
Abstract
Human noroviruses (HuNoVs) are the leading cause of viral foodborne outbreaks worldwide. To date, no available methods can be routinely used to detect infectious HuNoVs in foodstuffs. HuNoVs recognize Histo-Blood Group Antigens (HBGAs) through the binding pocket (BP) of capsid protein VP1, which promotes infection in the host cell. In this context, the suitability of human HBGA-binding assays to evaluate the BP integrity of HuNoVs was studied on GII.4 virus-like particles (VLPs) and GII.4 HuNoVs during natural ageing at 20 °C and heat treatments. Our results demonstrate that this approach may reduce the over-estimation of potential infectious HuNoVs resulting from solely using the genome detection, even though some limitations have been identified. The specificity of HBGA-binding to the BP is clearly dependent on the HGBA type (as previously evidenced) and the ionic strength of the media without disturbing such interactions. This study also provides new arguments regarding the ability of VLPs to mimic HuNoV behavior during inactivation treatments. The BP stability of VLPs was at least 4.3 fold lower than that of HuNoVs at 20 °C, whereas capsids of both particles were disrupted at 72 °C. Thus, VLPs are relevant surrogates of HuNoVs for inactivation treatments inducing significant changes in the capsid structure.
Similar content being viewed by others

Introduction
Human noroviruses (HuNoVs), responsible for acute gastroenteritis, are the leading cause of viral foodborne outbreaks in Europe and USA each year1,2. HuNoVs, which belong to the family Caliciviridae, are mainly genogroup I (GI) and II (GII) NoVs which are subdivided into several genotypes3. Genogroup II, genotype 4 (GII.4) NoV is the most prevalent, and has been causing for about 70% of outbreaks since 20024. Infected individuals release high concentrations of HuNoVs in their feces, ranging from 106 to 109 genome copies (gc)/gram of stools over a 4- to 8-week period5. The infectious dose for HuNoVs has not been clearly defined yet. For Norwalk virus (NV), the 50% human infectious dose (HID50) was estimated between 18 and 2,800 genomic equivalents for secretor individuals and by considering the viral aggregation6,7. Thus, high viral shedding in the environment combined with a relatively low HID50 contributes to promote HuNoV transmission to the human population. HuNoVs are transmitted through the fecal-oral route, either directly by close contact with infected people or indirectly through consumption of contaminated water and food8.
HuNoVs are non-enveloped viruses (about 40 nm in diameter) with a capsid of icosahedral symmetry, and a single-stranded positive-sense RNA (~7,500 nucleotides) containing three open reading frames (ORFs). ORF1 encodes non-structural proteins while ORF2 and ORF3 encode for the major (VP1) and the minor (VP2) capsid proteins, respectively. The capsid is composed of 180 copies of VP1 assembled into 90 dimers and few copies of VP2. VP1 is organized into two domains, the shell domain (S) and the protruding domain (P) which is subdivided into P1 and P2 subdomains9.
The occurrence of HuNoV foodborne outbreaks underlines that these viruses can remain infectious for a substantial time in the environment10,11,12. The food vehicles most commonly involved in HuNoV foodborne outbreaks are raw and minimally processed foods (e.g. bivalve molluscan shellfish, fresh vegetables, drinking water). Even though no regulation exists in Europe for the management of HuNoVs, their detection in foodstuffs put on the market is on the rise for the last few years. This trend is related to the publication of the ISO 15216-1 standard in 2013, allowing the genome detection of HuNoVs in vulnerable foodstuffs13. Based on representative sampling, the prevalence of HuNoV genomes ranges from 9.0% to 71.6% in shellfish and from 5.0% to 28.2% in fresh produce14. Although its interest is unquestionable for viral outbreak investigations, the routine use of this ISO standard13 for surveillance in foodstuffs has limitations due to its inability to discriminate infectious from non-infectious particles when HuNoV genomes are detected15. This drawback is related to the fact that HuNoVs cannot be routinely cultivated in laboratories, despite recent advances for the in vitro replication of few strains of HuNoVs using human intestinal enteroids16. From our point of view, it is important to underline that even if the cellular models of HuNoVs were mastered by control laboratories over the coming years, this approach would not be suitable for routine food testing owing to its complexity, high cost, and time-consumption.
In this context, it becomes necessary to develop a suitable molecular method for discriminating infectious from non-infectious HuNoVs in food and water in order to (i) enable better management of the viral hazard and (ii) improve the effectiveness of risk management strategies in industries. To solve this critical issue, numerous strategies have been implemented over the past two decades. Firstly, a number of cultivable enteric viruses were used as HuNoV surrogates to assess the virucidal effect of disinfection treatments applied in food and water industries. Among HuNoV surrogates, feline calicivirus, murine NoV (MNV), Tulane virus (TV), and F-specific RNA bacteriophages (FRNAPH) are commonly used8,17,18. VLPs of HuNoVs are also used as well8,19,20. They are morphologically and immunologically similar to the native virions21,22.
Secondly, several methods have been proposed to discriminate infectious from non-infectious HuNoVs such as (i) the detection of the whole genome23, (ii) the use of enzymatic pre-treatments or intercalating dyes before genome detection24,25, and (iii) the detection of oxidative damages on the capsid26. The use of receptor binding to assess the capsid integrity is another promising approach27,28,29. Several studies have demonstrated that Histo-Blood Group Antigens (HBGAs) display a role of receptors or co-factors for HuNoVs which promote the infection in host cells30,31,32,33. HBGAs are complex carbohydrates linked to glycolipids or glycoproteins found on the red blood cells and mucosal epithelial cells. They can also be present as free antigens in biological fluids such as saliva34. The biosynthesis of HBGAs is governed by the ABO and the fucosyl-transferase FUT2 and FUT3 gene families34,35. The expression of active or non-active FUT2 defines the secretor or non-secretor status, respectively32,35.
The HuNoV-binding site to HBGAs is located in a region of the P2 subdomain of VP1 called “binding pocket” (BP)36. Interactions between HuNoVs and HBGAs have been described to be very specific with multiple binding patterns and variable relative affinities34,37,38. Thus, the human susceptibility to HuNoV infection is dependent on both polymorphic HBGA expression and HuNoV genotype30,33,39,40. According to these findings, some authors have made the assumption that only HuNoV particles able to bind the HBGAs would infect host cells, and thus potentially allowing discrimination infectious from non-infectious particles41.
The aim of this study was to investigate the ability of GII.4 VLPs to mimic the HuNoV behavior and the suitability of the HBGA-binding assays to avoid the over-estimation of potential infectious HuNoV number given by genome detection during natural ageing at 20 °C over time and heat treatments (50 °C, 60 °C, and 72 °C) by using the most prevalent HuNoV genotype worldwide (i.e. GII.4). First, the capsid integrity of GII.4 VLPs was studied during these inactivation treatments using receptor-binding enzyme linked immunosorbent assays (ELISA) and transmission electronic microscopy (TEM). The influence of the ionic strength of the medium on VLP binding to HBGAs was explored to assess the VLP stability and the nature of interactions between VLPs and HBGAs. Then, the HBGA-binding capacity of intact GII.4 HuNoVs extracted from human stools was evaluated using HBGA-binding followed by RNA amplification by quantitative RT-PCR (RT-qPCR) under the same conditions as those used with GII.4 VLPs. Finally, we compared the representativeness of using VLPs as HuNoV surrogates for a same genotype during inactivation treatments. We also discussed the benefits of using HBGA-binding assays to assess the capsid integrity and the suitability of this strategy to indicate the infectivity of HuNoVs after inactivation treatments.
Results
Binding profile of GII.4 VLPs to saliva samples
HBGA types were determined for the 27 human saliva samples by ELISA and were distributed as follows: non-secretor phenotypes (5/27; 18.5%), A Lewis-negative (ALe−) (1/27; 3.70%), A Lewis-positive (ALe+) (8/27; 29.6%), BLe− (1/27; 3.70%), BLe+ (4/27; 14.8%), and OLe+ (8/27; 29.6%). OLe− type was not found in the tested saliva samples. This distribution is consistent with the prevalence of 20% of non-secretor individuals in the Caucasian population37.
The binding profile of GII.4 VLPs to all saliva samples was performed using HBGA-binding ELISA (see Supplementary Fig. S1). Based on the optical density (OD450) values obtained under the same conditions, VLPs recognized all HBGAs from secretor individuals (22/27; 81.5%). Conversely, no VLP binding was observed for the five non-secretor saliva samples (positive/negative (P/N) ratio < 2). These results are consistent with published data22,37. Nevertheless, variable relative affinities were highlighted regarding interactions between VLPs and saliva samples from secretor individuals. This could be explained by the amount of HBGAs in each saliva37. The extent of VLP binding was particularly high for one secretor saliva corresponding to OLe+ type (OD450 = 0.625 at 1 µg/mL of GII.4 VLPs). Consequently, this sole saliva sample was used for all experiments performed in this study to evaluate the effect of natural ageing at 20 °C and heat treatments (50 °C, 60 °C, and 72 °C) on the NoV binding capacity.
Sensitivity and linearity of the HBGA-binding ELISA
The sensitivity and the linearity of the HBGA-binding ELISA method was investigated using OLe+ saliva and various concentrations of GII.4 VLPs ranging from 0.05 and 10.0 µg/mL in 10 mM and 150 mM PBS solutions (see Supplementary Fig. S2). The limit of detection (LOD) was estimated at 0.10 µg/mL of GII.4 VLPs because the P/N ratio was lower than 2 below this threshold value under the two ionic strength conditions. The relationship between OD450 and the VLP concentration was not linear in the whole range especially above a threshold value corresponding at 1.00 µg/mL, due to the concentration of anti-GII.4 VLP monoclonal antibody (MAb) which becomes limiting. In tested conditions for natural ageing and heat treatments (10.0 µg/mL of GII.4 VLPs), the first log10 reduction of HBGA-binding does not have a substantial impact on the OD450 values. Conversely, the first 2 log10 reductions lead to a decrease of OD450 values until the LOD. So, we considered that 2 log10 reductions (T99) were obtained when OD450 values were equal to the LOD.
Effect of natural ageing at 20 °C and heat treatments on the GII.4 VLP binding to human HBGAs and on the capsid integrity
The effect of natural ageing at 20 °C over 105 days under two ionic strength conditions (10 mM and 150 mM PBS solutions) on GII.4 VLP binding to OLe+ saliva is shown in Fig. 1. For each assay, a sodium periodate treatment on OLe+ saliva was added as a negative control to check the specificity of VLP-HBGA interactions. For all trials, no VLP binding was observed after sodium periodate treatment (OD450 < 0.200) due to the oxidative cleavage of saccharide rings42. According to the OD450 values, a progressive reduction in VLP binding to OLe+ saliva was observed over time until reaching a complete loss of binding capacity after 63 and 70 days under low (10 mM PBS) and high (150 mM PBS) ionic strength conditions, respectively (P/N ratio < 2). From these days, the LOD was reached, consequently, 2 log10 reductions of the OD450 values were obtained. Under the high ionic strength condition, no significant difference was observed after 70 days between untreated and sodium periodate-treated samples (paired sample t-test, p > 0.26). Under the low ionic strength condition, after 63 days, differences between untreated and sodium periodate-treated saliva samples appeared at the limit of significance (paired sample t-test, p < 0.0021). Based on the GII.4 VLP binding profiles, natural ageing at 20 °C affects gradually the BP integrity. However, changes in the magnitude of the binding profile were highlighted as a function of the ionic strength. For the first 63 days, the relative affinity of the VLP binding was significantly higher at high ionic strength compared to low ionic strength (unpaired sample t-test, p < 10−4). Between 70 and 98 days, the VLP binding was low and statistically different between the two ionic strength conditions (unpaired sample t-test, p < 10−3). After 105 days, no statistically significant difference was observed between the two ionic strength conditions (unpaired sample t-test, p > 0.25).

GII.4 VLP binding to OLe+ saliva after exposure to natural ageing at 20 °C under low (10 mM PBS) and high (150 mM PBS) ionic strength conditions. The OD450 values of purified GII.4 VLPs were obtained by HBGA-binding ELISA in 10 mM solution (solid line) and 150 mM PBS solution (dashed line). Squares (▪) and circles (•) represent the VLP binding to untreated and sodium periodate-treated OLe+ human saliva, respectively. Each data point represents the mean OD450 values of four replicates and error bars indicate standard deviations. Paired and unpaired Student’s t-test was used to compare groups.
To better evaluate the influence of the ionic strength on GII.4 VLP binding to OLe+ saliva, the ODt/OD0 ratio was determined in 10 mM and 150 mM PBS solutions (Fig. 2). Similar trends were observed regarding the reduction in VLP binding during natural ageing at 20 °C. No significant difference was observed between the two ionic strength conditions after 14, 35, 56, and 63 days (unpaired sample t-test, p > 0.07). For the others points, significant differences appeared (unpaired sample t-test, p < 0.01). The ionic strength of the medium seems to influence the magnitude of the binding profile but without disturbing the specificity of VLP-HBGA interactions. Consequently, the binding profiles of GII.4 VLPs to OLe+ saliva after inactivation by heating were only performed under the high ionic strength condition.

Decay of GII.4 VLP binding to OLe+ saliva after exposure to natural ageing at 20 °C under low and high ionic strength conditions. The solid and dashed lines represent purified GII.4 VLP binding to untreated saliva under low (10 mM PBS) and high (150 mM PBS) ionic strength conditions, respectively. The ODt/OD0 ratio was determined by dividing the mean OD450 values obtained by HBGA-binding ELISA at each time point (ODt) by the mean OD450 values obtained at day 0 (OD0). Each data point represents the mean OD450 values of four replicates and error bars indicate standard deviations. Unpaired Student’s t-test was used to compare the two groups.
The log10 reductions in GII.4 VLP binding to OLe+ saliva after exposure to heat treatments (50 °C, 60 °C, and 72 °C) for 30 min were represented by the log (ODt/OD0) at 450 nm (Fig. 3). A sodium periodate treatment was also introduced as a negative control for all assays (data not shown). No significant reduction in the VLP binding was observed after heating at 50 °C and 60 °C for 30 min (paired sample t-test, p > 0.05). The behavior of VLP binding was not statistically different for these two heat treatments (unpaired sample t-test, p > 0.2). Because of the non-linear relationship between OD450 values and GII.4 VLP concentration, it is only possible to consider that after 30 min at 50 °C and 60 °C, the reduction of the VLP binding to OLe+ saliva is lower than 1 log10. Conversely, a significant reduction in the VLP binding was observed at 72 °C for the first 3 min until reaching a complete loss of the OD450nm detection after 10 min (paired sample t-test, p < 10−5). After 2 min at 72 °C, 2 log10 reductions were achieved because OD450 values were equal to the LOD. According to the log10 reduction profiles, temperatures below 60 °C for 30 min affect slightly the BP integrity whereas 72 °C can be considered as a critical temperature which disturbs the BP from the early stage of treatment.

Decay of GII.4 VLP binding to OLe+ saliva after exposure to heat treatments under high ionic strength condition (150 mM PBS). Squares (▪, solid line), circles (•, dotted line), and triangles (▲, dot-and-dash line) represent purified GII.4 VLP binding to untreated OLe+ human saliva after heating at 50 °C, 60 °C, and 72 °C for 30 min, respectively. The log (ODt/OD0) values were determined by dividing the mean OD450 values obtained by HBGA-binding ELISA at each time point (ODt) by the mean OD450 values obtained at time 0 (OD0). Each data point corresponds to the mean OD450 values of triplicates and error bars indicate standard deviations. Paired and unpaired Student’s t-test was used to compare groups.
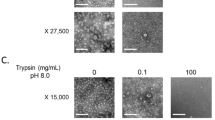
TEM observations were performed to evaluate the influence of heat treatments on the overall structure of GII.4 VLPs (Fig. 4). Similar structures were observed for native and treated VLPs after heating at 50 °C and 60 °C for 10 min, supporting structural integrity of the capsid. Conversely, no VLP was observed after heating at 72 °C for 10 min, suggesting disruption and/or denaturation of the capsid. Two different sizes (15 and 40 nm) of GII.4 VLP populations were distinguished at 50 °C and 60 °C. Particles of 15 and 40 nm are respectively formed of 60 and 180 copies of VP1 which are described to be morphologically and antigenically similar43. According to these data, TEM results are consistent with the log10 reduction profiles of the VLP binding after heat treatments.

TEM observations of native and heat-treated GII.4 VLP capsids after negative staining. (a) Native GII.4 VLPs, (b) GII.4 VLPs after exposure at 50 °C for 10 min, and (c) GII.4 VLPs after exposure at 60 °C for 10 min. No GII.4 VLP was observed by TEM after heating at 72 °C for 10 min. The scale bar is at the bottom left of each picture.
Effect of natural ageing at 20 °C and heat treatments on the GII.4 HuNoV binding to human HBGAs and on the viral genome
The decay curves of the GII.4 HuNoV genomes treated with ribonuclease A (RNase A) and the binding capacity of viral particles to OLe+ saliva with or without sodium periodate treatment after exposure to natural ageing at 20 °C for 294 days in 10 mM PBS solution are shown in Fig. 5. No significant reduction in the RNase-treated genomes of HuNoVs was observed during the whole treatment (<1.2 log10) (paired sample t-test, p > 0.05) suggesting the persistence of potentially intact capsids because the genomes were not accessible to RNase A. Based on linear regression, the T90 was estimated at 294 days. During the whole treatment, a fraction of purified HuNoVs (mainly ranging between the LOD and the limit of quantification (LOQ) of the method) were able to bind to OLe+ saliva treated with sodium periodate, suggesting non-specific interactions between capsids and denatured HBGAs. Concerning GII.4 HuNoV binding to untreated OLe+ saliva, no significant variation was observed for the first 154 days with genome detection rates ranging from 4.50 to 5.20 log10 (paired sample t-test, p > 0.05). A 1.2 log10 reduction in the binding capacity was shown between day 154 and day 294, achieving almost the LOQ determined at 1,000 gc/mL (paired sample t-test, p < 0.03). Based on linear regression over the whole time period, the T90 was estimated at 166 days.

Persistence of GII.4 HuNoV genomes and GII.4 HuNoV binding to OLe+ saliva after exposure to natural ageing at 20 °C under low ionic strength condition (10 mM PBS). Quantification of purified GII.4 HuNoV gc was obtained by HBGA-binding assays followed by RNA amplification by RT-qPCR. Squares (▪, solid line) represent GII.4 HuNoV gc/mL treated with RNase. Circles (•, dotted line) and triangles (▲, dot-and-dash line) represent GII.4 HuNoV gc/mL quantified after capsid binding to untreated and sodium periodate-treated OLe+ human saliva, respectively. Each data point is an average of two replicates and error bars indicate standard deviations. Arrows (↓) represent data below the LOQ but above the LOD. The LOD and LOQ were respectively 200 and 1,000 gc/mL of purified GII.4 HuNoVs. Paired Student’s t-test was used to compare groups.
As performed for GII.4 VLPs under the same conditions, the log10 reductions in GII.4 HuNoV binding to OLe+ saliva after exposure to heat treatments (50 °C, 60 °C, and 72 °C) for 30 min were determined using the log (C/C0) (Fig. 6). A sodium periodate treatment was also added as a negative control for all assays (data not shown). No significant reduction in the binding capacity was observed after heating at 50 °C and 60 °C for 30 min (paired sample t-test, p > 0.05). Conversely, a statistically significant reduction was shown at 72 °C after 2 min until a complete loss of the genome detection after 3 min (paired sample t-test, p < 10−3). As described for VLPs, temperatures below 60 °C for 30 min affect slightly the BP, whereas 72 °C seems to impair the BP integrity. Interestingly, the behavior of viral capsids of GII.4 VLPs and HuNoVs seems to be similar after heat treatments in the tested conditions (Figs 3 and 6). Depending on the contact time, a critical temperature above which the viral capsid is disrupted, could be given between 60 °C and 72 °C.

Decay of GII.4 HuNoV binding to OLe+ saliva after exposure to heat treatments under high ionic strength condition (150 mM PBS). Squares (▪, solid line), circles (•, dotted line), and triangles (▲, dot-and-dash line) represent purified GII.4 HuNoV binding to untreated OLe+ human saliva after heating at 50 °C, 60 °C, and 72 °C for 30 min, respectively. The log (C/C0) values were calculated by dividing the mean GII.4 HuNoV gc/mL values obtained by HBGA-binding assays followed by RNA amplification by RT-qPCR at each time point (Ct) by the mean GII.4 HuNoV gc/mL values obtained at time 0 (C0). Each data point is an average of three replicates and error bars indicate standard deviations. Arrows (↓) represent data below the LOQ but above the LOD. The LOD and LOQ were 200 and 1,000 gc/mL of purified GII.4 HuNoVs, respectively. Paired Student’s t-test was used to compare groups.
Comparison of the loss of GII.4 VLP and HuNoV binding to OLe+ saliva after inactivation treatments
Because of the absence of linear relationship between OD450 values and the content of GII.4 VLPs, the time to reach the first 2 log10 reductions of HBGA-binding (T99) was used to compare the loss of GII.4 VLP and HuNoV binding to OLe+ saliva after natural ageing at 20 °C and heating at 72 °C (Table 1). For VLPs, T99 values were estimated between T0 (10 µg/mL) and the first time point when the LOD was reached (0.1 µg/mL). For HuNoVs, T99 values were derived from the slope of the linear regression analysis. After natural ageing at 20 °C, the T99 value of HuNoVs (333 days) is at least 4.3 fold higher than that of VLPs (ranging between 63 and 77 days) under the low ionic strength condition, demonstrating a different behavior of GII.4 VLPs and HuNoVs. Considering the similarity of the T99 values of VLPs at 10 mM and 150 mM PBS solutions, this result reinforces the fact that the ionic strength of the medium does not disturb the specificity of VLP-HBGA interactions. After heating at 72 °C, the T99 values were similar for GII.4 HuNoVs and VLPs and estimated at 0.002 days and ranging between 0.001 and 0.002 days, respectively.
Discussion
For the past two decades, many studies have tried to develop the most reliable strategy to distinguish infectious from non-infectious HuNoVs in foodstuffs and in the environment with the aim to improve the management of HuNoV hazard but also to better assess the effectiveness of inactivation treatments. These efforts have increased even more since the publication of the ISO 15216-1 standard13 in 2013 because of its inability to provide information about HuNoV infectivity when their genome is detected in vulnerable foodstuffs in prospective approaches such as surveys. Among all those proposed, the strategy based on the receptor/co-factor (HBGAs) binding to the BP to assess the capsid integrity of HuNoVs appeared promising28,41. The first objective of this study was to evaluate the ability of VLPs to mimic the behavior of HuNoVs for a same genotype (i.e. GII.4) during inactivation treatments. The second objective was to highlight the benefits/limitations of HuNoV binding to human HBGAs to potentially discriminate infectious from non-infectious particles or to prove the capsid integrity.
Our study provides new arguments to discuss whether VLPs might be suitable models to represent the behavior of HuNoVs. GII.4 VLPs are only self-assemblies of VP1 that also involve electrostatic or hydrophobic interactions in the environment44. Such interactions are strongly affected by the ionic strength of the medium. The results indicate that VLPs are only weakly affected by the ionic strength range of 10 to 150 mM at pH 7.4 over a long period of time at 20 °C, as observed for other complete viruses45,46. In the same way, the capsids of GII.4 VLPs and HuNoVs are both disrupted after heating at 72 °C, as it was observed for GII.4 VLPs at 68 °C by Moore et al.41. Ausar et al.19 have also shown that the native quaternary structure of NV is maintained until 60 °C but irreversible capsid disruptions occur above 65 °C. Structural changes in the capsid proteins and/or loss of binding affinity to host cells could occur above a critical temperature, as demonstrated for MS2 phage at 72 °C47.
Depending on the contact time, a critical temperature range of 60 °C to 72 °C leading to the disruption of the capsid may be proposed for VLPs as well as for HuNoVs. Surprisingly, the exact same critical temperature range has also been suggested for MNV48. Further research should be conducted to determine the critical contact time in the 60–72 °C temperature range.
On the other hand, our study also underlines major behavioral differences between GII.4 VLPs and HuNoVs. One difference is the size of the particles because part of VLPs have a smaller size (15 nm) than that expected for HuNoVs of about 40 nm43. But the main critical difference may be the stability of VLPs at 20 °C, which is at least 4.3 fold lower than that of HuNoVs. As highlighted by the T99 values, the reduction in VLP binding to human HBGAs was substantially more rapid than for HuNoVs under the same conditions. To our knowledge, no study has described the effect of natural ageing at 20 °C on the BP integrity of HuNoVs. Some differences may also exist at higher temperatures (above 50 °C) but the duration of treatment was not sufficient to draw any conclusions. Our results are consistent with the literature data regarding the effect of heat on GII.4 VLPs27,28. Hence, lesser stability of VLPs can be noted, at least in the BP region. It may be related to the absence of the viral genome or the VP2 protein which may play a role on HuNoV stability44,49. Dika et al.50 have shown that MS2 VLPs, which lack the genome and the maturation protein, have a capsid that expresses a different charge from that of the native particle, thus suggesting a different capsid organization. To summarize, GII.4 VLPs may be suitable for mimicking the behavior of HuNoVs with the exception of particle size and stability of BP below 72 °C. Thus, VLPs can be considered as relevant surrogates of HuNoVs when inactivation treatments induce significant changes in the capsid integrity, as described for some disinfectants20,51 and high-pressure processing29.
Bearing in mind the aforementioned limitations of VLPs, it is possible to discuss the relevance of HBGA-binding assays to reduce the over-estimation of the number of potential infectious HuNoVs given by the genome detection method. Substantial advantages of HBGA-binding approach can be suggested. Interactions between HuNoVs and HBGAs are very specific for numerous genotypes of HuNoVs. Although their role is not clearly defined in the host-cell infection process, HuNoV-HBGA interactions could be correlated with infectivity in inactivation assays18. HBGA-binding to the BP provides evidence of the presence of HuNoV capsids as it was confirmed by the lack of action of RNase A on the viral genome, suggesting persistence of potentially intact capsids. Conversely, this approach has several limitations that should be highlighted. The first point concerns methodological aspects. The specificity of HuNoV-HBGA interactions is dependent not only on the HBGA type (as previously demonstrated39), but also on the ionic strength of the media even though it does not disturb these interactions. The impact of the ionic strength is critical when comparing two different environmental media. Different interactions may be explained by the global charge of the viral particle. Considering the isoelectric point of GII.4 VLPs estimated to be between pH 5 and 644, the global charge of the particles at pH 7.4 is negative under the tested conditions. By increasing the ionic strength, the electric double layer of viral particles could be compressed leading to a decrease in repulsive electrostatic interactions and an increase in hydrophobic interactions between VLPs and HBGAs. Dika et al.52 pointed out this complex interplay by showing the adhesion features of different viral surrogates (i.e. FRNAPH) to abiotic surfaces as a function of ionic strength.
The second point is related to a more rapid reduction in the number of HuNoVs able to bind to human HBGAs (T90 estimated at 166 days) compared to the whole encapsidated HuNoVs at 20 °C (T90 estimated at 294 days). This result indicates that at least two kinds of HuNoV particles are present, both having a capsid (as shown by the resistance to RNase A) and a genome but only one being capable of binding to HBGAs. Even though there is no evidence that the latter are really infectious, the use of HBGA-binding assays may reduce the over-estimation resulting from solely using the genome detection method. Our results are consistent with a previous study describing the persistence of GII HuNoV genomes in drinking water at 21 °C since the T90 was estimated at 298 days using a non-linear model10. Seitz et al.53 have also shown that the NV remained infectious in groundwater at room temperature in the dark for 61 days and the capsid remained intact for at least 427 days. Finally, HuNoV capsids may specifically bind to HBGAs when inactivated particles have a structural integrity of the capsid but an altered genome, or an intact genome with capsid damage(s) located in surrounding regions of the BP.
For a full evaluation of the HBGA-binding strategy, experimental data are required to correlate the loss of HuNoV binding to human HBGAs with the loss of infectivity after inactivation treatments when cellular models for the in vitro replication of HuNoVs will be available for routine application. The use of human HBGAs to evaluate the capsid integrity of HuNoVs according to the inactivation treatments needs more comprehensive studies for improving our understanding focused on HuNoV-HBGA interactions and the structural changes in the capsid proteins. Further researches are underway to explore the molecular modifications occurred on the BP but also on the overall capsid during these inactivation treatments.
Materials and Methods
Preparation of GII.4 VLP and GII.4 HuNoV samples
GII.4 VLPs (Cairo4 strain, a 2007-Osaka variant) were kindly provided by the National Reference Center for Gastroenteritis Viruses (NRCgev, Dijon, France). Production and purification of GII.4 VLPs were performed using the baculovirus-expressed VP1 system, as described previously37,54. For all experiments, purified GII.4 VLPs were prepared at a final concentration of 100 µg/mL in TNC buffer using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The working suspensions were kept at 4 °C before use.
GII.4 HuNoVs from human stools were genotyped55 and kindly provided by the NRCgev (Dijon, France). The purification of GII.4 HuNoVs was performed using a method described previously56, with significant modifications. Briefly, 1 g of fecal suspensions of GII.4 HuNoVs was added to 10 mL of 150 mM PBS solution at pH 7.4. Samples were clarified by addition of 1/3 (v/v) of chloroform, vortexed for 60 sec and centrifuged at 2,500 × g for 5 min. Supernatant was submitted to a second clarification step as described previously. The clarified samples were purified by dialysis using Float-A-Lyzer G2 with Biotech Cellulose Ester membranes (MWCO: 100 kD, volume: 1 mL, Spectra/Por®, Spectrum Laboratories, CA, USA). The membranes were dialyzed in a tank containing 10 L of 10 mM PBS at 4 °C overnight with gentle stirring. Dialysate samples were recovered and filtered using a 0.22 µm cellulose acetate membrane. After each step, the loss of HuNoV genomes was controlled. The viral genomes from 100 µL of purified HuNoV suspensions were extracted directly using the NucliSENS® easyMAG® kit (bioMérieux, Marcy-l’Etoile, France). The HuNoV genomes were detected using an RNA UltraSense One-Step quantitative RT-PCR system (Life Technologies, Carlsbad, CA, USA), according to the ISO 15216-1 standard recommendations13. Cq values were determined using CFX Maestro software (BioRad, Hercules, CA, USA). Quantification of HuNoV gc was performed using a standard curve of plasmids with a concentration range of 5 to 105 gc/reaction mixture. Until use, the purified HuNoV suspensions were stored at 4 °C in the dark at a final concentration of 2.106 gc/mL.
Preparation and determination of HBGA types in human saliva
Saliva samples from 27 healthy adult volunteers were collected in 50 mL sterile polypropylene tubes. The saliva samples used in this study were described previously following approval by the Nantes University Hospital Review Board (study number BRD02/2-P)37, and informed consent was obtained from all donors. All experiments using saliva were performed in accordance with relevant guidelines and regulations. After harvesting, saliva samples were boiled at 100 °C for 5 min and then centrifuged at 10,000 × g for 5 min. Supernatants were stored at −20 °C until use. The presence of A, B, O blood group antigens and Lewis antigens in saliva samples at a dilution of 1:1,000 in 150 mM PBS was determined by ELISA, as described previously22.
Natural ageing at 20 °C of GII.4 VLPs and HuNoVs
One mL of 10 µg/mL purified GII.4 VLPs in 10 mM and 150 mM PBS solutions and 1 mL of 2.106 gc/mL of purified GII.4 HuNoVs in 10 mM PBS solution were exposed to natural ageing at 20 °C. This treatment, corresponding to the application of circadian cycles with a 24 h rotation of natural light and obscurity between October 2018 and July 2019, was evaluated over 105 days and 294 days for VLPs and HuNoVs, respectively. To that end, 20 separate 1 mL aliquots of VLPs and HuNoVs were placed in 2 mL Protein LoBind tubes. These aliquots were derived from a unique stock solution of 20 mL of VLPs and HuNoVs. For each sample, four and two analyses were performed for VLPs and HuNoVs, respectively.
Heat treatments of GII.4 VLPs and HuNoVs
One mL of 10 µg/mL purified GII.4 VLPs and 1 mL of 2.105 gc/mL of purified GII.4 HuNoVs in 150 mM PBS solution were separately exposed at 50 °C, 60 °C, and 72 °C for 30 min. Before adding 100 µL of 100 µg/mL GII.4 VLPs or 2.106 gc/mL of GII.4 HuNoVs in borosilicate 4.9 glass tubes (thickness: 0.4 mm, diameter: 12 mm, Fiolax, Schott AG, Mitterteich, Germany), 900 µL of 150 mM PBS solution was pre-warmed in a glycol bath GR150 S5 (Grant Instruments, Cambridge, UK) to minimize the effect of the temperature rise. After heating, treated samples were immediately placed on ice to stop the reaction. The temperature was monitored in the glycol bath but also in a control tube containing 1 mL of 150 mM PBS solution using temperature probes (SSA-TF, E-Val™ Pro, ELLAB, Hillerød, Denmark) coupled to a data logger (Valsuite™ Basic software, ELLAB, Hillerød, Denmark). Experiments were performed in triplicate for VLPs and HuNoVs under the same conditions.
GII.4 VLP binding to saliva samples: HBGA-binding ELISA
HBGA-binding ELISA was performed using the method described previously22, with minor adjustments. Briefly, 100 µL of supernatant of saliva samples of known HBGA types were coated in 96-well microtiter plates at a dilution of 1:1,000 in 150 mM PBS at 4 °C overnight. Negative controls were systematically added using saliva-coated wells treated with sodium periodate as described previously42, with slight modifications. After blocking with 200 µL of 5% non-fat dried milk at 37 °C for 2 h, 100 µL of native and treated GII.4 VLPs diluted in PBS solutions was incubated at 37 °C for 1 h. The VLP binding to the saliva samples was detected with anti-GII.4 VLP MAbs kindly provided by the NRCgev (Dijon, France) at a 1:10,000 dilution in 150 mM PBS for 1 h at 37 °C, followed by the addition of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Chemicon, Merck Millipore, Burlington, MA, USA) at a dilution of 1:30,000 in 150 mM PBS for 1 h at 37 °C. Between each step, the wells were washed three times with 200 µL of 150 mM PBS containing 0.05% Tween 20 using a microplate washer (Elx50, BioTeK Instruments, Winooski, VT, USA). The enzyme activity was detected using the TMB kit (Pierce Biotechnology, Rockford, IL, USA). After incubation for 15 min at room temperature, the reaction was stopped by adding 100 µL of 1 M sulfuric acid. The OD values of the enzyme activity were read at 450 nm with an enzyme immunoassay spectrum reader (Elx800, BioTeK Instruments, Winooski, VT, USA). All samples were analyzed in duplicate. Inconsistent results were rejected if the gap between OD450 value duplicates was above 0.1. To give confidence, the P/N ratio was calculated by dividing the mean OD450 values of the sample by the mean OD450 values of the negative control. When P/N ratio was above 2, results were considered positive. When P/N ratio was below 2, results were considered negative57.
The sensitivity and the linearity of the HBGA-binding ELISA method were determined using the supernatant of OLe+ saliva sample and various contents of GII.4 VLPs (ranging from 0.05 and 10.0 µg/mL) diluted in 10 mM and 150 mM PBS solutions. The experiments were performed in four replicates.
GII.4 HuNoV binding to saliva samples: HBGA-binding assays followed by RNA amplification by RT-qPCR
This method was performed using a combination of approaches described previously22,56. Briefly, supernatant of OLe+ saliva sample was coated in 96-well microtiter plates as described above. Negative controls were also added using the same saliva sample treated with sodium periodate. After blocking with non-fat dried milk, 100 µL of native and treated GII.4 HuNoV suspensions, previously treated by RNase A58, was incubated at 37 °C for 1 h. Between each step, the wells were washed three times as described above. The bound capsid proteins of HuNoVs were lysed directly in the wells using 150 µL of a buffer containing 5 M of guanidium thiocyanate for 10 min at room temperature. For each condition (native or treated HuNoVs, negative controls), 10 wells were pooled on the same 96-well microtiter plate to analyze 1 mL of purified HuNoVs. The viral genomes were purified and detected as described previously. The HuNoV genomes were also detected after RNase A treatment. The LOD was determined at 200 gc/mL and the LOQ was determined at 1,000 gc/mL.
TEM observations
GII.4 VLPs exposed to heating were observed by TEM after negative staining with phosphotungstic acid. Briefly, 10 µL of heated GII.4 VLPs were absorbed to carbon coated copper grids by incubating at room temperature for 3 min. The excess was eliminated and the copper grids were negatively stained with 2% phosphotungstic acid by incubating at room temperature for 1 min. The stained copper grids were air dried for 3 min and then observed by TEM.
Statistical analysis
All statistical analyses were performed using R statistical software (Rx64 v.3.5.3). The Shapiro-Wilk test was performed to check normality of the data with alpha level of 0.05. If data set followed a normal distribution (p > 0.05), parametric tests were applied. A paired or unpaired sample Student’s t-test was performed for dependent or independent data following a normal distribution, respectively. Non-parametric tests were used for data sets having a non-normal distribution (p < 0.05). For dependent or independent data with non-normal distribution, a Wilcoxon signed-rank test or a Mann-Whitney U test was applied. For all tests, the significance level was set to 0.05.
Data availability
All data analyzed in this study are included in this published article and also its Supplementary Information files.
References
European Food Safety Authority (EFSA). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food‐borne outbreaks in 2017. EFSA J. 16 (2018).
CDC. https://www.cdc.gov/norovirus/trends-outbreaks/index.html (2018).
Zheng, D.-P. et al. Norovirus classification and proposed strain nomenclature. Virology 346, 312–323 (2006).
van Beek, J. et al. Molecular surveillance of norovirus, 2005–16: an epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 18, 545–553 (2018).
Atmar, R. L. et al. Norwalk virus shedding after experimental human infection. Emerg. Infect. Dis. 14, 1553–1557 (2008).
Atmar, R. L. et al. Determination of the 50% human infectious dose for Norwalk virus. J. Infect. Dis. 209, 1016–1022 (2014).
Teunis, P. F. M. et al. Norwalk virus: how infectious is it? J. Med. Virol. 80, 1468–1476 (2008).
Hirneisen, K. A. et al. Viral Inactivation in Foods: A Review of Traditional and Novel Food-Processing Technologies. Compr. Rev. Food Sci. Food Saf. 9, 3–20 (2010).
Donaldson, E. F., Lindesmith, L. C., Lobue, A. D. & Baric, R. S. Viral shape-shifting: norovirus evasion of the human immune system. Nat. Rev. Microbiol. 8, 231–241 (2010).
Kauppinen, A. & Miettinen, I. T. Persistence of Norovirus GII Genome in Drinking Water and Wastewater at Different Temperatures. Pathog. Basel Switz. 6 (2017).
Bellou, M., Kokkinos, P. & Vantarakis, A. Shellfish-borne viral outbreaks: a systematic review. Food Environ. Virol. 5, 13–23 (2013).
Blanco, A. et al. Norovirus in Bottled Water Associated with Gastroenteritis Outbreak, Spain, 2016. Emerg. Infect. Dis. 23, 1531–1534 (2017).
ISO. Microbiology of the food chain - Horizontal method for determination of hepatitis A virus and norovirus using real-time RT-PCR - Part 1: Method for quantification (2017).
Boudaud, N. & Gantzer, C. Viral Hazard Assessment in food matrices. Bull Epidemiol Santé Anim Aliment 68, 26–30 (2015).
Hartard, C. et al. F-Specific RNA Bacteriophages, Especially Members of Subgroup II, Should Be Reconsidered as Good Indicators of Viral Pollution of Oysters. Appl. Environ. Microbiol. 84 (2018).
Ettayebi, K. et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016).
Bosch, A. et al. Foodborne viruses: Detection, risk assessment, and control options in food processing. Int. J. Food Microbiol. 285, 110–128 (2018).
Manuel, C. S., Moore, M. D. & Jaykus, L.-A. Predicting human norovirus infectivity - Recent advances and continued challenges. Food Microbiol. 76, 337–345 (2018).
Ausar, S. F., Foubert, T. R., Hudson, M. H., Vedvick, T. S. & Middaugh, C. R. Conformational stability and disassembly of Norwalk virus-like particles. Effect of pH and temperature. J. Biol. Chem. 281, 19478–19488 (2006).
Sato, J. et al. Effects of disinfectants against norovirus virus-like particles predict norovirus inactivation. Microbiol. Immunol. 60, 609–616 (2016).
Jiang, X., Wang, M., Graham, D. Y. & Estes, M. K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66, 6527–6532 (1992).
Huang, P. et al. Noroviruses bind to human ABO, Lewis, and secretor histo-blood group antigens: identification of 4 distinct strain-specific patterns. J. Infect. Dis. 188, 19–31 (2003).
Kostela, J. et al. Amplification by long RT-PCR of near full-length norovirus genomes. J. Virol. Methods 149, 226–230 (2008).
Nuanualsuwan, S. & Cliver, D. O. Pretreatment to avoid positive RT-PCR results with inactivated viruses. J. Virol. Methods 104, 217–225 (2002).
Oristo, S., Lee, H.-J. & Maunula, L. Performance of pre-RT-qPCR treatments to discriminate infectious human rotaviruses and noroviruses from heat-inactivated viruses: applications of PMA/PMAxx, benzonase and RNase. J. Appl. Microbiol. 124, 1008–1016 (2018).
Sano, D., Pintó, R. M., Omura, T. & Bosch, A. Detection of oxidative damages on viral capsid protein for evaluating structural integrity and infectivity of human norovirus. Environ. Sci. Technol. 44, 808–812 (2010).
Moore, M. D. & Jaykus, L.-A. A plate-based histo-blood group antigen binding assay for evaluation of human norovirus receptor binding ability. Anal. Biochem. 533, 56–59 (2017).
Li, D. et al. Evaluation of methods measuring the capsid integrity and/or functions of noroviruses by heat inactivation. J. Virol. Methods 181, 1–5 (2012).
Lou, F. et al. High-pressure inactivation of human norovirus virus-like particles provides evidence that the capsid of human norovirus is highly pressure resistant. Appl. Environ. Microbiol. 78, 5320–5327 (2012).
Lindesmith, L. et al. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9, 548–553 (2003).
Hutson, A. M., Atmar, R. L., Graham, D. Y. & Estes, M. K. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 185, 1335–1337 (2002).
Marionneau, S. et al. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122, 1967–1977 (2002).
Rockx, B. H. G., Vennema, H., Hoebe, C. J. P. A., Duizer, E. & Koopmans, M. P. G. Association of histo-blood group antigens and susceptibility to norovirus infections. J. Infect. Dis. 191, 749–754 (2005).
Huang, P. et al. Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 79, 6714–6722 (2005).
Marionneau, S. et al. ABH and Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie 83, 565–573 (2001).
Tan, M. et al. Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: evidence for a binding pocket. J. Virol. 77, 12562–12571 (2003).
de Rougemont, A. et al. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 85, 4057–4070 (2011).
Shirato, H. et al. Noroviruses distinguish between type 1 and type 2 histo-blood group antigens for binding. J. Virol. 82, 10756–10767 (2008).
Tan, M. & Jiang, X. Histo-blood group antigens: a common niche for norovirus and rotavirus. Expert Rev. Mol. Med. 16, e5 (2014).
Nordgren, J. & Svensson, L. Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses 11 (2019).
Moore, M. D., Mertens, B. S. & Jaykus, L.-A. Alternative In Vitro Methods for the Determination of Viral Capsid Structural Integrity. J. Vis. Exp. JoVE, https://doi.org/10.3791/56444 (2017).
Maalouf, H. et al. Distribution in tissue and seasonal variation of norovirus genogroup I and II ligands in oysters. Appl. Environ. Microbiol. 76, 5621–5630 (2010).
White, L. J., Hardy, M. E. & Estes, M. K. Biochemical characterization of a smaller form of recombinant Norwalk virus capsids assembled in insect cells. J. Virol. 71, 8066–8072 (1997).
Da Silva, A. K., Kavanagh, O. V., Estes, M. K. & Elimelech, M. Adsorption and aggregation properties of norovirus GI and GII virus-like particles demonstrate differing responses to solution chemistry. Environ. Sci. Technol. 45, 520–526 (2011).
Moore, R. S., Taylor, D. H., Sturman, L. S., Reddy, M. M. & Fuhs, G. W. Poliovirus adsorption by 34 minerals and soils. Appl. Environ. Microbiol. 42, 963–975 (1981).
Gantzer, C., Quignon, F. & Schwartzbrod, L. Poliovirus‐1 adsorption onto and desorption from montmorillonite in seawater. Survival of the adsorbed virus. Environ. Technol. 15, 271–278 (1994).
Wigginton, K. R., Pecson, B. M., Sigstam, T., Bosshard, F. & Kohn, T. Virus inactivation mechanisms: impact of disinfectants on virus function and structural integrity. Environ. Sci. Technol. 46, 12069–12078 (2012).
Brié, A. et al. The Effect of Heat and Free Chlorine Treatments on the Surface Properties of Murine Norovirus. Food Environ. Virol. 9, 149–158 (2017).
Pogan, R., Dülfer, J. & Uetrecht, C. Norovirus assembly and stability. Curr. Opin. Virol. 31, 59–65 (2018).
Dika, C., Duval, J. F. L., Ly-Chatain, H. M., Merlin, C. & Gantzer, C. Impact of internal RNA on aggregation and electrokinetics of viruses: comparison between MS2 phage and corresponding virus-like particles. Appl. Environ. Microbiol. 77, 4939–4948 (2011).
Manuel, C. S., Moore, M. D. & Jaykus, L.-A. Efficacy of a disinfectant containing silver dihydrogen citrate against GI.6 and GII.4 human norovirus. J. Appl. Microbiol. 122, 78–86 (2017).
Dika, C., Chatain-Ly, M. H., Francius, G., Duval, J. F. L. & Gantzer, C. Non-DLVO adhesion of F-specific RNA bacteriophages to abiotic surfaces: Importance of surface roughness, hydrophobic and electrostatic interactions. Colloids Surf. Physicochem. Eng. Asp. 435 (2013).
Seitz, S. R. et al. Norovirus infectivity in humans and persistence in water. Appl. Environ. Microbiol. 77, 6884–6888 (2011).
Kamel, A. H. et al. Predominance and circulation of enteric viruses in the region of Greater Cairo, Egypt. J. Clin. Microbiol. 47, 1037–1045 (2009).
Ayouni, S. et al. Relationship between GII.3 norovirus infections and blood group antigens in young children in Tunisia. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 21(874), e1–8 (2015).
Langlet, J., Kaas, L. & Greening, G. Binding-Based RT-qPCR Assay to Assess Binding Patterns of Noroviruses to Shellfish. Food Environ. Virol. https://doi.org/10.1007/s12560-015-9180-x (2015).
Maalouf, H. et al. Strain-dependent norovirus bioaccumulation in oysters. Appl. Environ. Microbiol. 77, 3189–3196 (2011).
Brié, A., Bertrand, I., Meo, M., Boudaud, N. & Gantzer, C. The Effect of Heat on the Physicochemical Properties of Bacteriophage MS2. Food Environ. Virol. 8, 251–261 (2016).
Acknowledgements
The results of this study were obtained within the scope of OxyVir, a project funded by the “Fonds Européen pour les Affaires Maritimes et la Pêche” (FEAMP). This study was also supported by the Joint Technological Unit ACTIA VIROcontrol and the “Conseil Régional de Normandie”.
Author information
Authors and Affiliations
Contributions
N.B., C.G., and G.B. designed the study. M.R., M.C., and J.L. performed the experiments. M.R., M.C., G.B., A.R., and C.G. analyzed the data. M.R., C.G., and N.B. wrote the manuscript. M.R. and M.C. prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Robin, M., Chassaing, M., Loutreul, J. et al. Effect of natural ageing and heat treatments on GII.4 norovirus binding to Histo-Blood Group Antigens. Sci Rep 9, 15312 (2019). https://doi.org/10.1038/s41598-019-51750-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51750-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
