, 282 (2018).
Sherman, M. H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93 (2014).
Froeling, F. E. M. et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology 141, 1486–1497 (2011).
Lee, H.-M. et al. Drug repurposing screening identifies bortezomib and panobinostat as drugs targeting cancer associated fibroblasts (CAFs) by synergistic induction of apoptosis. Invest. N. Drugs 36, 545–560 (2018).
Ma, Y. et al. Extreme low dose of 5-fluorouracil reverses MDR in cancer by sensitizing cancer associated fibroblasts and down-regulating P-gp. PLoS ONE 12, e0180023 (2017).
Yao, H., Xu, K., Zhou, J., Zhou, L. & Wei, S. A tumor microenvironment destroyer for efficient cancer suppression. ACS Biomater. Sci. Eng. 6, 450–462 (2020).
Fang, J. et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int. J. Cancer 138, 1013–1023 (2016).
Li, L. et al. Photosensitizer-encapsulated ferritins mediate photodynamic therapy against cancer-associated fibroblasts and improve tumor accumulation of nanoparticles. Mol. Pharm. 15, 3595–3599 (2018).
Scott, A. M. et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 9, 1639–1647 (2003).
Watanabe, S. et al. Photoimmunotherapy for cancer-associated fibroblasts targeting fibroblast activation protein in human esophageal squamous cell carcinoma. Cancer Biol. Ther. 20, 1234–1248 (2019).
Welt, S. et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J. Clin. Oncol. 12, 1193–1203 (1994).
Zhen, Z. et al. Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic t cell infiltration and tumor control. Nano Lett. 17, 862–869 (2017).
Santos, A. M., Jung, J., Aziz, N., Kissil, J. L. & Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 119, 3613–3625 (2009).
Sharma, M. et al. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 218, e20200712 (2021).
Chiappori, A. A. et al. A phase I pharmacokinetic and pharmacodynamic study of s-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin. Cancer Res. 13, 2091–2099 (2007).
Wang, Z.-F. et al. Astragaloside IV inhibits pathological functions of gastric cancer-associated fibroblasts. World J. Gastroenterol. 23, 8512–8525 (2017).
Pietras, K., Pahler, J., Bergers, G. & Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 5, e19 (2008).
US National Library of Medicine. ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT01507545 (2021).
Reardon, D. A. et al. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: phase II study results. J. Clin. Oncol. 24, 115–122 (2006).
Takai, K., Le, A., Weaver, V. M. & Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 7, 82889–82901 (2016).
Zalcman, G. et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet 387, 1405–1414 (2016).
Qian, L. et al. Fusion of dendritic cells and cancer-associated fibroblasts for activation of anti-tumor cytotoxic T lymphocytes. J. Biomed. Nanotechnol. 14, 1826–1835 (2018).
Xiang, H. et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 8, 436–450 (2020).
Lan, Y. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 10, eaan5488 (2018).
Murphy, J. E. et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol. 5, 1020–1027 (2019).
Carapuça, E. F. et al. Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J. Pathol. 239, 286–296 (2016).
Hu, C. et al. Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials 144, 60–72 (2017).
Neesse, A. et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl Acad. Sci. USA 110, 12325–12330 (2013).
Ji, T. et al. Transformable peptide nanocarriers for expeditious drug release and effective cancer therapy via cancer-associated fibroblast activation. Angew. Chem. Int. Ed. Engl. 55, 1050–1055 (2016).
Su, S. et al. CD10GPR77 cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856 (2018). e816.
Hingorani, S. R. et al. HALO 202: randomized phase II study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 36, 359–366 (2018).
Duluc, C. et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 7, 735–753 (2015).
US National Library of Medicine. ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT03481920 (2021).
Cheteh, E. H. et al. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 8, e2848–e2848 (2017).
Amornsupak, K. et al. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer 14, 955 (2014).
Soon, P. S. H. et al. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr. Relat. Cancer 20, 1–12 (2013).
Tanaka, K. et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 36, 894–903 (2015).
Wang, L. et al. Cancer‐associated fibroblasts contribute to cisplatin resistance by modulating ANXA 3 in lung cancer cells. Cancer Sci. 110, 1609–1620 (2019).
Liao, J. K. et al. Cancer-associated fibroblasts confer cisplatin resistance of tongue cancer via autophagy activation. Biomed. Pharmacother. 97, 1341–1348 (2018).
Yan, H., Guo, B.-Y. & Zhang, S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem. Biophys. Res. Commun. 470, 947–954 (2016).
Broekgaarden, M. et al. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 222, 119421 (2019).
Gao, Q., Fang, X., Chen, Y., Li, Z. & Wang, M. Exosomal lncRNA UCA1 from cancer-associated fibroblasts enhances chemoresistance in vulvar squamous cell carcinoma cells. J. Obstet. Gynaecol. Res. 47, 73–87 (2021).
Li, Y., Rong, G. & Kang, H. Taxotere-induced elevated expression of IL8 in carcinoma-associated fibroblasts of breast invasive ductal cancer. Oncol. Lett. 13, 1856–1860 (2017).
Yu, T. et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 36, 2131–2145 (2017).
Gonçalves-Ribeiro, S. et al. Carcinoma-associated fibroblasts affect sensitivity to oxaliplatin and 5FU in colorectal cancer cells. Oncotarget 7, 59766–59780 (2016).
Ying, L. et al. Cancer associated fibroblast-derived hepatocyte growth factor inhibits the paclitaxel-induced apoptosis of lung cancer A549 cells by up-regulating the PI3K/Akt and GRP78 signaling on a microfluidic platform. PLoS ONE 10, e0129593 (2015).
Leung, C. S. et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Invest. 128, 589–606 (2018).
Yegodayev, K. M. et al. TGF-beta-activated cancer-associated fibroblasts limit cetuximab efficacy in preclinical models of head and neck cancer. Cancers 12, 339 (2020).
Eder, T. et al. Cancer-associated fibroblasts modify the response of prostate cancer cells to androgen and anti-androgens in three-dimensional spheroid culture. Int. J. Mol. Sci. 17, 1458 (2016).
Choe, C. et al. Crosstalk with cancer-associated fibroblasts induces resistance of non-small cell lung cancer cells to epidermal growth factor receptor tyrosine kinase inhibition. OncoTargets Ther. 8, 3665–3678 (2015).
Yoshida, T. et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin. Cancer Res. 21, 642–651 (2015).
Mao, Y. et al. Cancer-associated fibroblasts induce trastuzumab resistance in HER2 positive breast cancer cells. Mol. Biosyst. 11, 1029–1040 (2015).
Fernández-Nogueira, P. et al. Tumor-associated fibroblasts promote HER2-targeted therapy resistance through FGFR2 activation. Clin. Cancer Res. 26, 1432–1448 (2020).
Yuan, J. et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res. 17, 69 (2015).
Kharaziha, P. et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of Sorafenib against prostate cancer cells. Cell Death Dis. 3, e262–e262 (2012).
Hellevik, T. et al. Changes in the secretory profile of NSCLC-associated fibroblasts after ablative radiotherapy: potential impact on angiogenesis and tumor growth. Transl. Oncol. 6, 66–74 (2013).
Hwang, R. F. et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926 (2008).
Chu, T.-Y., Yang, J.-T., Huang, T.-H. & Liu, H.-W. Crosstalk with cancer-associated fibroblasts increases the growth and radiation survival of cervical cancer cells. Radiat. Res. 181, 540–547 (2014).
Zhang, H. et al. Cancer-associated fibroblast–promoted LncRNADNM3OS confers radioresistance by regulating DNA damage response in esophageal squamous cell carcinoma. Clin. Cancer Res. 25, 1989–2000 (2019).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 82071124, 82002884, and 81772898), Sichuan Science and Technology Program (Nos. 2021YFS0194, 2021YFH0143, and 2019YFS0361), and Science and Technology Program of Chengdu City (No. 2019YF0501151SN). Figures are created with BioRender.com.
Author information
Authors and Affiliations
Contributions
H.Z. conceived and designed the study. F.W. and J.Y. drafted the manuscript. J.L., Y.W., J.M., Q.Z., S.D., F.W., and J.Y. searched and reviewed the literatures, and made the figures and tables. All the authors critically reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, F., Yang, J., Liu, J. et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Sig Transduct Target Ther 6, 218 (2021). https://doi.org/10.1038/s41392-021-00641-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00641-0
This article is cited by
-
SRT-Server: powering the analysis of spatial transcriptomic data
Genome Medicine (2024)
-
Characterization of gastric cancer-stimulated signaling pathways and function of CTGF in cancer-associated fibroblasts
Cell Communication and Signaling (2024)
-
GPX8+ cancer-associated fibroblast, as a cancer-promoting factor in lung adenocarcinoma, is related to the immunosuppressive microenvironment
BMC Medical Genomics (2024)
-
The immune regulation and therapeutic potential of the SMAD gene family in breast cancer
Scientific Reports (2024)
-
Single-cell integrative analysis reveals consensus cancer cell states and clinical relevance in breast cancer
Scientific Data (2024)
