
Abstract
Based on first-principles density-functional theory (DFT) calculations, we report that the transition-metal bis-dithiolene, M3C12S12 (M = Mn and Fe), complexes can be a two-dimensional (2D) ferromagnetic insulator with nontrivial Chern number. Among various synthetic pathways leading to metal bis-dithiolenes, the simplest choice of ligand, Benzene-hexathiol, connecting metal cations to form a Kagome lattice is studied following the experimental report of time-reversal symmetric isostructural compound Ni3C12S12. We show sulfur and carbon-based ligands play the key role in making the complexes topologically nontrivial. An unusual topological quantum phase transition induced by the on-site Coulomb interaction brings a nearly flat band with a nonzero Chern number as the highest occupied band. With this analysis we explain the electronic structure of the class M3C12S12 and predict the existence of nearly flat band with nonzero Chern number and it can be a fractional Chern insulator candidate with carrier doping.
Similar content being viewed by others

Introduction
The quantum Hall effect (QHE) is observed in two-dimensional (2D) electron systems under the presence of strong external magnetic field as a result of the formation of well-defined Landau levels1,2. Considering that the dissipationless states at sample edges is a consequence of the topologically nontrivial ground state of the Landau quantization3,4, however, there have been numerous proposals to realize the QHE without applying any magnetic field. Haldane5 first pointed out that a topologically nontrivial electronic structure with broken time reversal symmetry (TRS) in a 2D insulator can induce such quantized anomalous Hall effect (QAHE), where the quantized Hall conductance is determined by the topological character of the band structure, i.e., Chern number. Later, the idea has been extended to magnetically doped topological insulator thin films6,7,8,9,10.
Following the theoretical proposals that the magnetic order induced by the doped transition metal elements can lead to a topological electronic structure with a finite Chern number in 2D thin films, various attempts have been made to realize magnetic topological insulators and the observations of QAHE were reported8,11,12,13. These magnetic topological insulators have attracted a lot of attention for their potential applications to the low energy consumption electronics and spintronics devices14,15. However, since the realization was limited to an extremely low temperature of about 30 mK due to the small bulk energy gap and ferromagnetic (FM) Curie temperature13, there is a high demand for high temperature QAHE materials for their future applications.
So far, most of theoretical proposals for novel magnetic topological insulators, namely, Chern insulators, are based on the honeycomb-lattice platform and utilize the half-metallic band structure with Dirac bands16,17,18,19,20. Also, the co-doping of exisiting topological insulators21,22, the bulk ferromagnetic BaFe2(PO4)223, the honeycomb structure stanene24, and niobate25 were suggested as candidates for Chern insulators. Further, the (111)-oriented growth of perovskite oxide heterostructures was proposed26,27,28. Apart from inorganic materials, however, Wang and coworkers suggested that a metal-organic compound, i.e., triphenyl-transition-metal (Mn2C18H12), in a 2D hexagonal lattice shows the QAHE with a nonzero Chern number29. Further, the successful synthesis of 2D metal-organic frameworks (MOFs) has opened a possiblity of numerous novel properties of 2D organic transition-metal chalcogen family30,31. This kind of two-dimensional system such as the honeycomb stanene32, arsenene33, and Bi/Sb(111) films34 could also host quantum spin Hall insulator phase. Also, there were proposals for the organometallic lattices with Fe, Cr and Co to exhibit half-metallic from first-principles calculations35. Although the single layer π-conjugated nickel-bis-dithiolene Ni3C12S12 was identified to have nontrivial topological states29,36, its realization still requires a precise control of oxidation state at the gapped Dirac point37. However, question remains on the choice of ligands and metal cations to carry out precise tuning of Fermi level to observe exotic properties.
A flat band, dispersionless band in the whole Brillouin zone, has attracted lots of attention since it can be an ideal playground to study strong correlation physics. Especially, a nearly flat band with a nontrivial Chern number was recently proposed as a promising candidate for the realization of fractional Chern insulators38,39,40,41,42. Kagome lattice with nearest hopping naturally provide flat bands due to the self-localization under destructive interference43,44. Then one also could expect a nearly flat band with nontrivial Chern number in the ferromagnetically ordered Kagome-lattice-based MOF.
The choice of transition metal cations in MOFs gives a subtle balance between strong correlation and spin-orbit coupling into the system and giving rise to a range of functionalized compounds with non-trivial topological electronic structure. The role of Mn and Fe is two fold, the time-reversal-symmetry breaking by rather large spin and giving rise to localized electronic bands with large spin-splitting near Fermi level. We propose a realization of Chern insulator in the Kagome-lattice-based MOF with the simplest choice of benzene-hexathiole (BHT) ligands with Mn and Fe as 3d transition metal cations for Chern insulators. We investigate the topological character of the band structures of 2D MOF kagome lattice of π-conjugated metal bis-dithiolene M3C12S12 (M = Mn and Fe), which can take an advantage of several important ingredients: the lattice symmetry, broken time-reversal symmetry, and reliable spin-orbit coupling. The flat band feature arising from the frustrated kagome lattice become a central part of the band structure across the Fermi level. Using density functional theoretical analysis we have shown that the 2D Kagome lattice of metal-bis dithiolene can show intrinsic anomalous Hall effect. The non-typical phase transition places the topologically nontrivial flat band just below the Fermi level in both Mn and Fe bis-ditholene complex. Both the complex show Ising type ferromanetic ordering with near room temperature magnetic Tc which could be very useful in spintronics application.
Results and Discussion
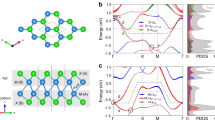
Figure 1(a) shows a schematic illurstration of the metal (M) bis-dithiolene unit which supports a network of M cations in a 2D kagome lattice as shown in Fig. 1(b). The synthesis of the complex nanosheet consisting of π-conjugated nickel (Ni) bis-dithiolene has already been reported by Kambe et al.30. The network of π-conjugated metal bis-dithiolene in a chemical formula of M3C12S12 (M = Mn and Fe) forms a triangular lattice with the unit vectors a1 and a2, as drawn in Fig. 1(b). The M atom is coordinated by four S atoms. The ligand Benzene-hexathiol connects the M atoms forming two motifs, honeycomb motif and triangle motif, which gives rise to Kagome lattice for M atoms.

(a) The schematics of reaction to produce metal bis-dithiolene complex unit that makes network of metal cations (M) as Kagome lattice. (b) Crystal structure of single layer metal bis-dithiolene kagome lattice (MOF). a1 and a2 being inplane lattice constants.
Figure 1(b) shows the schematic repeesenatation of the metal bis-dithiolene (M3C12S12). A full relaxation is carried out for the single-layer Manganese bis-dithiolene (MDT) Mn3C12S12 and iron bis-dithiolene (FDT) Fe3C12S12. In the primitive unit cell, there are three M atoms, twelve C atoms and twelve S atoms. The space group of the single-layer M3C12S12 is P6/mmm. The fully relaxed lattice constant is \(a=14.971\,{\rm{\AA }}\) for Mn and \(a=14.798\,{\rm{\AA }}\) for Fe, respectively. (The details of lattice constants and atom position coordinates are listed in the Supplementary Information). The relaxation is carried out the way to maintain C3 symmetry and inversion symmetry in the lattice. The plaquet of S around M atom is rectangular.
First we analyze the basic electronic and magnetic structures of M3C12S12 (M = Mn and Fe). Within the PBE + U method with U = 3 eV in vasp, the ground state of both systems is a half-metallic ferromagnet, Dirac cone at the Fermi level in the majority spin channel and insulating in the minority spin channel (see Fig. 2(a,b)). In the absence of spin-orbit coupling, the degeneracy at \(\Gamma \) point is preserved. The inclusion of spin-orbit coupling using opens a tiny gap at the Fermi level about 0.5 meV lifting degeneracy at the \(\Gamma \) point and the complex is an insulator (see Fig. 2(c)). A three-dimensional view of the electronic band structure of MDT is shown in Fig. 2(d)).

(a, b) PBE + U “fatband” and projected density of states of the Fe dxz, dyz and S pz, pπ states within the energy manifold near Fermi level plotted for majority (a) and minority (b) spin channel. The size of symbols is proportional to character of states. The projected density of states (PDOS) in unit of eV/states/atom for the Fe dxz, dyz and S pz, pπ states are plotted corresponding to both the spin channel representing relative positions of the states. (c) PBE + U + SOC band structure shows opening tiny band gap at \(\Gamma \) point (shown in the inset). (d) Three dimensional PBE + U + SOC band structure of Mn3C12S12 (MDT) monolayer.
The main orbital characters near Fermi level are from Fe dxz, dyz and S pz, pπ as projected within the energy manifold shown in the Fig. 2(a,b). The superexchange interaction between the two consequtive Fe atoms is mediated by the π-conjugated bonding due to molecular orbitals formed by Fe dxz, dyz and S pz orbitals (Fig. 2(a,b)). Whilst the band center of Fe dxz and dyz orbitals lies around −5 eV to −4 eV the tail part goes beyond Fermi level due to the large covalency with S p orbitals. The C pz orbitals mainly dominate energy range just below Fermi level. The Fe dz2 is fully occupied around −3 eV in the majority spin channel due to non-bonding along z direction of the unitcell. The S pπ has a occupied bonding character lying around −1 eV in the majority spin channel while unoccupied antibonding character lies around 2 eV. On the other hand, the Fe d orbitals are unoccupied in the minority spin channel. Two holes at S pz orbital in the minority spin channel contributes two electrons at the Fe site which makes the total spin at Fe to be \(S=1\) (\({d}^{6}{\underline{{\rm{L}}}}^{-2,\downarrow }\)). Here, the notation “\({d}^{6}{\underline{{\rm{L}}}}^{2\downarrow }\)” represents a local electronic configuration of six d–electrons (d6) of Fe and two down-spin (↓) holes (\({\underline{{\rm{L}}}}^{2}\)) of S ligand. With the same analysis total spin at Mn site in MDT appears to be \(S=\tfrac{3}{2}\) (\({d}^{5}{\underline{{\rm{L}}}}^{-2,\downarrow }\)).
The ferromagnetic ground state arises from the local moment of Mn and Fe. The formal oxidation state of Fe is 2+ with electronic configurations \({d}^{6}{\underline{{\rm{L}}}}^{-2,\downarrow }\). With the same analysis electronic configuration for Mn in MDT appears to be \({d}^{5}{\underline{{\rm{L}}}}^{-2,\downarrow }\). The d electron configuration of isolated Mn ion should be d5. Due to the molecular orbital formation with dithiolene ligands which contribute two “↓” spin electrons per connected Mn site the local electron spin at Mn site is \({d}^{5}{\underline{{\rm{L}}}}^{-2,\downarrow }\). The similar argument can be attributed for the electron configuration at Fe site \({d}^{6}{\underline{{\rm{L}}}}^{-2,\downarrow }\). It makes the molecular t2g orbital at Mn site half-filled, \(S=\tfrac{3}{2}\), which interacts with the nearest-neighbour Mn sites ferromagnetically through the antiferromagnetic interaction with dithiolene ligands to lift the spin-frustrated situation with out-of-plane orientation. This explanation would equally valid for FDT with spin at Fe S = 1. The calculated spin moments are 2.6 μB at the Fe site, −0.076 μB at the S site, −0.034 μB at the C site for FDT; 3.78 μB at the Mn site, −0.13 μB at the S site, −0.04 μB at C site for MDT.
The magnetic Curie temperature Tc is also calculated from DFT total energy comparison of various collinear magnetic configurations with considering nearest neighbor Heisenberg interaction. The effective Fe-Fe spin exchange interaction \({J}_{Fe-Fe}\) is computed to be 11.3 meV and Mn-Mn interaction \({J}_{Mn-Mn}\) is 4.2 meV. Using the values of exchange interactions and the total spin value of Fe \(S=1\) and of Mn \(S=3/2\) (both in classical spin limit), mean-field estimation of magnetic Tc is computed for FDT as \({T}_{c}^{{\rm{Fe}}}\sim 352\,{\rm{K}}\) and for MDT as \({T}_{c}^{{\rm{Mn}}}\sim 291\,{\rm{K}}\). We have also performed classical Monte-Carlo simulation to estimate Tc for both MDT and FDT by calculating specific heat and absolute magnetization as a function of temperature (see the Supporting Information). Our Monte Carlo simulation gives close but little less magnitude to the mean-field estimate of the Tc for MDT to be \({T}_{c}^{{\rm{Mn}}}\sim 234\,{\rm{K}}\) and for FDT to be \({T}_{c}^{{\rm{Fe}}}\sim 281\,{\rm{K}}\). The magnetic anisotroy energy is also calculated by comparing the energy of in-plane and out-of-plane collinear configurations. The out-of-plane configuration is 0.87 meV lower in energy compared to the in-plane order for FDT and the same for MDT is 1.9 meV.
To highlight the uniqueness of electronic structure in the 2D MOF kagome lattice, we focus on the FDT complex in the rest of the paper, but the main features of the Mn3C12S12 (MDT) system are similar except the differences in band gap and bandwidths. In Fig. 3(a), we have shown the evolution of band structure of majority spin by increasing on-site U. It is interesting to note that at U = 3 eV there are non-typical four bands near the Fermi level (EF), out of which top three bands displaying a typical kagome band dispersion. Among the four bands, the top and bottom bands are almost dispersionless flat bands and share only one band touching point at the \(\Gamma \) point. Further, the Fermi level is pinned right above the bottom flat band and the system becomes half-metallic semi-metal, when calculated without SOC. Interestingly, two conical bands and the nearly flat hole band near the zone center show typical Kane model type band alignment45,46. At U = 0 eV, the system shows typical three bands above Fermi level. At critical U, Kane fermion phase with triple degeneracy at \(\Gamma \) point arises. By increasing U more, the system shows band inversion thereby to form four bands and the Fermi level crosses the meeting point of the upper cone and the flat band.

Strong correlation driven band inversion in single layer Fe3C12S12. (a) The PBE + U spin-polarized majority (red line) and minority (black) spin channel) band evolution with U. The majority spin channel bands shows similar to low-energy Kane model behaviour. (b) The PBE + U (U = 0 eV) band structure with weight of eigenstates at \(\Gamma \) point, \(|{a}_{1},\Gamma \rangle \) (red color), \(|{e}_{1},\Gamma \rangle \) (green color) and \(|{e}_{2},\Gamma \rangle \) (blue color) projected along \(\Gamma -K\) direction in the BZ. (c) The shape of the molecular orbitals \(|{a}_{1},\Gamma \rangle \), \(|{e}_{1},\Gamma \rangle \) and \(|{e}_{2},\Gamma \rangle \) at \(\Gamma \) point. (d) The PBE + U (U = 3.5 eV) band structure with degenerate eigenstate exactly at the \(\Gamma \) point and flat band at the Fermi level. The color of the wights of the MOs are now inverted. (e) Chern number of flat band in PBE + U + SOC calculations with U = 0 and 3 eV.
To provide the microscopic picture of the transition, we analyzed a symmetry of three involving bands. In Fig. 3(b,d), the U = 0 and 3.5 eV band structures are shown along \(\Gamma -K\) direction in the Brillouin zone (BZ). At U = 0 eV the highest occupied flat band and second highest occupied conical band (Fig. 3(b)) have doubly degenerate eigenstates \(|{e}_{1},\Gamma \rangle \), \(|{e}_{2},\Gamma \rangle \) at \(\Gamma \) point below Fermi level while a singly degenerate unoccupied eigenstate \(|{a}_{1},\Gamma \rangle \) exist at \(\Gamma \) point just above Fermi level. The p–d hybridization between Fe-dxz,dyz and S-pz is significant near Fermi level in majority spin channel, at U = 0 eV, as can be seen from the partial density of states plot for Fe d and S p orbitals. The molecular orbitals corresponding to singly degenerate and doubly degenerate eigenstates are plotted in figure Fig. 3(c). The \(|{a}_{1},\Gamma \rangle \) has a node at Fe site with zero weight from dxz + dyz orbitals. On the other hand, molecular orbitals for doubly degenerate eigenstates \(|{e}_{1},\Gamma \rangle \) has finite weight from Fe dxz + dyz orbitals. With the increase in Hubbard U to 3.5 eV at Fe site, the p–d hybridization gets lowered due to the localization of Fe d states below Fermi level. That makes zero weight for Fe-dxz,dyz system above Fermi though S-pz weight remain same. It makes the band inversion at \(\Gamma \) point by making doubly degenerate eigenstates \(|{e}_{1},\Gamma \rangle \), \(|{e}_{2},\Gamma \rangle \) partially filled higher in energy and singly degenerate eigenstate \(|{a}_{1},\Gamma \rangle \) fully occupied lower in energy. Thereby, the flat band makes a pair with above Dirac band as shown in Fig. 3(d).
This band inversion is responsible for making the system nontrivial with the inclusion of spin-orbit coupling. The Hubbard U = 0 eV band structure is trivial with respect to total Berry curvature up to the occupied level. It is also checked by edge state calculation. Especially, Chern number of the flat band is zero while the dispersive conduction band carries the non-zero Chern number C = 1 (see Fig. 3(e)). For U = 0 eV, through the band inversion, the conduction band transfers its Chern number to the flat band for U = 3 eV. As a result, the Chern number of the flat band as well as whole valence bands become C = 1. Our band structure and topological properties of MDT with smaller U (<2.4 eV) is consistent with the previous report47.
To improve the estimation of a band gap, we further employed the HSE hybrid functional calculation as shown in Fig. 4. It opens a gap at the Fermi level as large as 22 meV for FDT complex and 23 meV for MDT complex (shown in Supplementary material). With this enhanced gap, we have used maximally localized Wannier functions (MLWFs) to calculate the anomalous Hall conductivity and Berry curvature by using the Wannier90 package48. The energy manifold chosen to accurately reproduce DFT bands (HSE06 + SOC) is set from −2.76 eV to 1.47 eV with respect to Fermi energy. The integration of the Berry curvature \({\Omega }_{z}(k)\) over the entire BZ gives anomalous Hall conductivity σz. The variation of anomalous Hall conductivity σz as a function of energy is shown in Fig. 4(b). It clearly show that the dispersive conical bands above Fermi level are trivial though The unoccupied flat band is nontrivial. When we focus on the Berry curvature up to Fermi level, as shown in Fig. 4(c), the Berry curvature \({\Omega }_{z}(k)\) is concentrated only at \(\Gamma \) point in the entire BZ. The quantized value of anomalous Hall conductivity shows Chern number C = 1 as Chern number is given by \(C=\frac{1}{2\pi }\,{\int }^{}\,{d}^{2}{k\Omega }_{z}(k)\). The system, therefore, is a Chern insulator with highest occupied non-trivial nearly flat band with Chern number C = 1.

(a) HSE06 + SOC band structure (violet line) shows large band gap at Fermi level. The Chern number (C) of each band is also marked in the figure. (b) The anomalous Hall conductivity σz in unit of quantum conductance is shown with variation of Fermi level. (c) The Berry curvature in the BZ. (d) The left (red) and right (blue) edge bands of FDT.
According to quantum transport theory, each edge current carries one e2/h conductivity. So, the direct confirmation of Chern insulating phase is to verify presence of chiral edge mode circulating at the boundary. The edge band structure was calculated using real space Hamiltonian in the basis of MLWFs and iterative Green’s function approach49. The edge band structure with left edge band (red) and right edge band (blue) connecting valence bulk band with conduction bulk bands is shown in Fig. 4(d). The number of chiral edge bands on one edge confirms the Chern number C = 1.
The presence of flat band in metal-organic framework based metal bis-dithiolene further pushes the organometallic system towards the direction of fractional quantum Hall effect. The redox control on these complexes are possible in practice as already reported in the previous literatures on Ni3C12S1230. So, with the proper control of electronic charge on highest occupied flat band exotic fraction Chern insulating phase can be realized in the future.
In summary we have shown that metal-organic framework based single layer metal bis-dithiolene complexes M3C12S12 (M = Mn and Fe) can be robust 2D ferromagnetic organic Chern insulator. From the DFT + U as well as hybrid-functional calculations, we confirmed that the Ising-type ferromagnetic state is the magnetic ground state of the single layer M3C12S12 with a gap as large as 22 meV. By controlling U, a topological quantum phase transition mediated by Kane fermion phase gives rise to a nearly flat band with a nonzero Chern number. To observe these phases in an experiment a wide band gap substrate would be necessary to avoid charge transfer between substrate and the compound near Fermi level. It would also be an interesting project to study the electronic and topological properties under the effect of strain. In addition, the presence of nontrivial flat band further pushes the boundary of metal-organic framework system towards realization of many-body phenomena like fractional Chern insulators by means of partial filling of the flat band with the help of any kind of hole doping.
Methods
To determine the crystal structures and the electronic structures of the single-layer M3C12S12, we carried out first-principles density-functional-theory (DFT) calculations by employing both the PBE + U and the Heyd-Scuseria-Ernzerhof screened hybrid functional (HSE06)50 as implemented in the vasp51,52. All the calculations were performed with a plane-wave cutoff of 600 eV on the 6 × 6 × 2 Gamma centered Monkhorst-Pack k-point mesh in vasp. The OpenMX code53 based on the linear combination of pseudo-atomic-orbital basis formalism was used to do analysis for molecular orbitals. The energy cut-off for OpenMX is 400 Ryd. The vacuum layer was set to be 15 Å thick to decouple neighbouring layers along the z-direction. The full relaxation was carried out with keeping the three-fold rotational symmetry and inversion symmetry inherent in the space group P6/mmm with forces smaller than 0.01 eV/Å. The analysis of topological properties was carried out using Wannier90 package48 to fit tight-binding Hamiltonian in the basis of maximally-localized Wannier function (MLWFs) to DFT band structures.
Data Availability
All data in this work are available from the corresponding author on reasonable request.
References
Klitzing, K. V., Dorda, G. & Pepper, M. New method for high-accuracy determination of the fine-structure constant based on quantized Hall resistance. Phys. Rev. Lett. 45, 494–497 (1980).
Willett, R. et al. Observation of an even-denominator quantum number in the fractional quantum Hall effect. Phys. Rev. Lett. 59, 15 (1987).
Laughlin, R. B. Quantized Hall conductivity in two dimensions. Phys. Rev. B 23, 5632–5633 (1981).
Thouless, D. J., Kohmoto, M., Nightingale, M. P. & den Nijs, M. Quantized Hall conductance in a two-dimensional periodic potential. Phys. Rev. Lett. 49, 405–408 (1982).
Haldane, F. D. M. Model for a quantum Hall effect without landau levels: Condensed-matter realization of the “parity anomaly”. Phys. Rev. Lett. 61, 2015 (1988).
Qi, X.-L., Wu, Y.-S. & Zhang, S.-C. Topological quantization of the spin Hall effect in two-dimensional paramagnetic semiconductors. Phys. Rev. B 74, 085308 (2006).
Liu, C.-X., Qi, X.-L., Fang, Z. & Zhang, S.-C. Quantum anomalous Hall effect in hg1ymnyte quantum wells. Phys. Rev. Lett. 101, 146802 (2008).
Liu, C., Hughes, T. L., Qi, X.-L., Wang, K. & Zhang, S.-C. Quantum spin Hall effect in inverted type-ii semiconductors. Phys. Rev. Lett. 100, 236601 (2008).
Yu, R. et al. Quantized anomalous Hall effect in magnetic topological insulators. Science 329, 61–64 (2010).
Nomura, K. & Nagaosa, N. Surface-quantized anomalous Hall current and the magnetoelectric effect in magnetically disordered topological insulators. Phys. Rev. Lett. 106, 166802 (2011).
Checkelsky, J. G., Ye, J., Onose, Y., Iwasa, Y. & Tokura, Y. Dirac-fermion-mediated ferromagnetism in a topological insulator. Nature Physics 8, 729 (2012).
Xu, S.-Y. et al. Hedgehog spin texture and berry’s phase tuning in a magnetic topological insulator. Nature Physics 8, 616 (2012).
Chang, C.-Z. et al. Experimental observation of the quantum anomalous Hall effect in a magnetic topological insulator. Science 340, 167–170 (2013).
Hasan, M. Z. & Kane, C. L. Colloquium: Topological insulators. Rev. Mod. Phys. 82, 3045–3067 (2010).
Novoselov, K. S., Mishchenko, A., Carvalho, A. & Castro Neto, A. H. 2d materials and van der waals heterostructures. Science 353 (2016).
Zhang, H. Z., Lazo, C., Blügel, S., Heinze, S. & Yuriy, M. Electrically tunable quantum anomalous Hall effect in graphene decorated by 5d transition-metal adatoms. Phys. Rev. Lett. 108, 056802 (2012).
Zhou, M. et al. Epitaxial growth of large-gap quantum spin Hall insulator on semiconductor surface. Proceedings of the National Academy of Sciences 111, 14378–14381 (2014).
Cai, T. et al. Single-spin dirac fermion and Chern insulator based on simple oxides. Nano Letters 15, 6434 (2015).
Wu, S.-C., Shan, G. & Yan, B. Prediction of near-room-temperature quantum anomalous Hall effect on honeycomb materials. Phys. Rev. Lett. 113, 256401 (2014).
van Miert, G., Ortix, C. & Smith, C. M. Topological origin of edge states in two-dimensional inversion-symmetric insulators and semimetals. 2D Materials 4, 015023 (2017).
Qi, S. et al. High-temperature quantum anomalous Hall effect in n − p codoped topological insulators. Phys. Rev. Lett. 117, 056804 (2016).
Crisostomo, C. P., Huang, Z.-Q., Hsu, C.-H., Chuang, F.-C. & Bansil, A. Chemically induced large-gap quantum anomalous Hall insulator states in iii-bi honeycombs. npj Computational Materials 3, 39 (2017).
Song, Y.-J., Ahn, K.-H., Pickett, W. E. & Lee, K.-W. Tuning ferromagnetic BaFe2(PO4)2 through a high Chern number topological phase. Phys. Rev. B 94, 125134 (2016).
Zhang, M. H., Zhang, C. W., Wang, P. J. & Li, S. S. Prediction of high-temperature Chern insulator with half-metallic edge states in asymmetry-functionalized stanene. Nanoscale 10, 20226–20233 (2018).
Zhang, S.-J. et al. Intrinsic dirac half-metal and quantum anomalous Hall phase in a hexagonal metal-oxide lattice. Phys. Rev. B 96, 205433 (2017).
Qi, X.-L. & Zhang, S.-C. Topological insulators and superconductors. Rev. Mod. Phys. 83, 1057–1110 (2011).
Si, L. et al. Quantum anomalous Hall state in ferromagnetic SrRuO3 (111) bilayers. Phys. Rev. Lett. 119, 026402 (2017).
Guo, H., Gangopadhyay, S., Köksal, O., Pentcheva, R. & Pickett, W. E. Wide gap Chern mott insulating phases achieved by design. npj Quantum Materials 2, 4 (2017).
Wang, Z. F., Liu, Z. & Liu, F. Quantum anomalous Hall effect in 2d organic topological insulators. Phys. Rev. Lett. 110, 196801 (2013).
Kambe, T. et al. π-conjugated nickel bis(dithiolene) complex nanosheet. Journal of the American Chemical Society 135, 2462–2465 (2013).
Ken, H. et al. Fabrication of dense and multilayered films of a nickel bis(dithiolene) nanosheet by means of the langmuirschäfer method. Chem. Lett. 43, 252–253 (2014).
Zhang, R.-W. et al. Ethynyl-functionalized stanene film: a promising candidate as large-gap quantum spin Hall insulator. New Journal of Physics 17, 083036 (2015).
Wang, Y.-P. et al. Two-dimensional arsenene oxide: A realistic large-gap quantum spin Hall insulator. Applied Physics Letters 110, 213101 (2017).
Li, S.-S., Ji, W.-X., Hu, S.-J., Zhang, C.-W. & Yan, S.-S. Effect of amidogen functionalization on quantum spin Hall effect in bi/sb(111) films. ACS Applied Materials & Interfaces 9, 41443–41453 (2017).
Mandal, B. & Sarkar, P. A new two-dimensional metalorganic framework with high spin-filtering efficiency. Phys. Chem. Chem. Phys. 17, 17437–17444 (2015).
Wang, Z. F., Su, N. & Liu, F. Prediction of a two-dimensional organic topological insulator. Nano. Lett. 13, 2842–2945 (2013).
Kambe, T. et al. Redox control and high conductivity of nickel bis(dithiolene) complex -nanosheet: A potential organic two-dimensional topological insulator. J. Am. Chem. Soc. 136, 14357 (2014).
Tang, E., Mei, J.-W. & Wen, X.-G. High-temperature fractional quantum Hall states. Phys. Rev. Lett. 106, 236802 (2011).
Sun, K., Gu, Z., Katsura, H. & Das Sarma, S. Nearly flatbands with nontrivial topology. Phys. Rev. Lett. 106, 236803 (2011).
Neupert, T., Santos, L., Chamon, C. & Mudry, C. Fractional quantum Hall states at zero magnetic field. Phys. Rev. Lett. 106, 236804 (2011).
Sheng, D. N., Gu, Z. C., Sun, K. & Sheng, L. Fractional quantum Hall effect in the absence of landau levels. Nature Communications 2, 389 (2011).
Regnault, N. & Bernevig, B. A. Fractional Chern insulator. Phys. Rev. X 1, 021014 (2011).
Bergman, D. L., Wu, C. & Balents, L. Band touching from real-space topology in frustrated hopping models. Phys. Rev. B 78, 125104 (2008).
Rhim, J.-W. & Yang, B.-J. Classification of flat bands according to the band-crossing singularity of bloch wave functions. Phys. Rev. B 99, 045107 (2019).
Orlita, M. et al. Observation of three-dimensional massless kane fermions in a zinc-blende crystal. Nature Physics 10, 233 (2014).
Wang, A. et al. Kane fermion in a two-dimensional -conjugated bis(iminothiolato)nickel monolayer. J. Phys. Chem. Lett. 9, 614 (2018).
Zhao, M., Wang, A. & Zhang, X. Half-metallicity of a kagome spin lattice: The case of a manganese bis-dithiolene monolayer. Nanoscale 5, 10404–10408 (2013).
Mostofi, A. A. et al. wannier90: A tool for obtaining maximally-localised wannier functions. J. Comput. Phys. Commun. 178, 685–699 (2008).
Wu, Q., Zhang, S., Song, H.-F., Troyer, M. & Soluyanov, A. A. Wanniertools: An open-source software package for novel topological materials. Computer Physics Communications 224, 405–416 (2018).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. The Journal of Chemical Physics 118, 8207–8215 (2003).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Kresse, G. & Furthmüllar, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 54, 11169 (1996).
Ozaki, T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 67, 155108 (2003).
Acknowledgements
We gratefully acknowledge Kee Hoon Kim for valuable discussions. This work is supported by Institute for Basic Science (IBS) in Korea (Grant No. IBS-R009-D1). J.Y. acknowledges the support by the National Research Foundation of Korea (NRF) (no. 2017R1A2B4007100) and the support and hospitality provided by the Max Planck Institute for the Physics of Complex systems during his visit to the institute.
Author information
Authors and Affiliations
Contributions
S.B. and S.K. performed the calculations under the supervision of C.H.K. and J.Y. All authors contributed to the discussion and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baidya, S., Kang, S., Kim, C.H. et al. Chern insulator with a nearly flat band in the metal-organic-framework-based Kagome lattice. Sci Rep 9, 13807 (2019). https://doi.org/10.1038/s41598-019-50163-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50163-7
This article is cited by
-
A p-orbital honeycomb-Kagome lattice realized in a two-dimensional metal-organic framework
Communications Chemistry (2023)
-
Field-controlled quantum anomalous Hall effect in electron-doped CrSiTe3 monolayer
npj 2D Materials and Applications (2023)
-
Correlation-induced magnetism in substrate-supported 2D metal-organic frameworks
npj Computational Materials (2022)
-
Magnetization plateaus and thermodynamic properties of a ferrimagnetic Kagome-like nanoparticle under an applied magnetic field
Journal of Materials Science (2022)
-
Kagome van-der-Waals Pd3P2S8 with flat band
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
