
Abstract
The complement system consists of a network of plasma and membrane proteins that modulate tissue homeostasis and contribute to immune surveillance by interacting with the innate and adaptive immune systems. Dysregulation, impairment or inadvertent activation of complement components contribute to the pathogenesis of some autoimmune neurological disorders and could even contribute to neurodegenerative diseases. In this Review, we summarize current knowledge about the main functions of the complement pathways and the involvement of complement in neurological disorders. We describe the complex network of complement proteins that target muscle, the neuromuscular junction, peripheral nerves, the spinal cord or the brain and discuss the autoimmune mechanisms of complement-mediated myopathies, myasthenia, peripheral neuropathies, neuromyelitis and other CNS disorders. We also consider the emerging role of complement in some neurodegenerative diseases, such as Alzheimer disease, amyotrophic lateral sclerosis and even schizophrenia. Finally, we provide an overview of the latest complement-targeted immunotherapies including monoclonal antibodies, fusion proteins and peptidomimetics that have been approved, that are undergoing phase I–III clinical trials or that show promise for the treatment of neurological conditions that respond poorly to existing immunotherapies.
Key points
-
Complement has an important physiological role in host immune defences and tissue remodelling.
-
The physiological role of complement extends to the regulation of synaptic development.
-
Complement has a key pathophysiological role in autoimmune neurological diseases and mediates the actions of pathogenic autoantibodies, such as acetylcholine receptor antibodies and aquaporin 4 antibodies.
-
For some autoimmune neurological diseases, such as myasthenia gravis and neuromyelitis optica spectrum disorders, approved complement-targeted treatments are now available.
-
Complement also seems to be of pathogenic relevance in neurodegenerative diseases such as Alzheimer disease, in which innate immune-driven inflammation is receiving increasing attention.
-
The field of complement-targeted therapeutics is rapidly expanding, with several FDA-approved agents and others currently in phase II and phase III clinical trials.
Similar content being viewed by others
Introduction
The human complement system is an effector of innate and adaptive humoral immunity. The system comprises soluble membrane-bound proteins (opsonins) that act collectively to recognize pathogens and non-self material and subsequently initiate opsonization and phagocytosis or lysis of pathogens. The proteolytic fragments that derive from complement activation can be targeted by white blood cells and endothelial cells, leading to extravasation and migration of immune cells at the sites of inflammation. Other physiological functions of complement include timely removal of altered and senescent self, tissue remodelling, cell lysis, chemotaxis, opsonization, inflammation and immune cell stimulation1,2,3,4.
Complement activation products link the innate and adaptive immune systems by acting directly on receptors on T cells and B cells or by modulating dendritic cell functions5,6,7,8. Disruption of the delicate coordination required for complement activation and control is fundamental in the pathogenic mechanism of several autoimmune neurological disorders, and is even emerging as a contributor in some neurodegenerative diseases. As a result, interest is increasing in targeting complement as a therapeutic approach to prevent ongoing tissue destruction in several difficult-to-treat neurological diseases.
In this Review, we provide an overview of the main components of the initial complement activation pathways, the lytic pathway and the factors associated with inappropriate complement activation or control in disease initiation and progression. We consider the role of complement in the tissue destruction that is responsible for the genesis of the most common autoimmune neurological diseases, including complement-mediated myopathies, myasthenia gravis, neuropathies and CNS disorders. We also discuss the role of complement in some neurodegenerative disorders, including Alzheimer disease (AD), amyotrophic lateral sclerosis (ALS), Huntington disease, traumatic brain injury and even schizophrenia. Finally, we discuss emerging complement-targeted therapies, such as monoclonal antibodies, fusion proteins and cyclic peptides, that inhibit the functions of individual complement proteins, especially therapies that disrupt the lytic pathway. Some of these therapeutic agents have been approved or are being tested in phase I–III clinical trials and promise to change the therapeutic armamentarium for treatment of neurological conditions that respond poorly to existing immunotherapies.
Main complement functions
Functionally, the complement system can be divided into two main parts: the enzymatic cascade and the lytic pathway. The enzymatic cascade generates the molecules needed to initiate the lytic pathway, in which the soluble proteins undergo conformational changes that enable their insertion into lipid bilayers and ultimately form the osmolytic membrane attack complex (MAC; also known as the C5b–918 complex)9. These pathways are described in more detail below.
With respect to the nomenclature of the complement proteins, in this Review we have adhered to that officially adopted by the International Complement Society (ICS) in 2014 (ref.10). Changes to this nomenclature have subsequently been proposed by experts of the ICS11 in an attempt to harmonize complement nomenclature; if universally adopted, these changes could help to improve precision.
The enzymatic cascade
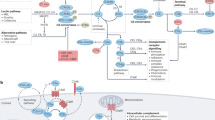
The main functional aspect of the enzymatic cascade is assembly of the convertases, which drive subsequent complement activation, amplification and opsonization. The cascade comprises three activation axes: the classical pathway, the lectin pathway and the alternative pathway (Fig. 1). In these cascades, the proteolytic enzyme complexes activate each other in a strict order. The lectin pathway and the alternative pathway are evolutionarily ancient — some of their components are found in invertebrates12,13 — whereas the classical pathway seems to have evolved alongside the immunoglobulins. The enzymatic cascades lead to generation of biologically highly active pro-inflammatory anaphylatoxins (C3a and C5a) and opsonins (C3b and C4b) that covalently bind to cell surfaces and that are involved in the clearance of invading organisms and altered or senescent self. These processes have a fundamental role in innate immunity and tissue homeostasis.

The complement pathway begins with the enzyme cascade, which involves de novo assembly of enzyme complexes known as convertases. The enzyme cascade can proceed via three different activation pathways: the classical pathway (CP), the lectin pathway (LP) and the alternative pathway (AP). The classical pathway begins with antibody-mediated activation of C1, which leads to formation of the C4bC2a complex, which is the C3 convertase. This C3 convertase cleaves C3 to produce C3b, which forms a complex with C4b and C2a. This complex is the C5 convertase, which cleaves C5 to produce C5a and C5b. The lectin pathway begins with signal recognition by oligomeric structures of mannose-binding lectin (MBL), ficolins and collectins, which activate mannan-binding lectin serine protease 1 (MASP1) and MASP2, which in turn mediate production of C4b. From this point, the lectin pathway follows the same steps as the classical pathway. In the alternative pathway, C3 interacts with factor B and factor D, leading to cleavage of further C3, and this process is perpetuated through an amplification loop. In the final step of this pathway, an additional C3b binds to the C3 convertase and forms a C5 convertase, which cleaves C5. All three pathways generate C5b, which initiates the second part of the system — the lytic pathway and membrane attack complex (MAC) formation. C5b associated with the surface-bound convertases sequentially accepts C6 and C7. For successful continuation of MAC formation, the C5b–7 complex must translocate to the outer part of the lipid bilayer. The translocation results in conformational changes and the complex becomes transmembrane, exposing lipophilic structures that enable association with C8 and C9. Further binding with up to another 17 C9 molecules widens the inner pore size, resulting in the osmolytic MAC. Molecules and processes that represent therapeutic targets are shown on the right along with drugs that are available or are in development. The complement protein nomenclature used is in accordance with Kemper et al.10. IVIg, intravenous immunoglobulin.
Classical pathway
The classical pathway begins with activation of the C1 complex, which consists of the sensing molecule C1q and two heterodimers formed by the zymogens C1r and C1s9,14,15. The primary targets of C1q are antigens that, when in complex with IgG1, IgG2 and IgG3 (but not IgG4) or IgM, enable multivalent C1q binding15. Even in the absence of pathogen-specific antibodies, C1q can initiate first-line defences by binding directly to the surfaces of pathogens. C1q can also interact with molecules such as actin, adiponectin, amyloid fibrils, cardiolipin, C-reactive protein, DNA, fibronectin, laminin, lipopolysaccharides, mucopolysaccharides, myelin, pentaxrin 3, phosphatidylserine and prion proteins, all of which can cause excess inflammation. C1q is also produced locally in the CNS and can play a role in modulating microglia and synaptic pruning13.
Binding of C1q to its target leads to conformational changes that generate enzymatically active forms of C1r and C1s13,14,15. The resultant C1qC1r2C1s2 enzyme complex mediates cleavage of native C4 followed by cleavage of C2 and the subsequent formation of the C3 convertase C4bC2a. The C3 convertase activates C3, causing dissociation of C3 into C3a and C3b. C3b then binds to the existing C3 convertase to form the C4bC3bC2a complex, which is a C5 convertase16 (Fig. 1).
Lectin pathway
The process of lectin pathway activation closely resembles that of classical pathway activation, but a wide array of carbohydrates can precipitate this pathway. The complexes of the lectin pathway that become activated have oligomeric structures similar to the pentamolecular C1 complex17. The pattern-recognition molecules within these complexes include mannose-binding lectin (MBL), ficolins and collectins, which sense danger signals from pathogens and senescent or apoptotic cells. Upon activation by these signals, the enzymes of the complexes — mannan-binding lectin serine protease 1 (MASP1) and MASP2 — mediate the formation of the C3 convertase C4bC2a18,19 (Fig. 1). C4bC2a mediates activation of the same downstream pathways as in the classical pathway19. Activity of the various sensing complexes of the classical and lectin pathways are controlled by C1-esterase inhibitor (C1-INH)20. In the classical and lectin pathways, C1-INH binds to the proteases of the active sensing complexes and these proteases cleave C1-INH, leading to formation of a covalent complex of C1-INH with C1r, C1s, MASP1 or MASP2.
The alternative pathway
The alternative pathway differs from the classical and lectin pathways in that it is spontaneously and continuously active at a low rate and its activation can be amplified. The pathway involves factor D, which is present predominantly in its active form in normal blood, and properdin, the only known positive regulator of the complement system. Initially, a thioester-hydrolysed form of C3 is spontaneously generated (C3(H2O)), which acts with factor B and factor D to form the fluid phase alternative pathway C3 convertase C3(H2O)Bb21,22 (Fig. 1). C3(H2O)Bb cleaves native C3 to generate C3a and C3b — the latter binds to its receptor on the lipid membrane. C3b at the membrane combines with factor B, which is cleaved by factor D to form the alternative pathway C3 convertase C3bBb. Via an amplification loop, fuelled by cyclical formation of C3b and C3bBb, the alternative pathway C3 convertases increase in number and generate abundant C3a and C3b (Fig. 1). The efficiency of the initial assembly of the alternative pathway C3 convertase depends on the density of terminal sugars within the cell walls of pathogens or the terminal sialic acid of self within the microenvironment of bound C3b21. Binding of additional C3b to the existing alternative pathway C3 convertase generates a C5 convertase that leads to production of C5a and C5b16.
Control of alternative pathway C3 and C5 convertases
The default setting of the alternative pathway is continuous activation. To evade homologous attack via the alternative pathway, tight control of C3 and C5 convertases is necessary. This control is achieved via two steps that ultimately generate complement degradation products that are important for elimination of pathogens or apoptotic cells. In the first step, the enzyme Bb is displaced from the convertases, but this step is not sufficient for complete convertase inactivation because C3b/C3bn still provides a platform for assembly with factor B. The second step is sequential cleavage by factor I of C3b into inactivated C3b (iC3b) and C3d, resulting in complete inactivation of C3 and C5 convertases16. iC3b and C3d are ligands for various complement receptors on phagocytic cells that remove altered and senescent self in a non-inflammatory manner16. In addition, the transmembrane protein CUB and Sushi multiple domains 1 (CSMD1), which is highly expressed in the human brain, might serve as a cofactor together with factor I-mediated degradation of C3b and C4b, thereby inhibiting downstream processes18.
Complement in development and plasticity
The two core effector molecules of the complement system are C3a and C5a, and their receptors play an important role in the developing brain and in neuroplasticity. The receptor for C3a is C3aR, and C5a has two receptors: C5aR1 (also known as CD88), a G protein-coupled pro-inflammatory receptor that is expressed on cells of myeloid origin, and C5aR2 (also known as C5L2 or GPR77). The C3a–C3aR axis has a substantial role in neuronal migration and the C5a–C5aR1 axis is critical during embryonic human brain neurogenesis as it regulates proliferation and differentiation of neural progenitor cells13. The role of C5aR2 has not been fully determined but it seems to have immunomodulatory properties23.
The lytic pathway
The molecules of the lytic pathway assemble to form osmolytic pores in cell membranes. C5 convertases cleave C5 into C5a and C5b, and formation of the MAC is initiated after the cleavage product C5b non-covalently binds to the cleaving C5 convertase and sequentially accepts C6 and C7 (Fig. 1). If the C5b–7 complex does not translocate to the outer part of the lipid bilayer then activation is aborted. However, if the complex translocates to the C5 convertase decorated cell membrane, it undergoes conformational changes, exposes lipophilic structures and becomes transmembrane, enabling its association with C8 and C9, which also undergo conformational changes before association and membrane insertion. Further association with up to 17 more C9 molecules widens the inner pore, forming the final osmolytic MAC24 (Fig. 1). Control of MAC assembly to ensure protection from autologous complement attack and MAC formation is mainly mediated by CD59, a glycosylphosphatidylinositol-anchored protein that is widely expressed and prevents C9 from binding to the C5b–8 complex.
Control by CD59 can never be absolute, so metabolically active cells have additional defence mechanisms to prevent MAC formation and ensure survival of the cells. In otherwise healthy host tissue, inadvertent MAC formation in nucleated cells can be kept sublytic by repair of mild membrane damage. This repair function leads to the release of arachidonic acid, the precursor of several inflammatory mediators25,26,27, or the induction of inflammasomes28. In tissue with slightly impaired CD59 control, such a repair mechanism can initiate or, over time, aggravate inflammation and tissue destruction.
Complement in neurological diseases
Dysfunction of the complement system — such as the absence or overconsumption of components, inappropriate control or misguidance by autoantibodies — is associated with several acute or chronic autoimmune and inflammatory neurological disorders29,30 (Table 1). Complement-associated neurological diseases can result from dysfunction of complement proteins or from aberrant complement activation.
Dysfunction of complement proteins
Dysfunction of complement activation predisposes to infections and to autoimmune-like phenomena that result from inappropriate removal of altered or senescent self, leading to prolonged exposure of structures that are normally inaccessible to the immune system. Neurological manifestations of such dysfunction are rare. The most severe of these manifestations arise from genetic deficiency of CD59 or mutations in its main anchor protein glycosylphosphatidylinositol31,32, which result in uncontrolled destruction of nerve tissue owing to uncontrolled autologous complement attack.
CD59 deficiency is mainly associated with paroxysmal nocturnal haemoglobinuria (PNH); secondary neurological manifestations are related to systemic infections, respiratory failure and haemolytic anaemia. Primary neurological disease has been associated with mutations in CD59, but have been described in only 13 patients to date32. The most commonly reported disease in these patients is an acute, predominantly motor, demyelinating neuropathy with conduction block that is exacerbated by recurrent episodes of acute areflexic paralysis and leads to secondary axonal damage, indistinguishable from recurrent Guillain–Barré syndrome (GBS) or chronic inflammatory demyelinating polyneuropathy (CIDP). Therapy with eculizumab (a humanized monoclonal antibody to C5, described in detail below), can be life-saving33. Recurrent aseptic meningitis has also been reported in one 71-year-old man with glycosylphosphatidylinositol deficiency; this patient experienced 121 episodes over a period of 16 years and subsequently developed PNH34. All symptoms stopped with eculizumab therapy.
Some conditions can impair the efficiency of CD59. For example, sustained hyperglycaemia in diabetes mellitus can lead to increased glycation and consequent functional impairment of proteins, including CD59 (ref.35). Hyperglycaemia itself also activates complement, and these effects might contribute to the vascular and neurological complications of diabetes36.
Inappropriate complement activation
Circulating autoantibodies — particularly those of the IgG1 subclass — can mobilize complement upon binding to their antigen, leading to tissue damage. Antibodies of the IgG4 subclass, however, cannot activate complement because their binding to C1q is inefficient. Nevertheless, pathogenic IgG4 antibodies can induce autoimmune pathology over time by competing with naturally occurring antibodies for C1q binding sites, and therefore delaying removal of altered and senescent self.
In physiological conditions, the blood–brain barrier (BBB) generally restricts the access of plasma proteins to the brain37. However, upon complement activation, IgG and complement components can enter the brain if the BBB becomes leaky38,39, as occurs with ageing, mitochondrial dysfunction and oxidative stress, leading to neurological manifestations. These conditions can also lead to the release of intracellular antigens that might be recognized by C1q and thereby activate complement. Complement can also be activated if C1q binds to the Fc portion of IgG and IgM antibodies that have neuronal antigens, such as gangliosides, glial fibrillary acidic protein or various neuronal channels or receptors.
Even when the BBB is intact, complement activation can occur in the brain; studies in animal models have shown that microglia and astrocytes can synthesize and secrete most complement proteins40. In physiological conditions, the roles of complement in the brain include clearance of surplus neurotransmitters, removal of aged proteins, synaptic pruning and modulation of adult neurogenesis, all functions with protective effects.
Diseases of roots and peripheral nerves
GBS and CIDP
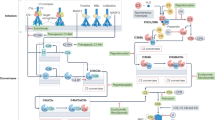
In various GBS variants, particularly acute motor axonal neuropathy, acute motor and sensory axonal neuropathy and Miller–Fisher syndrome, antibodies to gangliosides target myelinated fibres or axons, causing complement activation, MAC formation, disrupted expression of sodium channels and conduction block (Fig. 2). Deposition of C3b on the surface of Schwann cells can lead to myelin damage followed by infiltration with activated macrophages and T cells41. In passive transfer mouse models of ganglioside antibody-mediated neuropathy, ganglioside antibodies produce a GBS-like phenotype by damaging perisynaptic Schwann cells, but this effect only occurs in the presence of human complement42,43,44. In these models, eculizumab effectively prevented dysfunction and nerve damage45. In an in vitro study, quantitative assessment of C3 uptake showed that deposition of C3 fragments onto sensitized targets was greater with serum from patients with GBS than with serum from healthy controls, an observation that supports the idea that C3b and MAC deposition have a role in nerve damage46.

Once autoreactive B cells are activated by, for example, CD4+ T cells, they mature into plasma cells, which secrete autoreactive antibodies with pathogenic potential. The antibodies bind to their autoantigen and, through C1q binding, activate the complement cascade. Typical examples of sites at which complement damages neuronal structures include the neuromuscular junction via acetylcholine (ACh) receptor antibodies, the node of Ranvier via antibodies to gangliosides or some nodal proteins, and astrocyte end feet via antibodies to aquaporin 4 (AQP4). MHC, major histocompatibility complex; TCR, T cell receptor.
In CIDP, complement and IgG are deposited on myelinated fibres47 and serum levels of terminal complement activation components increase with disease severity48. However, complement-fixing antibodies to compact myelin, which would explain the active demyelination and conduction block, have not been identified in CIDP. Studies of the nodes of Ranvier have shown that up to 10% of patients with CIDP have antibodies to neurofascin 155, contactin 1 and contactin-associated protein 1, which are necessary for the maintenance of the nodal structure49,50, but these IgG4 antibodies do not cause the complement-fixing, macrophage-mediated demyelination that is seen in classical CIDP but instead block protein–protein interaction, resulting in dis-adhesion of structural nodal proteins without inflammation51,52. Most patients with these antibodies do not respond to intravenous immunoglobulin (IVIg), which inhibits C3, but do respond to rituximab, a monoclonal anti-CD20 antibody that targets B cells53,54,55.
Multifocal motor neuropathy
Multifocal motor neuropathy (MMN) is a distinct motor neuropathy characterized by distal, asymmetric, primarily upper limb weakness, without substantial sensory abnormalities but associated with conduction block and, in up to 50% of patients, the presence of IgM antibodies to ganglioside GM1, a glycolipid constituent of nerve fibres. These antibodies can activate the classical complement pathway; the extent of complement activity seems to be associated with disease severity56. In an induced pluripotent stem cell-derived model of MMN, the interaction of these antibodies with complement can interfere with motor neuron function, thereby causing cell damage and death57.
Paraproteinaemic neuropathies
In IgM paraproteinaemic neuropathy associated with antibodies to myelin-associated glycoprotein (MAG) and sulfoglucuronyl paragloboside, sural nerve biopsies have shown that the anti-MAG IgM is deposited with complement on myelinated fibres58, suggesting that activated complement is needed to induce demyelination. In addition, skin biopsies from these patients have shown that IgM anti-MAG and C3d are deposited in myelinated fibres, supporting the idea that complement has a role in splitting the myelin lamellae and inducing loss of nerve fibres59,60,61. Nevertheless, no complement-related therapies have been tested in paraproteinaemic neuropathies to date.
Neuropathic pain
Though still controversial, complement is thought to be involved in neuropathic pain, based on the assumption that inflammation initiated by the complement cascade can drive neuropathic pain62,63. The production of the anaphylatoxins C3a and C5a, particularly C5a, is probably the link between complement activation and pain. The binding of C5a to its receptor C5aR1 could trigger the release of cytokines that attract macrophages and neutrophils62. Drugs that prevent the binding of C5a to C5aR1, such as PMX53 (also known as AcF-OpdChaWR) reduced pain in animal models63,64,65; however, these results have not been translated into the clinic63 and should be interpreted with caution.
Diseases of the neuromuscular junction
Myasthenia gravis
Myasthenia gravis is the prototypic autoimmune disease. The condition is associated with autoantibodies to the acetylcholine receptor (AChR) in up to 85% of patients; in others, it is associated with antibodies to other synaptic proteins, such as muscle-specific tyrosine kinase (MuSK), and possibly LDL receptor-related protein 4 (LRP4) and agrin66. AChR antibodies reduce synaptic transmission by blocking the binding of acetylcholine to the receptor at the neuromuscular junction, crosslinking the receptor, which leads to its internalization, and activating complement upon binding to the receptor (Fig. 2), leading to in situ MAC formation. MAC formation causes direct damage to the postsynaptic membrane, thereby reducing the surface area and the number of AChR channels and voltage-gated sodium channels67. The neuromuscular junctions of patients with myasthenia gravis contain deposits of IgG, C3 and C9, and myasthenia gravis exacerbations are accompanied by increased consumption of complement68. Quantitative measurement of C3 uptake into sensitized targets in vitro has shown that uptake from the serum of people with myasthenia gravis is particularly high, indicating that C3b and MAC formation have a role in the disease46,69.
The role of AChR autoantibodies in promoting the fixing of complement has also been illustrated in animal models with active immunization or passive transfer, both of which lead to complement deposition at the neuromuscular junction that results in postsynaptic endplate destruction similar to that seen in human myasthenia gravis. In animals that are deficient in complement C3, C4, C5 or C6, the incidence of experimental autoimmune myasthenia gravis is substantially lower than in non-deficient animals, and IgG deposits at the neuromuscular junction do not lead to MAC formation70,71. Anti-C1q and anti-C6 antibodies effectively inhibited MAC formation and reduced symptoms of experimental autoimmune myasthenia gravis in active and passive transfer models72,73.
Unlike AChR antibodies, MuSK antibodies, which are of the IgG4 subclass, do not bind to complement but disrupt MuSK signalling by blocking the interaction of MuSK with agrin and LRP4. As for patients with CIDP associated with IgG4 anti-nodal antibodies, patients with MuSK-positive myasthenia gravis do not benefit from treatments that interfere with complement assembly, such as IVIg, but respond well to anti-B cell therapies such as rituximab74.
Lambert–Eaton myasthenic syndrome
Lambert–Eaton myasthenic syndrome (LEMS) has autoimmune and paraneoplastic subtypes, but, in both, antibodies target voltage-gated calcium channels at the presynaptic endplate. As in myasthenia gravis, these antibodies mobilize the MAC, which causes direct damage to the presynaptic part of the neuromuscular junction75. No formal complement-related therapies have yet been tried for the treatment of LEMS other than IVIg, for which efficacy has been demonstrated75.
Inflammatory muscle diseases
Inflammatory myopathies are categorized into five main groups: dermatomyositis, polymyositis, inclusion body myositis, necrotizing autoimmune myositis (NAM) and anti-synthetase syndrome-overlap myositis76. A role for complement has been predominantly demonstrated in dermatomyositis; complement and possibly some autoantibodies also have a role in NAM and, to a lesser extent, the anti-synthetase syndrome, making these conditions the most suitable for the use of anti-B cell or anti-complement therapies. However, necrotic muscle fibres from any cause, even muscular dystrophies, activate complement, which subsequently stimulates the recruitment of inflammatory infiltrates and macrophages77.
Dermatomyositis
In dermatomyositis, the primary disease target is the endothelium of the capillaries, which undergoes chronic attack by the MAC. The MAC is deposited on the endothelial cells of the endomysial capillaries before any destruction of muscle fibres or capillaries is evident, leading to endothelial cell necrosis, a reduced density of endomysial capillaries, and ischaemia or microinfarcts, which are most prominent at the periphery of the fascicles76,78,79. MAC activation triggers the release of pro-inflammatory cytokines, upregulates expression of adhesion molecules on endothelial cells and facilitates the migration of activated CD4+ T cells, macrophages, B cells and CD123+ plasmacytoid dendritic cells into the endomysium76.
The way in which the lytic pathway is activated in dermatomyositis is not clear. Deposition of C1q and C4 in the proximity of the MAC but without immunoglobulin deposits has been observed early in the disease, suggesting that C1 is directly activated by the diseased endothelium80 and that immunoglobulin-independent, C1q-mediated classical pathway activation occurs81. On the basis of an inverse correlation between MAC deposition and CD59 expression, the hypothesis that MAC deposition is a consequence of low CD59 expression has been proposed82 but has never been explored.
Despite uncertainty about the events that trigger complement activation, dermatomyositis is unambiguously a complement-mediated microangiopathy. Inhibition of C3b deposition with IVIg not only prevents MAC assembly in the tissues of patients but also leads to substantial clinical improvements, resolution of histopathological changes, and clearance of MAC from the muscle fibres83.
Necrotizing autoimmune myositis
Up to 65% of patients with NAM are positive for non-pathogenic antibodies against cytoplasmic signal recognition particle (SRP) and 3-hydroxy-3-methylglutarylcoenzyme A reductase (HMGCR)76,84,85,86. In this condition, necrotic muscle fibres are strongly invaded by macrophages and contain sarcolemmal deposits of the MAC, the extent of which correlates with the extent of fibre necrosis, as generally seen in necrotic fibres from any cause77,87,88. These observations, although suggestive of involvement of the classical pathway, are not NAM-specific and they do not indicate that anti-SRP or anti-HMGCR are complement-fixing antibodies because complement activation is inherently associated with muscle fibre necrosis from any cause, including muscular dystrophies77. Open-label studies have shown that NAM responds to IVIg or rituximab, but whether this benefit is related to complement inhibition or other immunomodulatory effects is unclear87,88,89.
Diseases of the CNS
Clinical evidence that complement is involved in CNS autoimmunity mainly comes from one antibody-mediated disease: anti-aquaporin 4-positive neuromyelitis optica90. However, complement is also known to be involved in multiple sclerosis (MS) and acute disseminating encephalomyelitis (ADEM), and might have a role in a subset of patients with autoimmune encephalopathies91. Emerging information about the role of complement in some neurodegenerative conditions is exciting even though the available data are still too preliminary to be of clinical value because understanding of the pathogenesis of these conditions is poor and effective therapies are generally lacking.
MS and ADEM
Demyelination in MS has traditionally been thought to be T cell-mediated, but evidence is mounting for roles of B cells, autoantibodies and complement. Several lines of evidence suggest that complement is involved. First, postmortem studies have revealed complement deposition in and around demyelinating plaques in the pattern II pathological MS type92. Second, increased cerebrospinal fluid (CSF) levels of C4a are associated with disease activity, indicating intrathecal complement activation via the classical pathway93. Third, binding of C1q to the targeted tissue and activation of C3 and the MAC has been observed in white matter lesions, implying that the complement classical pathway has a role in mediating demyelination. Fourth, C4d has been detected at the edges of intracortical lesions94,95,96,97,98,99. Fifth, increased expression of C1q and activation of C3 in hippocampi (in myelinated and demyelinated regions of chronic MS plaques) is associated with low synaptic density, indicating a role for complement in synaptic alterations. Whether this synaptic localization — particularly in chronic lesions — reflects a physiological role of complement in mediating synaptic pruning as occurs during development100 and normal ageing101 or whether it reflects direct involvement of complement in neurodegeneration via elimination of synapses102,103 remains unclear. Evidence from animal models suggests that, as in some neurodegenerative diseases, microglial synaptic engulfment and profound synapse loss occurs. In mice, synapse loss occurred independently of local demyelination and neuronal degeneration but coincided with gliosis and increased levels of complement component C3, but not of C1q, at synapses104. However, given that the main autoantigen and autoantibody in MS remain unknown, whether these findings represent an early stage in disease pathogenesis or are secondary to other inflammatory events remains uncertain.
In experimental autoimmune encephalomyelitis (EAE), a widely used rodent model of MS, C6-deficient animals are protected from demyelination, highlighting the role of MAC in acute demyelination105. In addition, systemic inhibition of MAC in an EAE model by use of an antisense oligonucleotide that targets C6 prevented disease relapse and protected the animals from axonal and synaptic damage106. These observations are especially relevant to ADEM, an acute demyelinating syndrome that is often associated with myelin oligodendrocyte glycoprotein (MOG) antibodies, because MOG is localized to the outer myelin lamellae and so is readily accessible to complement. In various experimental models, MOG antibodies induce demyelination with the MS or ADEM phenotype only in the presence of complement107.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders (NMOSD) include a spectrum of autoimmune inflammatory demyelinating disorders that affect the spinal cord, optic nerve and specific brain areas, such as area postrema, and in which complement clearly has a pathogenic role. NMOSD are caused by autoantibodies to the water channel aquaporin 4 (AQP4), a protein that is expressed in the end feet of astrocytes that line the BBB (Fig. 2). These antibodies are produced in the periphery, penetrate the BBB, bind to their antigen on astrocytes and cause astrocytic destruction via complement-mediated cytotoxicity108,109. The inflammatory events lead to further BBB damage, attraction of neutrophils and macrophages from the periphery and more destruction of myelin, oligodendrocytes and neurons110. Postmortem studies have shown that complement deposition is abundant at sites of pathology, and markers of complement activation (C5a and soluble MAC) can be detected in the plasma and CSF during active disease111. Furthermore, vasculocentric deposition of activated complement is prominent in NMOSD lesions112.
In mice, injection of AQP4 antibodies directly into the brain, either intracerebrally or as a continuous infusion into the CSF, does not cause disease, but in the presence of abundant complement, the animals develop NMOSD-like pathology113. Similarly, addition of AQP4 antibodies to cultured astrocytes causes no cellular destruction unless exogenous complement is added108. In mice, inactivation of CD59 or knockout of the gene that encodes this protein114,115 increased the extent of NMOSD pathology caused by co-injection of AQP4 antibodies and complement. The pathogenic role of complement in NMOSD is clinically substantiated by the beneficial effects of the C5 antibody eculizumab (see below).
Autoimmune encephalopathies
Patients with autoimmune encephalopathies harbour pathogenic antibodies to synaptic proteins, such as NMDA, GABAA, GABAB and AMPA receptors. These antibodies cross the BBB from the periphery but are also produced intrathecally116. Binding of the antibodies to their corresponding receptor results in functional blockade117. These antibodies are predominantly of the IgG1 or IgG3 subtypes, but no evidence has convincingly demonstrated that they fix complement91. However, complement can exacerbate antibody-mediated destruction in some patients with NMDA receptor autoantibodies118.
In other autoimmune encephalopathies, the role of complement is clearer. In progressive encephalomyelitis with rigidity and myoclonus, which affects inhibitory synapses, glycine receptor antibodies activate complement, leading to internalization of the glycine receptors to the lysosomes119. In CASPR2 antibody-associated encephalitis, complement-mediated inflammation and loss of brain tissue correlates with MRI-documented hippocampal and cerebral atrophy120. In all autoimmune encephalitides, factors that regulate BBB permeability are likely to permit complement constituents to enter the brain, resulting in high levels of complement that might facilitate the disruptive role of pathogenic antibodies121.
Neurodegenerative diseases
Neurodegenerative diseases are characterized by progressive loss of neurons and neuronal synapses. The most common is AD, the pathological hallmarks of which are neuronal loss, deposits of amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau protein. Several studies showed that complement activation products colocalize with cerebral Aβ deposits, including fibrillar and diffuse plaques that typically contain dystrophic neurites122,123. Many complement components, including C1q, C3 and C4, can be produced by glia, and the activation products of these components (C3b, C3c and C3d) surround amyloid plaques124. In addition, neurofibrillary tangles are immunopositive for C1q, C3d, C4d and the MAC. However, whether complement plays a primary role in initiating or perpetuating AD or these findings represent end-stage pathological events remains unclear125.
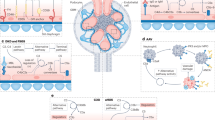
Studies in animal models of AD have also suggested that complement and microglia have a synaptotoxic effect in hippocampal regions (Fig. 3) and that early accumulation of C1q and C3 leads to synaptic deficits before plaque formation and neuroinflammation. Inhibition of C1q and C3 rescued this synaptic loss, suggesting that C1q — possibly increased by Aβ — triggers activation of the downstream classical pathway and contributes to neurodegeneration in AD126. In an AD mouse model in which Aβ plaques accumulate with age, knockout of complement C3 improved cognitive performance, reduced activation of astrocytes and microglia, and decreased age-dependent synaptic degeneration and neuronal loss at 16 months of age127.

Synaptic pruning eliminates weak synapses during normal brain development204. However, when neurons are stressed, as in neuroinflammatory disorders, complement components such as C1q are expressed by neurons or secreted by reactive astrocytes and can bind to synaptic proteins. C1q activation leads to C3 cleavage and C3b tagging of synapses. Microglial CR3 receptors recognize tagged neurons and eliminate synapses by phagocytosis (right). In diseases such as Alzheimer disease126 and multiple sclerosis104, this process might contribute to neurodegeneration.
These experiments have raised the controversial hypothesis that anti-complement therapeutic intervention could improve AD symptoms even if it does not halt the pathological process127. However, this issue is not settled because complement modulation in mouse models of AD have produced mixed effects. For example, in a C3-deficient amyloid precursor protein transgenic AD mouse model, all disease markers indicated worse pathology than in mice that expressed C3 (ref.128). Similarly, inhibition of C3 activation in another mouse model of AD increased Aβ deposition129. By contrast, in another mouse model of AD130, knockout of C5aR1 prevented microglia from becoming pro-inflammatory and reduced memory loss, supporting the hypothesis that C5aR1 antagonists could be a treatment for cognitive decline in AD. On this basis, the C5aR1 antagonist peptide PMX205 was tested in AD mouse models and reduced cognitive decline and attenuated microglial activation131. Therefore, if proven safe, such agents could be tested in patients with AD132.
Some evidence suggests that complement has roles in other neurodegenerative diseases. For example, neuroinflammation has been widely proposed as a contributing mechanism in ALS, and evidence indicates complement involvement. Deposition of C1q and C3d has been observed in motor neurons, astrocytes and microglia in ALS, and C1q and C4 are overexpressed in spinal cord glial cells, indicating local complement activation133. Similar deposits have been seen in the SOD1-G93A animal model, and oral administration of a C5aR1 antagonist in this model can slow disease progression and increase survival134,135. Similarly, in Huntington disease, neurons, astrocytes and myelin sheaths in the caudate and striatum are immunoreactive for C1q, C4, C3 and C3b136, and oral administration of a C5aR antagonist (PMX53 or PMX205) reduces neuronal loss and motor deficits in a rat model137. In traumatic human brain injury, expression of C3 and the MAC is increased in the brain and CSF138,139. In cortical contusion rat models, deposits of C3 and MAC are observed in combination with increased microglial and astrocytic synaptic activation (Fig. 3), findings that have led to exploratory studies of anti-complement therapies in this context140. Similar complement deposits have also been observed in animal models of spinal cord injuries141. Finally, in schizophrenia, one genetic risk variant is within the C4 complement locus142 and microglia-like cells from patients with this variant exhibit increased synapse engulfment and elimination143.
Of note, high C1q staining is observed in some healthy ageing brains (an increase of up to 330-fold compared with younger controls)101,144 and in the brains of people with MS95. This observation suggests that complement is involved in the broad process of neurodegeneration in a still undefined role.
Complement-targeted therapies
Therapeutic agents that target molecules in the complement activation cascade could arrest complement-mediated tissue damage, a possibility that has generated great interest and a series of ongoing clinical trials in several neurological disorders. Several anti-complement therapeutic strategies are possible: preventing initiation of the initial activation step; inhibiting convertase assembly or promoting convertase breakdown; and inhibiting C5 cleavage, thereby preventing C5a–C5aR1 interactions and intercepting formation of the MAC145,146. To date, targeting C5 cleavage has been the most successful strategy, and several emerging biologics have had remarkable benefits in difficult-to-treat autoimmune neurological diseases, leading to FDA approval for some.
In the following sections, we discuss the main anti-complement agents with immediate or indirect relevance to neurological diseases and their experimental models. The disorders in which complement has a major role and are therefore most amenable to complement-targeted therapeutics are myasthenia gravis and NMOSD; indeed, anti-complement therapies have already been approved for these conditions. However, anti-complement therapies might also be particularly promising for dermatomyositis, NAM, GBS, CIDP, MMN, LEMS and autoimmune CNS vasculitis. We do not discuss complement-targeted therapies that are in development for non-neurological disorders.
Targeting C3
C3 is at the convergence point of all the complement activation pathways and so is an attractive target for therapeutic modulation of the complement cascade. Several therapeutics affect or specifically target C3.
High-dose IVIg
IVIg preparations are made up of pooled donor IgG that neutralize autoantibodies, suppress T cell-activated and B cell-activated cytokines and inhibit complement147,148. The inhibition of complement by IVIg involves effects on multiple components of the complement cascade, although its main clinical effects are thought to be mediated via effects on C3. IVIg can bind to C1q and prevent pathogenic antibodies from triggering the complement cascade83,148,149. The F(ab’)2 region of IgG is involved in neutralization of activated complement proteins, including C3a and C5a, and high levels of IVIg inhibit uptake of C3b and C4b to the cell surface, thereby preventing complement-mediated tissue damage148,150.
The strongest evidence that IVIg has beneficial effects via complement inhibition suggests that the main mechanism involves C3. In vitro and in vivo findings in a controlled trial in patients with dermatomyositis showed that IVIg rapidly forms complexes with C3b, inhibits the amplifying C3 convertase151 and further C3 consumption as early as 2 days after infusion, and intercepts MAC formation by reducing assembly of C5 convertase83,152. These effects, observed in patients’ serum and repeated muscle biopsy samples, resulted in normalization of patients’ muscle strength and eliminated skin changes after three monthly infusions152. Analysis of muscle biopsy samples from patients who improved clinically revealed elimination of MAC deposition on the muscle capillaries, neovascularization and resolution of histological changes, including reversal of atrophic muscle fibres, elimination of inflammatory cells and downregulation of key cytokines and adhesion molecules152,153. This same mechanism might well be relevant in other neurological diseases that involve complement activation and in which IVIg has been effective in controlled trials; these diseases include GBS, CIDP, MMN, myasthenia gravis and myasthenic syndrome148,150.
In CIDP, clinical benefits of IVIg are seen whenever the disease is associated with putative antibodies of the IgG1 and IgG3 subclasses, which can activate complement154. By contrast, CIDP associated with specific antibodies to paranodal antigens, such as contactin 1, neurofascin or CASPR1, responds poorly to IVIg, presumably because these antibodies belong to the IgG4 subclass that does not activate complement154. The effects of IVIg on the complement system in CIDP were investigated in a study of 39 participants of the ICE trial, the largest trial of CIDP treatment to date that demonstrated the efficacy of IVIg versus placebo155,156,157. The serum and CSF levels of the complement activation products C3a, C5a and the soluble MAC remained unchanged in individuals who responded to IVIg therapy, suggesting that the therapeutic efficacy of IVIg in CIDP involves immunomodulatory mechanisms other than C3 complement inhibition155.
Experimental evidence suggests that IVIg could have beneficial effects in other conditions associated with complement activation. For example, in an ex vivo model of MOG-mediated demyelination, IVIg protected organotypic cerebellar cultures via interference with complement-mediated oligodendroglial damage and microglial activation158. Similarly, in animal models of acute brain dysfunction associated with sepsis, IVIg ameliorated neuronal dysfunction and behavioural deficits by reducing apoptotic cell death and glial cell proliferation159.
Compstatin
Compstatin is a generic name for a family of cyclic peptides that inhibit complement activation by binding to C3 and interfering with convertase function and C3 cleavage. On the basis of this mechanism, compstatins could be relevant for neurological therapeutics, although they have not yet been tested in this context160. They have, however, been tested as drug candidates in a plethora of other disease models that involve dysregulated or excessive complement activation, paving the way for a generation of more potent analogues that are currently in clinical trials such as one that is being tested in age-related macular degeneration161. One compstatin called APL-2 is currently in phase III trials for the treatment of PNH162, while a newer analogue of APL-2 called AMY-101 is now in early clinical development163.
Targeting C5
Eculizumab
Eculizumab is a fully humanized monoclonal antibody that comprises constant regions from human IgG2 and IgG4 sequences and murine complementarity-determining regions. It targets C5 in the plasma with high binding affinity and prevents cleavage of C5 to C5a and C5b, thereby inhibiting MAC assembly (Fig. 1). Eculizumab is approved by the FDA for the treatment of PNH, atypical haemolytic uraemic syndrome and, most recently, for refractory myasthenia gravis and NMOSD164,165,166,167,168,169,170,171,172.
Eculizumab was first tested in refractory anti-AChR-positive myasthenia gravis in a phase II, placebo-controlled, multicentre study that lasted 16 weeks and included 14 patients. From baseline to week 16, scores on the Quantitative Myasthenia Gravis and Myasthenia Gravis Activities of Daily Living scales improved in 86% of patients who received eculizumab versus 57% of patients who received placebo166. These promising data led to a phase III, randomized, placebo-controlled trial (REGAIN) of eculizumab that involved 125 patients with refractory myasthenia gravis. No significant difference in Myasthenia Gravis Activities of Daily Living score (the primary end point) was seen between the groups that received eculizumab and placebo, but preplanned and post hoc sensitivity analyses of the primary outcome and several secondary measures showed that eculizumab was beneficial. These results led to approval of eculizumab by the European Medicines Agency as the first and only treatment for refractory AChR-positive myasthenia gravis and by the FDA and the Japanese Pharmaceuticals and Medical Devices Agency for severe generalized myasthenia gravis167.
In the REGAIN extension study, 117 patients with refractory myasthenia gravis were treated with 1,200 mg eculizumab every 2 weeks for a mean period of 22.7 months. Treatment reduced exacerbation rates by 75% and maintained improvements in functional abilities throughout a 3-year period. In 56% of patients, minimal manifestation or pharmacological remission was attained. No incidents of meningococcal infection occurred, demonstrating long-term safety as well as sustained efficacy168.
The effects of eculizumab in myasthenia gravis are presumably brought about by a reduction in MAC formation at the neuromuscular junction and inhibition of endplate destruction. This hypothesis is supported by studies in animal models, which showed that administration of a C5 monoclonal antibody in a passive immunization model not only restored strength in rats but also inhibited C9 deposition, thereby preventing endplate damage169.
Eculizumab is also effective in NMOSD, in which relapses result in stepwise accumulation of disability, blindness, paralysis, or sometimes death. In a phase II trial of eculizumab in patients with active AQP4-seropositive NMOSD, 12 of the 14 patients who received eculizumab experienced no relapses for 12 months, the conditions of no patients worsened, and neurological disability remained stable or improved in 10 of the 14 patients. Within 12 months of eculizumab withdrawal, five patients experienced a total of eight relapses170. These results led to a phase III trial (PREVENT) of eculizumab in 143 patients. In this trial, the primary end point was met — eculizumab reduced the risk of relapse by 94.2% among patients who received eculizumab compared with the risk in patients who received placebo. At 48 weeks, 97.9% of patients who received eculizumab were free of relapses compared with 63.2% of patients who received placebo171. On this basis, the FDA approved eculizumab for the treatment of NMOSD172.
Eculizumab has also been tested in GBS in a multicentre, double-blind, placebo-controlled study that involved 34 non-ambulatory patients who were randomly assigned to receive IVIg and 900 mg eculizumab or IVIg and placebo. At week 4, 61% of patients who received eculizumab could walk independently versus 45% in the control group. This result was not sufficient to meet the primary end point. However, at week 24, treatment with eculizumab had improved motor function in patients with severe GBS173. Drawing conclusions from this study is difficult because many of the molecular mechanisms that are implicated in the GBS spectrum — including complement activation — precede clinical disease onset, whereas others might have been inhibited by the concurrent IVIg therapy rather than eculizumab. Nevertheless, the promising effects observed suggest that eculizumab has the potential to alter the disease course and that this could be demonstrated with a larger study with a modified protocol. This study is needed because GBS causes substantial disability and death despite the availability of two modestly effective therapies — IVIg and plasmapheresis.
Eculizumab was also life-saving in a study of patients with acute, predominantly motor, demyelinating neuropathy with conduction block and subacute CIDP due to p.Cys89Tyr substitution in CD59. Four patients who were homozygous for the p.Cys89Tyr CD59 substitution dramatically improved or did not relapse again with eculizumab33.
The clinical efficacy of eculizumab in MMN has also been investigated in a small, open-label study. The study showed that eculizumab was not effective — 90% of patients who also received IVIg continued to require IVIg treatment on termination of the study174. This outcome supports the notion that mechanisms independent of terminal complement inhibition contribute to conduction block in patients with MMN.
Ravulizumab
Ravulizumab (ALXN1210) is a humanized monoclonal antibody engineered from eculizumab and that is functionally similar. Ravulizumab specifically binds with high affinity to C5 and inhibits C5 enzymatic cleavage, thereby preventing generation of the pro-inflammatory and prothrombotic complement activation product C5a and the cytolytic and pro-inflammatory and prothrombotic MAC175,176. Ravulizumab provides sustained complement inhibition throughout a prolonged dosing interval and was designed to have a longer half-life than eculizumab so that less frequent dosing is required (once every 8 weeks rather than once every 2 weeks). In a phase III study of ravulizumab for PNH, the benefit-to-risk profile was good — even after the first loading dose, complete and sustained terminal complement inhibition was confirmed177. The drug has been approved by the FDA for treatment of PNH178. On this basis, two FDA-approved phase III studies of ravulizumab — one in myasthenia gravis and another in NMOSD — have been initiated and are currently ongoing179. The drug could also be applicable to the other complement-mediated disorders discussed, particularly dermatomyositis.
Zilucoplan
Zilucoplan is a synthetic, macrocyclic peptide that binds to C5 with subnanomolar affinity and that allosterically inhibits cleavage of C5 into C5a and C5b (Fig. 1). This prevention of C5b generation blocks assembly of the MAC. Zilucoplan is administered subcutaneously.
In a phase II trial designed to evaluate the safety, tolerability and preliminary efficacy of zilucoplan in generalized myasthenia gravis, 44 participants received 0.3 mg/kg zilucoplan daily, 0.1 mg/kg zilucoplan daily or a placebo over 12 weeks180. Among patients who received the higher dose, the Quantitative Myasthenia Gravis scores decreased by a mean of 6.0 points from baseline, whereas scores reduced by a mean of 3.2 points among patients who received placebo. Similarly, among patients who received the higher dose, Myasthenia Gravis Activities of Daily Living scores decreased by a mean of 3.4 points from baseline compared with 1.1 points among those who received placebo. No serious adverse effects of zilucoplan were observed180. The rapid, meaningful, and sustained improvements over 12 weeks implied that maximal complement inhibition is necessary for pronounced disease suppression. These findings have led to an ongoing phase III trial in myasthenia gravis.
Tesidolumab
Tesidolumab is a fully humanized monoclonal antibody to C5. Like eculizumab, it inhibits terminal complement activation. Tesidolumab is currently being tested for treatment of PNH181. In a phase II study the antibody did not halt progression of retinal atrophy182. Whether this drug will be of value in a neurological disease trial is unknown.
SKY59
SKY59 is a C5 monoclonal antibody with a long half-life owing to the fact that it is engineered to enable its recycling and recirculation from the lysosome183. This design allows for long-lasting neutralization of C5 so that infrequent subcutaneous dosing is required183.
In a clinical trial involving healthy volunteers, treatment-naive patients with PNH and patients who had previously been treated for PNH with eculizumab, low-volume subcutaneous SKY59 was well tolerated, had a good benefit-to-risk ratio and was efficacious in treatment-naive and eculizumab-treated patients with PNH. Accordingly, SKY59 has the potential to provide treatment for patients whose condition does not respond to eculizumab and to substantially reduce the treatment burden associated with chronic intravenous administration. On this basis, it could be useful in myasthenia gravis and NMOSD, but trials are needed.
Targeting the C1qC1r2C1s2 complex
Two monoclonal antibodies to the C1qC1r2C1s2 complex are being tested in humans. The first targets C1q and the other targets C1s. Both are of the IgG4 subclass and so do not activate the classical pathway.
Anti-C1q humanized antibody
C1q humanized antibody (known as ANX005) binds with high affinity (~10 pM) to C1q and inhibits activation of the classical complement pathway by inhibiting C1q function. ANX005 reduces deposition of C1q, C4 and C3 onto cold agglutinin-sensitized human red blood cells and blocks haemolysis184. The safety and tolerability of ANX005 is currently being evaluated in a phase I trial in 64 healthy volunteers184.
Inhibition of C1q can be applied therapeutically to a broad spectrum of diseases, including acute antibody-mediated autoimmune disease, such as some forms of GBS, and in chronic CNS disorders that involve complement-mediated neurodegeneration, such as AD. Beneficial effects of ANX005 have been observed in mouse models of GBS and AD owing to its functional inhibition of the classical pathway184, so the foundations have been laid for testing in these conditions in humans.
Sutimlimab
Sutimlimab (also known as BIVV009 or TNT009) is the humanized form of the mouse monoclonal antibody TNT003 to C1s. Humanized C1s antibodies are intended for treatment of the same broad spectrum of diseases as C1q antibodies. In contrast to ANX005, however, sutimlimab specifically inhibits the classical pathway and leaves intact the opsonic function of C1q, which removes altered and senescent self and captures free DNA185.
A phase I, double-blind, randomized, placebo-controlled, dose-escalation trial of single and multiple doses of sutimlimab versus placebo has been conducted186, followed by two phase Ib trials. In one of these phase Ib trials, the effect of sutimlimab in haemolytic anaemia associated with cold agglutinin disease was assessed; all six patients who received sutimlimab no longer needed blood transfusions187. In the other trial, sutimlimab had no effect in late antibody-mediated kidney allograft rejection, which involves alloantibody-triggered classical pathway activation188. Most recently, an in vitro study showed that sutimlimab prevents complement-enhanced activation of autoimmune human B cells188, an effect that could have therapeutic potential in some B cell-mediated autoimmune neurological diseases such as myasthenia gravis and NMOSD, but trials are needed.
Safety of complement-targeted therapies
Complement-targeted therapeutics are promising for neurological diseases, but caution is needed given the importance of complement in natural defences and its broader role in immune signalling. Prolonged complement suppression could increase susceptibility to bacterial infections owing to loss of the opsonic capacity of C3, as observed in primary C3 deficiency, or could result in adverse effects. These effects might be similar to those observed with other chronically used immunomodulators, such as susceptibility to bacterial or viral infections, even though no major events have been observed when appropriate prophylaxis is used.
The classical complement pathway is required for normal postnatal synaptic pruning via microglial destruction of low-activity synapses189 (Fig. 3). Similarly, migration of neurons during development also depends on the lectin pathway190. Given that there are considerable similarities in the mechanisms of proliferation and migration between the embryonic and regenerating brain, complement inhibition could also block the beneficial effects of complement in synaptic plasticity, neuroprotection and neurogenesis191.
In trials of eculizumab for PNH, myasthenia gravis and NMOSD146,192, safety profiles have been very good, but some aspects need attention. For example, eculizumab impairs host defences against Neisseria spp. and increases susceptibility to various infectious pathogens, particularly life-threatening meningococcal infections, so meningococcal vaccination is always necessary 2 weeks before the first dose of eculizumab193,194,195. Subsequent monitoring for early signs of meningococcal infection remains essential during treatment.
Infusion reactions also need to be acknowledged; these range from common headaches, nasopharyngitis, back pain, nausea, diarrhoea, upper respiratory infection, abdominal pain, urinary tract infections and pyrexia to rare anaphylactic or hypersensitivity reactions. In patients with myasthenia gravis, the most frequently reported adverse reaction was musculoskeletal pain, noted in ≥10% of patients167. In patients with NMOSD, upper respiratory tract infections and headaches were most common170,171. No cases of progressive multifocal leukoencephalopathy have been associated with eculizumab treatment.
Complement therapeutics and COVID-19
Although severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection has the potential to worsen pre-existing autoimmune conditions such as myasthenia gravis, as do most viruses, no evidence suggests that patients who are receiving common immunotherapies for these conditions have an increased susceptibility to coronavirus disease 2019 (COVID-19)196. If a patient is clinically stable and not lymphopenic, no compelling or data-driven reasons exist to change any of the immunosuppressive therapies they are receiving and risk disturbing clinical stability.
Indeed, data suggest that anti-complement therapies could have beneficial effects in COVID-19. Complement is an integral component of the innate immune response to viruses, and evidence suggests that C3 activation can exacerbate SARS-CoV-2-associated acute respiratory distress syndromes197. Furthermore, complement deposits are abundant in lung biopsy samples from patients with COVID-19 (ref.198). On this basis, complement inhibition was proposed as an intervention to alleviate the inflammatory complications of COVID-19 — trials of ravulizumab199 and eculizumab200 for acute respiratory distress syndromes are ongoing, and AMY-101 has been tested in some patients198,201. In patients with myasthenia gravis and NMOSD who are receiving therapy with eculizumab, the drug could even protect against severe COVID-19 (ref.202). Four patients with COVID-19 with severe pneumonia or acute respiratory distress syndrome have successfully recovered after treatment with eculizumab203.
Conclusions
The field of complement therapeutics is developing rapidly and holds promise for many neurological diseases in which complement is either a key or contributing factor in disease pathogenesis. The approval of eculizumab for myasthenia gravis and NMOSD and the ongoing phase III trials of ravulizumab and zilucoplan in myasthenia gravis are examples of success that point to an exciting future. Progress in our understanding of the role of complement in neurodegenerative disorders also opens new dimensions and raises guarded expectations and optimism for complement-targeted therapies in diseases for which no effective therapies are available, such as ALS, AD and traumatic brain injury. Complement-targeted therapies are unlikely to cure or reverse these diseases, but they have the potential to modify or slow the disease course by protecting the brain from ongoing autoinflammatory cascades or by promoting regeneration.
As for other biologic therapies, however, several challenges with complement-targeted therapeutics remain to be addressed. For example, monoclonal antibody delivery across the BBB to target CNS disorders is uncharted territory, and C3 and C5 activation and cleavage can occur within the BBB. The high cost of such therapies is also of concern, particularly given that their benefit in some of these disorders might be limited to modifying the disease course rather than inducing remission. Further understanding the role of complement in increasing or perpetuating chronic CNS or PNS damage will shed light on the molecular mechanisms that underlie disease progression in many neurological disorders and could lead to further novel therapeutic approaches.
References
Ricklin, D., Hajishengallis, G., Yang, K. & Lambris, J. D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 (2010).
Hawksworth, O. A., Coulthard, L. G. & Woodruff, T. M. Complement in the fundamental processes of the cell. Mol. Immunol. 84, 17–25 (2017).
Merle, N. S., Church, S. E., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part I — molecular mechanisms of activation and regulation. Front. Immunol. 6, 262 (2015).
Merle, N. S., Noe, R., Halbwachs-Mecarelli, L., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part II: role in immunity. Front. Immunol. 6, 257 (2015).
Arvieux, J., Yssel, H. & Colomb, M. G. Antigen-bound C3b and C4b enhance antigen-presenting cell function in activation of human T-cell clones. Immunology 65, 229–235 (1988).
Dempsey, P. W., Allison, M. E., Akkaraju, S., Goodnow, C. C. & Fearon, D. T. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271, 348–350 (1996).
Kemper, C. & Atkinson, J. P. T-cell regulation: with complements from innate immunity. Nat. Rev. Immunol. 7, 9–18 (2007).
Kemper, C., Mitchell, L. M., Zhang, L. & Hourcade, D. E. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc. Natl Acad. Sci. USA 105, 9023–9028 (2008).
Tenner, A. J., Stevens, B. & Woodruff, T. M. New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol. Immunol. 102, 3–13 (2018).
Kemper, C., Pangburn, M. K. & Fishelson, Z. Complement nomenclature 2014. Mol. Immunol. 61, 56–58 (2014).
Bohlson, S. S., Garred, P., Kemper, C. & Tenner, A. J. Complement nomenclature — deconvoluted. Front. Immunol. 10, 1308 (2019).
Nonaka, M. Evolution of the complement system. Subcell. Biochem. 80, 31–43 (2014).
Almitairi, J. O. M. et al. Structure of the C1r–C1s interaction of the C1 complex of complement activation. Proc. Natl Acad. Sci. USA 115, 768–773 (2018).
Mortensen, S. A. et al. Models of the complement C1 complex. Proc. Natl Acad. Sci. USA 115, E3866 (2018).
Sharp, T. H. et al. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl Acad. Sci. USA 116, 11900–11905 (2019).
Zwarthoff, S. A. et al. Functional characterization of alternative and classical pathway C3/C5 convertase activity and inhibition using purified models. Front. Immunol. 9, 1691 (2018).
Kjaer, T. R. et al. Oligomerization of mannan-binding lectin dictates binding properties and complement activation. Scand. J. Immunol. 84, 12–19 (2016).
Escudero-Esparza, A., Kalchishkova, N., Kurbasic, E., Jiang, W. G. & Blom, A. M. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 27, 5083–5093 (2013).
Nan, R. et al. Flexibility in mannan-binding lectin-associated serine proteases-1 and -2 provides insight on lectin pathway activation. Structure 25, 364–375 (2017).
Wong, N. K., Kojima, M., Dobo, J., Ambrus, G. & Sim, R. B. Activities of the MBL-associated serine proteases (MASPs) and their regulation by natural inhibitors. Mol. Immunol. 36, 853–861 (1999).
Blaum, B. S. et al. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat. Chem. Biol. 11, 77 (2014).
Chen, Z. A. et al. Structure of complement C3(H2O) revealed by quantitative cross-linking/mass-spectrometry and modelling. Mol. Cell Proteom. 15, 2730–2743 (2016).
Li, X. X., Lee, J. D., Kemper, C. & Woodruff, T. M. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J. Immunol. 202, 3339–3348 (2019).
Bayly-Jones, C., Bubeck, D. & Dunstone, M. A. The mystery behind membrane insertion: a review of the complement membrane attack complex. Philos. Trans. R. Soc. Lond. B 372, 20160221 (2017).
Hansch, G. M. et al. Macrophages release arachidonic acid, prostaglandin E2, and thromboxane in response to late complement components. J. Immunol. 133, 2145–2150 (1984).
Tradtrantip, L., Yao, X., Su, T., Smith, A. J. & Verkman, A. S. Bystander mechanism for complement-initiated early oligodendrocyte injury in neuromyelitis optica. Acta Neuropathol. 134, 35–44 (2017).
Yang, C., Yang, L. & Liu, Y. Soluble complement complex C5b-9 promotes microglia activation. J. Neuroimmunol. 267, 16–19 (2014).
Triantafilou, K., Hughes, T. R., Triantafilou, M. & Morgan, B. P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 (2013).
Carroll, M. C. The complement system in regulation of adaptive immunity. Nat. Immunol. 5, 981 (2004).
Alexander, J. J., Anderson, A. J., Barnum, S. R., Stevens, B. & Tenner, A. J. The complement cascade: Yin-Yang in neuroinflammation — neuro-protection and -degeneration. J. Neurochem. 107, 1169–1187 (2008).
Tabib, A., Karbian, N. & Mevorach, D. Demyelination, strokes, and eculizumab: lessons from the congenital CD59 gene mutations. Mol. Immunol. 89, 69–72 (2017).
Karbian, N. et al. Molecular pathogenesis of human CD59 deficiency. Neurol. Genet. 4, e280 (2018).
Mevorach, D. et al. Therapy with eculizumab for patients with CD59 p.Cys89Tyr mutation. Ann. Neurol. 80, 708–717 (2016).
Kawamoto, M., Murakami, Y., Kinoshita, T. & Kohara, N. Recurrent aseptic meningitis with PIGT mutations: a novel pathogenesis of recurrent meningitis successfully treated by eculizumab. BMJ Case Rep. bcr2018225910 (2018).
Acosta, J. et al. Molecular basis for a link between complement and the vascular complications of diabetes. Proc. Natl Acad. Sci. USA 97, 5450–5455 (2000).
Ghosh, P., Sahoo, R., Vaidya, A., Chorev, M. & Halperin, J. A. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr. Rev. 36, 272–288 (2015).
Carpanini, S. M., Torvell, M. & Morgan, B. P. Therapeutic inhibition of the complement system in diseases of the central nervous system. Front. Immunol. 10, 362 (2019).
Alexander, J. J. Blood–brain barrier (BBB) and the complement landscape. Mol. Immunol. 102, 26–31 (2018).
Jacob, A. & Alexander, J. J. Complement and blood–brain barrier integrity. Mol. Immunol. 61, 149–152 (2014).
Luchena, C., Zuazo-Ibarra, J., Alberdi, E., Matute, C. & Capetillo-Zarate, E. Contribution of neurons and glial cells to complement-mediated synapse removal during development, aging and in Alzheimer’s disease. Mediators Inflamm. 2018, 2530414 (2018).
Hafer-Macko, C. E. et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann. Neurol. 39, 625–635 (1996).
Goodfellow, J. A. et al. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J. Neurosci. 25, 1620–1628 (2005).
Halstead, S. K. et al. Anti-disialoside antibodies kill perisynaptic Schwann cells and damage motor nerve terminals via membrane attack complex in a murine model of neuropathy. Brain 127, 2109–2123 (2004).
Willison, H. J. et al. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain–Barré syndrome. J. Neuroimmunol. 201–202, 172–182 (2008).
Halstead, S. K. et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain 131, 1197–1208 (2008).
Basta, M., Illa, I. & Dalakas, M. C. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain–Barré syndrome, myasthenia gravis and dermatomyositis. J. Neuroimmunol. 71, 227–229 (1996).
Dalakas, M. C. & Engel, W. K. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch. Neurol. 37, 637–640 (1980).
Quast, I., Keller, C. W., Hiepe, F., Tackenberg, B. & Lunemann, J. D. Terminal complement activation is increased and associated with disease severity in CIDP. Ann. Clin. Transl Neurol. 3, 730–735 (2016).
Stathopoulos, P., Alexopoulos, H. & Dalakas, M. C. Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat. Rev. Neurol. 11, 143–156 (2015).
Querol, L., Devaux, J., Rojas-Garcia, R. & Illa, I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat. Rev. Neurol. 13, 533–547 (2017).
Manso, C. et al. Anti-neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J. Clin. Invest. 129, 2222–2236 (2019).
Uncini, A. & Vallat, J. M. Autoimmune nodo-paranodopathies of peripheral nerve: the concept is gaining ground. J. Neurol. Neurosurg. Psychiatry 89, 627–635 (2018).
Roggenbuck, J. J., Boucraut, J., Delmont, E., Conrad, K. & Roggenbuck, D. Diagnostic insights into chronic-inflammatory demyelinating polyneuropathies. Ann. Transl Med. 6, 337 (2018).
Roux, T. et al. Rituximab in chronic inflammatory demyelinating polyradiculoneuropathy with associated diseases. J. Peripher. Nerv. Syst. 23, 235–240 (2018).
Velardo, D. et al. Rituximab in refractory chronic inflammatory demyelinating polyradiculoneuropathy: report of four cases. J. Neurol. 264, 1011–1014 (2017).
Vlam, L. et al. Complement activity is associated with disease severity in multifocal motor neuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2, e119 (2015).
Harschnitz, O. et al. Autoantibody pathogenicity in a multifocal motor neuropathy induced pluripotent stem cell-derived model. Ann. Neurol. 80, 71–88 (2016).
Weiss, M. D., Dalakas, M. C., Lauter, C. J., Willison, H. J. & Quarles, R. H. Variability in the binding of anti-MAG and anti-SGPG antibodies to target antigens in demyelinating neuropathy and IgM paraproteinemia. J. Neuroimmunol. 95, 174–184 (1999).
Lombardi, R. et al. IgM deposits on skin nerves in anti-myelin-associated glycoprotein neuropathy. Ann. Neurol. 57, 180–187 (2005).
Ritz, M. F. et al. Anti-MAG IgM penetration into myelinated fibers correlates with the extent of myelin widening. Muscle Nerve 22, 1030–1037 (1999).
Dalakas, M. C. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther. Adv. Neurol. Disord. 11, 1756285617746640 (2018).
Li, M., Peake, P. W., Charlesworth, J. A., Tracey, D. J. & Moalem-Taylor, G. Complement activation contributes to leukocyte recruitment and neuropathic pain following peripheral nerve injury in rats. Eur. J. Neurosci. 26, 3486–3500 (2007).
Fritzinger, D. C. & Benjamin, D. E. The complement system in neuropathic and postoperative pain. Open Pain J. 9, 26–37 (2016).
Griffin, R. S. et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J. Neurosci. 27, 8699–8708 (2007).
Xu, M. et al. Pain and the immune system: emerging concepts of IgG-mediated autoimmune pain and immunotherapies. J. Neurol. Neurosurg. Psychiatry 91, 177–188 (2020). An insightful view of autoimmune markers and autoimmunity in patients with common neurological disorders dominated by pain.
Ruff, R. L. & Lisak, R. P. Nature and action of antibodies in myasthenia gravis. Neurol. Clin. 36, 275–291 (2018).
Cetin, H. & Vincent, A. Pathogenic mechanisms and clinical correlations in autoimmune myasthenic syndromes. Semin. Neurol. 38, 344–354 (2018).
Romi, F., Kristoffersen, E. K., Aarli, J. A. & Gilhus, N. E. The role of complement in myasthenia gravis: serological evidence of complement consumption in vivo. J. Neuroimmunol. 158, 191–194 (2005).
Howard, J. F. Jr. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann. NY Acad. Sci. 1412, 113–128 (2018).
Morgan, B. P. et al. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin. Exp. Immunol. 146, 294–302 (2006).
Chamberlain-Banoub, J., Neal, J. W., Mizuno, M., Harris, C. L. & Morgan, B. P. Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin. Exp. Immunol. 146, 278–286 (2006).
Kusner, L. L., Sengupta, M. & Kaminski, H. J. Acetylcholine receptor antibody-mediated animal models of myasthenia gravis and the role of complement. Ann. NY Acad. Sci. 1413, 136–142 (2018).
Souroujon, M. C., Brenner, T. & Fuchs, S. Development of novel therapies for MG: studies in animal models. Autoimmunity 43, 446–460 (2010).
Dalakas, M. C. Immunotherapy in myasthenia gravis in the era of biologics. Nat. Rev. Neurol. 15, 113–124 (2019).
Farrugia, M. E. & Vincent, A. Autoimmune mediated neuromuscular junction defects. Curr. Opin. Neurol. 23, 489–495 (2010).
Dalakas, M. C. Inflammatory muscle diseases. N. Engl. J. Med. 373, 393–394 (2015).
Dalakas, M. C. Myositis: are autoantibodies pathogenic in necrotizing myopathy? Nat. Rev. Rheumatol. 14, 251–252 (2018).
Kissel, J. T., Mendell, J. R. & Rammohan, K. W. Microvascular deposition of complement membrane attack complex in dermatomyositis. N. Engl. J. Med. 314, 329–334 (1986).
Emslie-Smith, A. M. & Engel, A. G. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann. Neurol. 27, 343–356 (1990).
Lahoria, R., Selcen, D. & Engel, A. G. Microvascular alterations and the role of complement in dermatomyositis. Brain 139, 1891–1903 (2016).
Nauta, A. J. et al. Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur. J. Immunol. 32, 1726–1736 (2002).
Sakuta, R. et al. Diagnostic significance of membrane attack complex and vitronectin in childhood dermatomyositis. J. Child. Neurol. 20, 597–602 (2005).
Basta, M. & Dalakas, M. C. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J. Clin. Invest. 94, 1729–1735 (1994). A clinico-immunopathological study that demonstrates the beneficial effect of IVIg in complement-mediated damage in dermatomyositis.
Watanabe, Y. et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J. Neurol. Neurosurg. Psychiatry 87, 1038–1044 (2016).
Wang, L. et al. Myopathy with anti-signal recognition particle antibodies: clinical and histopathological features in Chinese patients. Neuromuscul. Disord. 24, 335–341 (2014).
Dalakas, M. C. Necrotising autoimmune myopathy (NAM): antibodies seem to be specific markers in aiding diagnosis. J. Neurol. Neurosurg. Psychiatry 87, 1037 (2016).
Moghadam-Kia, S., Aggarwal, R. & Oddis, C. V. (2017). Biologics for idiopathic inflammatory myopathies. Curr. Opin. Rheumatol. 29, 645–651 (2017).
Dalakas M. C. in Contemporary Neurology Series (eds Bielekova, R., Birnbaum, R. & Lisak, R.) 177–192 (Oxford University Press, 2020).
Glaubitz, S., Zeng, R. & Schmidt, J. New insights into the treatment of myositis. Ther. Adv. Musculoskelet. Dis. 12, 1759720X19886494 (2020).
Fujihara, K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr. Opin. Neurol. 32, 385–394 (2019).
Dalmau, J. & Graus, F. Antibody-mediated encephalitis. N. Engl. J. Med. 378, 840–851 (2018).
Lucchinetti, C. et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717 (2000).
Ingram, G., Hakobyan, S., Robertson, N. P. & Morgan, B. P. Elevated plasma C4a levels in multiple sclerosis correlate with disease activity. J. Neuroimmunol. 223, 124–127 (2010).
Prineas, J. W. et al. Immunopathology of secondary-progressive multiple sclerosis. Ann. Neurol. 50, 646–657 (2001).
Ingram, G. et al. Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol. Commun. 2, 53 (2014).
Barnett, M. H., Parratt, J. D., Cho, E. S. & Prineas, J. W. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 65, 32–46 (2009). An important study of human postmortem tissue that shows the potential pathogenic role of complement in multiple sclerosis.
Breij, E. C. et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann. Neurol. 63, 16–25 (2008).
Schwab, C. & McGeer, P. L. Complement activated C4d immunoreactive oligodendrocytes delineate small cortical plaques in multiple sclerosis. Exp. Neurol. 174, 81–88 (2002).
Brink, B. P. et al. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J. Neuropathol. Exp. Neurol. 64, 147–155 (2005).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). A very important study that demonstrates the physiological role of complement in synapse formation.
Stephan, A. H. et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474 (2013).
Rosen, A. M. & Stevens, B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv. Exp. Med. Biol. 703, 75–93 (2010).
Howell, G. R. et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Invest. 121, 1429–1444 (2011).
Werneburg, S. et al. Targeted complement inhibition at synapses prevents microglial synaptic engulfment and synapse loss in demyelinating disease. Immunity 52, 167–182 (2020).
Tran, G. T. et al. Attenuation of experimental allergic encephalomyelitis in complement component 6-deficient rats is associated with reduced complement C9 deposition, P-selectin expression, and cellular infiltrate in spinal cords. J. Immunol. 168, 4293–4300 (2002).
Michailidou, I. et al. Systemic inhibition of the membrane attack complex impedes neuroinflammation in chronic relapsing experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 6, 36 (2018).
Peschl, P. et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J. Neuroinflammation 14, 208 (2017).
Alexopoulos, H. et al. Anti-aquaporin-4 autoantibodies in systemic lupus erythematosus persist for years and induce astrocytic cytotoxicity but not CNS disease. J. Neuroimmunol. 289, 8–11 (2015).
Soltys, J. et al. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Invest. 129, 2000–2013 (2019).
Marignier, R., Cobo Calvo, A. & Vukusic, S. Neuromyelitis optica and neuromyelitis optica spectrum disorders. Curr. Opin. Neurol. 30, 208–215 (2017).
Wang, H. et al. Increased soluble C5b-9 in CSF of neuromyelitis optica. Scand. J. Immunol. 79, 127–130 (2014).
Guo, Y. et al. Pathogenic implications of cerebrospinal fluid barrier pathology in neuromyelitis optica. Acta Neuropathol. 133, 597–612 (2017).
Wu, Y., Zhong, L. & Geng, J. Neuromyelitis optica spectrum disorder: pathogenesis, treatment, and experimental models. Mult. Scler. Relat. Disord. 27, 412–418 (2019).
Yao, X. & Verkman, A. S. Complement regulator CD59 prevents peripheral organ injury in rats made seropositive for neuromyelitis optica immunoglobulin G. Acta Neuropathol. Commun. 5, 57 (2017).
Yao, X. & Verkman, A. S. Marked central nervous system pathology in CD59 knockout rats following passive transfer of neuromyelitis optica immunoglobulin G. Acta Neuropathol. Commun. 5, 15 (2017).
Alexopoulos, H. & Dalakas, M. C. The immunobiology of autoimmune encephalitides. J. Autoimmun. 104, 102339 (2019).
Malviya, M. et al. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann. Clin. Transl Neurol. 4, 768–783 (2017).
Martinez-Hernandez, E. et al. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 77, 589–593 (2011).
Carvajal-Gonzalez, A. et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 137, 2178–2192 (2014).
Kortvelyessy, P. et al. Complement-associated neuronal loss in a patient with CASPR2 antibody-associated encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2, e75 (2015).
Platt, M. P., Agalliu, D. & Cutforth, T. Hello from the other side: how autoantibodies circumvent the blood–brain barrier in autoimmune encephalitis. Front. Immunol. 8, 442 (2017).
Stoltzner, S. E. et al. Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am. J. Pathol. 156, 489–499 (2000).
Eikelenboom, P., Hack, C. E., Kamphorst, W. & Rozemuller, J. M. Distribution pattern and functional state of complement proteins and alpha 1-antichymotrypsin in cerebral beta/A4 deposits in Alzheimer’s disease. Res. Immunol. 143, 617–620 (1992).
Eikelenboom, P. & Stam, F. C. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 57, 239–242 (1982).
Morgan, B. P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 40, 113–124 (2018).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). A paper that illustrates in mouse models how complement affects synaptic loss in Alzheimer disease.
Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl Med. 9, eaaf6295 (2017).
Maier, M. et al. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J. Neurosci. 28, 6333–6341 (2008).
Wyss-Coray, T. et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl Acad. Sci. USA 99, 10837–10842 (2002).
Hernandez, M. X. et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener. 12, 66 (2017).
Fonseca, M. I. et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375–1383 (2009).
Tenner, A. J. Complement-mediated events in Alzheimer’s disease: mechanisms and potential therapeutic targets. J. Immunol. 204, 306–315 (2020).
Sta, M. et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol. Dis. 42, 211–220 (2011).
Woodruff, T. M. et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J. Immunol. 181, 8727–8734 (2008).
Lee, J. D. et al. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 174, 689–699 (2017).
Singhrao, S. K., Neal, J. W., Morgan, B. P. & Gasque, P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp. Neurol. 159, 362–376 (1999).
Woodruff, T. M. et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 20, 1407–1417 (2006).
Kossmann, T., Stahel, P. F., Morganti-Kossmann, M. C., Jones, J. L. & Barnum, S. R. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 73, 63–69 (1997).
Stahel, P. F. et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood–brain barrier dysfunction in patients with traumatic brain injury. J. Neurotrauma 18, 773–781 (2001).
Bellander, B. M., von Holst, H., Fredman, P. & Svensson, M. Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J. Neurosurg. 85, 468–475 (1996).
Anderson, A. J., Robert, S., Huang, W., Young, W. & Cotman, C. W. Activation of complement pathways after contusion-induced spinal cord injury. J. Neurotrauma 21, 1831–1846 (2004).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Sellgren, C. M. et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 22, 374–385 (2019).
Zanjani, H. et al. Complement activation in very early Alzheimer disease. Alzheimer Dis. Assoc. Disord. 19, 55–66 (2005).
Mohebnasab, M. et al. Current and future approaches for monitoring responses to anti-complement therapeutics. Front. Immunol. 10, 2539 (2019).
Zelek, W. M., Xie, L., Morgan, B. P. & Harris, C. L. Compendium of current complement therapeutics. Mol. Immunol. 114, 341–352 (2019).
Lunemann, J. D., Quast, I. & Dalakas, M. C. Efficacy of intravenous immunoglobulin in neurological diseases. Neurotherapeutics 13, 34–46 (2016).
Dalakas, M. C. Mechanistic effects of IVIg in neuroinflammatory diseases: conclusions based on clinicopathologic correlations. J. Clin. Immunol. 34, S120–S126 (2014).
Dalakas, M. C. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA 291, 2367–2375 (2004).
Lunemann, J. D., Nimmerjahn, F. & Dalakas, M. C. Intravenous immunoglobulin in neurology — mode of action and clinical efficacy. Nat. Rev. Neurol. 11, 80–89 (2015).
Lutz, H. U. et al. Intravenously applied IgG stimulates complement attenuation in a complement-dependent autoimmune disease at the amplifying C3 convertase level. Blood 103, 465–472 (2004).
Dalakas, M. C. et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N. Engl. J. Med. 329, 1993–2000 (1993).
Raju, R. & Dalakas, M. C. Gene expression profile in the muscles of patients with inflammatory myopathies: effect of therapy with IVIg and biological validation of clinically relevant genes. Brain 128, 1887–1896 (2005).
Appeltshauser, L., Weishaupt, A., Sommer, C. & Doppler, K. Complement deposition induced by binding of anti-contactin-1 auto-antibodies is modified by immunoglobulins. Exp. Neurol. 287, 84–90 (2017).
Keller, C. W., Quast, I., Dalakas, M. C. & Lunemann, J. D. IVIG efficacy in CIDP patients is not associated with terminal complement inhibition. J. Neuroimmunol. 330, 23–27 (2019).
Davidson, A. I. et al. Inhibition of complement in Guillain–Barré syndrome: the ICA-GBS study. J. Peripher. Nerv. Syst. 22, 4–12 (2017).
Hughes, R. A. et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 7, 136–144 (2008).
Winter, M. et al. Dose-dependent inhibition of demyelination and microglia activation by IVIG. Ann. Clin. Transl Neurol. 3, 828–843 (2016).
Esen, F., Ozcan, P. E., Tuzun, E. & Boone, M. D. Mechanisms of action of intravenous immunoglobulin in septic encephalopathy. Rev. Neurosci. 29, 417–423 (2018).
Mastellos, D. C., Ricklin, D. & Lambris, J. D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 18, 707–729 (2019).
Ricklin, D., Reis, E. S., Mastellos, D. C., Gros, P. & Lambris, J. D. Complement component C3 — the “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 274, 33–58 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03500549 (2020).
Reis, E. S. et al. Safety profile after prolonged C3 inhibition. Clin. Immunol. 197, 96–106 (2018).
Socie, G. et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br. J. Haematol. 182, 297–310 (2019).
Collongues, N., Ayme-Dietrich, E., Monassier, L. & de Seze, J. Pharmacotherapy for neuromyelitis optica spectrum disorders: current management and future options. Drugs 79, 125–142 (2019).
Howard, J. F. Jr et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 48, 76–84 (2013).
Howard, J. F. Jr et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 16, 976–986 (2017). The study of eculizumab in myasthenia gravis on which approval of the drug is based.
Muppidi, S. et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve 60, 14–24 (2019).
Zhou, Y. et al. Anti-C5 antibody treatment ameliorates weakness in experimentally acquired myasthenia gravis. J. Immunol. 179, 8562–8567 (2007).
Pittock, S. J. et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 12, 554–562 (2013).
Pittock, S. J. et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N. Engl. J. Med. 381, 614–625 (2019). The study of eculizumab in NMOSD on which approval of the drug is based.
Duchow, A., Paul, F. & Bellmann-Strobl, J. Current and emerging biologics for the treatment of neuromyelitis optica spectrum disorders. Expert Opin. Biol. Ther. 20, 1061–1072 (2020).
Misawa, S. et al. Safety and efficacy of eculizumab in Guillain–Barré syndrome: a multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 17, 519–529 (2018). A study that shows the potential for a therapeutic effect of eculizumab in GBS.
Fitzpatrick, A. M. et al. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J. Peripher. Nerv. Syst. 16, 84–91 (2011).
Lee, J. W. et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood 133, 530–539 (2019).
Stern, R. M. & Connell, N. T. Ravulizumab: a novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Therapeutic Adv. Hematol. 10, 2040620719874728 (2019).
Peffault de Latour, R. et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase 3 randomised, multicentre studies. Br. J. Haematol. https://doi.org/10.1111/bjh.16711 (2020).
Kulasekararaj, A. G. et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood 133, 540–549 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03920293 (2020).
Howard, J. F. Jr. et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 77, 582–592 (2020). This phase II trial shows that daily subcutaneous injection of zilucoplan in patients with generalized myasthenia gravis leads to rapid, meaningful and sustained improvements in Quantitative Myasthenia Gravis and Myasthenia Gravis Activities of Daily Living scores over 12 weeks compared with placebo.
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02534909 (2020).
Kassa, E., Ciulla, T. A., Hussain, R. M. & Dugel, P. U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin. Biol. Ther. 19, 335–342 (2019).
Sampei, Z. et al. Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. PLoS ONE 13, e0209509 (2018).
Lansita, J. A. et al. Nonclinical development of ANX005: a humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int. J. Toxicol. 36, 449–462 (2017).
Paidassi, H. et al. The lectin-like activity of human C1q and its implication in DNA and apoptotic cell recognition. FEBS Lett. 582, 3111–3116 (2008).
Bartko, J. et al. A randomized, first-in-human, healthy volunteer trial of sutimlimab, a humanized antibody for the specific inhibition of the classical complement pathway. Clin. Pharmacol. Ther. 104, 655–663 (2018).
Jager, U. et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood 133, 893–901 (2019).
Eskandary, F. et al. Anti-C1s monoclonal antibody BIVV009 in late antibody-mediated kidney allograft rejection — results from a first-in-patient phase 1 trial. Am. J. Transpl. 18, 916–926 (2018).
Nonaka, S. & Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 707, 134317 (2019).
Gorelik, A. et al. Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 8, 15096 (2017).
Coulthard, L. G., Hawksworth, O. A. & Woodruff, T. M. Complement: the emerging architect of the developing brain. Trends Neurosci. 41, 373–384 (2018).
Hillmen, P. et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 162, 62–73 (2013).
Benamu, E. & Montoya, J. G. Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr. Opin. Infect. Dis. 29, 319–329 (2016).
Clancy, M. et al. Disseminated cryptococcosis associated with administration of eculizumab. Am. J. Health Syst. Pharm. 75, 1018–1022 (2018).
Crew, P. E. et al. Unusual Neisseria species as a cause of infection in patients taking eculizumab. J. Infect. 78, 113–118 (2019).
Dalakas, M. C. Guillain–Barré syndrome: the first documented COVID-19-triggered autoimmune neurologic disease: more to come with myositis in the offing. Neurol. Neuroimmunol. Neuroinflamm. 7, e781 (2020). An opinion paper that highlights and defends the view that patients with myasthenia gravis who are receiving common immunotherapies and who are clinically stable do not have increased susceptibility to COVID-19.
Hatzidionysiou, K., Svenungsson, E. & Faustini, F. Could severe COVID-19 be considered a complementopathy? Lupus Sci. Med. 7, e000415 (2020).
Risitano, A. M. et al. Complement as a target in COVID-19? Nat. Rev. Immunol. 20, 343–344 (2020). A highly topical opinion paper about how complement could be a target in COVID-19.
Smith, K., Pace, A., Ortiz, S., Kazani, S. & Rottinghaus, S. A phase 3 open-label, randomized, controlled study to evaluate the efficacy and safety of intravenously administered ravulizumab compared with best supportive care in patients with COVID-19 severe pneumonia, acute lung injury, or acute respiratory distress syndrome: a structured summary of a study protocol for a randomised controlled trial. Trials 21, 639 (2020).
Giudice, V. et al. Combination of ruxolitinib and eculizumab for treatment of severe SARS-CoV-2-related acute respiratory distress syndrome: a controlled study. Front. Pharmacol. 11, 857 (2020).
Mastaglio, S. et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 215, 108450 (2020).
Dalakas M. C. Progress in the therapy of myasthenia gravis: getting closer to effective targeted immunotherapies. Curr. Opin. Neurol. 33, 545–552 (2020).
Diurno, F. et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience Eur. Rev. Med. Pharmacol. Sci. 24, 4040–4047 (2020).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Author information
Authors and Affiliations
Contributions
All authors contributed to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Opsonization
-
The molecular mechanism by which antigens, microorganisms or apoptotic cells are targeted by antibodies and complement breakdown products so that attraction to the cell surface receptors of phagocytes and natural killer cells increases; when the antigen is coated in opsonins, binding to immune cells is greatly increased.
- Anaphylatoxins
-
Small pro-inflammatory oligopeptide fragments, such as C3a and C5a, that are produced during activation of the complement system and have important functions in the immune response and host defence.
- Zymogens
-
Inactive precursors of enzymes that requires a biochemical change to become an active enzyme; also called pro-enzymes.
- Quantitative Myasthenia Gravis
-
A physician-administered scale that assesses the severity of myasthenia gravis, ranging from 0 to 39, with higher scores representing more severe impairment.
- Myasthenia Gravis Activities of Daily Living
-
A patient-reported index of daily living assessment, where a score >6 suggests that patients are moderately or severely affected by myasthenia gravis.
Rights and permissions
About this article

Cite this article
Dalakas, M.C., Alexopoulos, H. & Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol 16, 601–617 (2020). https://doi.org/10.1038/s41582-020-0400-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-020-0400-0
This article is cited by
-
Assessment of serum complement level in a sample of patients of idiopathic childhood epilepsy
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2024)
-
Associations of cerebrospinal fluid complement proteins with Alzheimer’s pathology, cognition, and brain structure in non-dementia elderly
Alzheimer's Research & Therapy (2024)
-
Different inflammatory signatures based on CSF biomarkers relate to preserved or diminished brain structure and cognition
Molecular Psychiatry (2024)
-
Increased infiltration of CD4+ T cell in the complement deficient lymphedema model
BMC Immunology (2023)
-
Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation
Nature Reviews Drug Discovery (2023)
