
Abstract
Malaria fever has been pervasive for quite a while in tropical developing regions causing high morbidity and mortality. The causal organism is a protozoan parasite of genus Plasmodium which spreads to the human host by the bite of hitherto infected female Anopheles mosquito. In the course of biting, a salivary protein of Anopheles helps in blood feeding behavior and having the ability to elicit the host immune response. This study represents a series of immunoinformatics approaches to design multi-epitope subunit vaccine using Anopheles mosquito salivary proteins. Designed subunit vaccine was evaluated for its immunogenicity, allergenicity and physiochemical parameters. To enhance the stability of vaccine protein, disulfide engineering was performed in a region of high mobility. Codon adaptation and in silico cloning was also performed to ensure the higher expression of designed subunit vaccine in E. coli K12 expression system. Finally, molecular docking and simulation study was performed for the vaccine protein and TLR-4 receptor, to determine the binding free energy and complex stability. Moreover, the designed subunit vaccine was found to induce anti-salivary immunity which may have the ability to prevent the entry of Plasmodium sporozoites into the human host.
Similar content being viewed by others


Introduction
Malaria still remains one of the most devastating and deadly infectious disease, which is characterized by the intermittent high fevers and it’s another form namely cerebral malaria, leads to the neurological complications such as brain injury and coma1,2. As per the World Health Organization (WHO) latest estimate, almost 212 million cases of malaria along with 42,900 deaths were reported in December 2016 among 91 countries, worldwide3. The high risk of developing malarial infection among a population group depends upon the several factors including the presence of children under the age of 5 years4, a patient with HIV co-infection4, pregnant woman4, mobile population4,5, and travelers6. The causal organism of malaria is a protozoan parasite belongs to the genus Plasmodium and it spreads by the bite of hitherto infected female Anopheles mosquito7. There are mainly four species of Plasmodium namely P. falciparum, P. ovale, P. malariae and P. vivax that are responsible for the disastrous diseased condition among human being. Recently, P. knowlesi was considered as the fifth species with the ability to cause human malaria infection in Southeast Asian countries mainly Malaysia8. If we talk about the severity of infection and distribution of disease, P. falciparum is the most severe form of a parasite than other leads to most of the deaths whereas P. vivax is the most widely distributed human parasite outside the sub-Saharan region of Africa and cause huge morbidity. The life cycle of malaria parasite consisting of four stages namely liver stage, blood stage, and transmission stage in human being while the last one is mosquito stage and keeping in mind the end goal to destroy the ailment, each stage ought to be considered for treatment9. Artemisinin combination therapy is the first-line treatment for P. falciparum infection in the region where chloroquine resistance has been evolved. Along with the fast onset of action, artemisinin and its derivatives have rapid clearance from the human body, which needs the combination of the slow-clearing drug to increase the drug efficacy and kills the remaining parasites. The most widely used combination therapies are Artemether-lumefantrine (commercial name: Coartem) and amodiaquine-artesunate (commercial name: Coarsucam)10. While the recently approved combination therapies are artesunate-pyronaridine (commercial name: Pyramax) and dihydroartemisinin-piperaquine (Euartesim)10. The recent emerging resistance against artemisinin urges to develop some new strategy to prevent the malaria diseases condition11. Therefore, in this study, we applied a novel immunoinformatics approach to design multi-epitope based subunit vaccine that may prevent the disease by maintaining the host hemostasis by the inhibition of anticoagulant and anti-inflammatory proteins present in mosquito saliva. It will also inhibit the entry of parasite within the host body by a similar mechanism. Apart from this, if any way parasite enters into the host body, vaccine candidate will stop the salivary protein-mediated induction of parasitic growth.
Among different anopheline vectors, Anopheles stephensi is a sub-tropical species that most abundantly present in the Indian subcontinent and also distributed across the Middle East and South Asia region12. A. stephensi transmit the malarial infection by injecting various Plasmodium species to the human host typically via bites. Salivary gland of mosquitos implement various functions for the survival of the vector and complement their feeding behavior by producing a large array of a biochemically active molecule that has immunomodulatory, anti-coagulant and anti-inflammatory properties that disable the host hemostatic response for successful blood feeding13,14. Salivary proteins are antigenic and immunogenic in nature which helps the infectivity of parasite14,15. D7 protein and salivary apyrase are two different salivary proteins that help in binding, and inhibition of the platelets aggregation, respectively. Salivary peroxidase helps in heme binding and peroxidase activity while Putative TIL domain polypeptide functions as trypsin inhibitor16. Recently, it was reported that hamadarin is a 16 kDa protein present in Anopheles stephensi saliva which inhibits the activation of plasma contact system17 and ultimately blood coagulation. Another protein from Anopheles gambie saliva namely gSG6 plays essential blood feeding18,19. Recently, Vijay S. et al. reported that salivary proteins might be utilized to develop novel antimalarial control strategies via innate immune protection against malaria16. These proteins could likewise evoke a host IgG response in natural conditions20,21. Mosquito salivary gland surface (SGS) proteins are the prevalent immunogenic component present in saliva having ability to induce immunogenic responses22.
This is the reason why we chose the A. stephensi salivary proteins from the National Center for Biotechnology Information (NCBI) and subjected to design multi-epitope subunit vaccine. Allergenicity, antigenicity and physiochemical properties were also obtained for the vaccine protein. Moreover, tertiary structure prediction followed by refinement was performed to get a refined 3D model having a higher number of residues in the favored region of Ramachandran plot. Molecular docking and molecular dynamics simulation of vaccine constructs with TLR4 were also performed to check the binding energy and complex stability. Finally, disulfide engineering and in silico cloning was performed to increase the stability of vaccine construct and ensuring its effective expression in the microbial system, respectively. This study finally represents a novel approach to develop malaria vaccine using salivary protein instead of parasitic protein, which could be helpful to prevent the Plasmodium infection to human host.
Results and Discussion
Sequence retrieval of salivary protein and assurance of antigenic conduct
In order to design an immunogenic multi-epitope subunit vaccine, the sum of 33 A. stephensi salivary protein sequences was retrieved from the NCBI protein database. Major proteins name is salivary lysozyme, a salivary protein precursor, salivary galectin, salivary lipase, anti-thrombin anopheline, salivary protein SG3, salivary apyrase, salivary secreted serine protease inhibitor, salivary defensin and salivary cecropin. Among 33 salivary protein sequences, only 14 proteins were found to be antigenic as predicted by ANTIGENpro. These 14 sequences were selected based on their score obtained for the probability of antigenicity and all these proteins having a score of ≥0823. Obtained score for antigenicity probability clearly denoting the antigenic nature of selected protein sequences which can be used for the subunit vaccine designing24.
CTL epitope prediction and immunogenicity assessment
Cytotoxic T-lymphocytes are a CD8+ subset of T-cell responses to kill those target cells having intracellular viral, bacterial or protozoan infection25. During infection, whenever they encounter to the MHC-I mounted antigen specific to their receptor, they enter the cell cycle and perform several mitotic divisions followed differentiation into the effector cells26. Here, we tried to predict the CTL receptor specific immunogenic epitopes using the NetCTL 1.2 server and total 83 CTL epitopes of 9mer length were obtained for the input of 14 salivary protein sequences27. In the next step, the immunogenicity of epitopes was determined and as per the instruction of IEDB28, higher score indicate greater probability to elicit an immune response; therefore total 21 CTL epitopes with high immunogenicity score were selected and subjected to the vaccine designing (Table 1).
HTL epitope prediction
Helper T-lymphocyte is the key player of both humoral and cell-mediated immune response29. Therefore, HTL receptor specific epitopes are probably going to be a crucial part of the prophylactic and immunotherapeutic vaccine30. All 14 salivary protein sequences were subjected to IEDB MHC-II epitope prediction module and 8751 epitopes of 15mer length were obtained. In order to become highest immunogenic epitopes, they must have a lower percentile rank and IC50 value24. Only 14 epitopes with lowest percentile rank ranging from 0.03–0.3 were selected for the vaccine designing (Table 2). Their IC50 value ranging from 368–959 denoting that out of 14 epitopes with lowest percentile rank, 7 epitopes have intermediate affinity while remaining to have low affinity for the HTL epitopes. On the other side, all 14 epitopes were found to have IFN-γ inducing capability that was obtained from their positive score on the IFNepitope server output23,31 (Supplementary Table 1). All these 14 epitopes were used for the vaccine construction.
Construction of multi-epitope subunit vaccine
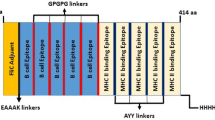
A final vaccine construct of 541 amino acid residues was designed using 21 CTL and 14 HTL epitopes as described elsewhere23,24 (Supplementary Figure 1). In order to attain maximum immune response TLR-4 agonist (RS09) was used as an adjuvant at the N-terminal site of the vaccine construct32. Each joint was occupied by the suitable linkers as described by Nezafat et al.32, for example, adjuvant and CTL epitopes were combined together by EAAAK linker, intra-CTL and intra-HTL epitopes joint by AAY and GPGPG linker, respectively. Finally, vaccine construct was obtained having adjuvant, linker, CTL, and HTL epitopes in a sequence moving from N-terminal to C-terminal. As this designed subunit vaccine consisting of immunogenic CTL and TTL epitopes along with suitable adjuvant and linker, it may have the ability to inhibit the entry of malaria parasite within the human host body23,24.
B-cell epitope mapping
B-cells are a key player of humoral immunity. An epitope corresponding to the B-cell receptor plays an important role in vaccine design following antibody production33. Therefore, BCPREDS server was used to reliably predict the linear B-cell epitopes where BCPRED was the selected prediction method23. Total 14 B-cell epitopes of 20mer length were predicted among the primary input sequence of final vaccine construct. Among them, only 11 epitopes were selected and finalized because of their high score of 1.0 (Fig. 1A). Due to the selection of highest scoring B-cell epitope among designed subunit vaccine, our vaccine may have the ability to enhance humoral immunity as well as cell mediated immunity23. Discontinuous epitopes of 60 amino acids long were also predicted from the final 3D model of vaccine construct with the probability scoring of 0.791 (Fig. 1B). The obtained probability score also confirming the immunogenic behavior of the designed subunit vaccine34.

Humoral epitope predictions for subunit vaccine. (A) Showing the linear B-cell epitopes (red color) among the 3D structure of final vaccine construct (golden color). (B) Conformational B-cell epitopes (magenta color) showing the sequence subunits composed of antigenic epitopes that will come in direct contact with immune receptor.
Antigenicity and allergenicity prediction of designed vaccine
A vaccine given to human host must be immunogenic in nature and capable to trigger significant humoral immune response which ultimately leads to the memory cell formation against the pathogenic epitopes. The antigenicity of designed vaccine construct was determined by using an alignment-free ANTIGENpro server and found that it has the antigenicity probability of 0.86, which represent the antigenic nature of vaccine construct24. The antigenicity score obtained for this vaccine construct is comparable with the antigenicity of subunit vaccine reported elsewhere24.
Allergy is an overreaction by our immune system to the previously encountered, ordinarily harmless substance that results in sneezing, wheezing, skin rash, and swelling of the mucous membrane35. Allergenicity of predicted vaccine construct was determined using AllerTOP online server and found that the vaccine protein is nonallergic in nature and safe for the human use23,35.
Physiochemical properties assessment
The physiochemical properties of vaccine construct were characterized by using ProtParam server and evaluated for seven parameters. The molecular weight of vaccine protein was found to be 58 kDa which will favor the antigenicity of the vaccine construct23. The theoretical pI was found to be 5.61 showing its slightly acidic nature while the total numbers of negative and positive charge residues were 48 and 40, respectively24. The estimated half-life in mammalian reticulocytes was 4.4 hours, in vitro; while 20 and 10 hours in yeast and E. coli, in vivo. The extinction coefficient was found to be 119530 M-1 cm-1, at 280 nm measured in water, assuming that all cysteine residues are reduced. The score obtained for instability index was 29.56, showing the stable nature of vaccine construct. The value of the aliphatic index and Grand average of hydropathicity (GRAVY) was 71.24 and −0.276, respectively. The estimated value of aliphatic index represents the thermostable nature of designed subunit vaccine because higher the value of aliphatic index, greater will be the thermo stability24. While, negative value of GRAVY for the input subunit vaccine represents the hydrophilic nature of vaccine24. Conclusively, the designed vaccine is immunogenic, thermostable and hydrophilic in nature.
Tertiary structure prediction, refinement, and validation
The tertiary structure was predicted by using the RaptorX server and 3D model was obtained as described elsewhere24 (Fig. 2A). The best template used for the homology modeling was crystal structure of a Legionella phosphoinositide phosphatase (PDB ID: 4FYE). Total 541 amino acid residues were modeled as a single domain with 1% disorder. Secondary structure information resulting in the presence of 53% helix, 4% Beta sheet, and 41% coiled structure. P-value is a parameter of homology modeling where low P-value defines the good quality of modeled structure23. The P-value obtained for the modeled structure was 6.14e-04 which is low and significant.

Tertiary structure prediction and validation of vaccine construct. (A) Tertiary structure predicted for the primary sequence of subunit vaccine construct showing helix, sheet and coiled region. (B) Ramachandran plot formation to validate the 3D modeled structure showing 92.4% residues in the favored region.
Further, protein refinement using GalaxyRefine leads to the increase in a number of residues in the favored region24. Initially, 87% of residues were in the Rama-favored region while after refinement the number of residues in the Rama-favored region reached to 92.4%. The refinement output was also validated by plotting Ramachandran plot and found the same that 92.4% residue in Rama-favored region, 5.4% residues in allowed region and only 2.2% residues in outlier region (Fig. 2B).
Disulfide engineering for vaccine stability
In order to stabilize the modeled structure of final vaccine constructs disulfide engineering was performed using Disulfide by design v2.036 and found that there are total 63 pairs of residues that can be used for the purpose of disulfide engineering. But after evaluation on other parameters like energy and Chi3 value, only four pairs of residues were finalized because their value comes under the allowed range i.e. the value of energy should be less than 2.2 and Chi3 should be in between −87 and +97 degree37. Therefore, total 8 mutations were created at the residues pairs namely Ala95-Gln414, Tyr96-Gly422, Trp133-Glu173, and Gly135-Phe255 (Fig. 3).

Disulfide engineering to improve protein stability. Showing total 4 mutated residues pairs in magenta and gray color. These residues were selected based on their energy, chi3 value, and B-factor.
Codon adaptation and in silico cloning
The main purpose of in silico cloning was to express the vaccine protein epitope of Anopheles mosquito origin into E. coli expression system23. Therefore, it was necessary to adapt the codon respective to subunit vaccine construct as per the codon usage of E. coli expression system. We adapted the codons as per E. coli K12 strain using JCAT server and found that the GC-content of the improved sequence was 58.16% while the value of codon adaptive index was 0.97 which is near to 1.0 that was satisfactory23. Later on, XhoI and NdeI restriction sites were created and cloned into the pET28a(+) vector (Fig. 4). The target sequence in the clone is represented in blue color in between aforementioned restriction sites23. The target sequence is also enclosed between 6-histidine residues on both ends that will be helpful to the purification purposes. The total length of the clone was 6.9kbp.

In silico cloning for adapted vaccine sequence into pET28a(+) vector showing the region of choice in blue color surrounded between XhoI (158) and NdeI (1788) while the vector has shown in black lines.
Molecular docking of vaccine constructs with TLR4
Molecular docking of subunit vaccine protein and TLR-4 receptor was performed using the ClusPro 2.0 and total 30 models were generated38. Among them, only that model was selected which properly occupied the receptor and having lowest energy score and found that model number 0.00 fulfill the desired criteria that’s why selected as the best-docked complex (Fig. 5). The energy score obtained for the model 0.00 was found to be −1187 which is lowest among all other predicted docked complex showing highest binding affinity.

Docked complex of vaccine protein and TLR-4 receptor here vaccine construct is shown in the sphere form, docked within the TLR-4 receptor
Molecular dynamics simulation for Vaccine-TLR4 complex
Molecular dynamics simulation was performed using Gromacs 5.1.5 using a GROMOS9643a1 force field39. The potential energy obtained for the complex was −9.9 KJ/mol, while the value of temperature, pressure, and density was obtained as 299.77 K, 2.54 bar and 1016.4 kg/m3 (Supplementary Figure 2A–C). The radius of gyration obtained for the docked complex showing that the distance in rotating complex from the center of mass is 4.3 nanometers that decreases up to 4.25nanometers at the time duration of 10 nanoseconds (supplementary Figure 2D). The RMSD value of protein backbone was 0.4 nanometers (Fig. 6A) while RMSF score obtained for the protein side chain was found to be 0.2 nanometers (Fig. 6B). Both these scores are satisfactory showing strong complex stability23,24.

Molecular dynamics simulation output of vaccine protein-TLR-4 docked complex. (A) RMSD obtained for the complex backbone showing that initially, RMSD increased up to 8 nanoseconds after that become stable at 0.5 nanometers, while (B) showing an average RMSF of 0.2 for the side chain residues of the docked complex.
Conclusion
Malaria is severe infection characterized by the high fever with irregularity but may also lead to brain injury and coma. It affects 212 million people from 91 countries, worldwide. Lack of effective vaccine and emergence of resistance against artemisinin created a disastrous condition among the people living in the endemic zone. Therefore, it’s the need of time to search for the new options to tackle this severe problem. The vector for malaria is the Anopheles stephensi in the Indian subcontinent leads to the transfer of malaria parasite. Literature survey reveals that salivary proteins of Anopheles mosquito not only supports the pathogenesis but are also immunogenic in nature. Therefore, this study was designed to reach a step ahead in the path of vaccine development. We used the primary amino acid sequence of Anopheles mosquito salivary protein to design a subunit vaccine construct. The constructed vaccine has CTL, HTL and BCL epitopes of varying length. It has antigenic properties in the absence of allergenic properties. It was stable and having a good binding affinity for the TLR-4 receptor. Collectively, this study applied a series of immunoinformatics tools in a sequential manner to find an effective vaccine that may fight against the malaria infection. However, this study needs experimental validation to prove this computational work. The experimental work may include the synthesis of designed subunit vaccine followed by the in vitro and in vivo analysis to determine the immunogenicity and safety concern of the same.
Methodology
Anopheles stephensi salivary protein sequence retrieval and assurance of antigenic conduct
Anopheles stephensi mosquito is the vector of malaria transmission to the human being in the Indian subcontinent. While the salivary gland proteins of Anopheles mosquito was reported for their role in parasite pathogenesis and having the ability to induce IgG response in the natural host20,21. Therefore, total 33 salivary proteins of A. stephensi were obtained from the national center for biotechnology information (NCBI) protein database (Retrieval date 25/08/2017) and subjected to multi-epitope vaccine designing. As the main purpose of vaccination is to induce an immunogenic response within the host body, all the retrieved protein sequences were subjected to their antigenicity prediction using ANTIGENpro. Based on the antigenicity result, only 14 proteins were found to have an antigenic probability of ≥0.8 were selected and used in the next step.
Cytotoxic T-lymphocyte (CTL) epitope prediction and immunogenicity assessment
CD8+ cytotoxic T-lymphocyte were shown to inhibit malaria parasitic growth and development, inside the hepatocytes cells40. To get an immunogenic CTL epitopes having the ability to elicit cell-mediated immunity and form the memory cells, all 14 salivary protein sequences were subjected to the NetCTL 1.2 server41. NetCTL 1.2 is an online web server intended for predicting CTL epitopes among input protein sequences based on the training dataset. NetCTL was selected to predict the CTL epitopes because of its higher prescient execution on all execution parameters as compared to the recently developed servers namely MHC-pathway, MAPPP, and EpiJen. All the salivary protein sequences were submitted in the FASTA format to predict the CTL epitope at the threshold score of 0.75 (default). Those epitopes having a combined score of greater than 0.75 were selected as CTL epitope and further subjected to the Immune Epitope Database (IEDB) MHC class I immunogenicity prediction module. Predicted CTL epitopes for each salivary protein was used as an input sequence and the result was obtained in the form of the score, where higher score determines that greater will be the probability of eliciting an immune response.
Helper T-lymphocyte (HTL) epitope prediction
Helper T- cell response is the major part of cell-mediated immunity and helps in pathogen clearance by the help of various cytokines and immune cells42,43. They have the ability to induce both CTL and humoral immune response by the secretion of lymphokines like IL-2, IL-4, IL6, Granulocyte-macrophage colony-stimulating factor (GM-CSF), and IFNγ. In view of that, we can say that HTL epitopes mainly of Th1 type are most likely going to be a crucial part of the prophylactic and immunotherapeutic vaccine. Therefore, IEDB MHC-II epitope prediction module was used to predict the HTL epitopes for all 14 Anopheles salivary protein sequences44. The available parameters were kept default except for allele selection where the nominated alleles were H2-IAb, H2-IAd, and H2-IEd. Output epitopes were ranked based on their percentile rank score where lower percentile rank representing that greater will be the binding affinity for HTL receptor. Secondly, to prove our work that the predicted HTL epitopes will have ability to activate Th1 type immune response followed by the IFN-γ production, top 14 predicted HTL epitopes were subjected to the IFN epitope server using predict option. All 14 epitopes were submitted in the FASTA format followed by the approach selection and model of prediction. Motif and SVM hybrid was selected as the approach and IFN-gamma versus other cytokine as model of prediction.
Designing of multi-epitope subunit vaccine
So as to plan an appropriate vaccine candidate, it must have the ability to induce CTL and HTL immune response. In other words subunit vaccine must contain both CTL and HTL epitopes along with suitable linkers. Keeping in mind the end goal to effectively activate both innate and adaptive immune response, subunit vaccine must consist of a strong immunostimulatory adjuvant. In the previous decades, there is a huge headway in the adjuvant engineering, for instance, Toll-like receptor (TLR) agonists have made its contribution as a part of peptide-based subunit vaccine as a functional option for present-day immunotherapy45. Recently, Junqueira et al. have shown that CpGs oligodeoxynucleotides (CpG ODNs) and Glycoinositolphospholipids (GIPL) gotten from Trypanosome cruzi having the ability to activate TLR-4 and TLR9 leads to actuate potent pro-inflammatory reaction46. Secondly, proteo-glycolipid complex (P8GLC) derived from Leishmania parasite has shown its affinity for the TLR-4 receptor and recognized as ligand47. Moreover, TLRs having the capability to recognize the Plasmodium ligands, for example, Plasmodium falciparum primes the human TLR-4 response towards high proinflammatory cytokine profile48. Shanmugam A. et at. has reported that synthetic TLR-4 agonist namely RS-09 (Sequence: APPHALS) can be used as a novel class of adjuvant49, therefore, it was added as an adjuvant and linked with epitopes (CTL and HTL) by using EAAAK linker50. Linkers assume an imperative part in simulating the vaccine construct to work as an independent immunogen and producing higher antibody titer than that of single immunogen51. Total three linkers namely EAAAK, AAY, and GPGPG, were used to construct the final vaccine. AAY and GPGPG linkers were added at the intra-epitope position to link the CTL and HTL epitopes, respectively.
B-cell epitope prediction
B lymphocytes, a type of white blood cells, are the key player of humoral immunity by antibody production. The identification of B-cell epitopes is an essential part in vaccine designing. BCPREDS server was used to predict the linear B-cell epitopes of 20 amino acids long. The amino acid sequence of final vaccine construct was used as an input sequence in plain format followed by the selection of fixed length epitope prediction method and length of the epitope. BCPREDS (default method) was selected as the prediction method for the epitope of 20 amino acids long52. The specificity threshold was set to be by default at 75% to get the result in a user-friendly format. While conformational epitopes were predicted using ElliPro server for the input of tertiary protein structure of vaccine construct.
Antigenicity and allergenicity prediction of designed vaccine
Antigenicity determines the ability of an antigen to binds with the B- and T-cell receptor that may lead to the immune response and memory cell formation. Therefore, the antigenic nature of predicted vaccine construct was determined to ensure its ability to interact with the immune receptor. ANTIGENpro is a sequence based, pathogen independent and alignment-free prediction method that was used to check the antigenic behavior of vaccine protein. It uses SVM classifier to summarize the probable antigenic or non-antigenic nature of proteins. ANTIGENpro uses the existing protein antigenicity microarray files of eight feature sets for five pathogens to construct two-stage architecture; among them, the first one is multiple representations of the primary protein sequence and the second one is five machine learning algorithms.
Allergenicity is the potential of a material to cause sensitization and allergic reactions associated with the IgE antibody response. Therefore, the predicted vaccine construct must be free from the allergenic nature. AllerTOP v. 2.0 was used to check the allergenicity of the vaccine construct based on the method that uses auto cross-covariance (ACC) transformation of protein sequences into uniform equal-length vectors. Input protein sequence of vaccine protein was classified by the k-nearest neighbor algorithm (kNN, k = 1) which is based on the training set of 2427 known allergen from different species and 2427 nonallergen from similar species.
Physiochemical properties assessment
The main purpose of vaccination is to induce an immune response after injecting the vaccine into the body. Therefore, it is necessary to define the physical and chemical parameters associated with the vaccine. ProtParam53 web server, a part of Expert Protein Analysis System (EXPASY), was used to define various physicochemical properties of predicted vaccine construct. The primary protein sequence of the vaccine was used to predict the various parameters including molecular weight (kDa), estimated half-life, theoretical pI, aliphatic index, grand average of hydropathy (GRAVY) and so on.
Tertiary structure prediction
Protein molecule achieves maximum stability in its lowest energy state by proper bending and twisting to form a tertiary structure. It is the interaction between the amino acids side chain residue which is responsible to stabilize the protein structure. The 3-dimensional structure of predicted vaccine construct was obtained by utilizing RaptorX structure prediction server. RaptorX is a pure ab initio method that can be used to build a 3D model in a template-free manner.
Refinement of 3D vaccine model and validation
It is the degree of likeness between the target and available template structure that determines the quality of protein model structure created by contemporary protein structure prediction techniques39,54. Therefore, it was necessary to improve the template based predicted model beyond the accuracy by utilizing the template information. To fulfill this thought, output model of RaptorX server was subjected to the GalaxyRefine web server55, which is based on the CASP10 tested refinement method. GalaxyRefine performs rehashed structure perturbation followed by overall structural relaxation by performing molecular dynamics simulation.
Disulfide engineering for vaccine stability
Before proceeding to the next step, it was necessary to improve the stability of refined protein model. Disulfide bonds are covalent interactions that emulate the stabilizing molecular interaction and provide a considerable stability to protein model by confirming precise geometric conformations. Disulfide engineering is a novel approach for creating disulfide bonds into the target protein structure. Therefore, the refined model of final vaccine construct was subjected to the Disulfide by Design 2.036 to perform disulfide engineering. Initially, the refined protein model was uploaded and run for the residue pair search that can be used for the disulfide engineering purpose. Total 4 residue pairs were selected to mutate them with cysteine residue using create mutate function of the Disulfide by Design 2.0 server.
Codon adaptation and In silico cloning
Codon adaptation is a way to attain major expression rate of foreign genes in the host when the codon usage of the host differs from that of the organism where the gene stems from. Unadapted codon may lead to the minor expression rate in the host. Therefore, the primary sequence of vaccine protein was submitted to the Java Codon Adaptation Tool (JCAT) to adapt their codon usage to most sequenced prokaryotic organisms (E. coli K12)23. CAI value and GC content of the adapted sequence was also obtained. Later on, the adapted nucleotide sequence corresponding to the designed vaccine construct was cloned into the E. coli pET28a(+) vector by using the restriction cloning module of SnapGene tool24.
Molecular docking of vaccine constructs with TLR4
Molecular docking is a computational method used to predict the preferred orientation of ligand molecule to the receptor molecule in their stable complex form56. It can be also used to predict the binding affinity between these two molecules in terms of scoring function. As mentioned in the previous section, TLRs having the capability to recognize the Plasmodium ligands and P. falciparum primes the human TLR-4 response towards high proinflammatory cytokine profile48. Therefore, TLR-4 was selected as receptor and its PDB file (PDB id: 4G8A) was obtained from RCSB-Protein Data Bank while the refined model of vaccine protein was used as a ligand. Protein-protein docking was performed using the ClusPro 2.0: protein-protein docking server, to check the binding affinity between them57.
Molecular dynamics simulation for Vaccine-TLR4 complex
Molecular dynamics simulation is a widely accepted computational approach which is used to determine the stability of protein-ligand complex at the microscopic level58. The protein-protein docked complex output of ClusPro was used as an input to perform the molecular dynamics simulation using Gromacs v5.1.5. Initially, the crystal water of complex was removed followed by the topology generation using a GROMOS9643a1 force field. In the next step, protein complex was centered in a cubic boundary box and filled by water molecule using simple point charge (SPC) water model and chloride ion was used for the charge neutralization of complex. Moreover, energy minimization followed by canonical equilibration (NVT ensemble) and isothermal-isobaric (NPT ensemble) was performed for a time duration of 100 ps. Finally, molecular dynamics simulation was executed for the time duration of 10ns59. The root mean square deviation (RMDS) for backbone and root mean square fluctuation (RMSF) for side chain was determined.
References
Idro, R., Marsh, K., John, C. C. & Newton, C. R. J. Cerebral Malaria; Mechanisms Of Brain Injury And Strategies For Improved Neuro-Cognitive Outcome. Pediatr. Res. 68, 267–274 (2010).
Pandey, R. K. et al. Exploring dual inhibitory role of febrifugine analogues against Plasmodium utilizing structure-based virtual screening and molecular dynamic simulation. J. Biomol. Struct. Dyn. 35, 791–804 (2017).
World Health Organization. World Malar. Rep. 2016. 1-186; http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ (2016).
Schumacher, R. F. & Spinelli, E. Malaria in Children. J. Hematol. Infect. Dis. 4, e2012073, https://doi.org/10.4084/MJHID.2012.073 (2012).
Flannery, E. L., Chatterjee, A. K. & Winzeler, E. A. Antimalarial drug discovery - approaches and progress towards new medicines. Nat. Rev. Microbiol. 15, 572, https://doi.org/10.1038/nrmicro.2017.88 (2017).
Chaves, T. D. S. S., Monteiro, W. M., Alves, J. R., Lacerda, M. & Lopes, M. H. Pre-travel malaria chemoprophylaxis counselling in a public travel medicine clinic in São Paulo. Brazil. Malar. J. 16, 64, https://doi.org/10.1186/s12936-017-1713-3 (2017).
Biamonte, M. A., Wanner, J. & Le Roch, K. G. Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett. 23, 2829–2843 (2013).
Singh, B. et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 363, 1017–1024 (2004).
Crompton, P. D., Pierce, S. K. & Miller, L. H. Advances and challenges in malaria vaccine development. J. Clin. Invest. 120, 4168–4178 (2010).
Anthony, M. P., Burrows, J. N., Duparc, S., Moehrle, J. J. & Wells, T. N. The global pipeline of new medicines for the control and elimination of malaria. Malar. J. 11, 316, https://doi.org/10.1186/1475-2875-11-316 (2012).
O’Brien, C., Henrich, P. P., Passi, N. & Fidock, D. A. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 24, 570–577 (2011).
Dash, A., Adak, T., Raghavendra, K. & Singh, O. The biology and control of malaria vectors in India. Curr. Sci. 92, 1571–1578 (2007).
Ribeiro, J. M. & Arca, B. From sialomes to the sialoverse: an insight into salivary potion of blood-feeding insects. Adv. In. Insect. Phys. 37, 59–118 (2009).
Ribeiro, J. M., Mans, B. J. & Arca, B. An insight into the sialome of blood-feeding Nematocera. Insect. Biochem. Mol. Biol. 40, 767–784 (2010).
Titus, R. G. & Ribeiro, J. M. Salivary gland lysates from the sand fly Lutzomyia longipalpis enhance Leishmania infectivity. Science 239, 1306–1308 (1988).
Vijay, S., Rawat, M. & Sharma, A. Mass spectrometry based proteomic analysis of salivary glands of urban malaria vector Anopheles stephensi. BioMed. Res. Int. 2014, 686319, https://doi.org/10.1155/2014/686319 (2014).
Isawa, H., Yuda, M., Orito, Y. & Chinzei, Y. A mosquito salivary protein inhibits activation of the plasma contact system by binding to factor XII and high molecular weight kininogen. J. Biol. Chem. 277, 27651–27658 (2002).
Lombardo, F. et al. The Anopheles gambiae salivary protein gSG6: an anopheline-specific protein with a blood-feeding role. Insect. Biochem. Mol. Biol. 39, 457–466 (2009).
Pandey, R. K. & Prajapati, V. K. Molecular and immunological toxic effects of nanoparticles. Int. J. Biol. Macromol. 0141, 33394–9, https://doi.org/10.1016/j.ijbiomac.2017.09.110 (2017).
Waitayakul, A. et al. Natural human humoral response to salivary gland proteins of Anopheles mosquitoes in Thailand. Acta. Trop. 98, 66–73 (2006).
Armiyanti, Y. et al. Identification of antigenic proteins from salivary glands of female Anopheles maculatus by proteomic analysis. Asian. Pac. J. Trop. Biomed. 6, 924–930 (2016).
King, J. G., Vernick, K. D. & Hillyer, J. F. Members of the salivary gland surface protein (SGS) family are major immunogenic components of mosquito saliva. J. Biol. Chem. 286, 40824–40834 (2011).
Khatoon, N., Pandey, R. K. & Prajapati, V. K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 7, 8285, https://doi.org/10.1038/s41598-017-08842-w (2017).
Ali, M., Pandey, R. K., Khatoon, N., Narula, A. & Mishra, A. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 7, 9232, https://doi.org/10.1038/s41598-017-09199-w (2017).
Jordan, K. A. & Hunter, C. A. Regulation of CD8(+) T Cell Responses to Infection With Parasitic Protozoa. Exp. Parasitol. 126, 318–325 (2010).
Moseman, E. A. & McGavern, D. B. The great balancing act: regulation and fate of antiviral T-cell interactions. Immunol. Rev. 255, 110–124 (2013).
Pradhan, D. et al. Discovery of T-cell Driven Subunit Vaccines from Zika Virus Genome: An Immunoinformatics Approach. Interdiscip. Sci. 9, 468–477 (2017).
Calis, J. J. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 9, e1003266, https://doi.org/10.1371/journal.pcbi.1003266 (2013).
Pross, S. & Lefkowitz, D. in xPharm: The Comprehensive Pharmacology Reference 1–4 (Elsevier, 2007).
Nezafat, N., Ghasemi, Y., Javadi, G., Khoshnoud, M. J. & Omidinia, E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. J. Theor. Biol. 349, 121–134 (2014).
Dhanda, S. K., Vir, P. & Raghava, G. P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 8, 30, https://doi.org/10.1186/1745-6150-8-30 (2013).
Li, M., Jiang, Y., Gong, T., Zhang, Z. & Sun, X. Intranasal Vaccination against HIV-1 with Adenoviral Vector-Based Nanocomplex Using Synthetic TLR-4 Agonist Peptide as Adjuvant. Mol. Pharm. 13, 885–894 (2016).
Chan, J. et al. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin. Immunol. 26, 588–600 (2014).
Shi, J. et al. Epitope-Based Vaccine Target Screening against Highly Pathogenic MERS-CoV: An In Silico Approach Applied to Emerging Infectious Diseases. PLoS One 10, e0144475, https://doi.org/10.1371/journal.pone.0144475 (2015).
Dimitrov, I., Flower, D. R. & Doytchinova, I. AllerTOP - a server for in silico prediction of allergens. BMC Bioinformatics 14, S4–S4, https://doi.org/10.1186/1471-2105-14-S6-S4 (2013).
Craig, D. B. & Dombkowski, A. A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics 14, 346, https://doi.org/10.1186/1471-2105-14-346 (2013).
Rana, A. & Akhter, Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 221, 544–557, https://doi.org/10.1016/j.imbio.2015.12.004 (2016).
Pandey, R. K., Sharma, D., Bhatt, T. K., Sundar, S. & Prajapati, V. K. Developing imidazole analogues as potential inhibitor for Leishmania donovani trypanothione reductase: virtual screening, molecular docking, dynamics and ADMET approach. J. Biomol. Struct. Dyn. 33, 2541–2553 (2015).
Chander, S. et al. Molecular docking and molecular dynamics simulation based approach to explore the dual inhibitor against HIV-1 reverse transcriptase and Integrase. Comb. Chem. High Throughput Screen. 20, 1–13, https://doi.org/10.2174/1386207320666170615104703 (2017).
Tsuji, M. & Zavala, F. T cells as mediators of protective immunity against liver stages of Plasmodium. Trends Parasitol. 19, 88–93 (2003).
Larsen, M. V. et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8, 424, https://doi.org/10.1186/1471-2105-8-424 (2007).
Pandey, R. K., Sundar, S. & Prajapati, V. K. Differential Expression of miRNA Regulates T Cell Differentiation and Plasticity during Visceral Leishmaniasis Infection. Front. Microbiol. 7, 206, https://doi.org/10.3389/fmicb.2016.00206 (2016).
Ahluwalia, P. K., Pandey, R. K., Sehajpal, P. K. & Prajapati, V. K. Perturbed microRNA Expression by Mycobacterium tuberculosis Promotes Macrophage Polarization Leading to Pro-survival Foam Cell. Front. Immunol. 8, 107, https://doi.org/10.3389/fimmu.2017.00107 (2017).
Wang, P. et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 11, 568, https://doi.org/10.1186/1471-2105-11-568 (2010).
Black, M., Trent, A., Tirrell, M. & Olive, C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev. Vaccines 9, 157–173, https://doi.org/10.1586/erv.09.160 (2010).
Junqueira, C. et al. Trypanosoma cruzi adjuvants potentiate T cell-mediated immunity induced by a NY-ESO-1 based antitumor vaccine. PloS One 7, e36245, https://doi.org/10.1371/journal.pone.0036245 (2012).
Gupta, G., Oghumu, S. & Satoskar, A. R. Mechanisms of immune evasion in leishmaniasis. Adv. Appl. Microbiol. 82, 155–184 (2013).
McCall, M. B. et al. Plasmodium falciparum infection causes proinflammatory priming of human TLR responses. J. Immunol. 179, 162–171 (2007).
Shanmugam, A. et al. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PloS One 7, e30839, https://doi.org/10.1371/journal.pone.0030839 (2012).
Lee, S. J. et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One 9, e104351, https://doi.org/10.1371/journal.pone.0104351 (2014).
Pentel, P. R. & LeSage, M. G. New directions in nicotine vaccine design and use. Adv. Pharmacol. 69, 553–580 (2014).
EL‐Manzalawy, Y., Dobbs, D. & Honavar, V. Predicting linear B‐cell epitopes using string kernels. J. Mol. Recogn. 21, 243–255 (2008).
Wilkins, M. R. et al. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 (1999).
Pandey, R. K., Prajapati, P., Goyal, S., Grover, A. & Prajapati, V. K. Molecular Modeling and Virtual Screening Approach to Discover Potential Antileishmanial Inhibitors Against Ornithine Decarboxylase. Comb. Chem. High Throughput. Screen. 19, 813–823 (2016).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384–388 (2013).
Pandey, R. K. et al. Febrifugine analogues as Leishmania donovani trypanothione reductase inhibitors: binding energy analysis assisted by molecular docking, ADMET and molecular dynamics simulation. J. Biomol. Struct. Dyn. 35, 141–158 (2017).
Kozakov, D. et al. The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 (2017).
Pandey, R. K. et al. High-throughput virtual screening and quantum mechanics approach to develop imipramine analogues as leads against trypanothione reductase of leishmania. Biomed. Pharmacother. 83, 141–152, https://doi.org/10.1016/j.biopha.2016.06.010 (2016).
Pandey, R. K., Kumbhar, B. V., Sundar, S., Kunwar, A. & Prajapati, V. K. Structure-based virtual screening, molecular docking, ADMET and molecular simulations to develop benzoxaborole analogs as potential inhibitor against Leishmania donovani trypanothione reductase. J. Recept. Signal Transduct. Res. 37, 60–70 (2017).
Acknowledgements
R.K.P. is thankful to Department of Science and Technology for providing INSPIRE fellowship. V.K.P. is thankful to the Central University of Rajasthan for providing computational facility. We acknowledge the scientific help of Ms. Rani Soni from Department of Biotechnology, Central University of Rajasthan for this manuscript.
Author information
Authors and Affiliations
Contributions
Protocol designed by R.K.P., T.K.B., V.K.P. Methodology performed by R.K.P., V.K.P. Manuscript was written by R.K.P., T.K.B.,V.K.P.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pandey, R.K., Bhatt, T.K. & Prajapati, V.K. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci Rep 8, 1125 (2018). https://doi.org/10.1038/s41598-018-19456-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19456-1
This article is cited by
-
Development of a novel multi‑epitope vaccine against the pathogenic human polyomavirus V6/7 using reverse vaccinology
BMC Infectious Diseases (2024)
-
Identification and prioritisation of potential vaccine candidates using subtractive proteomics and designing of a multi-epitope vaccine against Wuchereria bancrofti
Scientific Reports (2024)
-
Employing computational tools to design a multi-epitope vaccine targeting human immunodeficiency virus-1 (HIV-1)
BMC Genomics (2023)
-
An immunoinformatics approach to epitope-based vaccine design against PspA in Streptococcus pneumoniae
Journal of Genetic Engineering and Biotechnology (2023)
-
In silico designing of a novel polyvalent multi-subunit peptide vaccine leveraging cross-immunity against human visceral and cutaneous leishmaniasis: an immunoinformatics-based approach
Journal of Molecular Modeling (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
