
Abstract
The electronic structure, structural stability, mechanical, phonon, and optical properties of Zr2GaC and Hf2GaC MAX phases have been investigated under high pressure using first-principles calculations. Formation enthalpy of competing phases, elastic constants, and phonon calculations revealed that both compounds are thermodynamically, mechanically, and dynamically stable under pressure. The compressibility of Zr2GaC is higher than that of Hf2GaC along the c-axis, and pressure enhanced the resistance to deformation. The electronic structure calculations reveal that M2GaC is metallic in nature, and the metallicity of Zr2GaC increased more than that of Hf2GaC at higher pressure. The mechanical properties, including elastic constants, elastic moduli, Vickers hardness, Poisson’s ratio anisotropy index, and Debye temperature, are reported with fundamental insights. The elastic constants C11 and C33 increase rapidly compared with other elastic constants with an increase in pressure, and the elastic anisotropy of Hf2GaC is higher than that of the Zr2GaC. The optical properties revealed that Zr2GaC and Hf2GaC MAX phases are suitable for optoelectronic devices in the visible and UV regions and can also be used as a coating material for reducing solar heating at higher pressure up to 50 GPa.
Similar content being viewed by others

Introduction
MAX phase materials (with general formula Mn+1AXn n = 1–3) are the transition metal ternary carbides and nitrides, which provides the bridge between metals and ceramics in terms of properties, and have been attracted significant attention of the scientific community since they were discovered1. ‘M’ is denoted as early transition metals, ‘A’ represents the IIIA or IVA elements in the periodic table, and ‘X’ is either carbon or nitrogen. The MAX phases crystallize in P63/mmc hexagonal structure, having the combination of strong covalent M–X bonds and relatively weak metallic M–A bonds, which are responsible for their hybrid properties2,3. The unit cell of the MAX phase consists of edge-shared M6X octahedra is sandwiched by an A-atom sheet. To be specific, these compounds are machinable, thermal and electrical conductor, damage and irradiation tolerant, corrosion and oxidation resistant, possess high strength and stiffness at high temperature, and having low densities4,5,6,7,8. These outstanding attributes make MAX phase materials potential candidates applicable for wear, oxidation, and corrosion resistant coating materials9,10,11,12,13, superconducting materials14, and the cladding material in a nuclear reactor15. Moreover, the MXenes are the 2D derivatives of MAX phases possessing useful application in Li-ion and sodium-ion batteries and supercapacitors16,17,18.
In order to take the full advantages of MAX phase materials in technological applications, a series of experimental and theoretical calculations have been done so far. For example, T. Lapauw et al. synthesized Zr2AlC and Hfn+1AlCn (n = 1, 2) experimentally, and their lattice parameter was in good agreement with first-principle investigations19,20 and Hu et al.21 fabricated Nb4AlC3 by using the spark plasma sintering (SPS) technique and investigated the thermal expansion and electrical conductivity. In addition, Petruhins et al.22 predicted the phase stability, and the magnetic state of Cr2GaC and thin film of Cr2GaC was also prepared using the magnetron sputtering technique, and Hoffman et al.23 investigated the neutron irradiation tolerance behavior of Tin+1ACn (A = Al, Si, n = 1, 2) MAX phases and found that their radiation-hard is similar to that of SiC which is mostly used material in nuclear reactors. Moreover, it is experimentally proved that Al-based MAX phase materials are excellent oxidation resistant as bulk material and thin-film form because a protective Al2O3 layer formed, which further hinders the oxidation of core material9,24. The theoretical research on the MAX phase material is mainly based on density functional theory (DFT)25 calculations, and numerous studies can be found in literature26,27.
The theoretical studies related to MAX phase materials are enormous, and it's growing abruptly compared to experimental work. Recently, the electronic, elastic, thermodynamic, and vibrational properties of M2GaC MAX phases with M = Ti, Mo, V, Nb, Mn, and Cr have been studied theoretically14,22,28,29,30,31. Romeo et al. investigated the structural, elastic, and electronic properties of Nb2AC (where A = Sn, In, and S) under pressure range from 0 to 10 GPa and found the linear compressibility of unit cell along the c axis compared with the increase in pressure32,33. Similarly, Bouhemadou et al. investigated the structural, electronic, and elastic of a wide range of M2AX phases34,35,36, and pressure effect on structural and elastic properties was comparatively studied37,38,39. Among numerous computed investigations, the M2GaC (M = Zr, Hf) MAX phases are the least studied 211-type MAX phases26,40,41. Recently, the structural stability, elastic, phonon, and thermodynamic properties of Zr2GaC and Hf2GaC have been studied42 and computed elastic properties are in excellent agreement with the available data43. However, the properties of Zr2GaC and Hf2GaC MAX phases need to be studied under high pressure, and the stability of these compounds with respect to their competing phases should be explored further.
These above mentioned experimental and theoretical studies motivated us to investigate the electronic structure, mechanical, dynamical, and optical characteristics of Zr2GaC and Hf2GaC MAX phases under high pressure. In the present work, the structural stability, electronic, mechanical, phonon, and optical properties of the M2GaC (M = Zr, Hf) MAX phase materials have been investigated under pressure ranging from 0 to 50 GPa using the first-principles plane-wave pseudopotential DFT within the generalized gradient approximation (GGA). The results showed that the M2GaC MAX phases are electronically, elastically, and optically anisotropic in nature and suitable for high-temperature application, coating material, and optoelectronic devices. The paper is organized as follows: detailed computational methods is second section, the obtained results and discussion of M2GaC are presented in third section, and in fourth section summary of this research is given.
Computational details
The Density Functional Theory (DFT) is an ideal quantum mechanical tool to determine the ground state properties and electronic structures of the molecules and solid materials44. The calculations presented in this work were performed using the Cambridge Serial Total Energy Package (CASTEP) based on DFT. The exchange–correlation was treated within the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)45 with the plane-wave ultrasoft pseudopotential code. Exchange correlation in GGA is a function of charge density ρ(r) and spatial gradient and empirically written as; \(E_{XC}^{GGA} = \int {\varepsilon_{XC} F\left( {\rho ,\nabla \rho } \right)} d^{3} r\). The accuracy of calculations depends on two parameters, i.e., first, the kinetic energy cut-off, which determines the number of plane waves in the expansion, and second, the special k-points used for the Brillouin zone (BZ) integration. For both M2GaC (M = Zr, Hf) MAX phases, plane waves cut off energy was set at 600 eV, and the Monkhorst-pack46 scheme of 15 × 15 × 3k-points was employed, respectively. The ground state structural parameters were determined using Broyden–Fletcher–Goldfarb–Shanno (BFGS)47 minimization technique while the convergence tolerance were set as follows: total energy tolerance less than 5 × 10–6 eV/atom, stress component less than 0.02 GPa, maximum force tolerance 0.01 eV/Ǻ, and maximum displacement of the atom during the geometric optimization was less than 0.0005 Ǻ. Finally, Debye’s temperature, mean, transverse, and longitudinal sound velocities were calculated using elastic constants. For the dynamical stability of M2GaC MAX phases, the phonon dispersion was computed using a finite displacement method implemented in Material Studio48. The crystal orbital Hamilton population (COHP) calculations are performed using an open-source Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER) code to investigate the chemical bonding49,50,51.
The enthalpy of formation of M2GaC phase was calculated through the linear optimization procedure as:
where \(H_{211}\) is the enthalpy of the 211-M2GaC MAX phases and \(H_{{competing{ - }phases}}\) is the total enthalpy of the set of competing phases.
The energy of formation per atom \(\left( {\mathop E\nolimits_{for}^{{M_{2} GaC}} } \right)\) for M2GaC MAX phases can be calculated as52:
where \(E_{total}^{{M_{2} GaC}}\), \(E_{solid}^{M}\), \(E_{solid}^{Ga}\), and \(E_{solid}^{C}\) are the total energy of M2GaC MAX phase, M, Ga, and C atoms in the solid form, and x, y, z is the number of atoms for M, Ga, and C elements in the unit cell, respectively.
The distortion parameter for octahedra (\(o_{r}\)) and that of trigonal prism (\(p_{r}\)) can be defined as follows53,54:
The DOS at Fermi level is used to investigate the metallicity at ambient temperature using following expression55,56:
where \(n_{m}\) is the thermally excited number of electrons and \(n_{e}\) is the total number of valence electrons in the unit cell. \(k_{B}\) and \(N\left( {E_{F} } \right)\) are the Boltzmann constant, and value of DOS at Fermi level in unit states/eV/unit cell, respectively.
The Fermi energy of M2GaC MAX phases are used to estimate the velocity of electron (\(v_{F}\)) near the Fermi level:
Then use this value to estimate the conductivity (σ) as:
where \(\tau\), m, n, e, and l are the time between two collisions, mass of electron, number of electrons, electron’s charge, and mean free path of electron, respectively.
To calculate the bulk, shear, and Young’s modulus, the following equations used within the Voigt (V)57, Russ (R)58,59, and Voigt-Russ and Hill (VRH)60,61 approximation scheme:
where Bv, Gv, and Br, Gr are the bulk and shear modulus in terms of Voigt and Russ approximation, respectively. The Values of Young’s modulus and Poisson’s ratio obtained by:
The mechanical Anisotropy (A) calculated as follows:
To calculate the hardness, the semi-empirical method based on Pugh’s ratio, which is proposed by the Chen et al.62, was adopted as:
where k is the Pugh’s ratio (G/B), and G is the shear modulus. The Debye temperature determined by the Anderson method63:
The transverse sound velocity (Vt), longitudinal sound velocity (Vl), and mean sound velocity (Vm) calculated by:
where h is Planck’s constant, kB is Boltzmann’s constant, n represents the number of atoms per unit cell, Va is the atomic volume, respectively. The melting temperature (Tm) of MAX phase materials having hexagonal crystal structure was calculated from the elastic constants as follows64:
The 3D Young’s modulus surface was obtained using the following equation65:
where Sij and li represent the elastic compliance tensor of M2GaC MAX phases and direction cosine in the sphere coordination, respectively.
The value of the imaginary part can be calculated from the moment matrix element between the occupied and unoccupied electronic states as:
where e is the electronic charge, \(\omega\) is the light frequency, u is the vector defining the polarization of the incident electric field, \(\psi_{k}^{c}\) and \(\psi_{k}^{v}\) are the conduction and valence band wave function at k, respectively.
Results and discussion
Structural properties and compressibility
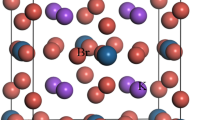
The considered M2GaC (M = Zr, Hf) MAX phase crystallizes in the hexagonal structure with a space group P63/mmc (No. 194) in which M6C (M = Zr, Hf) edge shared octahedron is interleaved by atomic layer of Ga atom. There are eight atoms per unit cell in MAX phase compounds, and the unit cell contains two formula units. The atoms in the M2GaC MAX phase are placed as follows: C atoms are at 2a (0, 0, 0), Ga atoms are at 2d (2/3, 1/3, 1/4), and M atoms are at 4f. (1/3, 2/3, zM), respectively where zM is known as the internal parameter. Figure 1 shows the optimized unit cell of the M2GaC MAX phase at 0 GPa pressure. The optimized parameters for each pressure up to 50 GPa with the pressure step of 10 GPa is tabulated in Table 1. With the increase in pressure, the lattice parameters, unit cell volume reduced while the internal parameter increases for both the MAX phases studied.

The unit cell of the M2GaC MAX phase (M = Zr, Hf). An edge shared [M6X] octahedra and a [M6A] trigonal prism are outlined.
The thermodynamic stability of M2GaC MAX phase materials is predicted in terms of formation enthalpy \(\left( {\Delta H_{cp} } \right)\) by comparing the total energy M2GaC MAX phase to the energy of non-MAX competing phases (single elements, binary and ternary compounds). Table 2 shows the most competing phases considered for M2GaC MAX phases determined by using linear optimization procedure66. This linear optimization procedure has been successfully used for predicting many MAX phases67,68 in which a phase is considered to be stable if \(\Delta H_{cp} < 0\). Based on identified competing phases, the Eq. (1) for Zr2GaC and Hf2GaC MAX phases can be rewritten as:
Both the Zr2GaC and Hf2GaC phases fulfill the criterion \(\Delta H_{cp} < 0\), indicating that the M2GaC MAX phases are thermodynamically stable and can be formed experimentally. Moreover, the calculated formation energy per atom for Zr2GaC and Hf2GaC at 0 GPa is − 7.59 eV/atom and − 7.45 eV/atom, respectively. There has been a increase in energy of formation for Zr2GaC and Hf2GaC as pressure is increased.
Figure 2 shows the effect of pressure on the normalized lattice parameters \(a/a_{0}\), c/\(c_{0}\) and volume V/V0 (\(a_{0}\), \(c_{0}\), and V0 are the lattice parameters and volume at the 0 GPa pressure). The compressibility for both the M2GaC MAX phase along the c-axis is more than that of the a-axis as pressure increases from 0 to 50 GPa32,33. Similarly, the volume change ratio gradually (V/V0) decreases with an increase in pressure, indicating that the compressibility of the M2GaC MAX phase system is strong. The compressibility results are in good agreement with other Zr and Hf based MAX phases37,39,69. Moreover, the V/V0 of Zr2GaC and Hf2GaC was reduced to 21.1% and 19.1%, respectively. In other words, external pressure has a more significant effect on Zr2GaC than the Hf2GaC MAX phase. Zhao et al.70 studied the properties of Ti3AC2 (A = Al, and Si) MAX phases under pressure in which the ratio of V/V0 of Ti3AlC2 and Ti3SiC2 was reduced to 18.7% and 16.9%, respectively. It is worth noticing that at high pressure, the volume ratio curve becomes steady, indicating that change in atomic distance is smaller, which results in stronger mutual repulsion as atoms come further closer; eventually, compression of the crystal becomes more difficult. Similarly, the reduction in lattice parameter ratios (\(a/a_{0}\), c/\(c_{0}\)) are in the order of Zr2GaC > Hf2GaC. Figure 3 exhibits the normalized bond lengths \({\raise0.7ex\hbox{${l_{1} }$} \!\mathord{\left/ {\vphantom {{l_{1} } {l_{10} }}}\right.\kern-\nulldelimiterspace} \!\lower0.7ex\hbox{${l_{10} }$}}\) and \({\raise0.7ex\hbox{${l_{2} }$} \!\mathord{\left/ {\vphantom {{l_{2} } {l_{20} }}}\right.\kern-\nulldelimiterspace} \!\lower0.7ex\hbox{${l_{20} }$}}\) (where \(l_{10}\) and \(l_{20}\) are the bond lengths of M–Ga and M–C at 0 GPa, respectively) M–Ga and M–C atoms within the M2GaC (M = Zr, Hf) MAX phase unit cell versus pressure. It can be noted that the bond length M–Ga (M = Zr, Hf) becomes steeper than that of M–C, indicating that the direction along the M–Ga is easily compressed compared to the M–C bond. These results agreed with a weaker metallic bond between M–Ga atoms in the unit cell, which defines the lattice parameter c. The bond lengths of Zr–Ga and Zr–C reduced more than Hf–Ga and Hf–C, exhibiting that the Zr2GaC MAX phase is more compressible than Hf2GaC along the Zr–Ga direction.

The pressure dependence of \({\text{a}}/{\text{a}}_{0}\), c/\({\text{c}}_{0}\) and volume V/V0 for M2GaC (M = Zr, Hf) MAX phase at T = 273 K.

Variation of normalized bond lengths between M–Ga and M–C atoms of M2GaC (M = Zr, Hf) MAX phase with pressure.
Furthermore, the c/a and the internal parameter z were used to calculate the distortion within the structure. As mentioned earlier that the crystal structure of the MAX phase is hexagonal and constituted by [M6X] octahedron and [M6A] trigonal prism. For an ideal structure, the octahedra and trigonal parameters should be equal to one (\(o_{r}\) = \(p_{r}\) = 1), the variation of \(o_{r}\) and \(p_{r}\) from 1 tells the distortion in these polyhedra. Moreover, Aydin et al. proposed that the smaller the distortion, the more stable the structure is71. In this work, the distortion parameters deduced from the optimized lattice parameters for M2GaC MAX are: \(o_{r}\) = 1.071 and \(p_{r}\) = 1.109 for Zr2GaC, and \(o_{r}\) = 1.058 and \(p_{r}\) = 1.128 for Hf2GaC, respectively. This indicates the distortion in the octahedra and trigonal prism in both proposed MAX phase structures and the distortion is small and similar because \(o_{r}\)/\(p_{r}\) = 0.93–0.97. This behaviour is explained as steric effect72. The reported \(o_{r}\) and \(p_{r}\) for Ti2GaC MAX phase are 1.088 and 1.081 with \(o_{r}\)/\(p_{r}\) = 1.0053. It can be seen that the octahedra and trigonal prisms’ distortion in Ti2GaC is smaller than that of Zr2GaC and Hf2GaC MAX phases. Thus, it is concluded that the structure of Ti2GaC is more stable than the M2GaC (M = Zr, Hf) MAX phases. The distortion parameters for IV-B and V-B group transition metal MAX phases are deduced from their optimized lattice parameters in the literature for comparison38,53. We found that, with increasing the valance of M element (Ti \(\to Zr \to Hf\), and V \(\to Nb \to Ta\)), \(o_{r}\) decreases towards value 1, but \(p_{r}\) increases to values higher than 1 in M2GaC (See Table 3). In other words, increasing the valance of M element in M2GaC MAX phases, distortion in octahedra decreases and tends to be stable while distortion in trigonal prisms increases. The distortion in [M6X] octahedron and [M6A] trigonal prisms can be observed in other MAX phases with different A-site elements26.
Electronic properties
The band structure for the M2GaC MAX phase at equilibrium lattice parameter within the GGA-PBE was calculated and discussed. Figure 4 shows the band structures from − 15 to 8 eV energy range along the high symmetry lines of the Brillouin zone (Γ–A–H–K–Γ–M–L–H) at 0, 30, and 50 GPa. The conduction bands overlap with the valance band at the Fermi level without having a bandgap in the vicinity of the Fermi level resulting in the metallic behavior of the M2GaC MAX phase compounds. Moreover, band structures' appearance resembles other metallic MAX phases, i.e., Cr2AlC73 and Ti2AlC74. The small energy dispersion along the K–H and L–M directions indicates the strong anisotropic behavior. In other words, the conductivity of MAX phase compounds is lower along the c-axis than to their basal planes. There is no apparent difference in band associated with Zr and Hf atoms in terms of energy level because electronegativity for Zr (1.33) and Hf (1.30) is almost the same. At different pressure, the increase in bandwidth was observed within the mentioned pressure limit. We found that with the increase in pressure from 0 to 50 GPa, the bands become looser, i.e., the bands above the Fermi level move upward while the bands below the Fermi level move downward. Moreover, the bands at the Fermi level increases with the increase in pressure.

Band structure for Zr2GaC at a 0 pressure, b 30 GPa, and c 50 GPa, and for Hf2GaC at d 0 pressure, e 30 GPa, and f 50 GPa. Fermi level is set to 0 eV.
To explain the bonding behavior of M2GaC (M = Zr, Hf) MAX phases, we examine the density of states (DOS). The partial and total density of states (TDOS) for M2GaC MAX phase compounds at pressure 0 GPa, 30 GPa, and 50 GPa are depicted in Fig. 5. In our previous work42, the hybridization of M (4d, 4p, 5s), Ga (3d, 4p, 4s), and C (2p, 2s) orbitals for M2GaC is explained at 0 GPa. The TDOS at Fermi level at 0 GPa for Zr2GaC and Hf2GaC are 3.00 states/eV/unit and 2.47 states/eV/unit, respectively. In the Zr2GaC MAX phase, the lowest valance bands of TDOS is formed by C-s with Zr-d, Zr-p in the energy ranges from − 10.85 to − 9.11 eV. The states range from − 8.01 to − 5.1 eV, and − 5 to − 1.90 eV are formed by Ga-s states and strong hybridization of Zr-d and C-s states, respectively. The highest valance bands in the created by Zr-d and Ga-p hybridization, which is relatively weaker than of Zr-d and C-s states. These results are consistent with that of Cr2AlC MAX phase material, which has the maximum DOS at EF, i.e., 6.46 states/eV cell/unit75.

The total and partial density of states (PDOS) for Zr2GaC at pressure a 0 GPa, b 30 GPa, and c 50 GPa, and for Hf2GaC at pressure d 0 GPa, e 30 GPa, and f 50 GPa. Fermi level is set to 0 eV.
Moreover, the density of states at the Fermi level increased with increased pressure, and obtained results are tabulated in Table 1. It is observed that the increase in TDOS for the Zr2GaC MAX phase is more significant than that of Hf2GaC. The TDOS values for M2GaC illustrate that these MAX phase materials are metallic, and their metallicity is in the order of Zr2GaC > Hf2GaC in the given range of pressure. However, there is an increase in bandwidth, and correspondingly, the intensity decreased with the increase in pressure. The density of states on the right side of the Fermi level moves rightwards, whereas the density of states on the left side of the Fermi level moves leftwards under pressure (See Fig. 6), which is in good agreement with the analysis of band structure. It is worth noticing that the main contribution at the Fermi level is from the M-4d electrons in both MAX phases, which is not affected by the pressure. This implies that Zr-d and Hf-d electrons mainly contribute to the conduction properties of MAX phase materials under pressure.

Total density of states (DOS) from GGA-PBE for M2GaC MAX phase (M = Zr, Hf) at different pressure. Figure inserted showing the effect of pressure in TDOS at Fermi level. Fermi level is set to 0 eV.
To understand the chemical bonding of Zr2GaC and Hf2GaC MAX phase, we investigate the Crystal Orbital Hamilton Population (COHP) for M–C, M–Ga, and Ga–C bonds, respectively. The COHP method has been widely applied to investigate the bonding and antibonding analysis of many MAX phases76,77,78. Figure 7 shows the COHP at the ground state of Zr2GaC and Hf2GaC MAX phases. It is observed that the COHP curves for both MAX phases have well-defined bonding and antibonding regions with crossover points near the Fermi level, which is an indication of covalent bonding nature. There is no occupation of antibonding orbitals in the M–C bonds of Zr2GaC and Hf2GaC MAX phases. Moreover, the occupied region below the Fermi level is dominated by strong M–C interaction, while at the Fermi level, the M–Ga bonding character was dominant. There is almost zero bonding interaction for Ga–C atoms due to the absence of orbital overlapping. The COHP curves are similar to the most studied Al-containing MAX phases77. Furthermore, the calculated values for metallicity and Fermi energy in the pressure range 0–50 GPa are tabulated in Table 1. It is observed that the metallicity of both Zr2GaC and Hf2GaC phases increases with increasing pressure. According to Eq. (6), the conductivity mainly depends upon \({n \mathord{\left/ {\vphantom {n {v_{F} }}} \right. \kern-\nulldelimiterspace} {v_{F} }}\) ratio because e, l, and m are constants. In the given pressure range, the \({n \mathord{\left/ {\vphantom {n {v_{F} }}} \right. \kern-\nulldelimiterspace} {v_{F} }}\) ratio of M2GaC MAX phases is in order of \(\left( {{n \mathord{\left/ {\vphantom {n {v_{F} }}} \right. \kern-\nulldelimiterspace} {v_{F} }}} \right)_{{Hf_{2} GaC}} > \left( {{n \mathord{\left/ {\vphantom {n {v_{F} }}} \right. \kern-\nulldelimiterspace} {v_{F} }}} \right)_{{Zr_{2} GaC}}\), hence we may conclude that the conductivity of Hf2GaC > Zr2GaC.

The calculated crystal orbital Hamilton population (COHP) showing the pair interactions of M–C, M–Ga, and Ga–C in a Zr2GaC and b Hf2GaC. Positive values in the y-axis (–COHP) indicating the bonding states. Fermi level is set to 0 eV.
To insight the chemical bonding of M2GaC MAX phases under pressure, the charge density distributions mapping along the (100) plane at 0 GPa, 30 GPa, and 50 GPa are plotted in Fig. 8. The bonding character of MAX phases is essential to understand the chemical bonding of their 2D derivatives (MXenes)79,80. The preferential accumulation of charges (positive regions at the scale bar) between two atoms indicates the covalent bonds, while balancing the positive or negative (depleted regions) charges at atomic position exhibits the ionic bonding81. At 0 GPa, the strong charge accumulation regions were observed at C and M = Zr, Hf atoms, indicating the formation of a strong covalent bond between C–Zr and C–Hf atoms. The charge accumulation at these atomic positions increases with an increase in pressure due to the decrease in atomic distance and an increase in internal parameters (z) (See Fig. 3). Furthermore, there is a sign of charge balancing around the Zr and Hf atoms, with the C indicating the small degree of ionic bonding. It is also seen that another covalent bond is formed between the Ga–M = Zr, Hf atoms, which is comparatively weaker than that of C–M = Zr, Hf atoms. Therefore, the chemical bonding in the M2GaC MAX phase is predicted to be a mixture of covalent and ionic nature and degree of bonding increases with the increase in pressure.

Electronic charge density mapping along the (100) plane for Zr2GaC at a 0 pressure, b 30 GPa, and c 50 GPa, and for Hf2GaC at d 0 pressure, e 30 GPa, and f 50 GPa.
Mechanical stability and dynamical properties
The mechanical properties of material help to predict the material’s response under the application of load. The mechanical properties of MAX phase materials also contribute to predicting the usefulness in service and are critical in the fabrication process. The elastic constants (Cij) for M2GaC MAX phase materials calculated in the pressure range from 0 to 50 GPa are shown in Fig. 9, and calculated mechanical properties are listed in Table 4. As we know that the M2GaC MAX phase has the hexagonal crystal structure, and there are six stiffness constants (C11, C12, C13, C33, C44 = C55, and C66), but five of them are independent since \(C_{66} = {{\left( {C_{11} - C_{12} } \right)} \mathord{\left/ {\vphantom {{\left( {C_{11} - C_{12} } \right)} 2}} \right. \kern-\nulldelimiterspace} 2}\)82. Our results are consistent with other MAX phases.

Elastic constants for M2GaC MAX phase (M = Zr, Hf) in function of pressure.
The elastic moduli versus pressure for the M2GaC MAX phase are plotted in Fig. 10. It is found that the elastic constants and moduli increase monotonically with an increase in pressure, and the values of C11, C33, Young’s modulus (E), and bulk modulus (B) increased significantly compared to other elastic constants. Contrary, the values of C66 and shear modulus (G) vary slowly. It can also be noticed that C11 and C66 for the Zr2GaC MAX phase increases uniformly up to 40 GPa and then decreases when pressure is exceeded to 50 GPa. A similar trend can be seen for Young’s and shear modulus of Zr2GaC. On the other hand, the Hf2GaC MAX phase shows a linear trend, consistent with the other MAX phase studies under pressure55,83. The mechanical stability of M2GaC MAX phases are predicted from the Born stability criteria84 i.e., C11 > 0, C11 − C12 > 0, C44 > 0, C66 > 0, (C11 + C12) C33 − 2\(C_{13}^{2}\) > 0. Both MAX phases satisfy the mechanical stability criteria in the mentioned range of pressure. The elastic constant C33 for Zr2GaC and Hf2GaC increases by up to 462 GPa and 506 GPa, while the values for C66 for both materials increased only by 112 GPa and 169 GPa, respectively. The rapid increase in C33 and moderate C66 infers the increasing insensitivity of the compression strain along the c axis, not the shear strain.

Variation of elastic moduli of M2GaC MAX phase (M = Zr, Hf) with pressure.
It is known that the moduli (B, G, and E) measure the resistance of the material to fracture, plastic deformation, and stiffness and are essential to understand the solid-state properties, i.e., structural stability, ductility, stiffness, and brittleness. In this work, the elastic moduli (B, G, and E) increased with an increase in pressure from 0 to 50 GPa, i.e., for Zr2GaC increase in moduli are B (122–302 GPa), G (94–126 GPa), and E (226–333 GPa) and that of Hf2GaC are B (142–347 GPa), G (112–168 GPa), and E (266–435 GPa), respectively. The elastic moduli for the M2GaC MAX phase are in the order of \(\left( {B,G,E} \right)_{{Hf_{2} GaC}} > \left( {B,G,E} \right)_{{Zr_{2} GaC}}\) in the given pressure range.
The brittle/ductile behavior of the M2GaC MAX phase is predicted from the Poisson’s ratio (σ). It is the ratio between the transverse strain to longitudinal strain under tensile stress. It is an important tool to quantify the failure state in the solids. Frantsevich et al.87 proposed a borderline value σ ~ 0.26, which separates the ductile and brittle materials. For the brittle materials, this value is small, whereas the material is considered to be ductile if Poisson’s ratio is greater than 0.26. Our calculated values for σ for Zr2GaC and Hf2GaC are 0.192 and 0.187 at 0 GPa, respectively. These values increase linearly with an increase in pressure, as shown in Fig. 11a. There is a sharp increase in σ noticed for the Zr2GaC MAX phase when pressure increase from 40 to 50 GPa. In the pressure range mentioned, the M2GaC MAX phase behaves brittle manner at 0 GPa pressure, similar to many other MAX phases28,88,89,90. Both MAX phases exhibit ductile nature when pressure is increased from 30 GPa. Moreover, the microscopic hardness model proposed by Chen et al.62 was used to calculate the Vickers hardness of M2GaC MAX phases, and the obtained results are listed in Table 4. The theoretical Vickers hardness for (Zr2GaC)Hv = 18.23 GPa < (Hf2GaC)Hv = 20.99 GPa at 0 GPa pressure, respectively, and Vickers hardness decreases with an increase in pressure69,85. It is worth noticing that the grain size of the material has an essential effect on hardness, yield strength, tensile, and fatigue strength according to the Hall–Petch relation because grain boundaries hinder the movement of dislocations91,92. The effect grain size on the compressive strength of bulk Ti2AlC MAX phase followed the Hall–Petch relation under the dynamic and quasi-static loads93. Moreover, the oxidation resistance and mechanical properties of MAX phase thin films can be improved by increasing the grain boundaries94,95.

Variation of a Poisson’s ratio (σ), and b Anisotropic Index (A) for the M2GaC MAX phase (M = Zr, Hf) in function of pressure. The horizontal line in (a) is the proposed borderline between brittle and ductile transition.
Figure 11b illustrates the anisotropic index (A) of the M2GaC MAX phase in the function of pressure. Typically, a material is called to be isotropic if anisotropic index A = 1 and the deviation from 1 indicate the anisotropic nature of the material. Figure 11b shows that the values of A do not satisfy the isotropic criteria, and an increase in pressure results in a higher anisotropic index for both Zr2GaC and Hf2GaC MAX phases suggesting the anisotropic nature of M2GaC MAX phases in the given pressure range. In other words, the properties for the M2GaC MAX is not identical in all directions. This fact is in good agreement with the anisotropic properties of M2GaC MAX phases, i.e., higher compressibility along the c-axis compared to other basal planes.
According to Pugh’s criteria, a material will behave ductile if the B/G > 1.75 and G/B < 0.57, otherwise it should be brittle62,96. For 0 GPa these ratios for M2GaC MAX phases are in the order (Hf2GaC)B/G = 1.26 < (Zr2GaC)B/G = 1.29 and (Hf2GaC)G/B = 0.78 > (Zr2GaC)G/B = 0.77, respectively. This indicates that the M2GaC phases behave in a brittle manner at 0 GPa: however, with an increase in pressure, M2GaC phases likely to be ductile (See Fig. 12). These results are consistent with studies available in the literature28. It is also worth noticing that the ductility of Zr2GaC phase increase abruptly when pressure is exceeded from 40 GPa and pressure has significant impact in term of brittle/ductile transition for Zr2GaC compared to that of Hf2GaC. Moreover, the ratio G/B can also be used to determine the chemical bond. For ionic materials, the value is G/B \(\approx\) 0.6, and that of covalent materials G/B \(\approx\) 1.1. In this work, G/B values change from 0.77 to 0.41 for Zr2GaC and 0.78–0.48 for Hf2GaC, suggesting that ionic bonding is crucial for M2GaC compounds.

The effect of pressure on Pugh’s ratio for M2GaC MAX phase (M = Zr, Hf). The horizontal line in (a) is the proposed borderline between brittle and ductile transition.
The Debye temperature is characteristic of solids that can evaluate many physical properties of the material, including thermal conductivity, thermal expansion, specific heat, and melting temperature. It can be calculated by numerous methods, among which the Anderson method is simple and widely used63. In this method, the average sound velocity (Vm) is used to calculate the Debye temperature. The calculated Debye temperature and sound velocities for M2GaC MAX phases are listed in Table 5. It is noticed that the density and Debye temperature increases with increasing the pressure, and at a given pressure, the Debye temperature is always in the order of (Zr2GaC)\(\theta\)D > (Hf2GaC)\(\theta\)D. The calculated melting point for the M2GaC MAX phase is tabulated in Table 5. The melting temperature for Zr2GaC and Hf2GaC MAX phases understudied are 1481 K and 1648 K at 0 GPa and increases with an increase in pressure. The higher melting temperature values indicate that M2GaC MAX phases are suitable for high-temperature applications.
For the dynamical stability of M2GaC MAX phases, phonon calculation was performed along the high-symmetry directions in the Brillouin zone. The calculated phonon dispersion curves at 0 GPa, 30 GPa, and 50 GPa are shown in Fig. 13. There are eight atoms per unit cell in the 211 family of MAX phases. So, 24 phonon branches are produced; three are acoustic, and the rest are for optical modes. The optical branches are situated at the upper part of the dispersion curves, responsible for the optical behavior of MAX phase materials. These optical modes originate from the out-of-phase oscillations of atoms in lattice when one atom goes to the left and its neighbor to the right. In contrast, the acoustic branches are located at the lower part of phonon dispersion curves and arise from the coherent vibration of atoms in a lattice outside their balance position. The absence of negative frequencies in the phonon dispersion curves within the whole Brillouin zone robustly indicates the dynamical stability of M2GaC MAX phases under normal and high pressure against mechanical perturbation. The phonon dispersion curves become loose with the increase in pressure, which is consistent with band structures. At point Γ, the values of transverse optical (TO) and longitudinal optical (LO) frequencies and the separation between TO and LO increases with an increase in pressure. At 0 GPa, the values of LO (lower dispersion curve) and TO frequencies at Γ are 14.58, 16.56 THz for Zr2GaC and 16.00, 18.32 THz for Hf2GaC, respectively. Moreover, at the center zone point (Γ), the acoustic mode frequency is zero for all pressures, which is another indication of the stability of M2GaC MAX phases within the given pressure range.

The phonon dispersion curve for Zr2GaC at pressure a 0 GPa, b 30 GPa, and c 50 GPa, and for Hf2GaC at pressure d 0 GPa, e 30 GPa, and f 50 GPa.
As mentioned earlier that the MAX phases compounds studied in this work have an elastic anisotropic nature, while the elastic anisotropy is not apparent. Figure 14 plots the 3D Young’s modulus surfaces obtained by an open-source software package (AnisoVis)97 of Zr2GaC (a, b, c) and Hf2GaC (d, e, f) at 0, 10, and 50 GPa, respectively. It can be observed that the shape begins to deviate from the sphere with an increase in pressure, and color varies in different regions, which indicates the elastic anisotropy of M2GaC MAX phases. The color variation (dark blue to yellow) exhibits that elastic modulus increases with an increase in pressure, and obtained values are mentioned at the top of each 3D plot, which agrees well with the previously calculated results (see Fig. 10). The Young’s modulus at 0 GPa pressure for Zr2GaC and Hf2GaC are 226.57 GPa and 266.68 GPa and rises to 333.38 GPa and 435.43 GPa when pressure is increased to 50 GPa, respectively. The pressure effect on the elastic anisotropy of Hf2GaC is more significant than that of the Zr2GaC MAX phase. According to the deviation degree of spherical shape, the anisotropy of Hf2GaC is bigger than Zr2GaC (See Fig. 11b).

3D plot of Young’s modulus (E) surface of M2GaC MAX phase (M = Zr, Hf) at various pressure.
Optical properties
The optical properties for the M2GaC MAX phase (M = Zr, Hf) were determined for the first time by frequency-dependent dielectric functions within the photon energies up to 20 eV. The optical properties for The MAX phases are optically anisotropic99,100. Thus, two polarization directions, <100> and <001>, were chosen to investigate the optical properties. The MAX phase compounds under this study are metallic in nature (see band structure) so, the term Drude (plasma frequency 3 eV and damping 0.05 eV) has been used with Gaussian smearing of 0.5 eV for all calculations. The frequency-dependent dielectric function \(\varepsilon \left( \omega \right) = \varepsilon_{1} \left( \omega \right) + i\varepsilon_{2} \left( \omega \right)\)(where \(\varepsilon_{1} \left( \omega \right)\) is the real part and \(\varepsilon_{2} \left( \omega \right)\) is the imaginary part of the dielectric function) has a close relation to the band structure. Once the imaginary part is known, the real part can be derived using the Kramers–Kronig equation. Later, all the optical properties can be obtained using the \(\varepsilon_{1} \left( \omega \right)\) and \(\varepsilon_{2} \left( \omega \right)\) 101, as shown in Figs. 15, 16, 17, 18 and 19.

Energy dependence of dielectric function a real part b imaginary part of M2GaC MAX phase (M = Zr, Hf).

Loss function of M2GaC MAX phase (M = Zr, Hf) for <100> and <001> polarization.

The optical absorption of M2GaC MAX phase (M = Zr, Hf) for <100> and <001> polarization.

Photoconductivity of M2GaC MAX phase (M = Zr, Hf) for <100> and <001> polarization.

Reflectivity of M2GaC MAX phase (M = Zr, Hf) for <100> and <001> polarization.
Dielectric constant
The real part of the dielectric constant \(\varepsilon_{1} \left( \omega \right)\) is essential for optoelectronic devices because it corresponds to the primitivity component that measures the stored energy. The imaginary part of the dielectric constant \(\varepsilon_{2} \left( \omega \right)\) gives the information about the optical system's energy reduction in the function of frequency. Figure 15a,b shows the real \(\varepsilon_{1} \left( \omega \right)\) and imaginary part \(\varepsilon_{2} \left( \omega \right)\) of dielectric constants calculated for the < 100 > and < 001 > polarization directions for M2GaC. It is observed that the real part of dielectric constant (\(\varepsilon_{1}\)) approaches to zero from below, while the imaginary part (\(\varepsilon_{2}\)) gets to zero from above, which implies that the M2GaC MAX phases are metallic in nature102. In the real part of the dielectric constant, the spectra within the infrared region (I.R ≤ 1.7 eV) has the highest dielectric constant for <100> polarization compared to < 001 > polarization due to intra-band transition of electron. The sharp peaks were observed in \(\varepsilon_{1} \left( \omega \right)\) for Zr2GaC and Hf2GaC phases along the <100> polarization at ∼1.01 eV and ∼1.04 eV, respectively. It is worth noticing that the spectra of \(\varepsilon_{1} \left( \omega \right)\) for different polarization directions exhibit different features in the photon energy range. There is no significant difference observed for both MAX phases in the \(\varepsilon_{2} \left( \omega \right)\) spectra for < 100 > and < 001 > polarization. Thus, we can deduce that the MAX phases studies here are optically anisotropic. Moreover, the value \(\varepsilon_{1} \left( \omega \right)\) approached zero from below for polarization \(< 100 >_{{M_{2} GaC}}\) at around 12.8–13.7 eV and for polarization \(< 001 >_{{M_{2} GaC}}\), reaches zero at approximately 13.9–15.6 eV. While the value for \(\varepsilon_{2} \left( \omega \right)\) reaches zero from above for polarization <100> and <001> at photon energy ranges from 12.5 to 16.7 eV.
Loss function
The loss function is peak corresponds to the bulk plasma frequency (\(\omega_{p}\)), which appears where \(\varepsilon_{2}\) < 1 and \(\varepsilon_{1}\) approaches to zero. It is the energy loss of the first electron traversing through a material, and bulk plasma frequency (\(\omega_{p}\)) is obtained from the loss function spectrum. The studied M2GaC MAX phases become transparent if the frequency of incident light is higher than that of plasma frequency. By analyzing, the energy-loss function peaks of Zr2GaC and Hf2GaC phases was occurred at around 12.79 eV and 13.7 eV, respectively, for <100> polarization: and corresponding 14.00 eV and 15.61 eV for <001> polarization as shown in Fig. 16. It is noticed that the plasma frequent (\(\omega_{p}\)) of M2GaC for the <001> polarization is larger than that for <100> polarization103. Moreover, energy loss spectra for M2GaC MAX phases show no peaks in the photon energy range of 0–10 eV due to large \(\varepsilon_{2} \left( \omega \right)\) (see Fig. 15b).
Absorption coefficient
The absorption coefficient gives the knowledge about the efficiency of the solar energy conversion, which is important for solar cell material. It corresponds to the amount of light of a specific wavelength into a solid before getting absorbed. Figure 17 depicts the energy-based absorption (α) spectra of M2GaC MAX phases. It is observed that the absorption spectra for both MAX phases are weak in the infrared region (I.R), increases monotonously in the visible region and dominant ultraviolet (UV) regions. The maximum value of α was observed for Zr2GaC and Hf2GaC at around 6.09 eV and 6.86 eV, respectively, for <100> polarization; and corresponding 6.34 eV and 6.97 eV for <001> polarization. Moreover, the light absorption of M2GaC in the <001> polarization direction is larger than that for <100> polarization. In other words, both Zr2GaC and Hf2GaC MAX phases absorb more light in the direction of <001> polarization compared to its counterpart <100> , indicating their optically anisotropic nature98. The rise in α was observed in the direction of the UV region, exhibits the high absorbent feature of the material. Based on the calculated absorption spectra of M2GaC MAX phases, it can be deduced that these materials are competing candidates for optoelectronic devices in both visible and UV regions.
Photoconductivity
Photoconductivity (σ) of material can be described as the increase in the electric conductivity due to absorbing photos. For M2GaC MAX phases σ is shown in Fig. 18. It is noticed that for both MAX phases under this study, photoconductivity increases exponentially when the photo energy goes to 0 eV as expected for metals because there is no band gap present in the M2GaC MAX phases. A sharp dip in photoconductivity of Zr2GaC and Hf2GaC for <100> polarization was observed at 0.37 eV and 0.39 eV, and that for polarization <001> was observed at 0.26 eV and 0.20 eV, respectively88. Peak heights for different polarization are different at various photon energies, and Zr2GaC <001> gives the highest peak at 4.98 eV. It is concluded that M2GaC MAX phases are photoconductive at near I.R, visible, and UV regions.
Reflectivity
Finally, reflectivity spectra of M2GaC MAX phases for <100> and <001> polarization, as a function of incident light are demonstrated in Fig. 19. The reflectivity for M2GaC MAX phases shows the highest reflectivity in the I.R region and visible region ranges from 4.4 to 13.10 eV and then approaches zero for both phases in the incident photon energy ranges from 19 to 22 eV. However, it is worth noticing that the reflectivity is almost constant for <100> polarization of Zr2GaC and Hf2GaC MAX phases within the visible region, and values are above 45% and should appear as a metallic gray color. It is known that materials having constant reflectivity in the visible regions with an average value of about 44% are capable of reflecting the solar light, which results in a reduction in solar heating in the visible light region104. So, it may be concluded that Zr2GaC and Hf2GaC MAX phases can be used as the coating material for the purpose of solar heating reduction. However, the variable reflectivity within the visible region of different polarization indicated the optical anisotropy M2GaC MAX phase105.
The dependence of reflectance on the pressure of M2GaC MAX phases was studied as well, and results for polarization <100> at a pressure range from 0 to 50 GPa is shown in Fig. 20. For the M2GaC MAX phase, the reflectance exhibits less change in the moderate range of the I.R region ranging from 0 to 0.48 eV at all pressures and show variable reflectivity in the rest I.R region. It is noticed that the reflectivity increases with an increase in pressure in the I.R region. However, the reflectivity of M2GaC decreased at higher pressure, but almost the same in the visible region then increases in the UV region more quickly and exhibits a higher value at 0 GPa. The reflectance at pressure range 0–50 GPa remains above 40% in the visible zone. Thus, it is concluded that M2GaC MAX phase materials are ideal for coating materials under high-pressure conditions to avoid solar heating in the <100> polarization direction.

Reflectivity of M2GaC MAX phases (M = Zr, Hf) for <100> polarization at 0–50 GPa pressure.
Conclusion
The effect of pressure on structural stability, mechanical, electronic, phonon, and optical properties of M2GaC MAX phases (M = Zr and Hf) in the pressure range from 0 to 50 GPa were calculated by using first-principles calculations. The formation of enthalpy with respect to its most competing phases showed that M2GaC MAX phases are thermodynamically stable. The band structure and total density of states exhibited that M2GaC MAX phases are metallic in nature, with an increase in bandgap at the Fermi level with an increase in pressure. The DOS at EF in the pressure range of 0–50 GPa are Zr2GaC = 2.96–4.27 states/eV/unit > Hf2GaC = 2.47–2.60 states/eV/unit, which implies that the metallicity of Zr2GaC increased more than that of Hf2GaC with increasing the internal pressure. According to COHP analysis, M–C bonds are stronger than that M–Ga in both Zr2GaC and Hf2GaC MAX phases. The volume ratio and lattice parameters Zr2GaC and Hf2GaC decrease with increasing pressure, and the compressibility of Zr2GaC is better than that of Hf2GaC. Besides, the normalized bond lengths show that crystals compressed more easily along the M–Ga (Zr, Hf) direction under pressure. The effect of pressure on the mechanical properties of M2GaC MAX phases is pronounced. Both the Zr2GaC and Hf2GaC MAX phases revealed the brittleness behavior at 0 GPa pressure and tended to ductile when pressure increased from 10 to 50 GPa. Moreover, there is a linear increase in elastic constants, elastic moduli, Poisson’s ratio, and a decrease in Vickers hardness was observed with the increase in pressure. The calculated Vickers hardness is found to be 18.23 GPa and 20.99 GPa for Zr2GaC and Hf2GaC, respectively. The phonon dispersion curves have confirmed the dynamical stability of compounds in the given pressure range. The optical properties of the MAX phase compound under this study reveals some interesting information. The absorption spectra of M2GaC increased to the maximum value in the visible region, and the UV region indicates its high absorbent capability and is suitable for optoelectronic devices in the visible and UV regions. Moreover, the reflectance curves show the constant values in the visible region with an average value above 44%. We conclude that these compounds can also be used as a coating material to avoid solar heating at even high pressure. To the author’s best knowledge, no study had been made to predict the mechanical, electronic, thermal, phonon, and optical properties of M2GaC MAX phases under pressure. Hence, these results can serve as a reference for future theoretical and experimental research.
References
Barsoum, M. W. The Mn+1 AXn phases and their properties. in Ceramics Science and Technology 2, 299–347 (Wiley-VCH, 2010).
Barsoum, M. W. MAX Phases: Properties of Machinable Ternary Carbides and Nitrides (Wiley-VCH, New York, 2013). https://doi.org/10.1002/9783527654581.
Eklund, P., Rosen, J. & Persson, P. O. Å. Layered ternary Mn+1AXn phases and their 2D derivative MXene: an overview from a thin-film perspective. J. Phys. D Appl. Phys. 50, 113001 (2017).
Wang, X. H. & Zhou, Y. C. Layered machinable and electrically conductive Ti2AlC and Ti3AlC2 ceramics: a review. J. Mater. Sci. Technol. 26, 385–416 (2010).
Eklund, P., Beckers, M., Jansson, U., Högberg, H. & Hultman, L. The Mn + 1AXn phases: materials science and thin-film processing. Thin Solid Films 518, 1851–1878 (2010).
Barsoum, M. W. & El-Raghy, T. The MAX phases: unique new carbide and nitride materials: tertiary ceramics are soft and machinable, yet heat-tolerant, strong and lighweight. Am. Sci. 89, 334–343 (2001).
Barsoum, M. W. & El-Raghy, T. Synthesis and characterization of a remarkable ceramic: Ti3SiC2. J. Am. Ceram. Soc. 79, 1953–1956 (1996).
Tzenov, N. V. & Barsoum, M. W. Synthesis and characterization of Ti3AlC2. J. Am. Ceram. Soc. 83, 825–832 (2004).
Hajas, D. E. et al. Oxidation of Cr2AlC coatings in the temperature range of 1230 to 1410°C. Surf. Coat. Technol. 206, 591–598 (2011).
Smialek, J. L. Oxidation of Al2O3 scale-forming MAX phases in turbine environments. Metall. Mater. Trans. A Phys. Metall. Mater. Sci. 49, 782–792 (2018).
Fu, J. et al. Oxidation and corrosion behavior of nanolaminated MAX-phase TilC film synthesized by high-power impulse magnetron sputtering and annealing. J. Nanomater. 2015, 1–12 (2015).
Gupta, S., Filimonov, D., Palanisamy, T. & Barsoum, M. W. Tribological behavior of select MAX phases against Al2O3at elevated temperatures. Wear 265, 560–565 (2008).
Hopfeld, M., Grieseler, R., Vogel, A., Romanus, H. & Schaaf, P. Tribological behavior of selected Mn+1AXnphase thin films on silicon substrates. Surf. Coat. Technol. 257, 286–294 (2014).
Shein, I. R. & Ivanovskii, A. L. Elastic properties of superconducting MAX phases from first-principles calculations. Phys. Status Solidi Basic Res. 248, 228–232 (2011).
Lee, W. E., Giorgi, E., Harrison, R., Maître, A. & Rapaud, O. Nuclear applications for ultra-high temperature ceramics and MAX phases. in Ultra-High Temperature Ceramics: Materials for Extreme Environment Applications 9781118700785, 391–415 (Wiley Blackwell, 2014).
Sun, D. et al. Structural transformation of MXene (V2C, Cr2C, and Ta2C) with O groups during lithiation: a first-principles investigation. ACS Appl. Mater. Interfaces 8, 74–81 (2016).
Lin, Z. et al. Capacitance of Ti3C2Tx MXene in ionic liquid electrolyte. J. Power Sources 326, 575–579 (2016).
Wang, Y. et al. Enhanced low-temperature Li-ion storage in MXene titanium carbide by surface oxygen termination. 2D Mater. 6, 045025 (2019).
Lapauw, T. et al. Synthesis of MAX phases in the Hf-Al-C system. Inorg. Chem. 55, 10922–10927 (2016).
Lapauw, T. et al. Synthesis of the new MAX phase Zr2AlC. J. Eur. Ceram. Soc. 36, 1847–1853 (2016).
Hu, C., Sakka, Y., Tanaka, H., Nishimura, T. & Grasso, S. Low temperature thermal expansion, high temperature electrical conductivity, and mechanical properties of Nb4AlC3 ceramic synthesized by spark plasma sintering. J. Alloys Compd. 487, 675–681 (2009).
Petruhins, A. et al. Phase stability of Crn+ 1GaCn MAX phases from first principles and Cr2GaC thin-film synthesis using magnetron sputtering from elemental targets. Phys. Status Solidi Rapid Res. Lett. 7, 971–974 (2013).
Hoffman, E. N. et al. MAX phase carbides and nitrides: properties for future nuclear power plant in-core applications and neutron transmutation analysis. Nucl. Eng. Des. 244, 17–24 (2012).
Wang, Q. M. et al. Fabrication and oxidation behavior of Cr2AlC coating on Ti6242 alloy. Surf. Coat. Technol. 204, 2343–2352 (2010).
Perdew, J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822–8824 (1986).
Bai, Y., Srikanth, N., Chua, C. K. & Zhou, K. Density functional theory study of M n+1 AX n phases: a review. Crit. Rev. Solid State Mater. Sci. 44, 56–107 (2019).
Sokol, M., Natu, V., Kota, S. & Barsoum, M. W. On the chemical diversity of the MAX phases. Trends Chem. 1, 210–223 (2019).
Pu, X. X., Long, X. J., Zhang, L. & Zhu, J. The structure, elastic and thermodynamic properties of Ti 2 GaC from first-principles calculation. Int. J. Mod. Phys. B 33, 1–17 (2019).
Thore, A., Dahlqvist, M., Alling, B. & Rosén, J. First-principles calculations of the electronic, vibrational, and elastic properties of the magnetic laminate Mn2GaC. J. Appl. Phys. 116, 103511 (2014).
Shein, I. R. & Ivanovskii, A. L. Structural, elastic, electronic properties and Fermi surface for superconducting Mo2GaC in comparison with V2GaC and Nb2GaC from first principles. Phys. C Supercond. Its Appl. 470, 533–537 (2010).
Qing-He, G. et al. Origin of the c-axis ultraincompressibility of Mo2GaC above about 15 GPa from first principles. J. Appl. Phys. 119, 015901 (2016).
Romeo, M. & Escamilla, R. Pressure effect on the structural, elastic and electronic properties of Nb2AC (A = S and In) phases; Ab initio study. Comput. Mater. Sci. 81, 184–190 (2014).
Romero, M. & Escamilla, R. First-principles calculations of structural, elastic and electronic properties of Nb 2SnC under pressure. Comput. Mater. Sci. 55, 142–146 (2012).
Bouhemadou, A., Khenata, R., Kharoubi, M. & Medkour, Y. First-principles study of structural and elastic properties of Sc2AC (A=Al, Ga, In, Tl). Solid State Commun. 146, 175–180 (2008).
Bouhemadou, A. Calculated structural and elastic properties of M2InC (M = Sc, Ti, V, Zr, Nb, Hf, Ta). Mod. Phys. Lett. B 22, 2063–2076 (2008).
Bouhemadou, A. Calculated structural, electronic and elastic properties of M 2GeC (M=Ti, V, Cr, Zr, Nb, Mo, Hf, Ta and W). Appl. Phys. A Mater. Sci. Process. 96, 959–967 (2009).
Bouhemadou, A., Khenata, R. & Chegaar, M. Structural and elastic properties of Zr 2AlX and Ti 2AlX (X = C and N) under pressure effect. Eur. Phys. J. B 56, 209–215 (2007).
Bouhemadou, A. & Khenata, R. Prediction study of structural and elastic properties under the pressure effect of M2 GaC (M=Ti, V, Nb, Ta). J. Appl. Phys. 102, 043528 (2007).
Bouhemadou, A. Structural and elastic properties under pressure effect of Hf2AlN and Hf2AlC. High Press. Res. 28, 45–53 (2008).
Music, D., Sun, Z., Ahuja, R. & Schneider, J. M. Electronic structure of M2AlC(0001) surfaces (M = Ti, V, Cr). J. Phys. Condens. Matter 18, 8877–8881 (2006).
Barsoum, M. W. & Radovic, M. Elastic and mechanical properties of the MAX phases. Annu. Rev. Mater. Res. 41, 195–227 (2011).
Qureshi, M. W., Ma, X., Tang, G. & Paudel, R. Structural stability, electronic, mechanical, phonon, and thermodynamic properties of the M 2 GaC (M = Zr, Hf) MAX Phase: an ab initio calculation. Materials 13, 5148 (2020).
Sun, Z., Music, D., Ahuja, R., Li, S. & Schneider, J. M. Bonding and classification of nanolayered ternary carbides. Phys. Rev. B Condens. Matter Mater. Phys. 70, 1–3 (2004).
Payne, M. C., Teter, M. P., Allan, D. C., Arias, T. A. & Joannopoulos, J. D. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys. 64, 1045–1097 (1992).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Fischer, T. H. & Almlöf, J. General methods for geometry and wave function optimization. J. Phys. Chem. 96, 9768–9774 (1992).
Materials Studio - PaNdata Software. https://software.pan-data.eu/software/86/materials-studio. Accessed 27 Oct 2020.
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 34, 2557–2567 (2013).
Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Huang, Z. W., Zhao, Y. H., Hou, H. & Han, P. D. Electronic structural, elastic properties and thermodynamics of Mg 17Al 12, Mg 2Si and Al 2Y phases from first-principles calculations. Phys. B Condens. Matter 407, 1075–1081 (2012).
Hug, G. Electronic structures of and composition gaps among the ternary carbides Ti2 MC. Phys. Rev. B Condens. Matter Mater. Phys. 74, 184113 (2006).
Hug, G., Jaouen, M. & Barsoum, M. W. X-ray absorption spectroscopy, EELS, and full-potential augmented plane wave study of the electronic structure of Ti 2AlC, Ti 2AlN, Nb 2AlC, and (Ti 0.5Nb 0.5) 2AlC. Phys. Rev. B Condens. Matter Mater. Phys. 71, 024105 (2005).
Huang, D., Qiu, R., Mo, C. & Fa, T. Structural, mechanical, dynamical and electronic properties and high-pressure behavior of Mo2GeC: a first-principles study. Comput. Mater. Sci. 137, 306–313 (2017).
Li, Y. et al. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds. J. Alloys Compd. 502, 28–37 (2010).
Mouhat, F. & Coudert, F. X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B Condens. Matter Mater. Phys. 90, 224104 (2014).
Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. ZAMM J. Appl. Math. Mech./Zeitschrift für Angew. Math. und Mech. 9, 49–58 (1929).
Brazhkin, V. V. High-pressure synthesized materials: treasures and hints. High Press. Res. 27, 333–351 (2007).
Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 65, 349–354 (1952).
Johnson, D. D. Electronic basis of the strength of materials. Phys. Today https://doi.org/10.1063/1.1801871 (2004).
Chen, X. Q., Niu, H., Li, D. & Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 19, 1275–1281 (2011).
Anderson, O. L. A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solids 24, 909–917 (1963).
Fine, M. E., Brown, L. D. & Marcus, H. L. Elastic constants versus melting temperature in metals. Scr. Metall. 18, 951–956 (1984).
Dobson, P. J. Physical properties of crystals—their representation by tensors and matrices. Phys. Bull. 36, 506–506 (1985).
Dahlqvist, M., Alling, B., Abrikosov, I. A. & Rosén, J. Phase stability of Ti2 AlC upon oxygen incorporation: a first-principles investigation. Phys. Rev. B Condens. Matter Mater. Phys. 81, 024111 (2010).
Chen, Q., Li, X., Ye, B. & Xiong, L. First-principle prediction of phase stability, electronic and elastic properties study of the Mn+1ANn (A = Al, Si, M = Ti, Zr, Hf). J. Phase Equilibria Diffus. https://doi.org/10.1007/s11669-020-00851-z (2020).
Dahlqvist, M., Alling, B. & Rosén, J. Stability trends of MAX phases from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 81, 220102 (2010).
Luo, F., Guo, Z. C., Zhang, X. L., Yuan, C. Y. & Cai, L. C. Ab initio predictions of structural and thermodynamic properties of Zr2AlC under high pressure and high temperature. Chin. J. Chem. Phys. 28, 263–268 (2015).
Zhao, Y. et al. First-principle investigation of pressure and temperature influence on structural, mechanical and thermodynamic properties of Ti3AC2 (A = Al and Si). Comput. Mater. Sci. 154, 365–370 (2018).
Aydin, S., Tatar, A. & Ciftci, Y. O. Some new members of MAX family including light-elements: nanolayered Hf2XY (X= Al, Si, P and YCombining double low lineB, C, N). Solid State Sci. 53, 44–55 (2016).
Kanoun, M. B., Goumri-Said, S. & Jaouen, M. Steric effect on the M site of nanolaminate compounds M2SnC (M = Ti, Zr, Hf andNb). J. Phys. Condens. Matter 21, 045404 (2009).
Cui, S., Wei, D., Hu, H., Feng, W. & Gong, Z. First-principles study of the structural and elastic properties of Cr 2AlX (X=N, C) compounds. J. Solid State Chem. 191, 147–152 (2012).
Tan, J. et al. A comparative first-principles study of the electronic, mechanical, defect and acoustic properties of Ti2AlC and Ti3AlC. J. Phys. D Appl. Phys. 47, 215301 (2014).
Lofland, S. E. et al. Elastic and electronic properties of select M 2AX phases. Appl. Phys. Lett. 84, 508–510 (2004).
Ohmer, D., Opahle, I., Singh, H. K. & Zhang, H. Stability predictions of magnetic M2AX compounds. J. Phys. Condens. Matter 31, 405902 (2019).
Music, D., Houben, A., Dronskowski, R. & Schneider, J. M. Ab initio study of ductility in M 2 Al C ( M = Ti, V, Cr). Phys. Rev. B 75, 174102 (2007).
Khazaei, M. et al. Insights into exfoliation possibility of MAX phases to MXenes. Phys. Chem. Chem. Phys. 20, 8579–8592 (2018).
Sun, Z. & Ahuja, R. Ab initio study of the Cr2 AlC (0001) surface. Appl. Phys. Lett. 88, 10–13 (2006).
Guo, Z., Zhu, L., Zhou, J. & Sun, Z. Microscopic origin of MXenes derived from layered MAX phases. RSC Adv. 5, 25403–25408 (2015).
Ernst, F. & Rühle, M. High-resolution imaging and spectrometry of materials. Mater. Today 6, 57 (2003).
Li, H. et al. Phonon and electronic properties of Ti2SiC from first-principles calculations. Solid State Commun. 204, 37–40 (2015).
Pang, M., Zhan, Y., Wang, H., Jiang, W. & Du, Y. Ab initio study of AlCu2M (M = Sc, Ti and Cr) ternary compounds under pressures. Comput. Mater. Sci. 50, 2930–2937 (2011).
Schäfer, K. Duane C. Wallace: Thermodynamics of Crystals. XVIII u. 484 S. mit 57 Abb. John Wiley & Sons Ltd. Chichester, New York, Sydney, Tokio, Mexiko 1972. Preis: £ 8,85. Berichte Bunsenges. Phys. Chem. 76, 1211–1211 (1972).
Surucu, G. Investigation of structural, electronic, anisotropic elastic, and lattice dynamical properties of MAX phases borides: an ab-initio study on hypothetical M2AB (M = Ti, Zr, Hf; A = Al, Ga, In) compounds. Mater. Chem. Phys. 203, 106–117 (2018).
Cover, M. F., Warschkow, O., Bilek, M. M. M. & McKenzie, D. R. A comprehensive survey of M2AX phase elastic properties. J. Phys. Condens. Matter 21, 305403 (2009).
Frantsevich, I. N., Voronov, F. F. & Bakuta, S. A. Handbook on Elastic Constants and Moduli of Elasticity for Metals and Nonmetals (Naukova Dumka, Kiev, 1982).
Sultana, F. et al. First principles study of M2InC (M = Zr, Hf and Ta) MAX phases: the effect of M atomic species. Results Phys. 11, 869–876 (2018).
Aryal, S., Sakidja, R., Barsoum, M. W. & Ching, W. Y. A genomic approach to the stability, elastic, and electronic properties of the MAX phases. Phys. Status Solidi Basic Res. 251, 1480–1497 (2014).
Hadi, M. A. New ternary nanolaminated carbide Mo2Ga2C: a first-principles comparison with the MAX phase counterpart Mo2GaC. Comput. Mater. Sci. 117, 422–427 (2016).
Taha, A. S. & Hammad, F. H. Application of the Hall–Petch relation to microhardness measurements on Al, Cu, Al-MD 105, and Al–Cu alloys. Phys. Status Solidi 119, 455–462 (1990).
Benitez, R. et al. Mechanical properties and microstructure evolution of Ti2AlC under compression in 25–1100 °C temperature range. Acta Mater. 189, 154–165 (2020).
Naik Parrikar, P., Benitez, R., Gao, H., Radovic, M. & Shukla, A. The effect of grain size on deformation and failure of Ti2AlC MAX Phase under thermo-mechanical loading. Exp. Mech. 57, 675–685 (2017).
Chen, X. et al. Enhancing the high temperature oxidation behavior of Cr 2 AlC coatings by reducing grain boundary nanoporosity. Mater. Res. Lett. 9(3), 127–133 (2020).
Wang, Z., Kang, H., Chen, R., Ke, P. & Wang, A. Enhanced mechanical and tribological properties of V–Al–C coatings via increasing columnar boundaries. J. Alloys Compd. 781, 186–195 (2019).
Pugh, S. F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 45, 823–843 (1954).
Nordmann, J., Aßmus, M. & Altenbach, H. Visualising elastic anisotropy: theoretical background and computational implementation. Contin. Mech. Thermodyn. 30, 689–708 (2018).
Hadi, M. A. et al. Insights into the physical properties of a new 211 MAX phase Nb2CuC. J. Phys. Chem. Solids 149, 109759 (2021).
Hadi, M. A., Vovk, R. V. & Chroneos, A. Physical properties of the recently discovered Zr2(Al1−xBix)C MAX phases. J. Mater. Sci. Mater. Electron. 27, 11925–11933 (2016).
Hadi, M. A., Panayiotatos, Y. & Chroneos, A. Structural and optical properties of the recently synthesized (Zr3−xTix)AlC2 MAX phases. J. Mater. Sci. Mater. Electron. 28, 3386–3393 (2017).
Saha, S., Sinha, T. P. & Mookerjee, A. Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3. Phys. Rev. B Condens. Matter Mater. Phys. 62, 8828–8834 (2000).
Hadi, M. A. Superconducting phases in a remarkable class of metallic ceramics. J. Phys. Chem. Solids 138, 109275 (2020).
Chowdhury, A. et al. Predicted MAX phase Sc2InC: dynamical stability, vibrational and optical properties. Phys. Status Solidi Basic Res. 255, 1–9 (2018).
Li, S., Ahuja, R., Barsoum, M. W., Jena, P. & Johansson, B. Optical properties of Ti3SiC2 and Ti 4AlN3. Appl. Phys. Lett. 92, 221907 (2008).
Roknuzzaman, M. et al. Physical properties of predicted Ti2CdN versus existing Ti2CDC MAX phase: An ab initio study. Comput. Mater. Sci. 113, 148–153 (2016).
Acknowledgements
The authors express their sincere gratitude to Mohammad Khazaei from the Computational Materials Science Research Team, RIKEN Advanced Institute for Computational Science (AICS)-Japan, to provide access to the VASP code.
Funding
This work is supported by the National Science and Technology Major Project (No. 2017-VII-0003-0096), and the National Natural Science Foundation of China (NSFC No. 51971084 and No. 52031003).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.W.Q. and X.M.; methodology, M.W.Q and R.P.; software, X.M.; validation M.W.Q., X.M., G.T., and R.P.; formal analysis, M.W.Q., X.M., and R.P.; investigation, M.W.Q., G.T.; writing—original draft preparation, M.W.Q., and X.M.; writing—review and editing, M.W.Q., X.M., and R.P.; supervision, X.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qureshi, M.W., Ma, X., Tang, G. et al. Ab initio predictions of structure and physical properties of the Zr2GaC and Hf2GaC MAX phases under pressure. Sci Rep 11, 3260 (2021). https://doi.org/10.1038/s41598-021-82402-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82402-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
