
Abstract
We report a patient with profound congenital hypotonia, central hypoventilation, poor visual behaviour with retinal hypopigmentation, and significantly decreased mitochondrial respiratory chain complex I activity in muscle, who died at 7 months of age having made minimal developmental progress. Biallelic predicted truncating P4HTM variants were identified following trio whole-genome sequencing, consistent with a diagnosis of hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy and eye abnormalities (HIDEA) syndrome. Very few patients with HIDEA syndrome have been reported previously and mitochondrial abnormalities were observed in three of four previous cases who had a muscle biopsy, suggesting the possibility that HIDEA syndrome represents a primary mitochondrial disorder. P4HTM encodes a transmembrane prolyl 4-hydroxylase with putative targets including hypoxia inducible factors, RNA polymerase II and activating transcription factor 4, which has been implicated in the integrated stress response observed in cell and animal models of mitochondrial disease, and may explain the mitochondrial dysfunction observed in HIDEA syndrome.
Similar content being viewed by others


Introduction
The differential diagnosis for neonatal hypotonia is wide as it may result from abnormality at any level of the nervous system, and the underlying basis includes both environmental and genetic causes [1, 2]. Very few causes are clinically recognisable and achieving a diagnosis requires a systematic, usually multidisciplinary, approach. Access to panel-based genetic testing and next-generation sequencing of both mitochondrial and nuclear genomes has greatly increased the potential to achieve a molecular genetic diagnosis. However, owing to the number of candidate sequence variations often yielded when multiple genes are sequenced, rigorous phenotyping is still important for the interpretation of genetic variants [3, 4].
We describe a patient presenting with a distinctive combination of persistent congenital hypotonia, central hypoventilation, abnormal visual behaviour, and minimal developmental progress. Through trio whole-genome sequencing (WGS), he was found to have novel compound heterozygous predicted truncating variants in P4HTM (Prolyl 4-Hydroxylase, Transmembrane). Biallelic loss of function variants in P4HTM have recently been reported in association with the syndrome of hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy and eye abnormalities (HIDEA, OMIM # 618493 [5]. The findings in the patient we report support hypopigmented fundi and mitochondrial respiratory chain deficiencies in skeletal muscle as recurrent features of this syndrome that are not encapsulated in the acronym HIDEA.
Materials and methods
Informed consent for WGS was obtained in accordance with approval from the HRA Committee East of England–Cambridge South (REC ref. 14/EE/1112). Written, informed consent was taken for publication from the patient’s legal guardians. Detailed phenotyping was undertaken by the authors of this study.
Trio (each parent and child) WGS was performed through the Genomics England 100,000 Genomes Project as previously described [6]. From the raw dataset, rare variants (de novo or biallelic) predicted to have a protein altering affect were reviewed [7]. Alamut-v.2.6 (https://www.interactive-biosoftware.com/alamut-visual/) was utilised in variant interpretation. Variants were confirmed by Sanger sequencing. Variants and phenotypic data were submitted to the Decipher database, available at https://www.deciphergenomics.org, accession number 429076.
Results
Clinical phenotype
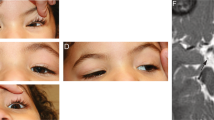
The patient was the third child born to healthy unrelated Caucasian parents. The pregnancy was unremarkable with normal antenatal ultrasound scans, liquor volume, and fetal movements. Normal delivery at 38 weeks’ gestation followed induction of labour for fetal bradycardia. Birth weight was 3.97 kg and head circumference 36.5 cm (both 75th to 91st centile). He did not require any resuscitation but was noted to have a right-sided Erbs palsy, left-sided positional talipes and torticollis, which all resolved with physiotherapy. He had small medial epicanthic folds, asymmetric ears with a left extended crus helix and a small capillary haemangioma of the right thigh but otherwise his appearance was normal. He was floppy and struggled to breastfeed from birth, necessitating intermittent nasogastric feeds from 24 h of age. Although subsequently established on bottle-feeding, he fed extremely slowly and by 6 weeks his weight had dropped to the 0.4th centile. He slept for long periods, had poor tone and head control and a weak cry. His developmental milestones were globally delayed. At 10 weeks he was hospitalised for investigation of faltering growth. He was found to be in type 2 respiratory failure and was intubated and transferred to the paediatric intensive care unit where he received invasive ventilation and inotropes for 4 days. A sleep study revealed central hypoventilation with low baseline oxygen saturation (mean 93%) during quiet sleep (normal 95–100%), bradypnoea with a mean respiratory rate 16/min (normal for 6–9 weeks 27–49 breaths/min), a mixture of central and obstructive events during sleep (total Apnoea-Hypopnoea Index 3.3 events/h) and hypercapnia (>6.7 kPa/50 mmHg) during 20% of quiet sleep (Supplementary Fig. 1). He was therefore commenced on nocturnal BiPAP (Bilevel positive airway pressure ventilation). He had severe central hypotonia and head lag, right-sided ptosis, poor suck, and antigravity movements only. Knee jerks were brisk with extensor plantar responses and ankle clonus bilaterally. He passed his newborn hearing screen. He was noted to have roving eye movements, and ophthalmological assessment revealed structurally normal eyes with moderate hypermetropia, pale optic nerves and blonde fundi. Flash visual evoked potentials (VEPs) were small and suggestive of bilateral generalised visual pathway dysfunction. There was no evidence of facial weakness or tongue fasciculation. Echocardiogram, ECG, diaphragmatic ultrasound, and MRI brain were all unremarkable. The patient’s clinical features are summarised in Table 1.
A trial of pyridostigmine and salbutamol had minimal effect. By 5.5 months of age, he was smiling responsively but had made no motor progress. No seizure activity was noted. His roving eye movements and profound hypotonia remained unchanged.
At 6 months he was readmitted in respiratory failure secondary to a viral lower respiratory tract infection. He decompensated rapidly and was admitted to intensive care where, despite a brief period tolerating BiPaP, he subsequently failed repeated attempts at extubation. Care was redirected to a palliative approach and he died aged 7 months.
Neurometabolic investigations
Extensive neurometabolic investigations in blood (acid maltase, amino acids, ammonia, biotinidase, blood spot carnitines, caeruloplasmin, copper, creatine kinase, full blood count, glucose, lactate, liver function tests, sterols, transferrin electrophoresis, urea and electrolytes, creatinine, bone profile, vacuolated lymphocytes, very long chain fatty acids), urine (glycosaminoglycans, organic acids) and cerebrospinal fluid (amino acids, glucose, lactate, lactate/pyruvate ratio, 5-methyltetrahydrofolate, neurotransmitters, pterins, pyridoxal phosphate), and maternal acetylcholine receptor antibody testing gave results within our laboratory normal reference ranges (values available upon request).
Initial electromyography and nerve conduction studies were interpreted as suggestive, although not diagnostic, of a myasthenic disorder with the degree of jitter abnormality outside the limits expected for immaturity. A muscle biopsy performed at 6 months of age revealed mild myopathic changes including fibre size variation and a few fetal myosin positive fibres reflecting immaturity. Of note, there were no ragged red fibres or fibres staining negatively for cytochrome c oxidase (complex IV, COX). Spectrophotometric analysis of mitochondrial respiratory chain function identified significantly reduced complex I activity (nicotinamide adenine dinucleotide [NADH]: ubiquinone oxidoreductase) to less than 30% of the mean control at 0.054 (reference range 0.104–0.268), normal complex II and III activity at 0.078 (0.040–0.204) and marginally reduced complex IV activity at 0.012 (0.014–0.034).
Genomic investigations
Genetic tests for common causes of neonatal hypotonia (spinal muscular atrophy, Prader–Willi syndrome, myotonic dystrophy), congenital myasthenic syndrome and central hypoventilation syndrome (PHOX2B polyalanine expansion mutations) were normal. There was no evidence of any detectable structural mitochondrial DNA (mtDNA) rearrangements, or mtDNA depletion, and sequencing of mtDNA and a large panel of nuclear genes associated with primary mitochondrial disease did not reveal any loss of function variants.
In trio WGS, no candidate variants were identified in ‘diagnostic grade’ genes listed on the applied panels (Mitochondrial disorders v1.66 and Intellectual disability v2.111), through standard 100,000 Genome Project analysis pipelines. Through careful reanalysis, focusing on rare variants with predicted protein impact, we identified compound heterozygous variants in P4HTM. This gene had not been included in either of the applied panels as there was insufficient evidence of gene:disease association at the point of panel curation, which was prior to 2019 (Fig. 1). The maternally inherited frameshift, P4HTM NM_177938.2: c.[569_579del], p.(Gln190LeufsTer9) and paternally inherited stop gain, P4HTM NM_177938.2: c.[659G>A], p.(Trp220Ter) variants were confirmed by Sanger sequencing. Both variants were novel, absent from gnomAD v2.1.1 and predicted to lead to loss of function (due either to protein truncation with loss of both the EF-hand and the P4H alpha subunit domains or more likely due to nonsense mediated decay) in keeping with previously reported variants in patients with HIDEA syndrome. Details of genomic investigations undertaken and P4HTM variant interpretation using ACMG guidelines can be found in Supplementary Tables 1 and 2 respectively [7].

The proband’s compound heterozygous variants are shown in bold with filled triangles (red). Previously reported variants are shown with unfilled triangles. All variants were rare or absent in population databases and predicted to result in loss of function of P4HTM. The numbers represent amino acid position. TMD transmembrane domain.
Discussion
Biallelic loss of function P4HTM variants have been associated with HIDEA syndrome in 20 patients from 9 families previously. Of note, the P4HTM NM_177938.2 c.[1073G>A] variant has been reported in two Finnish families and appears to have an allele frequency 20 times higher in the Finnish population than non-Finnish Europeans in gnomAD [5, 8]. The P4HTM NM_177938.2 c.[949delG] variant has now been reported in two Syrian families [5, 9] suggesting a possible founder effect.
Table 1 [5, 8, 9] summarises the findings in the previously reported cases and phenotypic overlap with the patient we report. The predominance of males is presumed to be by chance, given the autosomal recessive nature of the condition. The clinical picture appears to be characterised by moderate to severe neonatal hypotonia, severe to profound intellectual disability, hypoventilation that was documented to be central in origin in 6 of the patients, recurrent pneumonias, strabismus, abnormal visual behaviour, and a high incidence of seizures (48%), but without significant structural brain or ocular abnormalities. Only nine of the eighteen patients that survived beyond infancy learned to walk, at between 1.5 and 5 years of age. Of the 16 for whom information was available, 8 never developed speech and 5 were reported to have poor or absent speech, while 3 were able to communicate using simple sentences.
Three patients, including the patient we report, died in infancy, following intercurrent respiratory infections in two. No cause was specified in the third. A further three patients died in childhood, two during febrile respiratory illnesses at 5 years and 8 years respectively, and one at 8 years of age from a respiratory arrest. The oldest reported patient died aged 61 years with acute pneumonia.
Evidence of hypoventilation was found in eight of the patients reported to date and in those who had formal sleep studies, it appeared to be central in origin. Hypoventilation in patients with neuromuscular disorders is often due to respiratory muscle weakness. Central hypoventilation is not usually associated with hypotonia. Most commonly it results from heterozygous polyalanine expansion mutations in PHOX2B [10], which may not be reliably detected in exome based testing. Specific testing for PHOX2B polyalanine expansion mutations in this patient was normal and no other variants in PHOX2B or other genes known to be associated with central hypoventilation were identified by WGS. Obstructive sleep apnoea requiring nocturnal BiPAP was also a common finding in the reported patients with HIDEA. Four of the previously reported patients were suspected to have a sleep behaviour disorder [5, 8].
In addition to refractive errors, strabismus and amblyopia (all common findings in children with neurological disorders and the general paediatric population), poor visual behaviour with roving or rotary eye movements was noted in 57% of the HIDEA patients. No consistent VEP or electroretinogram (ERG) abnormalities have been reported, but where findings were documented, fundal hypopigmentation was noted in 44% (7/16); temporal pallor in a further patient; and pale optic nerves or optic atrophy in 12% (2/17). Visual dysfunction has been reported frequently in patients with mitochondrial complex I deficiency [11].
Mitochondrial abnormalities have been documented in three of four previously reported patients with HIDEA who had muscle biopsies (Table 2). Muscle histology was normal in one (P1) who had low complex II + III activity (31% of control); but non-specifically abnormal (including ultrastructural mitochondrial abnormalities) in a second (P2) who had low activity of complexes I–III (41%) and IV (38%). COX-negative fibres were observed at post-mortem in one (P13). A fourth patient (P5) had myopathic histological features. Respiratory chain activities were not reported in these latter two patients [5].
Muscle histology in the patient we report showed only mild myopathic fibre size variation. There were no ragged red or COX-negative fibres, hallmark features of mitochondrial disease that are rarely observed in childhood, even in genetically confirmed mitochondrial disorders [12]. However, respiratory chain enzyme assays revealed decreased complex I (NADH:ubiquinone oxidoreductase) activity at <30% of mean control, fitting published diagnostic criteria for a mitochondrial disorder [13]. In addition, complex IV activity was marginally decreased. The residual levels of mitochondrial respiratory chain complex I activity in the patient we report are similar to those seen in patients with primary mitochondrial disease caused by loss of function variants in subunits and assembly factors of complex I [14,15,16]. Together with the observation of mitochondrial dysfunction in previously reported patients with HIDEA syndrome, these findings lead us to postulate that P4HTM deficiency should be considered a mitochondrial disorder, although the precise molecular mechanisms leading to mitochondrial dysfunction remain to be unravelled. The distinction between primary and secondary mitochondrial disorders is the subject of much debate [17] but a widely accepted definition of a primary mitochondrial disorder is a genetic disorder leading to dysfunction of oxidative phosphorylation or other disturbances of mitochondrial structure, ultrastructure or function [18]. An increasing number of genetic disorders have been linked to secondary respiratory chain deficiency [17, 19].
The exact function of P4HTM is not known but it has been annotated as an endoplasmic reticulum (ER) transmembrane prolyl 4-hydroxylase (P4H) [20]. Prolyl 4-hydroxylation is a critical post-translational modification needed for the function of a diverse array of proteins [21]. Two groups of P4Hs are recognised: those that hydroxylate proline residues in collagen, and a second group of P4Hs that hydroxylate proline residues in hypoxia inducible factors (HIFs) [20]. HIFs are needed for the cellular response to hypoxia and 4-hydroxylation of critical proline residues targets HIFs for polyubiquitination and proteasomal degradation. Abnormal HIF-1α levels have recently been reported in some primary genetic mitochondrial disease [22, 23]. Targeting HIF P4Hs has been suggested as a strategy for neuroprotection [24, 25], but the presence of severe disease related to P4HTM deficiency suggests that caution should be exercised in exploring such a therapeutic approach. Other possible targets of P4HTM include the large subunit of RNA polymerase II and activating transcription factor 4 (ATF4) [20]. RNA polymerase II is needed for the transcription of all nuclear-encoded mRNAs, including the >1100 nuclear genes whose products are targeted to the mitochondrion [26]. Impaired function of RNA polymerase II as a result of abnormal prolyl 4-hydroxylation may lead to aberrant levels of hundreds of proteins needed for normal mitochondrial function. ATF4 has been implicated in the mitochondrial stress response in cellular and mouse models of mitochondrial disease [27, 28]. Thus, impaired prolyl 4-hydroxlation of HIF, RNA polymerase II or ATF4 could all plausibly account for the mitochondrial dysfunction observed in HIDEA patients and warrants further investigation. A final possibility is that, given its location in the ER membrane, P4HTM deficiency may cause mitochondrial dysfunction by perturbation of the mitochondria-associated membranes (MAMs), as is the case in MEGDEL syndrome, a form of Leigh syndrome caused by impaired lipid remodelling in the MAMs [29].
In summary, HIDEA syndrome is an important consideration in the investigation of profoundly hypotonic infants, particularly those with associated hypoventilation, poor visual behaviour or fundal hypopigmentation. Whilst management currently is principally supportive, a molecular diagnosis can inform counselling about prognosis and provide options for prenatal or preimplantation genetic diagnosis in subsequent pregnancies. Further research is warranted to elucidate the molecular basis, particularly of the mitochondrial dysfunction, and potential for specific disease-modifying therapies.
References
Sparks SE. Neonatal hypotonia. Clin Perinatol. 2015;42:363–71.
Madhok SS, Shabbir N. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021.
Cipriani V, Pontikos N, Arno G, Sergouniotis PI, Lenassi E, Thawong P, et al. An improved phenotype-driven tool for are mendelian variant prioritization: benchmarking exomiser on real patient whole-Exome Data. Genes (Basel). 2020;11:460. https://doi.org/10.3390/genes11040460.
Rahman J, Rahman S. The utility of phenomics in diagnosis of inherited metabolic disorders. Clin Med . 2019;19:30–36.
Rahikkala E, Myllykoski M, Hinttala R, Vieira P, Nayebzadeh N, Weiss S, et al. Biallelic loss-of-function P4HTM gene variants cause hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy, and eye abnormalities (HIDEA syndrome). Genet Med. 2019;21:2355–63.
Taylor RL, Arno G, Poulter JA, Khan KN, Morarji J, Hull S, et al. Association of steroid 5alpha-reductase type 3 congenital disorder of glycosylation with early-onset retinal dystrophy. JAMA Ophthalmol. 2017;135:339–47.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kaasinen E, Rahikkala E, Koivunen P, Miettinen S, Wamelink MM, Aavikko M, et al. Clinical characterization, genetic mapping and whole-genome sequence analysis of a novel autosomal recessive intellectual disability syndrome. Eur J Med Genet. 2014;57:543–51.
Maddirevula S, Ben-Omran T, AlMureikhi M, Eyaid W, Arabi H, Alkuraya H, et al. Further delineation of HIDEA syndrome. Am J Med Genet A. 2020;182:2999–3006.
Mei M, Yang L, Lu Y, Wang L, Cheng G, Cao Y, et al. Congenital central hypoventilation syndrome in neonates: report of fourteen new cases and a review of the literature. Transl Pediatr. 2021;10:733–45.
Fahnehjelm KT, Olsson M, Naess K, Wiberg M, Ygge J, Martin L, et al. Visual function, ocular motility and ocular characteristics in patients with mitochondrial complex I deficiency. Acta Ophthalmol. 2012;90:32–43.
Forny P, Footitt E, Davison JE, Lam A, Woodward CE, Batzios S, et al. Diagnosing Mitochondrial Disorders Remains Challenging in the Omics Era. Neurol Genet. 2021;7:e597. https://doi.org/10.1212/NXG.0000000000000597.
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–11.
Fassone E, Duncan AJ, Taanman JW, Pagnamenta AT, Sadowski MI, Holand T, et al. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum Mol Genet. 2010;19:4837–47.
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–90.
Fassone E, Taanman JW, Hargreaves IP, Sebire NJ, Cleary MA, Burch M, et al. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet. 2011;48:691–7.
Parikh S, Karaa A, Goldstein A, Bertini ES, Chinnery PF, Christodoulou J, et al. Diagnosis of ‘possible’ mitochondrial disease: an existential crisis. J Med Genet. 2019;56:123–30.
Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560–74.
Hui J, Kirby DM, Thorburn DR, Boneh A. Decreased activities of mitochondrial respiratory chain complexes in non-mitochondrial respiratory chain diseases. Dev Med Child Neurol. 2006;48:132–6.
Koivunen P, Tiainen P, Hyvarinen J, Williams KE, Sormunen R, Klaus SJ, et al. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. 2007;282:30544–52.
Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–24.
Boutoual R, Meseguer S, Villarroya M, Martin-Hernandez E, Errami M, Martin MA, et al. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARgamma-UCP2-AMPK axis. Sci Rep. 2018;8:1163.
Mukaneza Y, Cohen A, Rivard ME, Tardif J, Deschenes S, Ruiz M, et al. mTORC1 is required for expression of LRPPRC and cytochrome-c oxidase but not HIF-1alpha in Leigh syndrome French Canadian type patient fibroblasts. Am J Physiol Cell Physiol. 2019;317:C58–C67.
Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. a target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–43.
Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–80.
Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021;49:D1541–7.
Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016;5:e10575.
Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, et al. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017;26:419–28 e415.
Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44:797–802.
Acknowledgements
The authors would like to thank the family members for their consent and support of this article. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Funding
No funding assistance was received in support of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
Awarded to the 100,000 Genomes Project in 2015 by a Health Authority Research Ethics Committee for the main phase of the Programme.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hay, E., Wilson, L.C., Hoskins, B. et al. Biallelic P4HTM variants associated with HIDEA syndrome and mitochondrial respiratory chain complex I deficiency. Eur J Hum Genet 29, 1536–1541 (2021). https://doi.org/10.1038/s41431-021-00932-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00932-8
This article is cited by
-
Fond farewell to clinical utility gene cards
European Journal of Human Genetics (2021)
