
Abstract
While engineering the phase and structure of electrocatalysts could regulate the performance of many typical electrochemical processes, its importance to the carbon dioxide electroreduction has been largely unexplored. Herein, a series of phase and structure engineered copper-tin dioxide catalysts have been created and thoroughly exploited for the carbon dioxide electroreduction to correlate performance with their unique structures and phases. The copper oxide/hollow tin dioxide heterostructure catalyst exhibits promising performance, which can tune the products from carbon monoxide to formic acid at high faradaic efficiency by simply changing the electrolysis potentials from −0.7 VRHE to −1.0 VRHE. The excellent performance is attributed to the abundant copper/tin dioxide interfaces involved in the copper oxide/hollow tin dioxide heterostructure during the electrochemical process, decreasing the reaction free-energies for the formation of COOH* species. Our work reported herein emphasizes the importance of phase and structure modulating of catalysts for enhancing electrochemical CO2 reduction and beyond.
Similar content being viewed by others


Introduction
The increased CO2 level in the atmosphere has caused many climatic concerns, such as global warming, melting glaciers, and more disastrous weather1. The conversion of CO2 into renewable energy sources has attracted extensive attention; this conversion not only decreases the CO2 concentration in the atmosphere but also stores renewable energy2,3,4,5. Recently, the electroreduction of CO2 has attracted particular attention5,6, mainly due to its feasible operating conditions and the potential of sustainable direct integration with renewable resources. Many studies have reported the electrochemical CO2 reduction reaction (CO2RR) by using different catalysts6,7. For example, noble catalysts, such as Au8,9, Ag10,11, and Pd12, are generally reported to reduce CO2 into CO, whereas main group metals In13, Sn14, Pb15, and Bi16 favor the generation of formic acid (HCOOH) during the CO2RR. Unlike noble metal and main group metal catalysts, Cu is active to electroreduce CO2 into hydrocarbons or oxygenates17,18,19, but it also generates common products, such as CO, HCOO− and H220,21. Owing to the unique behavior of Cu in the CO2RR, substantial research efforts have been devoted to improve the activity and selectivity of Cu catalysts for the CO2RR22,23. Nevertheless, the achieved faradaic efficiency (FE) for the useful products is unsatisfactory and usually <40%. To this end, increasingly more researchers focus on combining other metals with Cu to achieve bimetallic Cu-based catalysts13,24, which could bring extra synergistic effects or stabilize the reaction intermediates to improve the activity and selectivity of the CO2RR25,26,27,28. Recently, relevant works, such as the synthesis of CuAu25, CuPd26, and CuSn27,28 bimetallic catalysts, etc. have been intensively reported, showing the enhanced performance for the CO2RR25,26,27,28. However, it should be pointed out that, while the majority of these previous studies focus on improving the CO2RR selectivity for only one product, the selectivity for different useful products at high levels cannot be tuned. In addition, most of these reports are limited to studying the composition (ratio of two metals) effect for the CO2RR, while the structure and phase effects of bimetallic catalysts on the CO2RR are largely unexplored.
It is well known that the bimetallic-based catalyst structures (e.g., core–shell structures, heterostructures, alloying structures, etc.) or phases (e.g., metal/metal phases, metal/oxide phases, metal/sulfide phases, etc.) usually have significant influences on their electrochemical performance29. Such observations have been demonstrated in several electrochemical processes, such as the hydrogen evolution reaction (HER) and the oxygen reduction reaction30,31,32, while limited studies have revealed the structure and phase effects on the CO2RR. Herein, by taking advantage of the intrinsic chemical reactivity differences of the bimetallic components, we successfully develop three distinct Cu–SnO2 bimetallic catalysts with different structures and phases: the CuO/hollow SnO2 heterostructure of CuSn nanoparticles (NPs)/C-A, the Cu41Sn11@SnO2 core–shell structure of CuSn NPs/C-H, and the Cu NPs/hollow SnO2 Janus structure of CuSn NPs/C-AH through a simple yet efficient post-annealing treatment of CuSn core–shell NPs at controlled conditions. These Cu–SnO2 bimetallic catalysts are thoroughly exploited for the electrochemical CO2RR, and the CO2RR performance is correlated with their unique structures and phases. This reveals that the optimized CuSn NPs/C-A exhibits promising activity and selectivity for the electrochemical CO2RR, which can tune the product from CO to HCOOH at high FE values by simply changing the electrolysis potential from −0.7 to −1.0 VRHE. Moreover, it also exhibits the highest CO2RR activity in all the Cu–SnO2 bimetallic catalysts, where the partial current densities of CO at −0.7 VRHE are 5.9 and 5.3 times higher and of HCOOH at −1.0 VRHE are 33.2 and 3.4 times higher than those of the individual Cu NPs/C and SnO2 nanoshell/C (SnO2 NSL/C), respectively. We attribute the excellent CO2RR performance to its abundant Cu/SnO2 interfaces involved in the CuO/hollow SnO2 heterostructure during the electrochemical process, which decrease the reaction free-energies (ΔG) for the formation of COOH* species. Furthermore, under a long-term durability test, these Cu–SnO2 bimetallic catalysts can endure at least 10 h with small activity and selectivity decays.
Results
Preparation and characterization of CuSn NPs
The CuSn NPs were obtained via the co-reduction of copper (II) acetylacetonate (Cu(acac)2), and dibutyltin bis(2,4-pentanedionate) (C18H32O4Sn) at 180 °C for 3 h with ascorbic acid (AA) as the reducing agent and oleylamine (OAm) as the solvent and surfactant. Transmission electron microscopy (TEM) (Fig. 1a) and high-angle annular dark-field scanning TEM (HAADF-STEM) images (Fig. 1b) clearly show that the CuSn NPs are highly dispersive. The diameter of the CuSn NPs was measured to be 23.0 ± 3.5 nm (Supplementary Fig. 1). The Cu/Sn ratio of the CuSn NPs was determined to be 80.7/19.3 by scanning electron microscopy energy-dispersive X-ray spectroscopy (SEM-EDS) (Supplementary Fig. 2). Powder X-ray diffraction (XRD) (Fig. 1c) was carried out to study the phase of the CuSn NPs; the diffraction peaks are perfectly indexed to face-centered cubic Cu, suggesting that the component of Sn are likely amorphous in CuSn NPs. The X-ray photoelectron spectroscopy (XPS) pattern was further carried out to confirm the surface environment of the CuSn NPs, where the Sn 3d5/2 and 3d3/2 peaks of CuSn NPs are located at 486.6 and 495.0 eV, respectively, which can be assigned to the 3d5/2 and 3d3/2 peaks of Sn4+ (Fig. 1d)28, confirming that the Sn species in CuSn NPs is present in the form of amorphous SnO2. To further investigate the distribution of amorphous SnO2 in CuSn NPs, high-resolution TEM (HRTEM) for a single CuSn NP was carried out. As shown in Fig. 1e, it is obvious that there is an amorphous SnO2 shell surrounding the crystalline Cu core. Figure 1f, g display the magnified images recorded from regions f and g marked in Fig. 1e, where the thickness of amorphous SnO2 shell is approximately 1.48 nm, consistent with the HAADF-STEM line scan result of 1.43 nm (Supplementary Fig. 3). The interplanar spacing of the lattice fringe was measured to be 0.209 nm, consistent with the (111) plane of metallic Cu (Fig. 1g). The distributions of Sn and Cu in CuSn NPs were further confirmed by HAADF-STEM EDS elemental mappings (Fig. 1h), where Cu is limited to the core of the CuSn NPs, while Sn surrounds the Cu core. Hence, we can conclude that the prepared CuSn NPs have a core–shell structure with metallic Cu cores and amorphous SnO2 shells.

Structure and phase characterization of CuSn NPs. a Transmission electron microscopic (TEM) image and atom model, b high-angle annular dark-field scanning TEM (HAADF-STEM) image, c X-ray diffraction (XRD) pattern, and d Sn 3d X-ray photoelectron spectroscopy (XPS) pattern of CuSn NPs. e High-resolution TEM (HRTEM) image and f, g the magnified regions in e of CuSn NPs. h HAADF-STEM and energy-dispersive X-ray spectroscopy (EDS) elemental mapping images of CuSn NPs. Scale bars, 2 nm in f, g, 5 nm in e, 20 nm in a, h, 100 nm in b. Red, blue, and white spheres in the model represent O, Cu, and Sn atoms, respectively
Preparation and characterization of CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH
Considering that the chemical reactivities of SnO2 and Cu are quite different, the thermal annealing treatment is expected to be effective to induce phase and structure changes of the pristine CuSn NPs. We thus focus on the phase and structure engineering of CuSn NPs through annealing. To this end, the pristine core–shell CuSn NPs were loaded on a carbon (C, VC-X72) by sonication, denoted as CuSn NPs/C. The core–shell CuSn NPs are highly dispersive on C, without shape, size, phase, and composition changes after the sonication process (Supplementary Fig. 4). The CuSn NPs/C was then annealed at three controlled conditions. The CuSn NPs/C samples treated at 250 °C for 1 h in air and H2/N2 (5/95) were denoted as CuSn NPs/C-A and CuSn NPs/C-H, respectively. The CuSn NPs/C-A sample was further annealed in H2/N2 (5/95) at 250 °C for another 1 h, which was denoted as CuSn NPs/C-AH. As shown in Supplementary Fig. 5, the EDS results reveal that the Cu/Sn ratios in CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH are 80.3/19.7, 79.3/20.7, and 79.2/20.8, respectively, suggesting that the thermal annealing treatment hardly induced composition changes. XRD was further carried out to analyze the structures after different annealing treatments. It is confirmed that the primary diffraction peaks of Cu largely decreased in the CuSn NPs/C-A with the appearance of distinct diffraction peaks of CuO (Fig. 2a), suggesting that the phase of CuSn NPs/C-A was mainly transformed into CuO after annealing in air. As presented in Supplementary Fig. 6, the CuSn NPs/C-H was transformed into the Cu41Sn11 alloy, as observed in the enlarged XRD pattern between 37.0° and 47.0°. By further reannealing the CuSn NPs/C-A at 250 °C for 1 h in H2/N2 (5/95) (CuSn NPs/C-AH), the CuO phase was transformed into metallic Cu. It is worth mentioning that the diffraction peaks of SnO2 do not appear throughout the different annealing treatments, suggesting that such annealing treatment cannot crystallize the amorphous SnO2.

Structure and phase characterizations of CuSn NPs/C with controlled annealing treatment. a X-ray diffraction (XRD) patterns and inset transmission electron microscopic (TEM) images of aI CuSn NPs/C-A, aII CuSn NPs/C-H, and aIII CuSn NPs/C-AH. b, d, f High-angle annular dark-field scanning TEM (HAADF-STEM) images and atom models and c, e, g HAADF-STEM images and energy-dispersive X-ray spectroscopy (EDS) elemental mappings of b, c CuSn NPs/C-A, d, e CuSn NPs/C-H, and f, g CuSn NPs/C-AH. Scale bars, 50 nm in b, d, f, 20 nm in aI–aIII, c, e, g. Red, blue, and white spheres in the models represent O, Cu, and Sn atoms, respectively
Besides the phase structure, the chemical states of CuSn NPs/C, CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH were further investigated by XPS and Auger spectra. Supplementary Fig. 7a shows the XPS pattern of Cu 2p, in which the peaks located at 952.2 and 932.3 eV belong to Cu0,1+ species, whereas the peaks at 953.8 and 933.8 eV belong to Cu2+ species28. The Cu in the initial CuSn NPs/C is in both Cu0,1+ and Cu2+ states. By being heated in air or H2/N2 (5/95), Cu0,1+ species in CuSn NPs/C-A disappeared, while the proportion of the Cu0,1+ species increased in CuSn NPs/C-H. After reannealing the CuSn NPs/C-A at 250 °C in H2/N2 (5/95) for another 1 h, Cu0,1+ species reappeared in the CuSn NPs/C-AH. The Auger spectra of Cu L3M45M45 was further carried out to distinguish the Cu0 and Cu1+ in these catalysts (Supplementary Fig. 8). In general, the characteristic peak of Cu0 for Cu L3M45M45 is located from 918.2 to 918.6 eV, whereas for Cu1+ is between 916.0 and 916.4 eV and the peak for the standard Cu2+ is from 917.6 to 917.8 eV33. The CuSn NPs/C-A shows a peak at 917.6 eV, confirming that there is Cu2+ in the CuSn NPs/C-A. The CuSn NPs/C-H and CuSn NPs/C-AH all have peaks at 916.8 eV and 918.4 eV, suggesting that both Cu0 and Cu1+ are present in the CuSn NPs/C-H and CuSn NPs/C-AH. The binding energy of Sn 3d for these catalysts was also explored (Supplementary Fig. 7b); the chemical state of Sn for all catalysts was mainly SnO2, while the binding energy of Sn in CuSn NPs/C-H and CuSn NPs/C-AH shifted to the high level of 487.0 and 486.9 eV compared to the initial CuSn NPs/C and CuSn NPs/C-A (486.6 eV), likely due to the charge transfer of SnO2 by the reduction of H2. Therefore, we can conclude that the phase of CuSn NPs/C has been readily tuned by thermal annealing in controlled conditions.
The electron microscopy characterizations were further utilized for investigating the structure change of CuSn NPs/C after thermal annealing at different conditions. Fig. 2aI, b show the TEM and STEM images of CuSn NPs/C-A. Interestingly, the initial solid CuSn NPs are transformed into hollow structures, with the size increasing to 26.5 ± 3.5 nm (Supplementary Fig. 9a-b). The HAADF elemental mappings further reveal that the Sn matches well with the hollow shell accompanied by Cu on its surface (Fig. 2c), indicating that the interior Cu diffuses outside to adhere to the hollow SnO2 shell due to the different diffusion rates of Cu and Sn at the core/shell boundary under the annealing treatment in air34. The HRTEM image of CuSn NPs/C-A further shows that the lattice fringe of the section adhering on the SnO2 shell is 0.251 nm, which is the (−111) plane of CuO (Supplementary Fig. 10a, b). Hence, the CuSn NPs/C-A has evolved into a CuO/hollow SnO2 heterostructure. In contrast, the structure of CuSn NPs/C-H is largely maintained compared to the initial CuSn NPs/C (Fig. 2aII, d, e and Supplementary Fig. 9c, d), while the interplanar spacing of the core of CuSn NPs/C-H is 0.212 nm, which is the (660) plane of intermetallic Cu41Sn11 (Supplementary Fig. 10c, d), revealing that the CuSn NPs/C-H has a core–shell structure. Impressively, after reannealing the CuSn NPs/C-A at 250 °C in H2/N2 (5/95) for another 1 h, H2 transformed CuO into metallic Cu conjoined with the hollow SnO2 shell to form a Janus structure with the increased size of 32.5 ± 4.5 nm (Fig. 2aIII, f and Supplementary Fig. 9e, f). The HRTEM and HAADF elemental mappings further confirm the Janus structure of CuSn NPs/C-AH, with the Cu NP as one side and the hollow SnO2 as the other side (Fig. 2g and Supplementary Fig. 10e, f). Therefore, it can be concluded that, after the phase and structure engineering by the simple thermal annealing treatment in controlled conditions, we have successfully transformed the initial core–shell CuSn NPs into three distinct catalysts with different phases and structures: the CuO/hollow SnO2 heterostructure of CuSn NPs/C-A, the Cu41Sn11@SnO2 core–shell structure of CuSn NPs/C-H, and the Cu NPs/hollow SnO2 Janus structure of CuSn NPs/C-AH. All the structures are presented by their corresponding models in the inset in Fig. 2b, d, f.
CO2RR performances of CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH
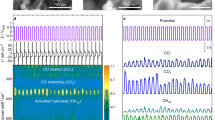
Considering that Cu and Sn are believed to be the active components in the CO2RR14,17,18,19,20,21, the structure and phase effect of the CuSn NPs/C on the CO2RR deserves further investigation. To this end, the CO2RR performances of the CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH were fully evaluated in 0.1 M KHCO3 (Fig. 3). To reveal the bimetallic effect, the Cu NPs/C and SnO2 NSL/C were prepared as references via a solvothermal method and chemical etching of CuSn NPs/C-A, respectively (Supplementary Fig. 11-12). The CO2RR performance was measured by the chronoamperometry method under different potentials (Supplementary Fig. 13). The products in gas and liquid phase were analyzed via gas chromatography (GC) and nuclear magnetic resonance (NMR), respectively (Supplementary Fig. 14-15). Figure 3a shows the polarization curves of CO2RR using linear sweeping voltammetric methods at 5 mV s−1. Compared to the SnO2 NSL/C and Cu NPs/C, all the Cu–SnO2 bimetallic catalysts with different phases and structures exhibit relative high activity from −0.7 to −1.1 VRHE, suggesting that the combination of Cu and Sn enhances the electrocatalytic response. Figure 3b, f show potential-dependent FEs of the major products for CO2RR by using different catalysts. The electrocatalytic process mainly produces H2, CO, and HCOOH from −0.7 to −1.1 VRHE as well as CH4 and C2H4 at −1.0 and −1.1 VRHE for all catalysts except SnO2 NSL/C (Supplementary Fig. 16). The Cu–SnO2 bimetallic catalysts exhibit lower FEH2 below −0.7 VRHE compared with Cu NPs/C and SnO2 NSL/C, revealing that the Cu–SnO2 bimetallic catalysts are propitious to depress the HER during the CO2RR. Moreover, the product FEs of Cu–SnO2 bimetallic catalysts present a trend as the potentials change from −0.7 to −1.0 VRHE; FEHCOOH gradually increases along with FECO and FEH2 decrease, which is different from that of Cu NPs/C or SnO2 NSL/C, revealing that the combination of Cu and SnO2 affects the product selectivity of the CO2RR.

Electrochemical CO2 reduction performance of various Cu–SnO2 catalysts, Cu NPs/C and SnO2 NSL/C. a Linear sweeping voltammetric curves of electrochemical CO2 reduction measured in 0.1 M KHCO3 for different catalysts. Scan rate: 5 mV s−1. Reduction potential-dependent faradaic efficiencies (FEs) of H2, CO, and HCOOH for b CuSn NPs/C-A, c CuSn NPs/C-H, d CuSn NPs/C-AH, e SnO2 NSL/C, and f Cu NPs/C
To further determine the correlation between CO2RR performance and their structures and phases, we summarized the FEs of H2, CO, and HCOOH for these five catalysts at −0.7 and −1.0 VRHE in Fig. 4a, b, respectively. It clear shows that the main product of the CO2RR was CO under the potential of −0.7 VRHE for CuSn NPs/C-A (70.1% FECO) and CuSn NPs/C-AH (45.1% FECO), which largely surpassed the CuSn NPs/C-H (20.1% FECO), Cu NPs/C (13.5% FECO), and SnO2 NSL/C (8.4% FECO). Moreover, at −0.7 VRHE the CuSn NPs/C-A reached the maximum partial current density of CO (1.66 mA cm−2, Supplementary Fig. 17), which was 1.9, 7.9, 5.9, and 33.2 times higher than the values of the CuSn NPs/C-AH (0.86 mA cm−2), CuSn NPs/C-H (0.21 mA cm−2), Cu NPs/C (0.28 mA cm−2), and SnO2 NSL/C (0.05 mA cm−2), respectively (Fig. 4c). The FE and partial current density for the CO under −0.7 VRHE of the CuSn NPs/C-A surpass the reported oxidized-derived copper catalysts6,33,35, the home-made oxidized-derived copper (CuO NPs/C and Cu2O NPs/C catalysts) (Supplementary Fig. 18 and Supplementary Fig. 19), and even comparable to those reported Au8, Ag11, and Pd12 catalysts. When the potential applied was −1.0 VRHE, the FEs of CO and H2 sharply decreased, with HCOOH as the main product for all catalysts except for Cu NPs/C. Particularly, the CuSn NPs/C-A still achieved the highest FEHCOOH of 71.5%, successfully realizing the tuning of the product FE from CO to HCOOH at ~70% by simply changing the electrolysis potential. Such tunable selectivity of CO and HCOOH at high levels surpassed most reported Cu-based bimetallic CO2RR catalysts (Supplementary Table 1). Meanwhile, at −1.0 VRHE the CuSn NPs/C-A also exhibited the highest partial current density of HCOOH (12.6 mA cm−2, Fig. 4d), which exceeded the CuSn NPs/C-AH (10.8 mA cm−2) and CuSn NPs/C-H (10.5 mA cm−2) and was far better than the Cu NPs/C and SnO2 NSL/C. Finally, the long-term stabilities of CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH were also investigated using chronoamperometry at −0.7 VRHE. As shown in Supplementary Fig. 20, all the above catalysts achieved stable current densities in the span of 10 h. Meanwhile, the product FE values of these catalysts could also be maintained at the initial value, suggesting that the CO2RR performances of these Cu–SnO2 bimetallic catalysts is stable. Furthermore, the hollow SnO2 shell and heterostructure of CuSn NPs/C-A were largely maintained after the stability test, revealing the stable structure of CuSn NPs/C-A (Supplementary Fig. 21).

Comparison of the faradaic efficiencies (FEs) and partial current densities of the electrochemical CO2 reduction products for various catalysts under specific potential. Histograms of CO, H2 and HCOOH FEs at a −0.7 VRHE and b −1.0 VRHE for different catalysts. Histograms of current densities of c CO at −0.7 VRHE and d HCOOH at −1.0 VRHE for different catalysts
Discussion
It is generally believed that the chemical state and structure of catalysts would gradually undergo changes during the CO2RR process, which can largely determine their ultimate performance. To this end, the TEM images and XPS characterizations of CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH after the 15 min chronoamperometry test at −0.7 VRHE (denoted as CuSn NPs/C-A-ACP, CuSn NPs/C-H-ACP, and CuSn /C-AH-ACP) were carried out to investigate their evolved surface states and structures. As shown in Fig. 5a, while the CuSn NPs/C-A-ACP still maintained the hollow structure during the CO2RR, the lattice fringes measured in the enlarged image are 0.209 and 0.180 nm, corresponding to the Cu (111) and Cu (200) planes, respectively, being consistent with the XRD result (Supplementary Fig. 22a), suggesting that the oxidized Cu2+ have been reduced into metallic Cu during the CO2RR process (Fig. 5b, c). The chemical state changes of Cu can also be confirmed by the XPS, where the ratio of Cu0,1+/Cu2+ in the CuSn NPs/C-A-ACP increased to 2.7/1, in contrast with 0 in the CuSn NPs/C-A (Supplementary Fig. 23). As opposed to Cu, Sn was still in the form of the amorphous SnO2 combined with the small composition loss in the CuSn NPs/C-A-ACP (Fig. 5b, c and Supplementary Fig. 24a). The element mapping further reveals that the amorphous SnO2 was mixed with Cu in the CuSn NPs/C-A-ACP, which can produce numerous Cu/SnO2 interfaces (Fig. 5d). In contrast, the CuSn NPs/C-H-ACP maintains the Cu41Sn11@SnO2 core–shell structure of the initial CuSn NPs/C-H with small composition change, as confirmed by the HRTEM image, HAADF EDS elemental mappings, XRD, and EDS results (Fig. 5e–h, Supplementary Fig. 22b, and Supplementary Fig. 24b), indicating that there are hardly any Cu/SnO2 interfaces exposed. The structure of CuSn NPs/C-AH-ACP also undergoes a significant change, where the original hollow amorphous SnO2 shrinks into a solid particle combined with the Cu in the CuSn NPs/C-AH-ACP due to the element corrosion and migration during the CO2RR process (Fig. 5i, Supplementary Fig. 22c, and Supplementary Fig. 24c). The enlarged HRTEM and element mapping images further show that the newly evolved structure of CuSn NPs/C-AH-ACP has a few interface between Cu and SnO2 (Fig. 5j–l, Supplementary Fig. 25). Moreover, the surface composition of the above catalysts was also confirmed by XPS results (Supplementary Table 2). The ratios of Cu/Sn in CuSn NPs/C-H-ACP and CuSn NPs/C-AH-ACP calculated by the XPS results are lower than those by EDS results, suggesting that the Sn is mainly located on the surface in these catalysts, being consistent with the results of element mapping. Moreover, according to the XPS results the ratio of Cu/Sn in CuSn NPs/C-A-ACP is similar with the CuSn NPs/C-AH-ACP, indicating that the composition effect on the FECO improvement of CuSn NPs/C-A-ACP and CuSn NPs/C-AH-ACP is negligible. Thus, with the surface state and structure evolutions of different catalysts, the greatest difference among them is the density of the newly formed Cu/SnO2 interfaces, which follows the sequence: CuSn NPs/C-A-ACP > CuSn NPs/C-AH-ACP > CuSn NPs/C-H-ACP. Therefore, it is reasonable to conclude that the newly produced Cu/SnO2 interfaces are the active sites for improving the catalytic selectivity in those Cu–SnO2 bimetallic catalysts and resulting in the different selectivity for the CO2RR. To further prove it, the mixed Cu NPs/C and SnO2 NSL/C with similar Cu/Sn ratio but limited Cu/SnO2 interfaces were used as the catalyst for the CO2RR (Supplementary Fig. 26). It is shown that the FECO of mixed Cu NPs/C and SnO2 NSL/C at −0.7 VRHE is 31.3% (Supplementary Fig. 27), which is between the FECO of CuSn NPs/C-H and of CuSn NPs/C-AH. Based on the TEM and HRTEM observation and the CO2RR results of various catalysts, we can conclude that the FEsCO of different catalysts are systematically controlled by the Cu/SnO2 interfaces (Supplementary Fig. 28), confirming the importance of Cu/SnO2 interfaces in boosting the product selectivity of CO2RR.

Structure and interface characterizations of CuSn NPs/C-A-ACP, CuSn NPs/C-H-ACP, and CuSn NPs/C-AH-ACP. a, e, i High-resolution transmission electron microscopy (HRTEM) images, b, c, f, g, j, k magnified HRTEM images, and d, h, l high-angle annular dark-field scanning TEM (HAADF-STEM) image and energy-dispersive X-ray spectroscopy (EDS) elemental mappings of a–d CuSn NPs/C-A-ACP, e–h CuSn NPs/C-H-ACP, and i–l CuSn NPs/C-AH-ACP. Scale bars, 10 nm in d, h, l, 5 nm in a, e, i, 2 nm in b, c, f, g, j, k
To further elucidate the Cu/SnO2 interfaces effect on product selectivity for Cu–SnO2 bimetallic catalysts, the density functional theory (DFT) calculations of Cu–SnO2 heterostructures were carried out. In general, the CO2 electroreduction usually initiates with the proton-coupled electron transfer process, then the C or O atom protonation would result in generating OCHO* or COOH* species, respectively. The adsorption free-energies of CO2RR intermediates and the ΔG of elementary steps for Cu (111) and SnO2 (110) surfaces were first calculated employing the computational hydrogen electrode (CHE) model23. As shown in Supplementary Fig. 29, the formation of OCHO* species (ΔG = 0.20 eV) is energetically more favorable than that of COOH* species (ΔG = 0.71 eV) on the Cu (111) surface. Similarly, the SnO2 (110) surface also favors HCOOH as the final product since the formation of OCHO* (ΔG = 1.48 eV) species is energetically much more favorable than that of COOH*species (ΔG = 2.32 eV) (Supplementary Fig. 30). Furthermore, the Cu/SnO2 interfacial effect on the CO2RR electrocatalytic performance of the Cu–SnO2 heterostructure were explored. The Cu/SnO2 interface was constructed by binding the Sn6O12 cluster onto the Cu (111) slab. Note that the surface-supported small oxide clusters are effective to describe catalytic activity of the complex36,37. As shown in Fig. 6a, in sharp contrast to individual Cu (111) and SnO2 (110) surfaces, the O atom rather than the C atom of the CO2 molecule prefers to be first hydrogenated at the Cu/SnO2 interface. Specifically, the ΔG for the COOH* species formation is 0.52 eV at Cu/SnO2 interfaces, being lower than those of 0.71 and 2.32 eV at the Cu (111) surface and SnO2 (110) surface, respectively. Meanwhile, the COOH* species formation is energetically more favorable than that of OCHO* species (ΔG = 0.85 eV) at Cu/SnO2 interfaces. The COOH* intermediate can be easily converted to a removed H2O molecule and an adsorbed CO* species with a ΔG of 0.43 eV. Finally, the CO* species can be desorbed easily with a ΔG of −0.13 eV to form the CO. The calculations vividly demonstrated that the construction of the Cu/SnO2 interface can essentially tune the catalytic products and enhance the activity of Cu–SnO2 bimetallic catalysts to generate CO, which are in good agreement with experimental observations. Thus owing to the more evolved Cu/SnO2 interfaces than that of the CuSn NPs/C-AH during the CO2RR process (Fig. 6b), the CuSn NPs/C-A shows a higher FECO at −0.7 VRHE. However, for the CuSn NPs/C-H, the exposed reactive site is only SnO2 (Fig. 6b), resulting in both high FEH2 and FEHCOOH values. Although Sarfaz et al. and Li et al.28 have also shown high FECO at lower overpotential for CO2RR by the CuSn complex structures, the active site for improving the CO2RR performance is vague. In fact, the biggest concept difference between our work and Sarfaz’s work/Li’s work is that the Sarfaz’s work/Li’s work mainly focused on controlling the Cu/Sn composition and SnO2 thickness to tune the CO2RR, while the unexplored phase/structure effects of Cu/Sn catalysts for enhancing CO2RR have been decoded in our work. This is also the main reason why we can generate both CO and HCOOH with high selectivity, while only high FECO can be obtained in Sarfaz’ work/Li’s work. By further combining the experimental characterizations with the DFT calculations, we have revealed the important role of Cu/SnO2 interfaces on enhancing the CO2RR, which makes a progress to understand the phase and structure effects for improving the CO2RR performance.

Density functional theory simulations of the CO2 reduction reaction on Cu/SnO2 interfaces. a Free energy profiles of two pathways for CO2 electroreduction on Cu/SnO2 interfaces. The upper and lower images are optimized geometric structures of various states (COOH*, OCHO*, and CO*) of the process on Cu/SnO2 interfaces, respectively. H, C, O, Cu, and Sn atoms are represented by green, gray, red, reddish brown, and dark gray spheres, respectively. b The models of CuSn NPs/C-A-ACP, CuSn NPs/C-H-ACP, and CuSn NPs/C-AH-ACP with different Cu/SnO2 interfaces
In summary, by simple thermal annealing of CuSn core–shell NPs in controlled conditions, we have created Cu–SnO2 catalysts with three distinct engineered structures and phases: the CuO/hollow SnO2 heterostructure of CuSn NPs/C-A, the Cu41Sn11@SnO2 core–shell structure of CuSn NPs/C-H, and the Cu NPs/hollow SnO2 Janus structure of CuSn NPs/C-AH. Benefitting from the abundant Cu/SnO2 interfaces involved in the CuO/hollow SnO2 heterostructure during the electrochemical process, the CuSn NPs/C-A exhibits promising electrocatalytic performance for CO2RR due to the decreased ΔG for the formation of COOH* species at Cu/SnO2 interfaces. It shows the highest activity toward CO2RR in all the Cu–SnO2 heterostructures, with partial current densities of CO at −0.7 VRHE over 5.9 and 5.3 times higher and of HCOOH at −1.0 VRHE over 33.2 and 3.4 times higher than those of the Cu NPs/C and SnO2 NSL/C, respectively. Particularly, the CuSn NPs/C-A can tune the products from CO to HCOOH with an FE at approximately 70% by simply changing the electrolysis potentials. These Cu–SnO2 bimetallic catalysts also exhibit excellent durability after the long-term chronoamperometry test at −0.7 VRHE. The present study highlights a phase and structure-engineering strategy for creating high-performance electrocatalysts for the CO2RR and beyond.
Methods
Chemicals
Copper (II) acetylacetonate (Cu(acac)2, 97%), AA (AR), and oleylamine (CH3(CH2)7CH=CH(CH2)7CH2NH2, 70%) were all purchased from Sigma-Aldrich. Potassium bicarbonate (KHCO3, AR) was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Dibutyltin bis(2,4-pentanedionate) (C18H32O4Sn) was purchased from Strem Chemical. Nitrogen (N2, 99.999%) and carbon dioxide (CO2, 99.999%) were purchased from WuGang Gas Co. Ltd. (Shanghai, China). All the above chemicals were directly used without further purification. The water (18 MΩ*cm) used in all experiments was prepared with an ultra-pure purification system.
Preparation of CuSn NPs
In a typical preparation of CuSn NPs, 13.3 mg Cu(acac)2, 6 μL C18H32O4Sn, 27 mg AA, and 5 mL oleylamine were mixed and ultrasonicated for approximately 0.5 h in a sealed vial (30 mL). Then the vial was transferred into an oil bath and heated at 180 °C for 3 h. The resulting colloidal products in the vial were collected by centrifugation and washed with an ethanol/cyclohexane mixture to obtain CuSn NPs.
Preparation of CuSn NPs/C, CuSn NPs/C-A, CuSn NPs/C-H, and CuSn NPs/C-AH
In a typical preparation of CuSn NPs/C, CuSn NPs were mixed with VC-X72 carbon in 10 mL cyclohexane and sonicated for 1 h to deposit them on carbon. The products were separated by centrifugation and washed with cyclohexane/ethanol to generate CuSn NPs/C. The CuSn NPs/C was annealed in air at 250 °C for 1 h and 5% H2 +95% N2 mixed gas at 250 °C for 1 h to obtain CuSn NPs/C-A and CuSn NPs/C-H, respectively. The CuSn NPs/C-A sample was further annealed in 5% H2 +95% N2 at 250 °C for another 1 h to generate CuSn NPs/C-AH. The mass loadings in all the catalysts were fixed to be 10%+0.5%, as analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES).
Preparation of Cu NPs and Cu NPs/C
The preparation of Cu NPs was similar to that of CuSn NPs but without adding C18H32O4Sn. The resulting products were collected by centrifugation and washed with an ethanol/cyclohexane mixture. The obtained Cu NPs were then mixed with VC-X72 carbon in 10 mL cyclohexane and sonicated for 1 h to deposit them on carbon. The products were separated by centrifugation and washed with cyclohexane/ethanol to generate Cu NPs/C.
Preparation of SnO2 NSL/C
In a typical preparation of SnO2 NSL/C, 10 mg CuSn NPs/C-A were mixed with 8 mL ethanol and 2 mL acetic acid and ultrasonicated for 1 h to remove the CuO. The products were separated by centrifugation and washed with ethanol to generate SnO2 NSL/C.
Characterization
TEM and HAADF-STEM were conducted on an FEI Tecnai F20 transmission electron microscope with acceleration voltage of 200 kV. The samples were prepared by dropping samples dispersed in cyclohexane or ethanol onto carbon-coated copper TEM grids using pipettes and were dried under ambient conditions. XRD patterns were collected on the X’Pert-Pro MPD diffractometer (Netherlands PANalytical) with a Cu Kα X-ray source (λ = 1.540598 Å). The concentrations of catalysts were determined by ICP-AES (710-ES, Varian). XPS was performed with an SSI S-Probe XPS Spectrometer. The carbon peak at 284.6 eV was used as a reference to correct for charging effects.
Electrochemical measurements
The electrochemical measurements of CO2RR was performed by a three-electrode system with a carbon paper (Toray, 1 × 1 cm2) as working electrode, a micro Ag/AgCl electrode (4.0 M KCl) as reference electrode, and a Pt wire as the reference electrode. In the preparation of working electrode, 8 mg catalyst was ultrasonicated with 380 µL of ethanol, 20 µL of water, and 5 µL of a 5 wt% Nafion solution for 1 has catalyst ink. Then 100 µL of the catalyst ink was dropped onto carbon paper and aired to obtain the working electrodes. The electrochemical measurements was conducted in an H-cell reactor with an anion exchange membrane (Nafion 117) on CHI660 (Chenhua, Shanghai) electrochemical workstation. Each chamber of the H-cell contained 60 mL of 0.1 M KHCO3 aqueous solution with an ~40 mL headspace. For the electrochemical measurements, the CO2 was delivered into the cathodic compartment (directly connected to the gas chromatograph (GC Agilent 7890B)) at a constant rate of 20 sccm and was allowed to purge for 30 min prior to the beginning of experiments. The gas-phase composition was analyzed by a GC equipped with a PLOT MolSieve 5A column and a Q-bond PLOT column every 15 min with different potentials applied. Liquid products were analyzed by 1H NMR on Agilent 600 MHz DirectDrive2 spectrometers. All potentials were given against the reversible hydrogen electrode (RHE), calculated using the Nernst equation, and the readouts were recorded with 90% Ohmic iR drop correction. The FE for the products was calculated as follows:
where e is the number of electrons transferred for different products, Q is the total charge, n is the total amount of different product (in moles), and F is the Faraday constant.
Computational method
DFT calculations were performed using the plane-wave technique implemented in the Vienna ab initio simulation package38,39. The ion–electron interaction was described using the projector-augmented plane wave approach40. The generalized gradient approximation expressed by the Perdew–Burke–Ernzerhof functional41 and a 360-eV cutoff for the plane-wave basis set were adopted in all the computations. To simulate the Cu/SnO2 interfaces, we placed a Sn6O12 cluster on a 5×5×3 Cu (111) surface with a 19-Å vacuum between the slabs. The Sn6O12 cluster was built based on rutile SnO2 bulk crystal and resembles the SnO2 (110) surface structure. For comparison, a 4×4×3 Cu (111) slab with a 3×3×1 k-point sampling was used. The SnO2 (110) surface was modeled by a six atomic layer, and the top three atomic layers were relaxed while the other layers were fixed at the bulk lattice position. For Cu–SnO2 and the Cu (111) slab, the upper most layer (including the Cu (111) slab and Sn6O12 cluster) were allowed to relax, except for those of the bottom two layers in Cu (111), which were fixed at the bulk lattice position. The Gaussian smearing method with σ value of 0.1 eV was used in this system. The convergence threshold was set as 10−4 eV in energy and 0.08 eV Å−1 in force. A (2 × 2 × 1) gamma centered k-point was chosen for the slab calculation. The solvent effect on adsorbates was simulated using the Poissson–Boltzmann implicit solvation model with a dielectric constant of 8042. The effects of dipole correction for adsorbates were also included.
The CHE method has been applied to determine the Gibbs free energy of the reaction species. In this method, the electrochemical potential of an electron–proton pair (H+ + e−) is equal to the free energy of half of hydrogen (1/2 H2) at standard pressure. The CO2RR involving a 2e pathway can be described by:
where * represents the adsorption site, while OCHO*, COOH*, and CO* represent reaction intermediates, respectively. The free energy (G) of each intermediate estimated at T = 298 K was expressed as:
where EDFT, EZPE, S, and Cp were the electronic energy, zero point energy, entropy, and heat capacity, respectively. For adsorbed intermediates, EZPE, S, and Cp were determined by vibration frequency calculations via standard methods, where all 3N degrees of freedom were treated as harmonic vibration motions with neglecting contributions from the slab; for molecules, however, those values were taken from the NIST database43. The contribution of zero point energy, entropy corrections, and heat capacity to G is provided in Supplementary Table 3.
Data availability
All relevant data are available from the authors on request.
References
Letcher, T. Climate Change: Observed Impacts on Planet Earth (Elsevier, Amsterdam, 2009).
Pacala, S. & Socolow, R. Stabilization wedges: solving the climate problem for the next 50 years with current technologies. Science 305, 968–972 (2004).
Appel, A. M. et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658 (2013).
Jhong, H.-R. M., Ma, S. & Kenis, P. J. A. Electrochemical conversion of CO2 to useful chemicals: current status, remaining challenges, and future opportunities. Curr. Opin. Chem. Eng. 2, 191–199 (2013).
Aresta, M., Dibenedetto, A. & Angelini, A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. technological use of CO2. Chem. Rev. 114, 1709–1742 (2014).
Zhu, D. D., Liu, J. L. & Qiao, S. Z. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 28, 3423–3452 (2016).
Costentin, C., Robert, M. & Saveant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 42, 2423–2436 (2013).
Zhu, W. et al. Monodisperse Au nanoparticles for selective electrocatalytic reduction of CO2 to CO. J. Am. Chem. Soc. 135, 16833–16836 (2013).
Liu, M. et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 537, 382 (2016).
Lu, Q. et al. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 5, 3242 (2014).
Liu, S. et al. Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates. J. Am. Chem. Soc. 139, 2160–2163 (2017).
Gao, D. et al. Pd-containing nanostructures for electrochemical CO2 reduction reaction. ACS Catal. 8, 1510–1519 (2018).
He, J., Dettelbach, K. E., Salvatore, D. A., Li, T. & Berlinguette, C. P. High-throughput synthesis of mixed-metal electrocatalysts for CO2 reduction. Angew. Chem. Int. Ed. 56, 6068–6072 (2017).
Lei, F. et al. Metallic tin quantum sheets confined in graphene toward high-efficiency carbon dioxide electroreduction. Nat. Commun. 7, 12697 (2016).
Lee, C. H. & Kanan, M. W. Controlling H+ vs CO2 reduction selectivity on Pb electrodes. ACS Catal. 5, 465–469 (2015).
Han, N. et al. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 9, 1320 (2018).
Kuhl, K. P., Cave, E. R., Abram, D. N. & Jaramillo, T. F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012).
Loiudice, A. et al. Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 55, 5789–5792 (2016).
Hori, Y., Murata, A. & Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 185, 2309–2326 (1989).
Raciti, D., Livi, K. J. & Wang, C. Highly dense Cu nanowires for low-overpotential CO2 reduction. Nano Lett. 15, 6829–6835 (2015).
Dai, L. et al. Ultrastable atomic copper nanosheets for selective electrochemical reduction of carbon dioxide. Sci. Adv. 3, e1701069 (2017).
Calle-Vallejo, F. & Koper, M. T. M. Theoretical considerations on the electroreduction of CO to C2 species on Cu (100) electrodes. Angew. Chem. Int. Ed. 52, 7282–7285 (2013).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Norskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Lee, C. W. et al. Defining a materials database for the design of copper binary alloy catalysts for electrochemical CO2 conversion. Adv. Mater. 30, 1704717 (2018).
Kim, D., Resasco, J., Yu, Y., Asiri, A. M. & Yang, P. Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold-copper bimetallic nanoparticles. Nat. Commun. 5, 4948 (2014).
Ma, S. et al. Electroreduction of carbon dioxide to hydrocarbons using bimetallic Cu-Pd catalysts with different mixing patterns. J. Am. Chem. Soc. 139, 47–50 (2017).
Huo, S. et al. Coupled metal/oxide catalysts with tunable product selectivity for electrocatalytic CO2 reduction. ACS Appl. Mater. Interfaces 9, 28519–28526 (2017).
Li, Q. et al. Tuning Sn-catalysis for electrochemical reduction of CO2 to CO via the core/shell Cu/SnO2 structure. J. Am. Chem. Soc. 139, 4290–4293 (2017).
Gilroy, K. D., Ruditskiy, A., Peng, H. C., Qin, D. & Xia, Y. Bimetallic nanocrystals: syntheses, properties, and applications. Chem. Rev. 116, 10414–10472 (2016).
Ding, J. et al. Morphology and phase controlled construction of Pt-Ni nanostructures for efficient electrocatalysis. Nano Lett. 16, 2762–2767 (2016).
He, D. et al. Amorphous nickel boride membrane on a platinum-nickel alloy surface for enhanced oxygen reduction reaction. Nat. Commun. 7, 12362 (2016).
Wang, P., Jiang, K., Wang, G., Yao, J. & Huang, X. Phase and interface engineering of platinum-nickel nanowires for efficient electrochemical hydrogen evolution. Angew. Chem. Int. Ed. 55, 12859–12863 (2016).
Sarfraz, S., Garcia-Esparza, A. T., Jedidi, A., Cavallo, L. & Takanabe, K. Cu-Sn bimetallic catalyst for selective aqueous electroreduction of CO2 to CO. ACS Catal. 6, 2842–2851 (2016).
Jana, S., Chang, J. W. & Rioux, R. M. Synthesis and modeling of hollow intermetallic Ni-Zn nanoparticles formed by the Kirkendall effect. Nano Lett. 13, 3618–3625 (2013).
Li, C. W. & Kanan, M. W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234 (2012).
Graciani, J. et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 345, 546–550 (2014).
Yang, X. et al. Low pressure CO2 hydrogenation to methanol over gold nanoparticles activated on a CeO(x)/TiO2 interface. J. Am. Chem. Soc. 137, 10104–10107 (2015).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. J. Phys. Rev. B Condens. Matter Mater. Phys. 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. J. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, K. T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Computational Chemistry Comparison and Benchmark Database. http://cccbdb.nist.gov/ (2018)
Acknowledgements
This work was financially supported by Ministry of Science and Technology (2016YFA0204100, 2017YFA0208200), the National Natural Science Foundation of China (21571135), Young Thousand Talented Program, Jiangsu Province Natural Science Fund for Distinguished Young Scholars (BK20170003), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the start-up supports from Soochow University.
Author information
Authors and Affiliations
Contributions
X.H. conceived and supervised the research. X.H. and P.W. designed the experiments. X.H., Y.L., M.Q., and P.W. performed most of the experiments and data analysis. X.H., P.W., M.Q., Q.S., Y.P., Y.L., and X.Z. participated in various aspects of the experiments and discussions. Y.L. and M.Q. performed the DFT simulations. X.H. and P.W. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, P., Qiao, M., Shao, Q. et al. Phase and structure engineering of copper tin heterostructures for efficient electrochemical carbon dioxide reduction. Nat Commun 9, 4933 (2018). https://doi.org/10.1038/s41467-018-07419-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07419-z
This article is cited by
-
Strongly Coupled Ag/Sn–SnO2 Nanosheets Toward CO2 Electroreduction to Pure HCOOH Solutions at Ampere-Level Current
Nano-Micro Letters (2024)
-
Rational design of bimetallic catalysts for electrochemical CO2 reduction reaction: A review
Science China Chemistry (2023)
-
Wet-chemical synthesis and applications of amorphous metal-containing nanomaterials
Nano Research (2023)
-
Unraveling the interfacial effect of PdBi bimetallic catalysts on promoting CO2 electroreduction to formate
Nano Research (2023)
-
Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
