
Abstract
Respiratory failure is the leading cause of death in patients with severe SARS-CoV-2 infection1,2, but the host response at the lung tissue level is poorly understood. Here we performed single-nucleus RNA sequencing of about 116,000 nuclei from the lungs of nineteen individuals who died of COVID-19 and underwent rapid autopsy and seven control individuals. Integrated analyses identified substantial alterations in cellular composition, transcriptional cell states, and cell-to-cell interactions, thereby providing insight into the biology of lethal COVID-19. The lungs from individuals with COVID-19 were highly inflamed, with dense infiltration of aberrantly activated monocyte-derived macrophages and alveolar macrophages, but had impaired T cell responses. Monocyte/macrophage-derived interleukin-1β and epithelial cell-derived interleukin-6 were unique features of SARS-CoV-2 infection compared to other viral and bacterial causes of pneumonia. Alveolar type 2 cells adopted an inflammation-associated transient progenitor cell state and failed to undergo full transition into alveolar type 1 cells, resulting in impaired lung regeneration. Furthermore, we identified expansion of recently described CTHRC1+ pathological fibroblasts3 contributing to rapidly ensuing pulmonary fibrosis in COVID-19. Inference of protein activity and ligand–receptor interactions identified putative drug targets to disrupt deleterious circuits. This atlas enables the dissection of lethal COVID-19, may inform our understanding of long-term complications of COVID-19 survivors, and provides an important resource for therapeutic development.
Similar content being viewed by others


Main
Globally, the pandemic of COVID-19, which results from infection with SARS-CoV-2, has led to more than 145 million cases (32 million in the USA) and 3.1 million deaths (570,000 in the USA; figures as of 26 April 2021)1. Approximately 15% of infected individuals develop severe disease, which can manifest as acute respiratory distress syndrome (ARDS) and is associated with substantial morbidity and mortality2,4.
Previously, single-cell RNA sequencing (scRNA-seq) analyses of healthy individuals have revealed the tissue distribution of host receptors that are required for SARS-CoV-2 entry5,6,7, and examination of bronchoalveolar lavage fluid and blood from patients with COVID-19 of varying severity has identified the effects of SARS-CoV-2 infection on immune responses and cytokine dysregulation8,9,10,11,12. However, owing to the practical limitations of accessing patient tissues, the effects of SARS-CoV-2 at the level of the lung tissue remain unclear. A series of autopsy studies that examined formalin-fixed, paraffin-embedded (FFPE) tissue sections from individuals who died of COVID-19 extended our understanding of virus organotropism, but these studies were limited in their discovery potential by low-plex assays (for example, immunohistochemistry) and/or prolonged post-mortem intervals (PMIs), which adversely affect RNA quality13,14,15.
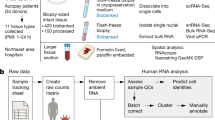
We established a rapid autopsy program and, under Institutional Review Board approved protocols, collected snap-frozen organ specimens from individuals with COVID-19 within hours of death. We performed single-nucleus RNA-seq (snRNA-seq) on lung samples from individuals who died from COVID-19 and control individuals to build an atlas that provides insight into the pathophysiology of COVID-19 and provides a key resource for further investigation.
The lung cellular landscape in COVID-19
The COVID-19 cohort consisted of 19 patients (12 males and 7 females) who died at a median age of 72 years (range, 58 to more than 89) (Supplementary Table 1, Extended Data Fig. 1a) and underwent rapid autopsy with a median post-mortem interval (PMI) of 4 h (range, 2–9 h). All had underlying co-morbidities that are associated with increased risk of severe COVID-1916 (Supplementary Table 1). The control cohort comprised 7 individuals (4 males and 3 females) with a median age of 70 years (range, 67 to 79 years) who underwent lung resection or biopsy in the pre-COVID-19 era (Supplementary Table 1).
Using snRNA-seq17 and an integrated quality control pipeline (see Methods), we generated a lung atlas that profiled 116,314 nuclei, including 79,636 from COVID-19-infected lungs and 36,678 from control lungs (Fig. 1a). We used a three-pronged approach for cell-type identification: unbiased identification of cluster markers, discovery of cell types using signatures from reported atlases, and manual curation to sub-stratify cell populations and cell states using expert knowledge (see Methods). We report cell-type assignment with three levels of granularity: major cell types, intermediate granularity, and fine granularity (Supplementary Table 2). We visualized data with dimensionality reduction using uniform manifold approximation and projection (UMAP) (Fig. 1b, c, Extended Data Fig. 1b–d). We identified nine major cell types: epithelial cells (n = 30,070 cells), myeloid cells (n = 29,632), fibroblasts (n = 22,909), endothelial cells (n = 5,386), T and natural killer (NK) lymphocytes (n = 16,751), B lymphocytes and plasma cells (n = 7,236), neuronal cells (n = 2,017), mast cells (n = 1,464), and antigen-presenting cells (APCs; primarily dendritic cells) (n = 849). At the most granular level, we identified 41 different cell types (Supplementary Table 2).

a, Overview of study design. b, Major clusters and respective cell-type assignments in UMAP. c, Origins of cells with same embedding as in b. d, Fraction of major cell types in control (n = 7) and COVID-19 lungs (n = 19). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × the interquartile range (IQR). Wilcoxon rank-sum test.
We found significant differences in cell fractions between COVID-19 and control lungs both globally (Fig. 1d) and within the immune and non-immune compartments (Extended Data Fig. 2a-c). There was a reduction in the epithelial cell compartment, due to loss of both alveolar type II (AT2) and type I (AT1) cells, and an increase in monocytes/macrophages, fibroblasts, and neuronal cells; these observations were independent of donor sex (Extended Data Fig. 3a, b).
We found no major differences in the expression of ACE2, CD147 (also known as BSG), NPR1, TMPRSS2, FURIN or CTSL between COVID-19 and control lungs (Extended Data Fig. 3c–f). This indicates that changes in cell-type proportions were unrelated to the expression of receptors or putative proteases that are important for viral entry, although we cannot exclude the possibility that virus-mediated cell death selectively depletes cells with high expression of these genes. We detected SARS-CoV-2 reads in two patients (Supplementary Table 3), one of whom had HIV/AIDS (CD4+ T cell count 29 per mm3 on hospital admission; 662 unique molecular identifiers detected in 28 cells), which suggests that viral reads can, in principle, be captured.
Aberrant activation of myeloid cells
Myeloid cells represented a major cellular constituent in COVID-19 lungs and were more prevalent there than in control lungs (Fig. 1d, Extended Data Figs. 2a, c, 4a). We identified monocytes (n = 3,176), monocyte-derived macrophages (MDMs; n = 9,534), transitioning MDMs (n = 4,203), and resident alveolar macrophages (AMs; n = 12,511), which were recovered as distinct trajectories in diffusion component (DC) analysis and were more frequent in COVID-19 lungs (Fig. 2a–c, Extended Data Fig. 4b–i, Supplementary Tables 2, 4, 5). Myeloid cells from individuals with COVID-19 were highly and aberrantly activated. For example, MDMs in COVID-19 lungs differentially expressed genes of activation (for example, CTSB, CTSD, CTSZ, PSAP) and two long non-coding RNAs, NEAT1 and MALAT1, that are associated with aberrant macrophage activation and impaired T cell immunity18 (Extended Data Fig. 5a, Supplementary Table 5). AMs, which arise from fetal monocytes and can self-renew19, were enriched and highly activated in COVID-19 lungs (Fig. 2c, Extended Data Fig. 5a). Notably, COVID-19 AMs showed strongly decreased mRNA and protein expression of the tumour-associated macrophage receptor AXL (Fig. 2d, Extended Data Fig. 5b, c), a receptor tyrosine kinase that is important for coordinated clearance of apoptotic cells (efferocytosis) and subsequent anti-inflammatory regulation during tissue regeneration20. These data suggest that myeloid cells are a major source of dysregulated inflammation in COVID-19.

a, UMAP projection highlighting immune cell clusters. b, Visualization of myeloid cells using the first three DCs. Inset indicates group assignment. c, Fraction of myeloid cells in control (n = 7) and COVID-19 lungs (n = 19). Middle line, median; box edges, 25th and 75th percentiles; whiskers: most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test. d. Representative immunofluorescence staining for CD169, AXL and DAPI (large image) in control and COVID-19 lung tissue; top, selected area with overlay; bottom, individual channels. Scale bar, 20 μm. e, f, Top 20 recurrently detected IGHV–IGLV combinations in COVID-19 (e) and corresponding group annotation (f). *Combination for previously described anti-RBD antibody21. g, UMAP of T/NK cells; inset, group assignments. h, i, RNA expression (log-normalized) of GZMB (h) and MKI67 (i) in the same embedding as g.
Plasma and T cell responses
To gain insights into humoral immunity against SARS-CoV-2 infection in the lung, we identified plasma cells (Extended Data Fig. 6a–c) and reconstructed immunoglobulins by determining mRNA co-expression of the variable heavy (IGHV) and light (IGLV) chains and isotypes on a per cell basis (see Methods; Extended Data Fig. 6d–k, Supplementary Table 6). IGHV1-18–IGLV3-20, which gives rise to a neutralizing antibody (S309)21 against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, was among the commonly identified IGHV–IGLV combinations, which suggests that a coordinated antibody response occurred (Fig. 2e, f, Extended Data Fig. 6l, m). In the T/NK cell compartment (Fig. 2g), we distinguished CD8+ T cells (n = 3,561), T regulatory (Treg) cells (n = 649), other CD4+ T cells (n = 7,586), and NK cells (n = 2,141). We found no significant increase in T cell abundances in COVID-19 lungs, and only modest upregulation of cytokines and programs associated with activation and tissue residency of T cells (Fig. 2g–i, Extended Data Fig. 7a–i). Although immune response patterns were highly variable (Extended Data Fig. 7j, k), these data suggest that an impaired T cell response might contribute to lethal outcomes in COVID-19 in the context of a principally preserved humoral immune response.
Impaired alveolar epithelial regeneration
Within the epithelial compartment, we identified alveolar epithelial cells (AT1 and AT2 cells; n = 20,949), airway epithelial cells (basal, ciliated, club, goblet, and mucous cells; n = 7,223), a cluster characterized by the expression of inflammatory and cell cycle genes, including IRF8, B2M, MKI67 and TOP2A (‘cycling epithelium’; n = 609), and a cluster showing high expression of the extracellular matrix (ECM) components COL6A3, COL1A2, and COL3A1 (‘ECMhigh epithelium’; n = 1,179) (Fig. 3a, b, Extended Data Fig. 8a–c, Supplementary Tables 2, 7).

a, b, UMAP of investigated alveolar and airway epithelial cells (a) and corresponding group assignments (b). c, Differential gene expression (log-normalized, scaled; see Methods) of AT1 and AT2 cells from COVID-19 and control lungs. Columns, single cells; rows, expression of top-regulated genes. Left bar, lineage markers for AT1 (purple) and AT2 (pink) cells. Colour-coded top lanes indicate expression strength of signatures (log-normalized; see Methods) and group assignment as indicated on the right. exp., expression. d, e, Violin plots of ETV5 and CAV1 mRNA expression (log-normalized) in AT2 and AT1 cells, respectively; Wilcoxon rank-sum test with Bonferroni correction. f, UMAP embedding of AT1 and AT2 cells and identified DATPs; inset indicates group assignments. g, Violin plots of DATP signature expression (log-normalized) in AT1 and AT2 cells. Wilcoxon rank-sum test. h, First three DCs showing main trajectories of AT2 and AT1 cells and DATPs, expression of DATP signature and group assignment (inset). i, Fractions of DATP and AT cells in control (n = 7) and COVID-19 lungs (n = 19). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test. j, Representative immunofluorescence staining for pro-SPC, KRT8 and DAPI in control and COVID-19 lung tissue; top, representative area with overlay; bottom, small images with individual channels of selected area. Scale bar, 50 μm. k, l, Tissue mass cytometric quantification of IL-1β (k) and IL-6 (l) in healthy lung tissue and samples from donors with different infectious aetiologies. Each dot represents quantification of IL-1β and IL-6 in a region of interest (ROI); two-sided Mann–Whitney U-test with Benjamini–Hochberg false discovery rate (FDR) adjustment.
AT2 cells serve as progenitors for AT1 cells during lung regeneration22. AT2 and T1 cells in control lungs formed distinct clusters (Fig. 3a, b) and demonstrated the expected changes in differential gene expression (DGE) analysis, including expression of the lineage markers SFTPC and SFTPB in AT2 cells, and CLIC5 and AGER in AT1 cells (Fig. 3c, Supplementary Table 7). By contrast, clustering of AT2 and AT1 cells in COVID-19 lungs was less discrete, with a substantial portion of cells not overlapping with their control counterparts (Fig. 3b). Both AT2 and AT1 cells from COVID-19 lungs showed decreased overall expression of defining markers (Fig. 3c). COVID-19 AT2 cells displayed decreased expression of ETV5 (Fig. 3d), a transcription factor that is required for maintaining AT2 cell identity. Decreased ETV5 expression is associated with differentiation towards AT1 cells23, indicating that AT2 cells had initiated a regeneration program (Fig. 3d, Extended Data Fig. 8d). CAV1, a marker of late AT1 maturation24, was expressed at significantly lower levels in AT1 cells from COVID-19 lungs (Fig. 3e). Overall, these data suggest incomplete transition of AT2 to AT1 cells in COVID-19 lungs.
Recent studies have shown that inflammation can induce a cell state that is characterized by failure to fully transition to AT1 cells; this has been termed ‘damage-associated transient progenitors’ (DATPs), ‘alveolar differentiation intermediate’ (ADI), or ‘pre-AT1 transitional cell state’ (PATS)25,26,27 (hereafter referred to as DATPs). We used expression of the DATP marker genes (KRT8, CLDN4 and CDKN1A)25 to develop a DATP signature (see Methods; Extended Data Fig. 8e–h, Supplementary Table 8) and found that alveolar epithelial cells from COVID-19 lungs scored significantly higher for expression of this signature than those from control lungs (Fig. 3f, g, Extended Data Fig. 8i). DC analysis separated a main trajectory from AT2 to AT1 cells, while DATPs were primarily localized between AT2 and AT1 cells (Fig. 3h, Extended Data Fig. 8j–n). Gene set enrichment analysis (GSEA) of DATPs compared to differentiated AT2 or AT1 cells showed enrichment for TNFα and p53 signalling, and for the hypoxia response via HIF-1α (Extended Data Fig. 8o), consistent with pathways that have been implicated in DATP in mouse models27. Consistent with overrepresentation of p53 signalling, the majority of DATPs did not undergo cell division (Extended Data Fig. 8p), suggesting that they arrest in the DATP cell state.
DATPs were more frequent in COVID-19 than control lungs (Fig. 3i). Immunofluorescence staining of corresponding tissues showed that the frequency of KRT8+ and CLDN4+ DATPs was higher in COVID-19 lungs (Fig. 3j, Extended Data Fig. 8r, s), and we observed progressive loss of AT1 cell abundance with increasing time from symptom onset to death (Extended Data Fig. 8t). Overall, these data suggest that, in addition to direct destruction of the alveolar epithelium by viral infection, lung-regenerative processes are impaired in individuals with COVID-19.
We next determined the sources of inflammation that contribute to the DATP cell state, and more generally, to the hyperinflammatory environment in COVID-19 lungs. Capture of the inflammatory cytokine interleukin (IL)-1β (and others) at an mRNA level may be limited, as the bioactive form of IL-1β, which has a major role in triggering DATPs25, is generated by cleavage from pro-IL-1β upon inflammasome activation; thus, protein-level assessment provides complementary information. For this purpose, we leveraged a recently released high-plex imaging mass-cytometry dataset that profiled 237 tissue regions from 23 individuals, including healthy controls; patients with influenza pneumonia, bacterial pneumonia, or ARDS; and ten patients who died from COVID-1928. IL-1β was more strongly expressed in monocytes and macrophages from individuals with COVID-19 than from healthy individuals or patients in the other disease groups (Fig. 3k, Extended Data Fig. 9a–c). IL-6, another key inflammatory cytokine invoked in the pathophysiology of COVID-19, was more abundant in epithelial cells from patients with COVID-19, but was not differentially expressed in macrophages from these patients compared to patients in other disease groups (Fig. 3l, Extended Data Fig. 9d–f). Finally, we found that the expression of type I interferons and interferon response genes in various cell types, including AT2 cells, monocytes, and macrophages, was stronger in patients with COVID-19 than in control donors (Extended Data Fig. 9g, h). Together, these data suggest that myeloid-derived IL-1β might be a distinguishing feature of COVID-19 compared to other viral or bacterial pneumonias and may contribute to the induction and maintenance of the DATP cell state.
Ectopic tuft-like cells in COVID-19
Among captured airway epithelial cells, we recovered four distinct trajectories: KRT5+TP63+ basal (n = 534), club (n = 1,232), and goblet cells (n = 1,757), and one trajectory with fewer cells (n = 110) that was primarily found in COVID-19 lungs, which we identify as putative tuft-like cells (Extended Data Fig. 10a–e). Tuft cells are involved in airway inflammation and intestinal tissue regeneration29, but their role in viral pneumonia remains unclear. The numbers of tuft cells (CHAT+ or POU2F3+) were increased threefold in the upper airways of individuals with COVID-19, and they were ectopically present in the lung parenchyma of COVID-19 but not control lungs (Extended Data Fig. 10f–k). To begin to elucidate a putative role of tuft cells in viral pneumonia, we infected both wild-type and Pou2f3−/− mice, which lack tuft cells, with PR8, a laboratory-adapted strain of H1N1 influenza virus (see Methods). Compared to controls, the lungs of Pou2f3−/− mice showed decreased infiltration of macrophages and decreased expression of chemotaxis genes (including Ccl3 and Ccl8) that are also involved in the recruitment of myeloid cells to the lungs of individuals who died of COVID-19 (Extended Data Figs. 9g, h, 11a–l). Although their role needs to be further examined, these ectopic tuft-like cells may contribute to the pathophysiology of COVID-19 (Supplementary Discussion).
Pathological fibroblasts and lung fibrosis
There were significantly more fibroblasts in COVID-19 lungs than in control lungs (Fig. 1d); immunohistochemistry staining for α-smooth muscle actin (α-SMA) validated this finding (Extended Data Fig. 12a–d). The degree of fibrosis (determined by a Sirius red fibrosis score, see Methods) was correlated with disease duration (Fig. 4a), indicating that lung fibrosis increases over time in COVID-19. We identified five fibroblast subtypes: alveolar (n = 4,670), adventitial (n = 3,773), pathological (n = 2,322), intermediate pathological (n = 8,779), and other (n = 1,099) (Fig. 4b, Extended Data Fig. 12e). The main driver of differences in the fibroblast cluster was the increased frequency of pathological or intermediate pathological fibroblasts (henceforth collectively referred to as pFBs) in COVID-19 lungs compared to control lungs (Fig. 4c, Extended Data Fig. 12f). pFBs strongly expressed CTHRC1, a recently described hallmark gene that defines these cells, and genes of pathological ECM3, including COL1A1 and COL3A1 (Extended Data Fig. 12e, Supplementary Table 9). pFBs are key drivers of lung fibrosis in mouse models and in patients with idiopathic pulmonary fibrosis (IPF) or scleroderma3. Their increased frequency suggests that pFBs promote rapidly evolving lung fibrosis in individuals with COVID-19.

a, Coefficient of determination (R2) of days from symptom onset to death and fibrosis score in COVID-19 samples (n = 16, see Methods). Error bands, 95% s.e. interval on the Pearson correlation. b, UMAP of fibroblast (FB) sub-populations; inset indicates group assignments. path., pathological. c, Fractions of pathological fibroblasts among all fibroblasts in control (n = 7) and COVID-19 lungs (n = 19). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test.
Given the importance of fibroblasts in remodelling of the lung ecosystem, we next investigated ligand–receptor interactions across all major cell types, including fibroblasts (see Methods). Among the enriched inferred ligand–receptor interactions across all cells were TGFβ1–TGFβ receptor 2 and BMP6–ACVR1 (Extended Data Fig. 12g–i, Supplementary Table 10), which belong to the TGFβ family and superfamily, respectively. TGFβ signalling has an important role in promoting lung fibrosis and has been implicated in fibroblast-mediated maintenance of the ADI27, which is closely related to the DATP cell state. To investigate potential therapeutic strategies directed against pFBs, we inferred protein activity from single-nucleus transcriptomes followed by comparison of pFBs with other fibroblasts. This analysis predicted that pFBs would show increased activity of JunB and JunD (Extended Data Fig. 12j, Supplementary Table 11), which induce lung fibrosis in mouse models via enhanced TGFβ and STAT3 signalling and are associated with increased production of IL-1β30. Finally, we inferred druggable targets in pFBs (see Methods) and identified MMP14 and STAT3 as potential targets to abrogate detrimental programs in pFBs (Extended Data Fig. 12j, Supplementary Table 11).
Discussion
We generated a single-cell transcriptome lung atlas of COVID-19 using short-PMI autopsy specimens and control lung samples. Our analysis provides a broad census of the cellular landscape, cell programs, and cell circuits of lethal COVID-19. The additional inference of protein activity and cell-to-cell interactions, and analysis of inflammatory cytokines across various cell types using imaging mass cytometry data, provide a granular perspective of the detrimental consequences of SARS-CoV-2 infection in the lung.
Our analyses suggest interactions among aberrantly activated monocytes/macrophages that produce IL-1β, inflammation-induced impairment of alveolar epithelial regeneration, and expansion of pathological fibroblasts that promote fibrosis and may impair regeneration (Extended Data Fig. 12f, k, Supplementary Discussion). In addition to these deleterious events, our data suggest that despite a potentially sufficient humoral immune response (Supplementary Discussion), there was an inadequate T cell response in the lungs of individuals who died of COVID-19. A recent study showed that impaired B cell function in patients with cancer who contracted COVID-19 was not associated with increased mortality31, but that lack of an adequate CD8+ T cell response (even in the presence of adequate humoral immunity) was associated with worse viral control and increased mortality31. Although our COVID-19 cohort did not include patients with cancer, these data suggest that whereas humoral immunity may be dispensable in the context of adequate T cell immunity against SARS-CoV-2, a lack of appropriate T cell responses in our patients is likely to have contributed to fatal outcomes.
Although our study provides insight into host responses to lethal SARS-CoV-2 infection, it is limited by a small sample size. However, through coordinated efforts, our work will contribute to a collection of studies, including the companion paper by T. M. Delorey et al.32, with streamlined protocols and harmonized metadata to enable integration and combined analyses, and will help to account for important co-variables. Furthermore, because our analysis is focused on lung tissue from patients who died of COVID-19, we have examined only a subset of potential disease phenotypes. Nonetheless, several observations, such as the rapid development of pulmonary fibrosis (Supplementary Discussion), are likely to be relevant for patients who survive severe COVID-19, and may inform our understanding of the long-term complications seen in these individuals33.
In conclusion, we have generated a molecular single-cell lung atlas from short-PMI tissue specimens and identified pathological circuits of lethal COVID-19. This atlas establishes an important resource for investigating host responses to SARS-CoV-2 and understanding potential long-term pulmonary sequelae resulting from COVID-19, and provides a basis for therapeutic development for severe disease.
Methods
Tissue collection
All tissue specimens from individuals with lethal COVID-19 (with SARS-CoV-2 infection confirmed by reverse transcription polymerase chain reaction (RT–PCR)) and control individuals were collected at New York Presbyterian Hospital or Columbia University Medical Center under IRB approved protocols (AAAB2667, AAAT0785, AAAS7370). Appropriate consent was obtained from patients or their next of kin. All procedures performed on patient samples were in accordance with the ethical standards of the IRB and the Helsinki Declaration and its later amendments. Samples were selected on the basis of pathological review of corresponding haematoxylin and eosin (H&E)-stained FFPE tissue slides showing pathological involvement of the selected biopsy region from donors with a post-mortem incision time of less than 10 h. The donor age was 59 to more than 89 years. Tissue samples of ~1 cm3 were snap-frozen embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek USA Inc., Torrance, CA) and stored at −80 °C until processing. For all decedents included in this study, affected lung tissues were removed, and additionally, for a subset of individuals, matching tissues from kidney and heart were collected32. Seven control lung samples were collected from patients without COVID-19. The dataset analysed and presented here focuses on lung specimens from 19 individuals who died of COVID-19 (profiled in 20 experiments) and 7 control (non-COVID-19) individuals.
Sample processing and preparation of single-nucleus suspensions
All samples were processed in a biosafety cabinet equipped to comply with Columbia University safety measures established for working with COVID-19 specimens. Samples were processed as described previously17 with the following specifications and modifications. For tissue dissociation we used Tween with salts and Tris (TST) buffer. For all wash steps we used salt and Tris (ST) buffer, and all buffers were supplemented with 40 U/ml RNase inhibitor (Thermo Fisher Scientific, Waltham, MA). All buffers were pre-chilled on ice and samples were kept on ice throughout the process to further prevent RNA degradation. In brief, a fraction of the OCT-embedded snap-frozen tissue was broken off and put into a pre-cooled 50-ml tube (Corning, NY) in a large volume of ice-cold phosphate buffered saline (PBS) and inverted until the OCT was fully dissolve. Tissue was then collected by centrifuging at 300g for 2 min at 4 °C. PBS was decanted, and the tissue was resuspended in 2 ml cold TST buffer, mechanically dissociated using fine scissors and pipettes with decreasing orifice size, and incubated on ice for 5–10 min. The TST was quenched with 8 ml ST buffer, and the suspension was filtered through a 70-μm cell strainer. The tissue/nucleus suspension was pelleted by centrifuging at 500g for 5 min at 4 °C. The supernatant was decanted, and the nuclei were resuspended in 200–1,000 μl ST buffer, filtered through a 40-μm cell strainer attached to a fluorescence-activated cell sorting (FACS) tube (Corning, NY), counted, and immediately processed for single-nucleus RNA sequencing.
Single-nucleus RNA library preparation and sequencing
Single-nucleus suspensions were counted using disposable counting chambers (Bulldog Bio, Portsmouth, NH) on a Leica DMi 1 microscope by a second investigator not involved in tissue processing. A total of 15,000–20,000 nuclei were loaded per channel on a Chromium controller using Chromium Next GEM Single Cell 3ʹ v3.1 reagents (10X Genomics, Pleasanton, CA) placed inside the bio-safety cabinet, and single-nucleus RNA-seq libraries were prepared per the manufacturer’s instructions (increasing the recommended initial cDNA amplification cycles by one to account for lower amounts of RNA from nuclei compared to whole cells). Single-nucleus RNA libraries were analysed and quantified using TapeStation D1000 screening tapes (Agilent, Santa Clara, CA) and Qubit HS DNA quantification kit (Thermo Fisher Scientific). Libraries were pooled equimolarly and quantified using quantitative PCR. Libraries were sequenced on a NovaSeq 6000 with S4 flow cell (Illumina, San Diego, CA) using paired-end, single-index sequencing with 28 cycles for read 1, 8 cycles for i7 index, and 91 cycles for read 2.
Generating single-nucleus gene expression matrices
Raw 3′ snRNA-seq data were demultiplexed using Cell Ranger (v5.0) ‘mkfastq’ followed by ‘count’ to align the sequencing reads and generate a counts matrix. Transcripts were aligned to the human GRCh38 reference genome, which was appended with the entire SARS-CoV-2 genome (severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome, GenBank MN908947.3) as an additional chromosome to the human reference genome. Subsequently, the customized ‘GRCh38_SARSCoV2’ reference genome was indexed using ‘cellranger_mkref’.
Removal of background noise in gene expression matrices
We used the ‘remove-background’ function of CellBender (v.0.2.0) to remove technical ambient RNA counts and empty droplets from the gene expression matrices34. Cell Ranger-generated ‘raw_feature_bc_matrix.h5’ files served as input for CellBender. The parameter ‘expected-cells’ was obtained from the Cell Ranger metric ‘Estimated Number of Cells’, while the parameter ‘total-droplets-included’ was set to a value between 18,000 and 24,000 to represent a point within the plateau of the barcode rank plot in all samples.
Quality control and filtering
The resulting expression matrices were processed individually in R (v.4.0.2) using Seurat (v.3.2.3)35. Filters were applied to keep nuclei with 200–7,500 genes, 400–40,000 unique molecular identifiers (UMIs), and less than 10% mitochondrial reads. In addition, Scrublet was applied to identify and remove doublets with an expected doublet rate ranging from 4 to 9.6% based on the loading rate36. Samples containing fewer than 1,000 nuclei after filtering were excluded from further analyses. Filtered gene–barcode matrices were normalized with the ‘NormalizeData’ function using ‘LogNormalize’ and the top 2,000 variable genes were identified using the ‘vst’ method in ‘FindVariableFeatures’. Gene expression matrices were scaled and centred using the ‘ScaleData’ function. Next, we performed principal component analysis (PCA) as well as UMAP using the first 30 principal components. UMAPs of individual samples were inspected before integration.
Integration of individual samples
Individual samples were integrated in Seurat using the reciprocal PCA (RPCA) pipeline to remove batch effects in large datasets. The ‘SelectIntegrationFeatures’ function was applied to choose the features ranked by the number of datasets they were detected in. Next, the ‘FindIntegrationAnchors’ function selected a set of anchors between different samples using the top 50 dimensions from the RPCA to specify the neighbour search space. Six samples were specified as a reference, including three controls (C51ctr, C52ctr, C53ctr) and three COVID-19 (L01cov, L12cov, L16cov) samples. ‘IntegrateData’ was then applied to integrate the datasets using the pre-computed anchors and the integrated dataset was scaled using ‘ScaleData’. PCA and UMAP dimension reduction based on the top 30 principal components were performed. Nearest-neighbour graphs using the top 30 dimensions of the PCA reduction were calculated and clustering was applied with a resolution of 0.8. Harmony37 was run on the PCA matrix above using default parameters with patient ID as the batch key and 10 iterations.
Cell-type identification
The main cell types were identified by manual annotation of differential gene expression (DGE) between clusters. The ‘FindAllMarkers’ function identified positive markers for each cluster with a minimal fraction of 25% and a log-transformed fold change threshold of 0.25. This initial labelling resulted in the identification of epithelial, endothelial, fibroblast, neuronal, myeloid, APC, mast, T/NK and B/plasma cell populations as well as one low-quality cluster, which we removed. Next, we split the Seurat object into subsets of the main labels and reran scaling, PCA, UMAP dimension reduction, clustering and DGE analysis on each subset. The resulting clusters were annotated manually or by using cell-type-specific single-cell signatures from respective cell atlases, and labels were added to the main object. In addition, cell cycle phases were scored in the subsets using the ‘CellCycleScoring’ function, adjusted for individual cut-offs and added to the main object. Within the myeloid subpopulation, two low-quality clusters (characterized by higher expression of mitochondrial reads) were observed and removed, leaving a total of 116,314 cells for downstream analyses (of 119,535 initial cells after QC). Signatures and canonical markers (Supplementary Table 4) to identify airway basal, club, ciliated, goblet, mucous, AT1, and AT2 cells were obtained from Travaglini et al.38. Alveolar macrophages were scored using a signature based on DGE obtained from Travaglini et al.38 and identified as AMs39 with a module score >0.15. A tuft-cell signature was obtained from Deprez et al.40. To further characterize the fibroblast population, fibroblast cells were selected using Seurat’s ‘subset’ function and reanalysed to identify the different fibroblast subtypes. The reanalysis included the standard Seurat workflow with ‘RunPCA,’ ‘FindNeighbours,’ ‘FindClusters,’ and ‘RunUMAP’ performed on the ‘integrated’ assay. The number of PCA dimensions used was 15, with a resolution parameter of 0.5. After the fibroblast cell clusters had been obtained, the DGE in each cluster was computed with ‘FindAllMarkers’ on ‘RNA’ assay (Supplementary Table 9). The fibroblast subtypes were identified by manually curating the cluster DGE with the reported literature, such as the single-cell lung atlas38, lung fibroblast atlas3, single-cell database PanglaoDB41, and Human Protein Atlas42,43,44. However, these resources were based on scRNA-seq or bulk studies. Therefore, the few reported fibroblast subtype markers were usually not specific or had low expression in snRNA-seq data. Therefore, we compared our subcluster DGE with the literature reported subtype DGE with shared high expression in snRNA-seq or scRNA-seq data. These manually curated lists of fibroblast-subtype-specific marker genes were used to identify fibroblast subtypes in our dataset (Supplementary Table 4). This procedure was used to identify alveolar fibroblasts, adventitial fibroblasts, pericytes, airway smooth muscle, vascular smooth muscle, and mesothelial fibroblasts. Cell clusters with high expression of COL1A1 and CTHRC1 were annotated as ‘pathological fibroblasts’ because they have been reported to contribute to the leading edge of fibrosis3. Clusters with lower expression of COL1A1 and CTHRC1 compared to pathological fibroblasts, but without any markers for other fibroblast subtypes in their DGE, were annotated as ‘intermediate pathological fibroblasts’. One cell cluster without distinct DGE was annotated as ‘other fibroblasts’. For visualization purposes, expression scores were plotted in UMAP embeddings or violin plots as log-normalized values (natural logarithm ln(1 + x)), and in dot plots as log-normalized values (natural logarithm ln(1 + x)) that were furthermore centred on 0 with a variance of 1 (scaled).
Cell-type frequency comparison
Unless otherwise noted, we calculated frequencies of cell types in each sample from COVID-19 and control lungs, and compared the medians of the two groups to identify differences in frequency. Significance was assessed using a Wilcoxon rank-sum test.
Module scores for feature expression
The ‘AddModuleScore’ function was applied to calculate the average expression levels of gene signatures on a single-cell level. Mouse-based signatures to identify DATPs and primed and cycling AT2 cells were obtained from Choi et al.25 and converted to human homologue genes. Three genes (CLDN4, KRT8, CDKN1A) comprised the initial DATP signature thus derived. AT1 and AT2 cells were subset from the main Seurat object and reintegrated using the Seurat standard integration with 30 dimensions and a k-neighbours filter of 60 in the ‘FindIntegrationAnchors’ function. First, all AT1 and AT2 cells were scored for the three-gene signature and cells with a module score >0.7 were preliminarily labelled as DATPs. Next, we used DGE to identify additional markers that define the DATP program. We then scored our resulting DATP signature, including 163 genes, to the AT1 and AT2 cells and labelled all cells with a module score of >0.4 as DATPs. T cell scores were obtained by using the Seurat implementation of gene set scoring with 50 bins and a control size equal to the number of genes in the set. Upregulation and downregulation programs (TRM, Tact, Tmem Texh), defined by K. S. P. Devi et al. (unpublished), were used to infer T cell phenotypes. The upregulation and downregulation signatures were scored separately, and the downregulation score was subtracted cell-wise from the upregulation score to obtain the composite score. Effect size was calculated using Cohen’s D (that is, the difference of means divided by the pooled standard deviation).
Diffusion component analysis
We applied diffusion maps as a nonlinear dimensionality reduction technique to examine the major components of variation across subsets of cells. We computed DCs using the ‘DiffusionMap’ function of the Destiny R-package (v3.3.0) with the top 30 principal components used in the k-nearest neighbours algorithm (k-NN)45. The epithelial subset consisting of airway basal, club, and goblet cells was reintegrated for the DC analysis using the Seurat standard integration with 30 dimensions and a k-neighbours filter of 50 in the ‘FindIntegrationAnchors’ function. Samples with <50 cells were excluded from reintegration, which removed a total of 10 samples (one control sample and nine COVID-19 samples). Tuft-like cells were identified as cells with DC1 values >0.015 based on an overlap with the tuft-cell signature in the diffusion trajectory that dominated the first DC.
Differential gene expression
DGE was identified by using the Seurat function ‘FindAllMarkers’ on normalized count data to identify positive (overexpressed) markers in each population. The Wilcoxon rank-sum test (two-sided) was used to identify differentially expressed genes between two groups of cells and the log-transformed fold change was set to 0.25. The parameter ‘min.pct’ was set to 0.25 to assure that genes were detected at a minimum fraction of 25% of cells in either of the populations. P values were adjusted using Bonferroni correction unless otherwise stated. Differentially expressed genes were plotted in violin plots using log-normalized expression values (natural logarithm ln(1 + x)). For heatmaps and dot plots, expression values were log-normalized (natural logarithm ln(1 + x)) and furthermore centred on 0 with a variance of 1 (scaled).
Differential expression of signature scores
To test differential expression of three immune pathway signatures (type I interferon abbreviated, inflammasome receptors, and chemotaxis, Supplementary Table 4), we obtained log-normalized expression values (ln(1 + x)) for each gene in the signatures, and summed them for each signature. We then used a two-sided Wilcoxon rank-sum test to test for differential expression of signatures in each cell type, and calculated log2(fold change).
Geneset enrichment
Geneset enrichment analyses were performed using the hypeR R-package46. The background population of genes was set to all detected genes. Geneset over-representation was determined by hypergeometric test.
B cell chain analysis
To analyse the distribution of heavy and light chains in B cells, the dataset was subset to include only B cells. For the identification of variable chain regions, we selected the highest expressed heavy and light chain gene of each cell that expressed both heavy (starting with IGHV) and light (starting with IGLV or IGKV) chain-encoding genes. Next, we identified the highest expressed constant chain region among expressed genes following the pattern ‘IGH[G, M, A, or E][number]’. The resulting pairs of heavy and light chains were visualized as a heatmap using average linkage for hierarchical clustering analysis and cross-referenced with previously described recurrently observed combinations47.
Master regulator analysis and drug target identification
The fibroblast regulatory network in this study was reverse-engineered from snRNA-seq data using the ARACNe-AP48,49 algorithm. We generated networks for each sub-cluster and integrated the networks by taking a union of the predictions of all networks. P values of Master regulator (MR)–target interactions predicted by the networks were integrated using Fisher’s method. The final fibroblast network contained predictions for 1,341 transcription factors regulating 9,770 target genes through 295,546 interactions. The relative activity of each transcription factor represented in the fibroblast network was inferred using the VIPER50,51 algorithm, available as a package through Bioconductor. Conceptually, the VIPER algorithm is similar to the master regulator inference algorithm (MARINA)49,52, which uses the MR targets inferred by the ARACNe48,49 algorithm to predict drivers of changes in cellular phenotypes. In addition to calculating the enrichment of ARACNe-predicted targets in the signature of interest, VIPER also considers the regulator mode of action, regulator–target gene interaction confidence, and the pleiotropic nature of each target gene’s regulation. Statistical significance, including P value and normalized enrichment score (NES), was estimated by comparison to a null model generated by permuting the samples uniformly at random 1,000 times. Druggable proteins with VIPER-predicted50,51,53 aberrant increases in activity were ranked by their −log10(Bonferroni adjusted P value).
Ligand–receptor interaction inference in individual samples
CellPhoneDB54 is a curated repository of ligand–receptor interactions along with their subunit architectures, integrated in a statistical framework to infer cell-type-enriched ligand–receptor interactions between cell types in single-cell or single-nucleus transcriptomics data. We used CellPhoneDB to identify ligand–receptor interactions between cell types in each individual control (n = 7) and COVID-19 (n = 19) snRNA-seq dataset. The ligand–receptor interactions were inferred in each patient separately, as by definition cell-to-cell interactions are biologically meaningful only within an individual. Moreover, separate inference also prevents spurious interactions from being inferred between patients with heterogeneous disease or health statuses. After identifying and annotating different cell types in our snRNA-seq datasets, we followed the recommended procedures for the preparation of input files for local implementation of CellPhoneDB v.2.0.054. In brief, for each individual sample, QC-filtered raw counts matrices were normalized to counts per 10,000 and metadata files were obtained from the respective cell-type annotations. CellPhoneDB analysis was performed with the ‘cellphonedb method statistical_analysis’ command with default parameters.
Cell–cell interaction differences between COVID-19 and control samples
CellPhoneDB analysis of each sample identified the number of ligand–receptor interactions between all nine major cell-types in that sample. We analysed these cell–cell interaction counts between control donors (n = 7) and individuals with COVID-19 (19 individuals, 20 samples) to identify the differences in cellular cross-talk between COVID-19 and control lungs. The median cell–cell interaction values from all the control samples formed the overall control lung cell–cell interaction counts. Similarly, the overall COVID-19 lung cell–cell interaction counts were the median from all the COVID-19 samples. The overall control and COVID-19 lung interaction counts were visualized as an interactome using the ‘igraph’ R package with circle layout, where the edge width between two cell types was proportional to the number of interactions between them and the size of a cell-type circle was proportional to its frequency in the snRNA-seq.
Differential enrichment of ligand–receptor interactions between COVID-19 and control samples
CellPhoneDB analysis of each sample identified the significantly enriched ligand–receptor interactions in that sample by computing a mean of the ligand and receptor gene expression for each ligand–receptor interaction together with a corresponding P value. To find ligand–receptor interactions that were differentially regulated between COVID and control conditions, we first identified the common interactions across all samples. In brief, we consolidated ligand–receptor expression for controls and COVID-19 separately by taking the median of ligand–receptor mean expressions from 7 control samples or 20 COVID-19 samples (from 19 donors). The minimum value of consolidated ligand–receptor expression in COVID-19 and control samples was set to 0.001 to prevent noise in low expression values from affecting the log(fold change) calculations. log2(control median expression) was subtracted from log2(COVID-19 median expression) to obtain the log2(fold change) of ligand–receptor expression in COVID-19. To compute the P value of the log2(fold change) for each interaction, we used an unpaired two-sided Wilcoxon rank-sum test for each interaction between COVID-19 and control samples. Adjusted P values were obtained using th eBenjamini–Hochberg procedure. Interactions with log2(fold change) ≥ |2| and FDR P < 0.1 were reported as the top differentially enriched interactions in COVID-19.
Tissue preparation and processing for imaging
Lung tissues (human and mouse) were fixed with 4% paraformaldehyde (PFA) at 4 °C overnight with rotation. For paraffin sections, tissues were dehydrated through a 70–100% ethanol gradient and then embedded in paraffin. For cryosections, tissues were sequentially incubated with 20% and 30% sucrose and subsequently embedded in OCT compound. We obtaind 8–10-μm-thick cryosections using a cryostat.
Microscopic imaging and quantification
Paraffin sections were dewaxed and rehydrated. Antigen retrieval was performed by high-pressure heating with a commercial antigen unmasking retrieval solution followed by blocking with 5% normal donkey serum. For immunofluorescence staining, the sections were then incubated with the primary antibodies listed in Supplementary Table 12 at 4 °C overnight. Cryosections were washed twice with PBS, and blocked with 5% normal donkey serum, followed by incubation with primary antibodies shown in Supplementary Table 12 at 4 °C overnight. Conjugated secondary antibodies (1:500) were added to the sections and incubated for 2 h at room temperature. Nucleus were stained with DAPI, and images were captured with a Zeiss LSM T-PMT confocal laser-scanning microscope (Carl Zeiss) and Zen 2012 SP1 (black edition) software (Zeiss). Immunohistochemistry for C4d was performed on a Leica Bond 3 automated staining platform. In brief, paraffin sections including both healthy control lung and COVID-19 lung tissues were treated with BOND Epitope Retrieval Solution 2 (Leica) for 20 min and they were incubated with a C4d antibody for 30 min. Immunohistochemistry signals were developed with the Bone Polymer Refine Detection kit (Leica) with treatment with post primary polymer for 20 min and DAB chromogen for 10 min. For quantification, cells were counted by a blinded investigator using tiled stitched 20× images from more than five sections per mouse and included at least three individual lobes or were from representative areas of at least three human control lungs and COVID-19 lungs. Images were processed and analysed using ZEN blue 2.3 (Zeiss) and Adobe Photoshop Creative Suite 6 (Adobe) software in a blinded fashion. DATPs were detected with co-immunostaining for pro-SPC and KRT8 or HTII-280 and CLDN4. DATP percentages were determined by counting KTR8hi pro-SPC+ cells over pro-SPC+ cells or CLDN4+ cells over HTII-280+ cells. Macrophages were quantified by counting the total number of CD45+CD64+ cells over CD45+ cells. CHAT+ tuft cells were quantified by counting the total number of CHAT+ cells over DAPI+ airway nuclei (for airway tuft cells) or per mm2 of lung parenchyma.
Multiplexed immunofluorescence
Multiplexed immunofluorescence staining of lung tissue from patients who died of COVID-19 and control individuals was performed using CD4, CD8, CD19, CD103, CD163 and granzyme B (GZMB) antibodies (Supplementary Table 12) with the Opal 7-colour IHC kit (Akoya Bioscience) on a Leica Bond RX automated stainer (Leica Biosystems). FFPE tissue sections (5 μm) were baked for 2 h at 60 °C, followed by automatic deparaffinization, rehydration, and antigen retrieval in BOND Epitope Retrieval Solution 2, pH 9 (Leica Biosystems) for 30 min at 95 °C. Immunofluorescence staining with Opal and tyramide signal amplification (TSA) were performed in six cycles. In each cycle, the tissue was incubated sequentially with a primary antibody for 30 min at room temperature, the secondary antibody conjugated to polymeric horseradish peroxidase (HRP), an Opal fluorophore in TSA buffer, and BOND Epitope Retrieval Solution 1, pH 6 (Leica Biosystems) for 20 min at 95 °C to strip the tissue-bound primary–secondary antibody complexes before the next staining cycle. After nuclear counterstaining with DAPI, slides were coverslipped with Vectrashield HardSet Antifade mounting medium (Vector Laboratories) and 12–15 areas per slide were imaged using the Vectra 3 automated multispectral microscope (Akoya/PerkinElmer) with Vectra 3.0.5 software. Regions of interest were chosen by the pathologist for multispectral imaging (MSI) at 20× magnification and spectral unmixing using the InForm v2.4.6 software (Akoya). Demultiplexed images were exported as 32-bit TIFF files for further analysis.
Multiplexed image analysis
All images were analysed and visualized using QuPath55. We used the highest resolution for all described steps. The QuPath project files and additional scripts are available at https://github.com/IzarLab/CUIMC-NYP_COVID_autopsy_lung/tree/main/code/Vectra_image_analysis. First, images were loaded, renamed and segmented using ‘WatershedCellDetection’ based on DAPI intensity with a cell expansion of 4 μm. Further parameter settings for these steps can be found in the ‘Load_and_segmentation.groovy’ script. Next, we created classes and the corresponding classifiers for each of the six markers of interest: CD4, CD19, GZMB, CD103, CD8 and CD163. The thresholds for the individual classifiers (‘ClassifyByMeasurementFunction’) were automatically calculated and adjusted for each patient on the basis of visual inspection of the mean marker expression. If no patient-specific classifier was created, the classifier with the ending ‘_04_A6.json’ was used. All classifiers can be found in the object classifiers folder as json files. Once performed for all images, the individual assignments for each single cell were exported to a CSV file for downstream analysis and boxplot visualization.
Imaging mass cytometry
Imaging mass cytometry data from post-mortem lung tissue of patients with lung infections and otherwise healthy donors was used28. The dataset comprised 237 images from 23 donors, containing 664,006 single cells for which cell-type identities were derived from the intensity of 36 markers. All analyses were conducted in Python v3.8.2 with the following programs: numpy v1.18.5, scipy v1.4.1, Tifffile 2020.6.3, Networkx v2.5, Scikit-image v0.17.2, Pingouin v0.3.7, and Scanpy v1.6.0. Single cells were labelled as positive for IL-6 or IL-1β based on their z-score of intensity using Gaussian mixture models (scikit-learn56, version 0.23.0) using model selection based on the Davies–Bouldin index57. The number of cells positive for a marker in each ablated region of interest (ROI) was normalized by its area, and mean values per disease group and cell type across all ROIs were visualized as bar charts. To assess the significance of changes across both disease groups and cell types, we used a two-sided Mann–Whitney U-test and adjusted P values with the Benjamini–Hochberg FDR adjustment using the pingouin package (version 0.3.9)58. Representative regions within the ROIs were displayed as false-colour images by normalizing the signal intensity to the unit scale after clipping the signal below and above the 3rd and 98th percentiles, respectively. Finally, a Gaussian filter with sigma of one pixel (one micrometre) was applied to the images.
Sirius red staining and fibrosis scoring
Paraffin-embedded lung sections were dewaxed, rehydrated and stained for 1.5 h with a picrosirius red solution (1.3% picric acid, 1% fast red and 1% fast green). Four or five fields at 4× magnification were taken using a polarized light filter on an Olympus IX71S1F-3 microscope with QCapture Suite Plus (v3.1.3.10) software. Images were quantified (percentage of Sirius red area/total area) using Adobe Photoshop (v 11.0). Pearson correlations between fibrosis score and days from symptom onset to death were calculated for 16 of 19 patients with COVID-19 for whom samples were available and time from symptom onset to death was reported.
αSMA immunohistochemistry
Antigen retrieval of dewaxed and rehydrated paraffin-embedded lung sections was performed with citrate pH 6, blocked with 3% BSA and incubated with anti-αSMA-FITC (Sigma, F3777) overnight at 4 °C. After incubation with a biotin-anti-FITC antibody (Abcam, ab6655), detection was performed using the Vectastatin Elite ABC-HRP kit (Vector Laboratories, SP-2001) with the DAB Peroxidase Substrate kit (Vector Laboratories, SK-4100), followed by counterstaining with haematoxylin. All reagents and dilutions are listed in Supplementary Table 12. All 7 control slides and 17 available slides from COVID-19 lungs were included in the analysis. Slides were scanned using a Leica SCN400 slide scanner with Leica Scanner Console software (v102.0.7.5) and quantified using the Leica Aperio ImageScope software (v12.4.3.5008) on at least five fields at 10× magnification.
Mice
Mouse studies were approved by the Columbia University Medical Center (CUMC) Institutional Animal Care and Use Committees (IACUC). The Pou2f3−/− mouse strain was described previously59. All mice were maintained on a C57BL/6 and 129SvEv mixed background and housed in the mouse facility at Columbia University according to institutional guidelines. The facility provides a 12-h light–dark cycle, 18–23 °C room temperature and 40–60% humidity. All animal studies used a minimum of three mice per group and sample size was based on pilot experiments and previous experience. Mice were randomized to experiments and 8–12-week-old animals of both sexes were used in equal proportions. The investigators were not blinded to allocation during experiments.
Influenza infection mouse model
A total of 260 plaque forming units (pfu) of influenza A/Puerto Rico/8/1934 H1N1 (PR8) virus (a gift from Dr. Jie Sun at Mayo Clinics, Cleveland) dissolved in 40 μl RPMI medium was pipetted onto the nostrils of anaesthetized mice, whereupon mice aspirated the fluid directly into their lungs. For all procedures, administration of the same volumes of vehicle (RPMI medium) was used as control.
Flow cytometry analysis
Fourteen days after infection, mice were euthanized and transcardially perfused with 10 ml cold PBS. The lungs were then perfused with 1 ml PBS with 2 mg/ml Dispase I and 0.5 mg/ml DNase I and incubated in 5 ml of the above buffer for digestion with gentle shaking for 60 min at room temperature. Lung lobes were removed and physically dissociated, followed by filtering through a 40-μm cell strainer. Cells were pelleted and resuspended in 1 ml lyse RBC buffer followed by incubation on ice for 5 min to remove red blood cells. After washing with FACS buffer (5% FBS, 0.2 mM EDTA in PBS), single cells were collected and immunostained with Fc blocking antibody (5 μg/ml) and a live/dead cell stain kit at room temperature for 10 min. Cells were then washed and incubated with the following antibodies for one hour: PE/cyanine7 anti-mouse CD45 (1:100), FITC anti-mouse CD64 (1:100), and APC anti-mouse F4/80 (1:100). Samples were analysed on LSR II (BD, Biosciences) with four lasers (405 nm, 488 nm, 561 nm, and 635 nm). Data were analysed using FlowJo software (Treestar).
Quantitative RT–PCR (qRT–PCR)
To quantitively measure the indicated cytokines, human lung tissue samples (three donors for both healthy and COVID-19 samples) or mouse lungs (a minimum of three mice per genotype) were individually homogenized in Trizol and total RNA was extracted using an RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s instructions. cDNA was synthesized using the Superscript-IV First-Strand Synthesis System (Invitrogen) and the gene-specific primers were mixed with cDNA templates and iTaq Universal SYBRR Green supermix (Bio-Rad). qPCR was carried out on a CFX Connect real-time PCR detection system (Bio-Rad) in a total volume of 20 μl. Three technical and biological replicates were performed. Relative fold change was determined by normalizing to Actb mRNA for mouse or to GAPDH mRNA for human. The primers for qPCR are listed in Supplementary Table 13.
Statistical analysis of imaging and qRT–PCR data
Imaging and qPCR data are presented as means with s.d. of measurements unless stated otherwise. Individual values are plotted and represent independent biological samples unless stated otherwise. Statistical differences between samples were assessed with unpaired Student’s t-test using GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA). P values below 0.05 are considered significant.
For multiplexed immunofluorescent images, cell fractions (percentage of total or percentage of parental population) were computed for each field of view individually using Excel 16.45 (Microsoft). After calculating the mean on a per sample basis, we plotted values using GraphPad Prism 9.0 (GraphPad Inc. San Diego, CA) and presented them as means with s.d. of measurements. Statistical differences between samples were assessed with unpaired Student’s t-test using GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA). P values below 0.05 are considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Processed data are available via the single-cell portal: https://singlecell.broadinstitute.org/single_cell/study/SCP1219. Processed data are also deposited in GEO with accession number GSE171524. Raw data are available on the Broad Data Use and Oversight System: https://duos.broadinstitute.org (study ID DUOS-000130). Source data are provided with this paper.
Code availability
Code is publicly available at https://github.com/IzarLab/CUIMC-NYP_COVID_autopsy_lung.
Change history
08 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-03921-5
References
Dong, Y. et al. Epidemiology of COVID-19 among children in china. Pediatrics 145, e20200702 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Tsukui, T. et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 11, 1920 (2020).
Bellani, G. et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. J. Am. Med. Assoc. 315, 788–800 (2016).
Muus, C. et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 27, 546–559 (2021).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e19 (2020).
Schulte-Schrepping, J. et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440.e23 (2020).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
Xu, G. et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 6, 73 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Puelles, V. G. et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 383, 590–592 (2020).
De Michele, S. et al. Forty postmortem examinations in COVID-19 patients. Am. J. Clin. Pathol. 154, 748–760 (2020).
Goyal, P. et al. Clinical characteristics of Covid-19 in New York City. N. Engl. J. Med. 382, 2372–2374 (2020).
Slyper, M. et al. A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. Nat. Med. 26, 792–802 (2020).
Hewitson, J. P. et al. Malat1 suppresses immunity to infection through promoting expression of Maf and IL-10 in Th cells. J. Immunol. 204, 2949–2960 (2020).
Hoeffel, G. et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678 (2015).
Doran, A. C., Yurdagul, A. Jr & Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 20, 254–267 (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Barkauskas, C. E. et al. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123, 3025–3036 (2013).
Zhang, Z. et al. Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc. Natl Acad. Sci. USA 114, 3903–3908 (2017).
Little, D. R. et al. Transcriptional control of lung alveolar type 1 cell development and maintenance by NK homeobox 2-1. Proc. Natl Acad. Sci. USA 116, 20545–20555 (2019).
Choi, J. et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27, 366–382.e7 (2020).
Kobayashi, Y. et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 22, 934–946 (2020).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Rendeiro, A. F. et al. The spatial landscape of lung pathology during COVID-19 progression. Nature https://doi.org/10.1038/s41586-021-03475-6 (2021).
Westphalen, C. B. et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 124, 1283–1295 (2014).
Cui, L. et al. Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat. Commun. 11, 2795 (2020).
Bange, E. M. et al. CD8 T cells compensate for impaired humoral immunity in COVID-19 patients with hematologic cancer. Preprint at https://doi.org/10.21203/rs.3.rs-162289/v1 (2021).
Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature https://doi.org/10.1038/s41586-021-03570-8 (2021).
Nalbandian, A. et al. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615 (2021).
Fleming, S. J., Marioni, J. C. & Babadi, M. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. Preprint at https://doi.org/10.1101/791699 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Mould, K. J., Jackson, N. D., Henson, P. M., Seibold, M. & Janssen, W. J. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 4, 126556 (2019).
Deprez, M. et al. A single-cell atlas of the human healthy airways. Am. J. Respir. Crit. Care Med. 202, 1636–1645 (2020).
Franzén, O., Gan, L. M. & Björkegren, J. L. M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, baz046 (2019).
Uhlén, M. et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016).
Federico, A. & Monti, S. hypeR: an R package for geneset enrichment workflows. Bioinformatics 36, 1307–1308 (2020).
Yuan, M. et al. Structural basis of a shared antibody response to SARS-CoV-2. Science 369, 1119–1123 (2020).
Basso, K. et al. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 37, 382–390 (2005).
Lachmann, A., Giorgi, F. M., Lopez, G. & Califano, A. ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235 (2016).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
Ding, H. et al. Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun. 9, 1471 (2018).
Lefebvre, C. et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 6, 377 (2010).
Alvarez, M. J. et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 50, 979–989 (2018).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protocols 15, 1484–1506 (2020).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Davies, D. L. & Bouldin, D. W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1, 224–227 (1979).
Vallat, R. Pingouin: statistics in Python. J. Open Source Softw. 3, 1026 (2018).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Acknowledgements
We are grateful to all donors and their families. This work is part of the Human Cell Atlas (www.humancellatlas.org/publications). We thank J. Bhattacharya, I. Tabas, A. Tall and S. Roth for discussions. B.I. is supported by National Institute of Health (NIH) National Cancer Institute (NCI) grants K08CA222663, R37CA258829 and U54CA225088, a FastGrant, the Burroughs Wellcome Fund Career Award for Medical Scientists and the Louis V. Gerstner, Jr. Scholars Program. J.Q. is supported by R01HL152293 and R01HL132996. H. Huang is supported by the Department of Defense (DoD) Discovery Award W81XWH-21-1-0196. A.R. is supported by an NCI T32CA203702 grant. O.E. is supported by Volastra, Janssen and Eli Lilly research grants, NIH grants UL1TR002384, R01CA194547, and Leukemia and Lymphoma Society SCOR 7012-16, SCOR 7021-20 and SCOR 180078-02 grants. R.E.S. is supported by NIH grants NCI R01CA234614, NIAID R01AI107301, NIDDK R01DK121072 and RO3DK117252, and is an Irma Hirschl Trust Research Award Scholar. D.S. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRQ-03-20). This research was funded in part through the NIH Support Grant S10RR027050 for flow cytometry analysis and the NIH/NCI Cancer Center Support Grant P30CA013696 at Columbia University Genetically Modified Mouse Model Shared Resource, Molecular Pathology Shared Resource and its Tissue Bank.
Author information
Authors and Affiliations
Contributions
B.I. provided overall supervision. J.C.M., H. Huang, J.Q. and B.I. conceived this project. J.C.M., H. Huang, A.D.A., A.F., Y.F., H.R., M.G.C., Y.B., X.V.G., M.R., S.W.C., P.H., A.E.K. and A.S.H performed experiments. J.C.M., J.B., H. Huang, Y.W., A.N., S.T., A.F.R., D.S., C.J.F., A.D.A., A.M.L. and G.A.A. performed analyses. I.K., A.B., J.H.L., C.M., S.M.L., A.D.P., E.Z., G.S.M., A.S. and H. Hibshoosh oversaw and performed tissue collection, and performed pathological review of tissues. E.J.T. facilitated rapid autopsy specimen collection. D.T.M., M.F.B., N.A., M.S.-F., S.F.B., R.E.S. and O.E. provided signatures, materials and data. R.F.S., R.E.S., O.E. and J.Q. performed coordination of specific analyses and experiments. R.F.S. oversaw fibroblast experiments and analyses. R.E.S. and O.E. oversaw tissue mass cytometry analysis. J.Q. oversaw in vivo studies. D.S., C.J.F. and A.M.L. contributed equally. J.C.M., H. Huang, A.D.A., J.Q. and B.I. wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.I. is a consultant for Merck and Volastra Therapeutics. O.E. is a scientific advisor and equity holder in Freenome, Owkin, Volastra Therapeutics and OneThree Biotech. R.E.S. is a member of the scientific advisory board of Miromatrix Inc. and is a speaker and consultant for Alnylam Inc. D.T.M. is a consultant for LASE Innovation, Inc. S.F.B. owns equity in, receives compensation from, and serves as a consultant for and on the Scientific Advisory Board and Board of Directors of Volastra Therapeutics Inc. The other authors declare no competing interests.
Additional information
Peer review information Nature thanks Christopher Mason, Michael Matthay and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Patient information and alternative batch correction.
a, Basic demographics of patients with COVID-19 and control donors. *Decedents with concurrently profiled heart and/or kidney tissue in companion study32. †Decedent with two independent lung specimens profiled. b, Effect of PMI on clustering. c, Cell-type labels overlaid on UMAP embedding resulting from the batch-corrected PCA matrix using Harmony (see Methods). d, Same embedding as in c with annotation of COVID-19 and control groups.
Extended Data Fig. 2 Changes in cellular composition.
a, Fraction of cell types in COVID-19 and control lungs across all cells (intermediate granularity). b, Fraction of cell types in COVID-19 and control lungs among non-immune cells only. c, Fraction of cell types in COVID-19 and control lungs among immune cells only. Control, n = 7 donors; COVID-19, n = 19 donors examined over 20 experiments. Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test.
Extended Data Fig. 3 Effect of sex on cellular composition and host receptor expression.
a, b, Cell fractions in female and male individuals for control (a; n = 7 donors) and COVID-19 lungs (b; n = 19 donors examined over 20 experiments). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test. c, d, log-normalized and scaled expression (see Methods) of selected receptors or putative receptors and proteases or putative proteases involved in SARS-CoV-2 entry in different cell types in control samples from female and male donors. Dot size indicates fraction of cells and colour indicates expression level. e, f, As in c, d for from COVID-19 lungs.
Extended Data Fig. 4 Global changes in myeloid cells.
a, Quantification of cells with CD163+ staining as percentage of all cells in a subset of control and COVID-19 samples (n = 4 donors per group). Mean ± s.d., t-test. b, c, UMAP embedding with myeloid cell-type assignment (b) and group assignment (c). d–f, Expression scores (log-normalized) for monocyte, macrophage and alveolar macrophage signatures in same UMAP embedding as b, c. g, First three DCs with annotation of control and COVID-19 lung samples. h, First three DCs with expression of the alveolar macrophage signature. i, Heatmap of top differentially regulated genes among indicated myeloid sub-populations. Left bar indicates genes that were differentially regulated in the respective cell types. Top lanes indicate cell type and group. Rows indicate log-normalized and scaled expression of genes (see Methods).
Extended Data Fig. 5 Differential gene expression in alveolar macrophages.
a, Heatmap of top differentially regulated genes (log-normalized and centred, see Methods) among indicated alveolar macrophages in COVID-19 and control samples. Top lane indicates cell type and group. Rows indicate expression of genes. b, Violin plot of AXL expression (log-normalized) in alveolar macrophages from controls and COVID-19 tissues. Wilcoxon rank-sum test with Bonferroni adjusted P value indicated on top. c, Expression of AXL (log-normalized) among major cell types. Expression of this gene was nearly exclusive to fibroblasts and myeloid and epithelial cells.
Extended Data Fig. 6 Inferred immunoglobulins in plasma cells.
a, b, UMAP embedding of cells within the B/plasma cell cluster (a) and corresponding group assignment (b). c, Selected genes that define cells within the B/plasma cell cluster. Dot size indicates fraction of cells and colour indicates log-normalized and scaled expression level (see Methods). d, Heatmap illustrating the number of cells with combinations of variable heavy (x-axis) and light (y-axis) chains recovered in plasma cells across all patients. Average linkage was used for hierarchical clustering analysis. The colour of each square indicates the number of cells detected for each specific pair (colour key). e, As in d, but indicating the number of control samples with each combination detected (Supplementary Table 6). f, As in e, but indicating isotype usage in control donors alone (Supplementary Table 6). g, As in e, but demonstrating isotype usage in patients with COVID-19 (corresponding to Fig. 3e, f; shown are the top 20 commbinations; complete list in Supplementary Table 6). h, Frequency (y-axis) of variable heavy chains (x-axis) in COVID-19 and control samples. i, As in h, but for variable light chain usage. j, Frequency (y-axis) of variable heavy chains (x-axis) on a per-donor basis. k, As in j, but for variable light chain usage. l, Exemplary H&E-stained image (n = 19 donors evaluated) with coloured outlines indicating different immune cell types. Scale bar, 100 μm. m, C4d immunohistochemistry in representative control (left) and COVID-19 (right) samples (n = 6 donors per group). Scale bar, 100 μm.
Extended Data Fig. 7 Activation, residency and dysfunction cell states in T cells.
a, Expression of selected genes in cells of the T/NK cell compartment. Dot size indicates fraction of cells and colour indicates expression level. b, Quantification of cells with CD4+ staining as percentage of all cells (y-axis) in control and COVID-19 lungs (n = 4 donors per group). c, As in b, but for CD8+ T cells. Mean ± s.d., t-test. d–g, Expression of different program scores (tissue residency memory program, activation score, memory score and exhaustion score, all from K. S. P. Devi et al. (unpublished); see Methods) in CD4+ T cells (left) and CD8+ T cells (right) among control donors and individuals with COVID-19. Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test. Cohen’s D is indicated between the whiskers for each comparison (COVID-19 versus control). h, Quantification of CD4+GZMB+ T cells as percentage of CD4+ T cells (y-axis) in control and COVID-19 lungs (n = 4 donors per group). i, As in h, but for CD8+ T cells. Mean ± s.d., t-test. j, k, Representative multiplexed immunofluorescence of lung tissue from a patient with COVID-19 with a pure myeloid infiltrate (j) or with a mixed myeloid and lymphoid infiltrate (k; n = 4 donors per group). Scale bars, 200 μm.
Extended Data Fig. 8 DATPs and lung regeneration.
a, Expression of selected, previously established cell-type-specific signatures (y-axis) in cell types defined in this study (x-axis). Dot size indicates fraction of cells and colour indicates expression level. b, c, Expression of selected genes (y-axis) in different cell types (x-axis), highlighting high expression of B2M in cycling epithelial cells (b) and collagen genes in ECMhigh epithelial cells (c). d, Fraction of KI67+ cells among pro-SPC+ cells in structurally preserved versus damaged areas (n = 3 distinct areas each) from a COVID-19 lung. Mean ± s.d., t-test. e–g, UMAP embedding of alveolar epithelium and expression of selected genes that define the DATP signature. h, Composite expression of the three-gene DATP signature. i, Expression of the refined DATP signature (see Methods). j–n, First three DCs showing group assignment (j), cell or cell-state assignment (k), expression of AT2 signature (l), AT1 signature (m; log-normalized, see Methods), and effect of PMI (n). o, Gene set enrichment analysis in DATPs (compared to AT1 and AT2 cells). Rows indicate pathways in descending order of enrichment or significance (see key); x-axis indicates FDR. p, Inference of G2/M and S phase of individual DATPs (dots) (see Methods). q, Representative immunofluorescence staining (DATP marker CLDN4 and AT2 cell marker HTII-280) in control and COVID-19 lung tissue sections. Dashed boxes indicate areas highlighted to the right of each image. Scale bar, 50 μm. r, s, Quantification of KRT8+ (r) and CLDN4+ (s) cells in a subset of tissue sections from control and COVID-19 lungs. Mean ± s.d., t-test. q–s, Control, n = 3 donors; COVID-19, n = 4 donors. t, Coefficient of determination (R2) of days from symptom onset to death and AT2/AT1 ratio. Error bands, 95% standard error interval on the Pearson correlation (n = 18 donors).
Extended Data Fig. 9 Cellular sources of inflammatory cytokines.
a, Average frequency of cell types expressing IL-1β across healthy and disease conditions. b, Quantification of IL-1β across cell types in healthy and disease conditions. Each dot represents a single region of interest (ROI). c, Quantification of IL-1β across healthy and disease conditions and cell types, including separation of patients with early death (within 14 days of onset of COVID-19 symptoms) and late death (within 30 days of onset of COVID-19 symptoms). d, Average frequency of cell types expressing IL-6 across healthy and disease conditions. e, Quantification of IL-6 across cell types in healthy and disease conditions. Each dot represents a single region of interest (ROI). f, Quantification of IL-6 across across healthy and disease conditions and cell types, including separation of patients with early death (within 14 days of onset of COVID-19 symptoms) and late death (within 30 days of onset of COVID-19 symptoms). g, Expression of selected manually curated gene sets of chemotaxis, inflammasome receptors and type I interferon (response) genes across different cell types (y-axis). Dot size indicates significance and colour indicates expression level (log2(fold change)). h, qRT–PCR comparing IFNA1, IFNA2, IFNB1, and IL-6 mRNA levels in COVID-19 and control lungs (n = 3 donors for each group). Mean ± s.d., t-test.
Extended Data Fig. 10 Identification of ectopic tuft-like cells.
a–c, First three DCs of airway epithelial cells with group annotation with cell-type assignment (a), group assignment (b) and indicating expression of tuft cell signature (c) in the same projections. d, Expression of previously established signatures identifying cell types in cell types assigned in this study. Dot size indicates fraction of cells and colour indicates expression level (log-normalized and scaled, see Methods). e, Expression of selected cell-type-specific signatures of airway and alveolar epithelium from previous studies in cells identified as tuft-like cells in this study. Signatures in descending order of enrichment or significance. Colour indicates significance. f, g, Representative immunofluorescence staining of control lungs (f; two areas) and COVID-19 (g; airway and parenchyma) for KRT5 and CHAT. Arrows indicate CHAT+ cells. Scale bar, 50 μm. h, Quantification of CHAT+ cells in the upper airway epithelium of control and COVID-19 lungs. Mean ± s.d., t-test. i, Quantification of CHAT+ cells in the alveolar epithelium of control and COVID-19 lungs. Mean ± s.d., t-test. j, k, Immunofluorescence staining for KRT5 and POU2F3 of control lungs (j) and COVID-19 lungs (k), including upper airway (left) and parenchyma (right). White arrows indicate POU2F3+ cells. Scale bars, 50 μm. f–k, n = 3 donors per group.
Extended Data Fig. 11 Role of tuft cells in macrophage infiltration in mouse viral pneumonia model.
a, Immunofluorescence staining for SCGB1A1 and DCLK1 of proximal (left) and distal (right) airway from wild-type (WT) mice at baseline. n = 3 mice per group. Arrow, DCLK1+ cell. Scale bar, 50 μm. b, As in a, but in wild-type (left) and Pou2f3−/− mice 14 days after infection with H1N1 (PR8). c, Quantification of tuft cells as percentage of DCLK1+ cells in Pou2f3−/− compared to wild-type mice. Mean ± s.d., t-test. b, c, n = 4 mice per group. d, Immunofluorescence staining for CD45 and CD64 of lung parenchyma from wild-type (left) and Pou2f3−/− (right) mice 14 days after infection with H1N1 (PR8). Arrows indicate CD45+CD64+ macrophages. Scale bar, 50 μm. e, Quantification (CD45+CD64+ cells among CD45+ cells) as percentage in Pou2f3−/− mice compared to wild-type mice 14 days after infection with H1N1. Mean ± s.d., t-test. d, e, n = 3 mice per group. f, Gating strategy to identify CD45+CD64+F4/80+ cells. g, Identification of CD64+F4/80+ cells (based on gating strategy in f) in wild-type (left) and Pou2f3−/− mice (right) 14 days after infection with H1N1. h, Quantification of flow-cytometric determination of CD45+CD64+F4/80+ cells as percentage of CD45+ cells in Pou2f3−/− relative to wild-type mice (n = 3 per group). Mean ± s.d., t-test. i, qRT–PCR comparing relative mRNA expression of indicated chemokines and cytokines in Pou2f3−/− and wild-type mice 14 days after infection with H1N1 (n = 3 per group). Mean ± s.d., t-test. j, As in i, but 44 days after infection with H1N1 (n = 3 per group). k, Exemplary immunofluorescence staining (n = 3 mice per group) for KRT5 and DCLK1 in wild-type mouse 90 days after infection. Arrows indicate DCLK1+ cells. Scale bar, 50 μm. l, As in i, j, but comparing expression of indicated chemokines and cytokines in control donors and patients with COVID-19 (n = 3 donors per group). Mean ± s.d., t-test.
Extended Data Fig. 12 Role of fibroblasts, potential drug targets and model of lethal COVID-19.
a, b, Exemplary αSMA immunohistochemical staining of tissue from control (a; sample C56; n = 7 donors) and COVID-19 samples (b; samples L05cov and L06cov; n = 17 donors). Scale bars, 500 μm. c, Percentage of α-SMA+ cells per total area (n as in a, b). Mean ± s.d., t-test. d, Exemplary Sirius red staining of control (left, n as in a) and COVID-19 (right, n as in b) samples. Scale bar, 600 μm. e, Detailed annotation of fibroblasts in this study and selected marker genes. Dot size indicates fraction of cells and colour indicates expression level (log-normalized and scaled). f, Fractions of cell types among all cells in COVID-19 (n = 19 donors examined over 20 experiments) and control lungs (n = 7 donors). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × IQR. Wilcoxon rank-sum test. g, h, Inferred cell-to-cell interactions among major cell types (indicated as circles connected by lines) in control (g) and COVID-19 (h) lung samples. The size of the circle corresponds to the frequency of the respective cell type and the thickness of the lines connecting circles indicates the absolute number of interactions. i, Differential enrichment (COVID-19 versus control samples) of specific ligand–receptor interactions (rows) between two different cell types (columns). Dot colour indicates log2(fold change) of inferred ligand–receptor expression in COVID-19 compared to control lungs (unpaired two-sided Wilcoxon rank-sum test); dot size is inversely correlated with Benjamini–Hochberg adjusted P (see Methods). j, Inferred protein activity (rows) among cells corresponding to pathological fibroblasts, intermediate pathological fibroblasts, and non-pathological fibroblasts (columns). Proteins with high activity in pathological fibroblasts are highlighted. k, Model summarizing potential mechanisms that contribute to morbidity and mortality in patients with COVID-19, focusing on impaired cellular regeneration and rapidly ensuing fibrosis.
Supplementary information
Supplementary Information
This file contains full legends for Supplementary Tables 1-13, a Supplementary Discussion and Supplementary References.
Supplementary Information
This file contains an overview of Supplementary Tables 1-13.
Supplementary Table 1
Clinical information of COVID-19 and control donors – see Supplementary Information document for full description.
Supplementary Table 2
Cell type assignment – see Supplementary Information document for full description.
Supplementary Table 3
SARS-CoV-2 reads in L19cov – see Supplementary Information document for full description.
Supplementary Table 4
Gene signatures – see Supplementary Information document for full description.
Supplementary Table 5
Macrophages differential gene expression – see Supplementary Information document for full description.
Supplementary Table 6
Immunoglobulin (IGHV/IGLV usage) – see Supplementary Information document for full description.
Supplementary Table 7
AT cells differential gene expression – see Supplementary Information document for full description.
Supplementary Table 8
Differential gene expression and signature DATP – see Supplementary Information document for full description.
Supplementary Table 9
Fibroblast cluster marker annotation and differential gene expression – see Supplementary Information document for full description.
Supplementary Table 10
Top ligand-receptor interactions – see Supplementary Information document for full description.
Supplementary Table 11
Inferred protein activity and oncotarget – see Supplementary Information document for full description.
Supplementary Table 12
List of antibodies, dilutions, and associated reagents used for immunofluorescent, immunohistochemistry, FACS, and multiplexed immunofluorescence experiments.
Supplementary Table 13
A list of qRT-PCR Primers for human and mouse tagets assessed by qRT-PCR.
Source data
Rights and permissions
About this article

Cite this article
Melms, J.C., Biermann, J., Huang, H. et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021). https://doi.org/10.1038/s41586-021-03569-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03569-1
This article is cited by
-
Organoids in virology
npj Viruses (2024)
-
Single-cell division tracing and transcriptomics reveal cell types and differentiation paths in the regenerating lung
Nature Communications (2024)
-
Dictionary learning for integrative, multimodal and scalable single-cell analysis
Nature Biotechnology (2024)
-
A single-cell atlas of lung homeostasis reveals dynamic changes during development and aging
Communications Biology (2024)
-
Severe pediatric COVID-19: a review from the clinical and immunopathophysiological perspectives
World Journal of Pediatrics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
