
Abstract
RNA molecules start assembling into ribonucleoprotein (RNP) complexes during transcription. Dynamic RNP assembly, largely directed by cis-acting elements on the RNA, coordinates all processes in which the RNA is involved. To identify the sites bound by a specific RNA-binding protein on endogenous RNAs, cross-linking and immunoprecipitation (CLIP) and complementary, proximity-based methods have been developed. In this Primer, we discuss the main variants of these protein-centric methods and the strategies for their optimization and quality assessment, as well as RNA-centric methods that identify the protein partners of a specific RNA. We summarize the main challenges of computational CLIP data analysis, how to handle various sources of background and how to identify functionally relevant binding regions. We outline the various applications of CLIP and available databases for data sharing. We discuss the prospect of integrating data obtained by CLIP with complementary methods to gain a comprehensive view of RNP assembly and remodelling, unravel the spatial and temporal dynamics of RNPs in specific cell types and subcellular compartments and understand how defects in RNPs can lead to disease. Finally, we present open questions in the field and give directions for further development and applications.
Similar content being viewed by others


Introduction
Proteins begin to interact with nascent RNAs as soon as transcription is initiated. The protein complement decorating an RNA molecule changes dynamically in space and time, orchestrating RNA processing and function in the nucleus and cytoplasm1. Ribonucleoprotein (RNP) complexes are key to every step of RNA processing and function, and understanding the roles that RNA-binding proteins (RBPs) play requires methods that identify the set of RNAs that they bind in cells during specific developmental stages, activities or disease states.
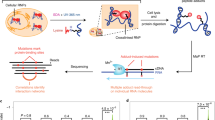
Numerous methods can characterize the RNA interactions that coordinate RNP assembly. These approaches can be protein-centric, describing the compendium of RNA sites bound by a specific RBP, or RNA-centric, identifying the RNA-bound proteome. The most common protein-centric strategies are based on the immunopurification of an RBP and its associated RNAs, and can be broadly categorized as RNA immunoprecipitation (RIP) or cross-linking and immunoprecipitation (CLIP) approaches. RIP approaches purify the RNA–protein complexes under native conditions2,3 or using formaldehyde cross-linking4. CLIP techniques are more widely used and rely on the irradiation of cells by UV light, which causes proteins in the immediate vicinity of the irradiated bases to irreversibly cross-link to the RNA by a covalent bond5 (Fig. 1). The covalent cross-links allow stringent purification of the RNA–protein complexes, which is followed by a series of steps to determine the interactions of a specific protein across the transcriptome. CLIP uses a limited RNase treatment of cross-linked RNPs to isolate RNA fragments occupied by the RBP and sequencing of these fragments can identify RBP binding sites, which allows inference of RBP function through determining the location of binding sites relative to, for example, other RBP binding sites or cis-acting elements (Box 1).

Schematic overview of the core steps common to most variants of the cross-linking and immunoprecipitation (CLIP) protocol. RBP, RNA-binding protein. Adapted with permission from ref.14, Elsevier.
The development of high-throughput sequencing of RNA isolated by CLIP (HITS-CLIP) has enabled a transcriptome-wide view of RNA binding sites6. CLIP techniques have been further developed to identify cross-link sites with nucleotide resolution, either through analysis of mutations in reads (photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP))7 or by capturing cDNAs that terminate at the cross-linked peptide during reverse transcription (individual-nucleotide resolution CLIP (iCLIP))8. The development of dedicated bioinformatics workflows has allowed the determination of binding sites and consensus motifs to better understand post-transcriptional regulation9.
This Primer focuses on experimental and computational aspects of CLIP methods that have been broadly adopted and have generated widely used data sets. We also cover the identification of RBP binding sites by tagging RBPs with enzymes that naturally act on RNA, where the resulting RNA modifications can be identified by high-throughput sequencing10, as well as the use of subcellular compartment-specific proximity labelling to study localized transcriptomes11. Finally, we discuss the applications of these techniques to obtain a systems-level view of RNP assembly and dynamics in multiple model organisms and review strategies for method optimization and quality assessment of the data. For discussion of additional protein-centric methods, we refer the readers to recent reviews12,13,14. Note that we do not extensively cover studies that identify the global RNA-bound proteome, as these have been reviewed elsewhere1; instead, we focus on methods that identify proteins bound to specific RNAs to discuss how their insights complement protein-centric methods, and outline how these integrative approaches can take us closer towards a comprehensive view of RNP assembly and remodelling.
Experimentation
Protein-centric methods
All CLIP-based methods for determining the binding landscape of RBPs on a transcriptome-wide scale share the following core workflow (Fig. 1). First, RNAs and interacting proteins are irreversibly cross-linked by UV light in intact cells (UVC at λ = 254 nm or UVA/B at λ = 312–365 nm for PAR-CLIP). The amount of UV cross-linking energy used needs to be adapted depending on whether cell monolayers, a suspension of dissociated tissue15, whole tissue or whole organisms such as worms16 and plants17,18 are used. For tissues that cannot easily be dissociated, such as most adult mammalian tissues, plants or post-mortem human tissues, frozen tissue can be ground in liquid nitrogen to a fine powder and cross-linked on dry ice18,19. After cross-linking, RNAs are trimmed to short fragments by RNase digestion and the cross-linked RNP of interest is stringently purified using immunoprecipitation or other methods14 (Box 2). RNPs are then further purified using denaturing polyacrylamide gel electrophoresis (SDS-PAGE) and cross-linked RNA fragments released by digestion of the RBP, usually by proteinase K. The yield of RNA fragments is typically in the low-nanogram range, and thus protocols optimized to work with a limited amount of short RNAs are used to convert the RNA into cDNA for high-throughput sequencing20,21. Sequenced reads are mapped to the genome and clusters of overlapping reads representing possible binding sites are computationally separated from the usually high levels of background7,22,23. In order to reveal sites that are likely to be functional, for example those conferring post-transcriptional gene regulatory effects, the list of binding sites can be sorted according to various criteria including the relative RBP occupancy, which describes the fraction of all instances of a binding site occupied by the RBP at the time of cross-linking24.
Each variant of CLIP uses a unique approach to one or more of the above-mentioned steps. We describe the differences among primary variants below, with further comparisons and additional variants being covered elsewhere14. We do not intend to advocate one variant over another, but the provided information can help researchers to make an informed choice of their preferred CLIP variant. Note that RBPs differ greatly in their cross-linking efficiencies depending on their mode of RNA binding and whether UVC, 4-thiouridine (4SU)-induced UVA/B or formaldehyde cross-linking is used25,26,27. However, further studies are needed to determine what factors influence these relative efficiencies.
Original CLIP and its adaptation to high-throughput sequencing
Cross-linking in original CLIP workflows is accomplished using UVC, which preferentially cross-links RBPs to uridines and, to a lesser extent, guanosines28,29,30. Following mild RNase digestion and purification of the selected RBP, RNA fragments are ligated to a 3′ adapter and radiolabelled to visualize and aid purification of the cross-linked RNP after SDS-PAGE and membrane transfer15. Cross-linked RNA fragments are recovered, ligated to a 5′ adapter, converted into cDNA by reverse transcription and amplified by PCR, similar to the standard protocols developed for microRNA (miRNA) characterization31. However, here the reverse transcriptase needs to read across the oligopeptide attached to the cross-linked nucleotide to reach the 5′ adapter. Premature termination results in a bias towards contaminating non-cross-linked sequences in resulting cDNA libraries; some computational tools for HITS-CLIP therefore take advantage of the low but consistent mutation signature at such events22,32,33. CLIP was adapted for next-generation sequencing in HITS-CLIP6 (Fig. 2a) by adding sequences required for Illumina sequencing to the PCR primers6. The related approach cross-linking and analysis of cDNAs (CRAC)32, originally developed for yeast RBPs, uses affinity-based purification under denaturing conditions as an alternative to immunoprecipitation.

a | Comparative schematic of methods for the primary cross-linking and immunoprecipitation (CLIP) variants, including high-throughput sequencing of RNA isolated by CLIP (HITS-CLIP), individual-nucleotide resolution CLIP (iCLIP), infrared CLIP (irCLIP), enhanced CLIP (eCLIP), photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP) and Proximity-CLIP. Top box: steps prior to immunoprecipitation, including treatment of cultured cells with 4-thiouridine (4SU) and cross-linking. Middle box: RNA manipulation steps. Bottom box: cDNA preparation, sequencing and peak calling steps. Note that PAR-CLIP is predominantly performed using 4SU as a photoreactive nucleoside, but 6-thioguanosine (6SG) can also be used and results in a G to A transition. Cross-linking and analysis of cDNAs (CRAC), which closely resembles HITS-CLIP, uses protein tags that allow denaturing purification. b | Targets of RNA-binding proteins identified by editing (TRIBE) workflow. The RNA-binding protein (RBP) of interest is expressed as a fusion protein with the catalytic domain of the RNA editing enzyme RNA-specific adenosine deaminase (ADARcd). Binding sites are detected by ADAR-mediated adenosine to inosine modifications revealed by scoring for A to G mutations in cDNA libraries prepared by standard long-read RNA sequencing (RNA-seq). 32P, 3′ ligation products detected using a phosphorous 32-labelled adapter; IR800, 3′ ligation products detected using an IR800-biotin dye-labelled adapter; x, step not applicable. aRBPs biotinylated in a compartment-specific manner.
Individual-nucleotide resolution CLIP, infrared CLIP and enhanced CLIP
iCLIP8, infrared CLIP (irCLIP)34 and enhanced CLIP (eCLIP)35 differ from original CLIP in their purification and cDNA library preparation strategies (Fig. 2a; Box 2). They take advantage of the tendency of reverse transcriptase to terminate at the cross-linked nucleotide, which yields cDNAs with a 5′ end mapping to the first nucleotide downstream of the cross-linking site and allows the identification of cross-link sites at nucleotide-level resolution. To introduce primer binding sites for cDNA library amplification, iCLIP uses a cDNA circularization approach similar to the ribosome footprinting protocol36; reverse transcription is primed with a long DNA oligonucleotide containing both PCR primer sites, and the cDNA products are circularized using thermostable RNA ligases that also act on DNA37. At least 18 variants of CLIP have adopted the approach to amplify truncated cDNAs14; some, such as irCLIP, use cDNA circularization approaches similarly to iCLIP, whereas others, such as eCLIP and iCLIP2 (ref.38), use highly concentrated T4 RNA ligase 1 to ligate a DNA adapter to the 3′ end of the cDNA.
Photoactivatable ribonucleoside-enhanced CLIP
In PAR-CLIP7,15,5, cultured cells are incubated with nucleosides modified with an exocyclic thione group, specifically 4SU or 6-thioguanosine (6SG), which are then incorporated into nascent RNAs (Fig. 2a). The exocyclic thione group increases the photoreactivity of the base, allowing cross-linking with a lower energy of UV light (UVA/B, 312 ≤ λ ≤ 365 nm) than that used in other CLIP methods. When using 4SU, cross-linked amino acids are attached to position 4 of the base — changing its base-pairing properties — whereas unmodified uridines cross-link at position 5, which leaves their Watson–Crick face intact39. Cross-linked 4SU preferentially pairs with guanosine during reverse transcription, resulting in a characteristic T to C transition in the sequenced cDNA (a G to A transition occurs when using 6SG)7. This may simplify data analysis as enrichment of such transitions at specific genomic regions indicates bona fide interaction sites and helps to determine the precise location and strength of the RNA–RBP interaction.
CLIP of RNA hybrids
Some RBPs, including Staufen proteins, or the Argonaute proteins at the heart of RNA silencing pathways, bind RNA at double-stranded sequence elements. Standard CLIP assays will only reveal one of the bound strands, thus losing information on the nature of the RNA–RNA interaction. All major CLIP variants have been adapted to include an additional step of intermolecular ligation after the limited RNase digestion, which maintains the proximity of the two RNA fragments bound to the RBP and allows the reconstruction of RNA–RNA hybrids interacting with the RBP of interest. Argonaute HITS-CLIP40, cross-linking and sequencing of hybrids (CLASH)41 and modified PAR-CLIP42 have been used to sequence miRNA–target chimeras, and RNA hybrid and iCLIP (hiCLIP)43 revealed a prevalence of long-range intramolecular RNA duplexes bound by human STAU1 protein. These are complementary to the many additional methods that profile RNA structures on a transcriptomic scale by chemical-based approaches or by mapping RNA–RNA contacts12. CLIP has recently been integrated with one such chemical-based approach, selective 2′-hydroxyl acylation analysed by primer extension (SHAPE), to reveal the hydrogen bonds at RNA–protein interfaces44.
Proximity-labelling based isolation of compartment-specific RNAs
Proximity-CLIP11 and the related technique APEX-seq45,46,47 allow the determination of RNA distribution to specific subcellular locations. Both techniques rely on the biotinylation of RNAs (exploited in APEX-seq) and proteins (exploited in Proximity-CLIP) by the engineered ascorbic acid peroxidase protein APEX2 (ref.48), a tool widely used to quantify the localized proteome49 (Supplementary Table 1). To allow subcellular compartment-specific biotinylation of RNA and proteins, APEX2 is typically fused to specific localization elements50. In the case of Proximity-CLIP, prior to protein biotinylation, nascent transcripts are labelled with either 4SU or 6SG and cross-linked to interacting RBPs with UV light of 312–365 nm (Fig. 2a). The compartment-specific proteome, including cross-linked RNPs, is then isolated on streptavidin beads and cross-linked RNA fragments are isolated and sequenced following mild RNase digestion. The characteristic mutations in the cDNA resulting from the use of photoreactive nucleosides reveal cross-linked sequences. A distinctive feature of Proximity-CLIP is that the sequencing of RBP-protected footprints allows for both the profiling of localized RNAs and the identification of protein-occupied, possibly regulatory, cis-acting elements on RNA. In contrast to APEX-seq, this approach provides a snapshot of regulatory elements on RNA that are occupied in the examined compartments.
Numerous other recently developed techniques are capable of performing compartment-specific labelling and analysis of RNA and/or proteins. Some approaches use genetically encoded photosensitizers localized to specific compartments, which mediate the oxidation of proximal guanosines by generating reactive oxygen species after irradiation with visible light51,52,53. Photosensitized guanosines can then be coupled with reactive amino group-containing probes to isolate and quantify localized RNA.
Targets of RNA-binding proteins identified by editing
Enzymatic tagging approaches can allow for transcriptome-wide identification of endogenous RBP interaction sites without requiring cross-linking, biochemical immunoprecipitation or cDNA library preparation steps. An example is targets of RBPs identified by editing (TRIBE)10, which is conceptually related to DNA adenine methyltransferase identification (DamID), a method that identifies chromatin protein-bound regions by fusing them to the Dam methyltransferase and identifying the methylation sites54. TRIBE relies on transgenic expression of the RBP of interest fused to the catalytic domain of double-stranded RNA-specific adenosine deaminase (ADARcd) — which catalyses adenosine to inosine conversions near the RBP interaction sites — or its hyperactive mutant (HyperTRIBE)55. These sites are revealed by excess A to G mutations in libraries that are prepared as standard RNA sequencing (RNA-seq) libraries (Fig. 2b). Among the distinct advantages of TRIBE over CLIP approaches are its minimal number of manipulation steps — which allows for the use of small numbers of cells — and the possibility of expressing the RBP–ADARcd fusion protein in a cell type-specific manner to reveal RBP interactomes in precisely defined subpopulations of cells in model organisms. A disadvantage is that very deep sequencing is necessary to capture sufficient editing signal (A to G mutations) to call interaction sites. Further, carboxy-terminal or amino-terminal fusions of ADARcd may compromise the localization and activity of some RBPs and their ectopic expression in vivo requires optimization to ensure proper cell-type specific expression patterns and avoid excessive levels of RBP–ADARcd fusion protein levels, which can obscure target sites and lead to toxicity caused by hypermodification of RNA. Recently, an approach termed surveying targets by APOBEC-mediated profiling (STAMP) has been developed where RBPs are tagged with APOBEC enzymes56. These enzymes access cytosine bases in single-stranded RNA and produce clusters of edits, giving increased coverage of mutations compared with TRIBE, which relies on ADAR-mediated editing of the relatively infrequent RNA duplexes containing a bulged mismatch10. This higher likelihood of encountering APOBEC1 cytosine substrates increases the sensitivity of STAMP and enables it to be coupled with single-cell capture.
RNA-centric methods
To unravel the composition of full RNPs assembling on a specific RNA, RNA-centric methods are needed to complement protein-centric approaches57. Such methods generally use either RNA affinity capture purification or proximity-based protein labelling.
RNA affinity proteome capture
RNA affinity proteome capture methods are mainly in vitro approaches based on either tagging the endogenous RNA or modifying in vitro-transcribed or synthesized RNA at the 3′, 5′ or both ends with biotin or similar small molecules58 and immobilizing them on solid surfaces such as streptavidin beads (Table 1). Cellular extracts are then added to the immobilized beads, the beads washed and proteins bound to the labelled probes eluted by boiling the beads in SDS elution buffer.
An alternative affinity capture approach is to tag an RNA of interest with aptamers derived from virus-derived heterogeneous RNA stem loops, such as MS2 (ref.59), PP7 (ref.60), S1 (ref.61), Cys4 (ref.62) and D8 (ref.63), or aptamers that mimic tobramycin64 or streptomycin65 (Table 1). When choosing the aptamer, one has to consider the binding affinity of the tag with the cognate ligand, keeping in mind that for highly enriched RNPs, a low binding affinity aptamer–ligand interaction can be sufficient to pull-down highly enriched interactors and will give less background with more specific elution. Lysates from cells expressing the tagged RNA of interest are passed through beads containing the respective substrates. These are stringently washed, which can include applying a competitive binder, and the proteins are eluted for mass spectrometry analysis.
Post-lysis reorganization of RNPs66 may result in the detection of false-positive associations of RBPs with specific RNA baits. To avoid this, several approaches cross-link RNPs in cultured cells by UV with or without photoreactive nucleosides or chemically with formaldehyde prior to cell lysis (Table 1). For example, capture hybridization of analysis of RNA targets (CHART) allows the mapping of interaction sites and proteins bound to the Drosophila RNA roX2 (ref.67) and RNA antisense purification (RAP) has been used to identify the interactome of the non-coding RNAs Xist68 and NORAD69. Comprehensive identification of RBPs by mass spectrometry70 (ChIRP-MS) also systematically identified Xist-interacting proteins in mice and in vivo interactions by pull-down of RNA (vIPR) studied proteins interacting with Caenorhabditis elegans gld-1 RNA71. During the recent COVID-19 public health emergency, RAP and ChIRP-MS were immediately applied to identify host and viral RBPs interacting with the SARS-CoV-2 RNA genome72,73.
RNA-directed proximity-based proteome labelling
RNA-directed proximity-based methods investigate the protein binding partners of a specific RNA in its native cellular context without the need for cross-linking, which is particularly useful for uncovering transient interactions and for studying RNPs from poorly soluble cellular compartments that are prone to precipitate during affinity capture methods, such as chromatin, peroxisomes or the Golgi body. In these methods, a labelling enzyme is recruited to a specific RNA to covalently modify the proteins located in the vicinity of the RNA (Table 2). The enzyme can be recruited to specific RNAs by expressing an aptamer on the RNA and a corresponding loop-binding protein tag on the labelling enzyme. RNA–protein interaction detection (RaPID) approaches use a plasmid expressing the RNA of interest flanked by BoxB stem loops and BASU — a mutant version of BirA*, engineered from Bacillus subtilis — fused to a BoxB stem loop-binding λN peptide13. The RNA of interest can also be tagged endogenously in approaches such as RNA-BioID74. Alternatively, a modified CRISPR–Cas system can be used to recruit an enzyme to an endogenous RNA by tagging the enzyme with an RNA-guided Cas variant and using guide RNAs that are antisense to the RNA of interest75. The excess pool of enzymes not docked to the tagged RNA can produce noise, but this can be reduced by using split proximity-based, RNA-assisted tools such as split APEX2, where two inactive APEX2 subunits are reconstituted to restore peroxidase activity upon physical co-localization76.
Results
Sources of background in CLIP
CLIP reads originate from a large number of RNAs, even when the RBP of interest is predicted to have few functional RNA partners. This could be because most reads reflect short-lived RBP–RNA interactions, whereas functional RNA partners tend to have a high total residence time on the RNA. Thus, binding regions that accumulate a high number of CLIP reads, either narrow or broad, are thought to be functionally relevant77, whereas the regions with few reads are viewed as ‘intrinsic’ background, reflecting transient interactions. There is no absolute distinction between stable and transient interactions, and the functionality of these modes of interaction differs between RBPs (Fig. 3a). For example, CLIP of the P granule protein MEG-3 in C. elegans showed that its function depends on interactions across the full transcripts that are not sequence-specific78. Thus, thought needs to be given to what may constitute an intrinsic background for different RBPs.

Sources of variability in cross-linking and immunoprecipitation (CLIP) experiments. a | Sources of intrinsic background. RNA-binding protein (RBP)–RNA interactions are dynamic, and therefore the probability of an RBP cross-linking to a cognate RNA site at the time of experiment is affected by multiple factors: synergistic or antagonistic interactions between RBPs on the same RNA region, the residence time of the RBP on the RNA (low on low-affinity sites and high on high-affinity sites) and the availability of the RBP and the cognate site, which is influenced by time-dependent stochastic fluctuations in expression and localization. b | After cross-linking, cells are lysed and the RNAs fragmented. Fragmentation is mediated by RNAses, most of which have some sequence bias. This, along with the duration of treatment, leads to fragments of variable length. An RBP-specific antibody is used to immunoprecipitate the protein along with cross-linked RNA fragments. Cross-reactivity or lack of antibody binding can lead to false or undetected sites (grey box). c | The cross-link constitutes a roadblock for reverse transcription, leading stochastically to different types of fragment: those that are accurately transcribed across the cross-link sites, those where reverse transcription stops at the cross-link site and those where mutations, deletions or insertions are introduced at the site of cross-link. Individual-nucleotide resolution CLIP (iCLIP) variants aim to capture the fragments that truncate at the cross-link position, whereas photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP) aims to capture fragments where read-through occurs. d | During PCR, individual fragments are amplified with variable efficiency. To recognize reads that resulted from the amplification of the same initial fragment, unique molecular identifiers (UMIs) are attached before the amplification step and identical reads (those with identical UMIs) are collapsed to a single read, representing the unique initial fragment. Adapters and UMIs are removed before read counts associated with individual genomic positions are tabulated.
Limited selectivity of the antibodies used to immunoprecipitate RBPs can lead to contamination of the sample with additional RBPs and their bound RNAs, and abundant RNAs may also be carried through sample preparation (Fig. 3b). The quality control and purification of the RBP–RNA complexes of interest on the SDS-PAGE gel are important in analysing and mitigating these two sources of ‘extrinsic’ background, and the way this step is implemented can vary between CLIP protocols (Box 2). It is advisable that control samples are prepared in parallel using IgG-bound or antibody-bound beads and RBP-knockout material, barcoded, pooled and sequenced, to compare with the experimental samples and assess their data specificity.
Quantification of CLIP reads can be complicated by the presence of PCR duplicates resulting from non-uniform amplification of different sequences. Aside from careful optimization of PCR cycle numbers79, the use of unique molecular identifiers (UMIs) for cDNAs produced by most current CLIP variants can mitigate introduction of these artefacts14 (Fig. 3c). UMIs are highly diverse barcodes composed of randomly incorporated nucleotides that are added to the RNA or cDNA fragments using adapters or reverse transcription primers before PCR amplification. As it is highly unlikely that the experiment produces two identical fragments that also ligate to two identical UMIs, the presence of multiple copies of a read with the same UMI will indicate PCR duplicates, which can be computationally collapsed to a single read. Computational tools, such as iCount8, expectation–maximization-based algorithms80 or UMI-tools81, take advantage of the presence of UMIs to quantify the number of unique cDNAs in the library even in the presence of sequencing errors.
CLIP analysis workflow
Peak identification
All CLIP variants aim to capture individual binding sites of RBPs with nucleotide-level resolution; however, the exact experimental approach determines the relationship of the reads to the cross-linked nucleotides on the RNAs and, consequently, the computational analysis that is necessary for revealing the binding sites. Workflows for CLIP data analysis generally cover the following main steps: preprocessing of CLIP reads; alignment of reads to the corresponding genome; peak identification; combined analysis of replicates to identify reproducible peaks; and meta-analysis to identify binding motifs, relationships between binding sites, their positioning relative to transcript landmarks and the functional consequences of binding. We provide a summary of recently introduced or updated tools for binding site identification and peak detection in Table 3. Software for finding motifs and predicting RBP binding sites and peak finding tools only applicable to specific sets of targets can be found in recent reviews9,82.
Peak identification is an important step that serves to identify regions of the RNA to which the RBP directly binds with high occupancy, thereby representing likely functionally relevant interactions (Fig. 4a,b). The primary goal of peak-calling is to identify RNA regions where the number of cross-link diagnostic features is significantly higher than expected based on background models. These features can be the number of reads mapping to these regions, as well as cross-linking-induced substitutions, insertions/deletions or truncations, depending on the experiment. cDNA mutation and/or truncation occur when the reverse transcriptase reads past the cross-linked nucleotides or truncates at them and are identified once the reads are aligned to the genome. Sites of high RBP occupancy on the RNA are revealed by their high density of reads or cross-linking-induced features relative to neighbouring regions of the same type (introns, coding sequence, 3′ untranslated region) that have similar expression within each gene (Fig. 4a,b). It is important to be aware that a gain in specificity through increased stringency of peak calling can lead to a drop in sensitivity, as discussed later.

Extraction of peaks from cross-linking and immunoprecipitation (CLIP) data. a | Following adapter and duplicate removal, inserts are mapped to the genome or transcriptome. As an example, tracks show density profiles in the region of the tubulin (TUBB) gene corresponding to samples obtained from K562 cells in the ENCODE project using enhanced CLIP (eCLIP) for Pumilio homologue 2 (PUM2 CLIP) and a size-matched input control (PUM2 SMI) and RNA sequencing (RNA-seq). Numbers 0–77, 0–20 and 0–33 correspond to the maximum read coverage in this region. Colours in the bands at the bottom indicate different nucleotides. The TGTA.ATA motif has been shown to be recognized by PUM2 and, thus, indicates the location of the true binding site. Various approaches are used to distinguish peaks of high RNA-binding protein (RBP) occupancy from background. Background models are constructed from regions neighbouring the putative peaks in the CLIP sample itself or from the same region as the peak in the SMI or RNA-seq samples (indicated by coloured brackets). b | Peaks defined as contiguous regions where the number of reads is significantly higher than expected based on the background models. Coloured dashed lines show the average coverage in different types of background regions/samples (indicated by brackets of matching colours in panel a). Some tools consider the number and type of cross-link diagnostic mutations. Grey shading on the peak indicates the region where most variation in peak shape is expected, depending on the CLIP variant. In individual-nucleotide resolution CLIP (iCLIP), mostly truncated cDNAs are sequenced, leading to an abrupt increase in read coverage starting right after the position of the cross-link.
Assessing background
Peak calling serves to computationally remove the intrinsic background generated by transient interactions. However, when the protein binds broadly along RNAs, without clear peaks of diagnostic features, estimates of the abundance of RNAs encountered by the RBP can improve the detection of these targets. The extrinsic background needs to be assessed experimentally during the quality control step of the size-separated protein–RNA complexes and possibly by obtaining additional data that identify the likely contaminating RNA fragments. In chromatin immunoprecipitation followed by sequencing (ChIP–seq), immunoprecipitation with beads lacking antibody is used to generate a background sample for peak calling. In CLIP experiments, however, it is more challenging to generate experimental background samples. When performing CLIP with beads lacking antibody, the signal on SDS-PAGE is negligible, yielding 100-fold fewer reads if sequenced, which is insufficient for extrinsic background modelling8. Instead, one can use RNA-seq to identify regions where a large number of CLIP reads are a result of high RNA abundance rather than high occupancy by the RBP (Fig. 4a). Outliers are identified with respect to a negative binomial distribution whose parameters are determined from the background sample. This distribution captures the fact that the variance in coverage is generally larger than the mean, contrary to what would be expected from sampling reads with constant probability along a genomic region9. A related approach to assess background experimentally has been taken in eCLIP, where a size-matched input (SMI) is generated by performing all steps of the protocol apart from immunoprecipitation35 (Fig. 4a). The importance of background samples was illustrated in eCLIP by the example of the stem loop-binding protein, where only 1.2% of the peaks identified from the foreground sample were enriched over the background SMI35.
Although approaches to remove background are expected to increase the proportion of functionally relevant binding sites among the called peaks, they can introduce new biases. The SMI sample in eCLIP is often dominated by RNAs cross-linked to abundant RBPs that may not be the same RBPs that contaminate experimental samples, owing to their interactions with the RBP of interest. Conversely, the SMI could be dominated by the RBP of interest itself, resulting in the foreground signal becoming erroneously assigned to the background, precluding the identification of relevant binding sites. RNA-seq may introduce bias depending on whether poly(A) selection or ribosomal RNA depletion was used, each of which yields somewhat different estimates of gene and transcript expression. Poly(A) selection enriches for fully processed RNAs, thereby depleting introns. Ribosomal RNA depletion requires enough sequencing depth to assess individual introns, as even within a gene the abundance of different introns can vary depending on the time taken for transcription, splicing and degradation of each intron. Moreover, the delay between transcription and co-transcriptional splicing leads to increased coverage towards the 5′ end of long introns83, which is common in genes expressed in the brain83,84,85. Such issues suggest that it will be important to obtain data that can accurately estimate the abundance of intronic regions in order to optimally detect enriched intronic CLIP peaks. Finally, most RBPs are localized to specific cellular compartments, where the abundance of RNAs may be quite different from the average abundance of the whole cell. Thus, it will be valuable to develop models based on the local abundance of RNAs that each RBP encounters, estimated based on RNA-seq from cellular subfractions, APEX-seq and/or Proximity-CLIP.
Characterizing RBP binding motifs
Once binding peaks have been identified, the immediate aim is to uncover the sequence and/or structure specificity of the protein. Traditionally, position-specific weight matrices (PWMs) have been used to represent the sequence specificity of nucleic acid-binding proteins, whether transcription factors or RBPs (Fig. 5). PWMs indicate the relative frequency with which individual nucleotides are observed among the binding sites of an RBP, which, in turn, can be related to the contribution of individual nucleotides in the binding site to the energy of interaction with the RBP and thus the affinity of this interaction. PWMs can be inferred from sequences obtained in CLIP experiments with readily available computational tools86,87,88. A key assumption of PWMs is that nucleotides in the binding site contribute independently to the energy of RBP–RNA interactions. This assumption started to be questioned as high-throughput binding data — for example, from protein microarrays — became available. It has been argued that parameter-rich models derived, for example, through machine learning approaches are necessary to quantify the affinity of protein–nucleic acid interactions89,90,91. However, other studies explicitly modelling confounding experimental factors concluded that PWMs are sufficient to quantitatively explain the binding data for the majority of transcription factors92.

Typically, peaks that are reproducibly identified in replicate experiments are extracted for further analyses. Here, the agreement between the peaks obtained in two replicates of enhanced CLIP (eCLIP) for Pumilio homologue 2 (PUM2 CLIP) is shown as a function of the number of top peaks selected from each replicate. Peaks are sorted by score, the top x peaks (x indicated by the x axis) are extracted, and the proportion of overlapping peaks is shown on the y axis. Two peaks are considered overlapping if they share at least one nucleotide. Reproducible peaks can be annotated with their location in different genomic regions, the types of RNA in which they occur or the region of protein-coding RNAs (5′ untranslated region (UTR), coding sequence (CDS), 3′ UTR) in which they reside. The sequences of the most enriched peaks (here, the top 1,000 from each of the two samples) are used to search for enriched motifs that point to the sequence preference of the RNA-binding protein (RBP). In this case, the motif identified from the top peaks is the recognition element of PUM2. Information content on the y axis summarizes the strength of the preference for a specific nucleotide at a given position in the binding site. CLIP, cross-linking and immunoprecipitation; lncRNA, long non-coding RNA; miRNA, microRNA; NMD, nonsense-mediated decay.
In the case of RBPs, PWMs are also used to explain both CLIP data and in vitro measured affinities of interaction with RNAs93,94. However, RNA–RBP interactions are likely more complex than the interactions of transcription factors with DNA. The accessibility of binding sites — modulated through an RNA secondary structure that depends on RNA modifications95 — plays an important role in RBP–RNA interactions. A detailed analysis of Gld-1 binding in C. elegans found that a biophysical model including the PWM-defined specificity of the Gld-1 RBP and the predicted structural accessibility of binding sites in RNAs was able to explain the relative enrichment of binding sites in CLIP, alleviating the need for a more parameter-rich model96. Examination of the secondary structure around CLIP binding sites demonstrated that the recognition of RBP binding motifs by RBPs often requires a specific structural context97,98 and led to models that simultaneously infer the sequence–structure preference of RBPs99,100,101 and allow the identification of sites that were missed in CLIP experiments owing to, for example, low RNA expression levels99. Similarly, machine learning approaches have increased the depth of miRNA binding site identification from Argonaute-CLIP data102. Biophysical approaches for the ab initio prediction of molecular interactions can pinpoint potential false negatives in CLIP experiments and provide insights into the interaction propensities that, ultimately, determine the location of binding sites in RNAs103. Conversely, CLIP data typically provide large data sets that can be used to infer biophysical models of RNA–RNA interactions in the context of RNP complexes, such as the ternary miRNA–mRNA–Argonaute protein complex104. These inferred models can predict affinity interactions measured in vitro with surprising accuracy105.
Many tools take into account cross-linking-induced mutations to call RBP binding sites and determine the sequence and structure specificity of the RBP28,100,106,107. Annotation of the putative location of binding sites with respect to various landmarks such as splice sites, the functional category of the gene as well as binding data for RBPs other than the RBP of interest can be further incorporated to improve the accuracy of binding site identification108,109. A drawback is that enforcing specific constraints without a mechanistic basis may lead to overlooking unusual binding sites. Furthermore, it is not always clear that the increase in accuracy justifies the potential for overfitting and reduced interpretability that comes with an increased number of parameters.
Regulatory grammar
The final step in deciphering CLIP data is uncovering the regulatory grammar of the RBP binding sites, including the spatial relationship of RBP binding sites to important transcript categories — such as coding/non-coding transcripts, repeats, small nucleolar RNAs and rRNAs — and landmarks such as exons, introns, exon/intron boundaries and translation start/stop sites110. Binding site data can be combined with data from knock-down and overexpression experiments to generate RNA maps reflecting the functional impact of binding sites located in different transcript regions111. Computational modelling of changes in the expression of transcript isoforms upon perturbation of individual RBPs provides complementary information regarding the RBP binding motifs that are involved, their location within transcripts and their functions in individual steps of RNA processing112. As the number of RBPs studied by CLIP continues to increase, direct comparisons of the binding site profiles in the genome are starting to reveal regulatory complexes and competition between RBPs. Both of these are reflected in multiple proteins binding to closely spaced sites in the RNA, whereas the data from perturbation experiments help resolve the nature of the interactions between RBPs110,113,114.
Assessing the specificity of CLIP
In contrast to RIP or ChIP-seq, CLIP has an in-built step for experimental control of specificity. Visualizing the size-separated protein–RNA complexes can allow estimation of the extrinsic background, which yields signals in negative control lanes or at unexpected sizes. From its initial publication, high standards were established for the specificity of CLIP, evident from the absence of a signal in the negative control and a >20-fold enrichment of binding motifs within Nova CLIP reads compared with the control5. Fusion of affinity tags to the studied RBP can further increase specificity by allowing even more stringent, denaturing purification conditions that maximize the removal of extrinsic background14. However, data specificity for the immunoprecipitation-based variants of CLIP can vary depending on the quality of the antibody and the degree of optimization; when studying a new RBP using CLIP, RNase fragmentation and immunoprecipitation conditions must be optimized for variations in RNase stocks, cross-linking efficiencies of RBPs, the stability of their interactions with other RBPs and the type of cells or tissue used15,115.
As optimizations are carried out to variable extents across laboratories employing CLIP, there is a need for computational assessment of CLIP data to facilitate integration of collected data sets. The first approach is to study the cross-link distribution across RNA types. Nuclear and cytoplasmic RBPs tend to have the most cross-links in introns and exons, respectively. In cases where the dominant RNA binding partners are known, these are expected to rank highly in the data. However, the most likely source of extrinsic background is RBPs that interact with the studied RBP, which often have similar localization patterns and RNA partners; therefore, analysis of RNA types offers only partial reassurance. The second approach is to compare the enrichment of sequence motifs in CLIP data with their affinities for the purified RBP as determined by biophysical methods. Systematic motif enrichment data are available from in vitro binding assays such as SELEX116,117, RNA Bind-n-seq118 and RNAcompete97. Often, in vivo-identified binding sites resemble the highest-affinity motifs derived from these methods. When they do not, the reason can either be the low specificity of the in vivo data or biases of in vitro assays. For example, these assays often examine the binding of individual domains rather than full proteins, which lack post-translational modifications and the context of other proteins. They also tend to study binding to short RNA sequences, whereas in vivo RBPs can assemble on long RNAs with complicated secondary structures. To distinguish whether the RNA features that are unique to the in vivo data reflect the specificity of the RBP or represent technical artefacts, it will be informative to examine the reproducibility of these features across multiple data sets produced by various laboratories or by various protein-centric methods for the same RBPs.
For many RBPs there is no in vitro binding information available to provide expected binding motifs. However, binding motifs can be identified de novo from the CLIP data and the extent of their enrichment provides some measure of data quality. For example, a comparison of publicly available data for polypyrimidine tract binding protein 1 (PTBP1) revealed that whereas all CLIP variants show enrichment of similar motifs, the extent of enrichment varies dramatically between variants, indicating major differences in data specificity115. There are several caveats to de novo motif discovery using CLIP, as factors unrelated to the studied RBP may result in enrichment of specific sequence motifs. Such factors include the nucleotide preferences of UV cross-linking or the sequence biases of the RNases and RNA ligases used to join adapters to the ends of RNA fragments22,29,79,115. One way to minimize the impact of these biases is by producing parallel data sets for diverse RBPs from the same type of biological material and then deriving motifs unique for each RBP after correcting for the features that are in common for different RBPs7,28,85,119.
A recent approach to assess the validity of de novo motifs involves the analysis of sites overlapping heterozygous single-nucleotide polymorphisms. A difference in the number of CLIP cDNAs mapping to the two alleles indicates that the single-nucleotide polymorphism affects cross-linking efficiency28, and therefore likely influences the affinity of the RBP of interest to the site. However, allelic imbalance is equally expected at motifs bound by co-purified RBPs that represent extrinsic background, and can also result from the nucleotide preferences of cross-linking, and therefore should be interpreted with caution.
Finally, enrichment of CLIP peaks around regulated elements, such as alternative exons, can be assessed using RNA maps to understand the ‘functional specificity’ of data, which can yield comparative assessment for multiple data sets of a specific RBP111. Such analysis requires that orthogonal data that examine functionality are available, such as RNA-seq of knockout or knock-down cells or tissues93. Finally, experiments to support the functionality of specific binding sites can be designed by perturbing such sites, such as through mutations of cis-acting elements in minigene reporters or CRISPR-mediated mutations of the endogenous gene, or by blocking them with antisense oligonucleotides.
Assessing the sensitivity of CLIP
The sensitivity of CLIP refers to its capacity to comprehensively identify the relevant RNA sites bound by the studied RBP. Such sensitivity depends on the complexity of the resultant cDNA library, that is, the number of unique cDNAs produced. This has increased by orders of magnitude with the adaptation of high-throughput sequencing and the increased efficiency of cDNA library preparation steps14. However, the capacity to prepare high-complexity libraries depends on RBP characteristics, particularly abundance and UV cross-linking efficiency. In addition to the cDNA complexity, the sensitivity of CLIP also depends on specificity because increased external background will decrease the proportion of signal for the RBP of interest. For example, CLIP libraries for PTBP1 of similar complexities showed different numbers of identified binding peaks115 and different capacities to identify binding sites around regulated exons as evident with RNA maps. The choice of peak-calling method strongly affected the functional sensitivity of the same PTBP1 CLIP data9. These points highlight the need for combined analysis of data specificity and sensitivity when assessing the pros and cons of the experimental variants of CLIP and of the various computational approaches to data analysis.
Applications
CLIP experiments have been carried out using various model organisms, including mammalian cell culture35, yeast32, mice6, flies120, worms16,121 and plants17,18 (Table 4). Below, we discuss applications of CLIP techniques in selected systems with distinctive considerations, advantages and disadvantages for various applications.
Cell culture models
Cultured cells (transformed cell lines, primary cells and stem cells) are the most widely used experimental model for CLIP, with more than 2,500 different CLIP data sets deposited on the Gene Expression Omnibus at the time of writing. Only ~7% of RBPs are either expressed in a tissue-specific manner or show strong tissue-specific expression bias, mainly in the germline and, to a lesser extent, neuronal tissues122,123, whereas the rest tend to be expressed across most cell types124, making cultured cells appropriate for the majority of cases with the caveat that some RBP targets may be absent. Cultured cells are easily genetically tractable, allowing for epitope tagging of RBPs for stringent purification, introduction of transgenically expressed cell type-specific RBPs or introduction of a clinically or functionally important mutation that could be lethal in an animal model. Cell culture also allows for multiple RBPs to be studied in a comparative manner in the context of the same transcriptome. The same principles apply to single-cell organisms such as yeast, although its lower cross-linking efficiency make it difficult to use in CLIP experiments32.
Although the use of cultured cells provides valuable insights into mechanisms of post-transcriptional regulation — even for ectopically expressed RBPs125 — certain key bound transcripts and interacting proteins may be expressed in a cell type-specific manner themselves. Further, the binding repertoire of RBPs regulating biological processes such as developmental transitions or circadian timekeeping may be best studied in an organismal context.
Model organisms
CLIP/HITS-CLIP5,6, iCLIP85, PAR-CLIP16,126 and eCLIP127 have all been successfully used in mouse, fly and worm models. These studies provided useful insights into the roles of RBPs in various aspects of mRNA biogenesis and regulation during neuronal development and function122, as well as specialized functions such as transposon silencing in human and mouse brain128 and the Piwi-interacting RNA (piRNA) pathway in mouse testes and fly embryos129,130,131. Animal models present unique challenges for the application of CLIP techniques. First, most tissues require mechanical dissociation of fresh or frozen tissue prior to UV cross-linking5,80. In the case of PAR-CLIP, modified nucleotides must be delivered to the cells of interest prior to cross-linking; this can be accomplished by injection or use of transgenic animals expressing uracil phosphoribosyltransferase in a cell type-specific manner to allow the conversion of thiouracil into thiouridine — a process known as TU tagging132. Second, lethal mutations can only be studied if introduced in a conditional manner. Last, if a specific antibody for immunoprecipitation of the RBP is not available, expression of an epitope-tagged version of the RBP in a transgenic animal is required. Nevertheless, by epitope tagging the RBP of interest in specialized cell types133, CLIP can be performed from a subset of cells, analogous to TRIBE10. This approach, employed by conditionally tagged CLIP (cTag-CLIP), revealed the interactome of Nova2, Pabpc1 and Fmrp in various cell types, including neuronal subsets of mouse brain134,135,136.
Plants
Investigating the RNP composition in higher plants is made difficult by several technical challenges. In contrast to mammalian cell cultures, plant cell cultures cannot be cultivated in monolayers and are of limited use for CLIP techniques; as a result, experiments have mostly been performed in transgenic Arabidopsis plants expressing epitope-tagged RBPs17,18. Although the presence of UV-absorbing pigments and secondary metabolites such as chlorophyll and flavonoids can inhibit cross-linking efficiency, UVC-based cross-linking has been successfully applied to whole plants17,18. Another obstacle in plants is the rigid cell wall that requires mechanical force and harsh denaturing conditions for efficient cell lysis137. Moreover, the large amounts of endogenous RNases present in the plant vacuole require the use of RNase inhibitors to prevent extensive RNA degradation during extract preparation (also reported for pancreatic tissue). To ensure a controlled RNase treatment to fragment RNA, RNase treatment is performed after immunoprecipitation of the RNA–protein complexes rather than on the lysate18.
Genome-wide binding data from HITS-CLIP have been obtained in Arabidopsis for HLP1, a protein with similarity to mammalian HNRNPA/B17. In the hlp1-knockout mutant, a shift from proximal to distal polyadenylation sites was observed for more than 2,000 transcripts. As HLP1 binds to approximately 20% of these aberrantly polyadenylated transcripts close to the polyadenylation site in vivo, it has been implicated in regulating their alternative polyadenylation; aberrant polyadenylation of transcripts involved in flowering time control may explain the delayed transition to flowering in the hlp1 mutant17.
The first plant iCLIP study was performed for the heterogeneous nuclear RNP (hnRNP)-like Arabidopsis thaliana glycine-rich RNA-binding protein 7 (AtGRP7)18, which revealed that AtGRP7 binds to U/C-rich motifs mainly in the 3′ untranslated regions of its targets. Among AtGRP7 binding partners were transcripts that are only expressed in inner cell layers of the leaf, demonstrating that UV light penetrates deep into the tissue. Cross-referencing RNA-seq data of mutants and overexpression lines revealed that AtGRP7 predominantly downregulates its binding partners, dampening the peak expression of circadian clock-regulated transcripts in line with its role as a slave oscillator transducing timing information from the circadian clock to rhythmic transcripts within the cell138.
Many protein candidates for CLIP have emerged from proteomic studies identifying proteins that UV cross-link to polyadenylated RNAs in Arabidopsis tissues. To increase the efficiency of UV cross-linking, these studies were performed in etiolated (dark-grown) seedlings to avoid the presence of chlorophyll139, as well as in leaf protoplasts, cells without a cell wall140, cell suspension cultures and leaves of adult plants141,142. These studies identified more than 1,100 candidate RBPs; only a few RBPs were identified by all studies142,143, potentially owing to the different developmental stages and tissues investigated and the different protocols and levels of stringency used. As in non-plant species144, a recurrent theme of these studies was that many proteins without known RNA-binding domains or without a link to RNA biology were identified139,140,141,142. Among these were photosynthesis-related proteins and photoreceptors with no known role in RNA-based regulation; it is imperative to validate their RNA-binding activity by methods such as CLIP143.
Development and disease
RBPs play many important roles in development and diseases1,124. The first applications of CLIP concerned brain-specific RBPs that regulate alternative splicing and are implicated in neurological diseases, such as Nova proteins122. The capacity of CLIP to define binding sites in low-abundant RNAs led to an unexpected finding that splicing regulators can have many thousands of high-affinity binding sites in introns5,6. Binding sites close to alternative exons coordinate splicing in a highly position-dependent manner that can be described by an RNA map6,111. Moreover, most binding sites are located far from annotated exons and these often repress splicing of cryptic exons such as those emerging from transposable elements145. CLIP of core spliceosomal components, such as PRPF8, can also be used to interrogate splicing mechanisms, such as the regulation of recursive splicing by the exon junction complex, which is particularly important for appropriate splicing in the brain146. Moreover, CLIP has been used to study a broad range of RBPs with roles in the regulation of RNA transport, stability and translation. For example, HITS-CLIP study of Fragile X mental retardation protein (FMRP) revealed its binding to a subset of transcripts across their entire coding length, which was suggested to result from its dual interactions with the ribosome and the mRNA that could be important for its regulation of local translation at the synapse80.
CLIP can be performed on post-mortem human tissues to interrogate pathology-related changes in protein–RNA interactions. For example, a study of brain tissue from patients with pathological aggregates of TDP43, an RBP implicated in multiple neurodegenerative diseases, demonstrated increased binding to the non-coding RNA NEAT1 (ref.147). NEAT1 assembles multiple RBPs, including TDP43, into biomolecular condensates called paraspeckles148. TDP43 in turn regulates the 3′ end processing of Neat1 RNA, which leads to cross-regulation between NEAT1 and TDP43 that contributes to exit from pluripotency in mouse embryonic stem cells149. Such cross-regulation between RNAs and RBPs is likely a common phenomenon; it is becoming clear that RNAs can act as regulators of their bound RBPs, as was shown for the case of vault RNA-dependent regulation of proteins involved in autophagy150.
CLIP is increasingly used in pathogen research, including in studies concerning the RNA interaction profiles of bacterial RBPs151 and viral remodelling of the host and viral RNA–RNP interactome. For example, miRNAs encoded by Kaposi’s sarcoma-associated herpesvirus (KSHV) may function by competing with host miRNAs for AGO2 (ref.152), and a later study using CLASH additionally identified more than 1,400 cellular mRNAs that are targeted and might be regulated by KSHV miRNAs153. Moreover, a study of the HIV-1 Gag protein uncovered dramatic changes in its RNA-binding properties that occur during virion genesis and contribute to viral packaging154, a study of APOBEC3 proteins showed how their RNA binding ensures their effective encapsidation into HIV-1 as part of the host’s defence155 and a study of poly(C)-binding protein 2 (PCBP2) provided support for its roles in hepatitis C virus-infected cells156. These studies also provided computational solutions for parallel analysis of human and user-definable non-human transcriptomes. Most recently, CLIP has been used to identify human RNAs that are bound by the proteins encoded by the SARS-CoV-2 genome, such as non-structural proteins157 and nucleocapsid protein158, which helped to show how these RBPs alter gene expression pathways to suppress host defences. Conversely, CLIP of host RBPs was used to identify their binding to SARS-CoV-2 RNAs, which contributes to host defence strategies73. Much more work remains to be done with CLIP and complementary approaches to understand how cross-regulation between the RBPs and RNAs of pathogens and their hosts modulates pathogenicity.
Complementary insights
Several studies combined protein-centric and RNA-centric approaches to gain complementary insights into RNP assembly and function. One example is the study of NORAD long non-coding RNA (lncRNA), where RNA-antisense purification coupled with mass spectrometry (RAP-MS) was used to identify its interaction with hnRNP G and several other proteins, the RNA binding sites of which were then mapped with CLIP. This showed how NORAD assembles an RNP that links proteins involved in DNA replication or repair69. Another example is the study of Xist lncRNA, where its bound RBPs were first identified through RNA-centric methods68,70 and later studied by CLIP to show how Xist seeds a heteromeric RNP condensate that is required for heritable gene silencing159. Most recently, host RBPs bound to SARS-CoV-2 RNAs were first identified by RAP-MS, and then studied further with CLIP to map their direct interactions with the SARS-CoV-2 RNA in infected human cells73. These studies show that complementary data from these approaches present an opportunity to build computational models that position each RBP at its bound cis-acting RNA elements along an RNA and thus understand how protein–RNA and protein–protein interactions act combinatorially to drive the assembly and remodelling of RNPs on full RNAs.
A question that is particularly pertinent to the field of RNA localization is how RNPs form dynamic condensates, often referred to as ‘RNP granules’, which regulate RNA transport and local translation in response to signalling160. Understanding RNP assembly and dynamics in RNP granules is particularly challenging as they are mediated by direct protein–RNA and protein–protein interactions and involve both structural domains and intrinsically disordered regions (IDRs). IDRs often form weak multivalent contacts that coordinate condensation of proteins into the granule161. Important questions are how the cis-regulatory sequence and structural elements on the RNA mediate the assembly of the full RNP in order to coordinate its selective transport, and how post-translational modifications of the IDRs mediate RNP remodelling in response to specific signals1. Performing both CLIP and RNA-centric methods under dynamic states will be essential for resolving how specific RBPs are released, rebound or repositioned on RNAs in response to stimuli. Comparisons between localized mRNAs may reveal whether they share a subset of core RBPs, and how these RBPs mediate mRNA recruitment to transport machineries and the translational apparatus. Finally, studies of RNA–RNA interactions in addition to protein–RNA and protein–protein contacts will be needed to fully disentangle the principles of RNP assembly160.
Such understanding of RNP remodelling is of paramount importance as it underlies many aspects of cellular remodelling, including cellular polarity and movement, axon guidance, synaptic plasticity and memory formation. Moreover, deregulated RNP dynamics can lead to formation of aberrant condensates and aggregates in many neurological diseases, such as amyotrophic lateral sclerosis and fragile X syndrome162. Combining RNA-centric and protein-centric methods in models of these diseases will be essential to understand how changes in RNP assembly contribute to the disease processes by affecting the biogenesis, transport, translation and degradation of specific RNAs.
Finally, to fully understand RNP assembly, it is also important to define sites on RBPs that bind to RNAs, which can be done through a combination of UV cross-linking, high-resolution mass spectrometry and a dedicated computational workflow to identify both cross-linked peptides and RNA oligonucleotides — an approach that can be RNA-centric or applied to the whole RBPome30. Recently, several additional approaches have been developed for high-throughput mapping of cross-linked peptides or amino acids within RBPs1. With the ever-increasing capacity of these complementary methods to monitor specific functions of RBPs, integrative approaches are bound to become increasingly informative.
Reproducibility and data deposition
Reproducibility of CLIP data
It is necessary to understand the reproducibility of CLIP data before one can proceed to studies of biological variation through comparisons of data sets produced across conditions, cell types, species and RBPs. Data have been obtained by multiple CLIP variants for many RBPs, and in some cases also by complementary methods such as RIP and TRIBE, yet such data remain to be comprehensively compared and integrated163,164. These comparisons are challenging partly because the metadata available from existing raw sequence archives are rarely sufficient. The minimal reporting standards appropriate for full annotation of CLIP and related methods are still to be consolidated, but our recommendation would be that the following should be reported with standardized nomenclature in a table format: name of the purified protein following official nomenclature, information on tags or mutations in the protein if present, the species, information on the biological material (name of cells or tissue), the essential description of its conditions (for example, treatment, genetic modification), the name of the protocol variant, the essential description of experimental conditions that complement the protocol (such as cross-linking, RNase conditions, the molecular weight range used for excision of the protein–RNA complex) and annotation of the experimental barcode and UMI (their sequence and position).
For comparisons between data sets documenting the same RBPs to be informative, technical and biological sources of variation need to be distinguished. Technical variation can be caused by differences between variant protocols in specific steps, such as cross-linking conditions, stringencies of lysis and washing steps, in use of different antibodies for immunoprecipitation or affinity purification for RBP purification and in cDNA library preparation. Moreover, even when the same CLIP variant is used, variation can arise from unintentional differences in implementation, such as in the density of cultured cells or RNase fragmentation conditions. Finally, even with optimal implementation, binding sites in lowly expressed RNAs are hard to reproduce due to stochastic variation in the low numbers of cDNA counts.
As discussed earlier, the most valuable indicator of CLIP data specificity is its cross-validation using orthogonal information, such as the motif enrichment in CLIP peaks, or enrichment of peaks around regulated events, as shown by RNA maps. Although a necessary indicator of data quality, reproducibility across replicate CLIP experiments is less informative than cross-validation. This is because cross-contamination from a co-immunoprecipitated RBP can be reproducible, as can technical biases of cross-linking, nuclease digestion and ligation. These reproducible biases can distort the data, potentially boosting the significance of otherwise low-occupancy sites. Therefore, performing comparative benchmarking of multiple data sets of the same RBPs and reconstructing comprehensive and accurate sets of binding sites are essential. For instance, although the peak identification methods mentioned above can yield tens of thousands of peaks for some well-characterized RBPs, it is informative to assess peak reproducibility for replicate samples within a laboratory, across laboratories and across CLIP variants35. For samples that assess biological variation, comparisons can be made between samples obtained from different animals6. A concern remains that reproducible peaks are more likely to be located in relatively abundant RNAs. Peaks in low-abundance RNAs may be less reproducible, although this can be partly compensated by predictive computational models99.
Data resources
Resources that provide CLIP data across studies are essential for compiling RBP interaction data and enabling comparisons across data sets. Raw sequencing data are made available upon publication from general public repositories such as the Sequence Read Archive165 or the European Nucleotide Archive, which enforce the tracking of metadata. However, full annotation of CLIP variants ideally requires annotation of additional metadata, as described in the previous section. Alignments of reads are provided as binary alignment map (bam) files that can be visualized with tools such as the Integrative Genomics Viewer166. Specialized databases such as doRiNA167, ENCORI (previously known as starBase)168 and POSTAR2 (ref.169) enable the exploration of processed CLIP peaks, along with additional information such as annotation of corresponding genes and gene expression. doRiNA also allows users to upload their binding site data for visualization. A tool called SEQing has been developed to visualize Arabidopsis iCLIP binding sites170, again in the context of gene expression data. Databases of RBP binding motifs have started to emerge; CISBP-RNA171 summarizes data on in vitro RBP–RNA interactions and ATtRACT contains curated data from various sources172, albeit without resolving discrepancies in motifs that are inferred for the same protein from different types of experiment.
Limitations and optimizations
RBP-specific data analysis challenges
RBPs differ in many aspects that can influence data analysis and interpretation. Perhaps the clearest are the characteristics of the RNA binding motifs. Some RBPs, such as the Pumilio family of proteins, primarily bind long, well-defined motifs that overlap with sharp cross-linking peaks7, whereas others recognize short (often only two to four nucleotides long) degenerate motifs, which often occur in multivalent clusters to drive in vivo binding173. Binding peaks for such RBPs can be dispersed over long clusters of motifs, as exemplified by RBPs binding to long interspersed nuclear element (LINE)-derived RNA elements that contain enriched motifs dispersed over hundreds of nucleotides174. RBPs with limited sequence preferences, such as FUS or SUZ12, show even broader cross-linking distributions across nascent transcripts85,175 In such cases, technical biases such as uridine cross-linking preferences can have a stronger impact on the positioning of identified peaks, which should therefore be considered with caution. Thus, strategies to assign binding sites from CLIP data ideally need to be adjusted to the binding characteristics of each RBP, although approaches for doing so are yet to be developed.
Many RBPs interact with large RNPs, and their RNA interactions are often dominated by one or a few abundant non-coding RNAs, such as small nuclear RNA (snRNA) for the spliceosome and rRNA for the ribosome. Nevertheless, even such RBPs can have additional moonlighting functions, as has been seen for ribosomal proteins176. Thus, one needs to be cautious not to automatically assign secondary binding to background. Moreover, even though the standard immunoprecipitation conditions of CLIP are quite stringent, stable RNPs may not fully disassemble and, in such cases, the RBP partners generate considerable extrinsic background in the resulting data. Such RBPs tend to bind to similar RNAs and perform shared functions, and in some cases CLIP experiments were designed to intentionally profile the RNA interactome of many RBPs that are associated with specific stable RNPs; for example, Sm proteins are immunoprecipitated in ‘spliceosome iCLIP’ to yield the RNA interactome of multiple RBPs associated with various snRNAs, thus revealing their interaction sites on snRNAs and pre-mRNAs, as well as the positions of intronic branch points177.
Challenges of RNA-centric methods
RNA affinity capture methods
The development of RNA-centric methods that are based on RNA affinity capture has greatly expanded our knowledge of RBPs bound to specific RNAs. However, an inherent limitation of these methods is the potential loss of transient and compartment-specific interactions and the possibility of co-purifying post-lysis, false-positive interactions66. The choice of lysis buffer and lysis method, and the addition of aptamers, can change the secondary structure, the half-life of the RNA and, thereby, the protein binding pattern on the RNA178,179. These issues can be partly addressed by maintaining the post-lysis integrity of the RNP with formaldehyde or UV cross-linking, followed by either biotin-labelled antisense oligo RAP180, peptide nucleic acid (PNA)-assisted affinity purification181,182 or 2′-O-methylated antisense RNA-mediated tandem RNA isolation (TRIP)183.
Proximity-based methods
Proximity-based methods can overcome limitations associated with affinity-based methods but are associated with limitations such as the need for sufficient available lysine or other electron-rich amino acids on the protein surface for efficient biotinylation. Moreover, free proximity biotinylation enzyme can biotinylate proteins in a non-specific manner. Background biotinylation can be partially corrected when analysing the data in a cell-specific or tissue-specific way, and general contaminants can be diminished from the data set by referring to the CRAPome database184. Various experimental approaches aimed at improving the signal to noise ratio are discussed in a recent review57.
Another consideration when using proximity biotinylation enzymes is their labelling range (10–20 nm). The enzymes differ in their labelling range and substrates, and can be broadly grouped into peroxidases and biotin ligases185 (Supplementary Table 1). Biotin ligases convert biotin and ATP into biotinoyl-5′-adenylate (bioAMP), which diffuses around the activation site and covalently bonds with nearby lysine residues186. In vitro, the BirA–bioAMP complex has a half-life of ~30 min; therefore, biotinylation of substrates also depends on the activity and diffusion speed of this complex in the cell. The efficiency of different proximity ligases also depends on the specific redox environment and proximal nucleophile concentrations, which might explain why BioID and TurboID are effective when tagged with a nuclear localization sequence, a mitochondrial targeting sequence or endoplasmic reticulum-targeting sequences, whereas miniTurboID is more effective in an open cytosolic environment than in membrane-enclosed organelles187.
miniTurboID can be used at a lower temperature (20–37 °C) than BioID (37 °C) and BioID2, which has an optimal temperature of 50 °C (refs187,188). However, it is concerning that constitutive expression of TurboID in the absence of exogenous biotin leads to decreased size and viability in Drosophila melanogaster187 and that incubation times greater than 6 h or use of excess biotin (50 µM) may result in non-specific biotinylation in the cell187. Deletion of the N-terminal region was found to decrease the stability of miniTurboID in C. elegans187. Recently, with the help of enzyme reconstruction algorithms and residue replacements on optimized biotin ligases, a new BirA enzyme, AirID (ancestral BirA for proximity-dependent biotin identification), has been developed and found to be less toxic than TurboID in Hek293 cells189.
Analysing RNA binding sites
Extracting RNA interaction parameters from CLIP data and interpreting the potential functions of these interactions can be challenging, and is an area of intense research. Defining cross-linking peaks of high occupancy is important; however, such peaks should not be directly equated to functionally relevant binding sites. Even though CLIP tends to detect binding events with high specificity, the functionality of these events depends on additional factors, such as the binding position relative to other functional elements and the total residence time of the protein173. Recently, femtosecond UV laser cross-linking followed by CLIP (KIN-CLIP) was shown to be capable of characterizing in vivo binding kinetics at individual sites and thus revealing the increased functionality of sites that are composed of clusters of motifs77, in agreement with insights from the studies of RNA maps111,190.
The assignment of RNA binding sites can be improved by combining CLIP data with analysis RNA sequences and structural motifs99. Further indication of the functional relevance of binding sites can be obtained by assessing their evolutionary conservation. However, many RNA sequences are not strongly conserved; for example, although the length and arrangement of lncRNAs and introns are under considerable evolutionary constraint, their sequences show weak conservation across species and rapid accumulation of repetitive elements, indicating weak functional constraint191. Nevertheless, even intronic repetitive elements can contain high-affinity binding sites that are under some selection, as demonstrated by the observation that many RBPs repress the inclusion of cryptic exons that are often present in these elements192.
To discern functionally relevant sites, it is valuable to integrate CLIP data with orthogonal transcriptomics data from RBP perturbation experiments5,7,190,193. On the one hand, such integration identifies CLIP peaks that likely mediate the regulation of specific elements, and, on the other, it distinguishes the RNAs detected by RNA-seq that are directly regulated by the RBP from those that likely change owing to off-target effects of RBP perturbation, feedback loops via other RBPs or other types of cellular compensation. When analysis leads to sensitive and specific positional patterns observed by an RNA map, it also provides a valuable measure of the quality of CLIP and RNA-seq data that are being integrated9. In addition to integration with RNA-seq for studies of RNA processing, CLIP-derived binding sites have also been integrated with additional types of orthogonal data sets to study 3′ end RNA processing6,194, RNA methylation14, stability7,164, translation80,136 and localization195,196.
Outlook
There is no one size fits all guideline for the design and analysis of CLIP experiments. It is important to be aware of the steps that can be taken for quality control and optimization in order to tailor the experimental and computational steps according to the RBP that is studied, the input material and the type of questions that are asked.
We expect many new applications of CLIP to be developed in coming years, with increasing integration of CLIP with data from methods based on enzymatic tagging and RNA-centric approaches. These complementary methods have not yet been used in combination, but we hope that this Primer will encourage their integrative use. Cross-method comparisons will be valuable to better understand the advantages of each method and correct for technical biases. Integration of CLIP data that detect direct protein–RNA interactions with approaches that also detect RNA-proximal proteins will help to understand which proteins are recruited to RNAs primarily through direct recognition of specific RNA elements versus protein–protein interactions with other RBPs. Another valuable application will be to study specific RBPs in subcellular compartments with complementary methods to provide insights into the assembly properties of RBPs at organelles or biomolecular condensates161. For example, such methods could be applied to chloroplasts, which rely heavily on post-transcriptional mechanisms for controlling the expression of their genome197.
Important questions in RNP remodelling and combinatorial assembly can be answered when CLIP and complementary methods are used under comparative scenarios. For example, CLIP of one RBP from cells lacking another RBP can reveal how individual RBPs compete for binding to overlapping sites113 or how larger RNPs compete, such as how the exon junction complex blocks access of the splicing machinery to regions around exon–exon junctions in spliced RNAs146. The competitive and combinatorial assembly principles can be further unravelled using ‘in vitro CLIP’ experiments, in which recombinant RBPs with varying concentrations are incubated with long transcripts, followed by modelling and machine learning198. Moreover, CLIP can be performed with purified RNPs in specific states, for example to define helicase–RNA contacts in specific spliceosomal states by purified spliceosome iCLIP (psiCLIP)199. A long-term challenge will be to understand how RNA regulatory networks are remodelled on various timescales, for example during cellular signal response, development, ageing, mutation-driven changes in cancer and other diseases, and over the course of organismal evolution. These questions are starting to be addressed by studies across species or in response to disease mutations27,200. It will be important to understand how variations in IDRs, which tend to evolve faster than structured domains and are hotspots of disease-causing mutations and post-translational modifications1, might affect the RNA binding and regulatory functions of RBPs.
Two emerging applications of transcriptomic techniques not covered in this Primer are mapping of RNA structure and RNA modifications genome-wide, as the topic has been comprehensively covered elsewhere12,201,202,203. Integration of protein–RNA interactions with information on RNA structure and RNA–RNA spatial interactions will help understand the roles of RNA molecules in organizing RNP assembly12,43,203,204,205. Recently, an RNA pull-down method was used to identify proteins bound to 186 RNA structures conserved across yeast species206. This approach enables the study of dozens of short RNA fragments to uncover RBPs that tend to bind similar RNA structures or other types of similar RNA motifs from a group of RNAs, offering a valuable complement to the RNA-centric or global RNA interactome approaches.
More than 100 RNA modifications have been described; most affect the assembly of protein–RNA complexes and therefore should be integrated into studies of protein–RNA interactions. Interestingly, mutations of certain methyltransferases can stabilize covalently linked protein–RNA catalytic intermediates, thus enabling CLIP to be performed without the need for UV cross-linking, as has been done for m5C-miCLIP207. Most methods to date have been developed for transcriptomic studies of m6A, the type of modification that is most common in mRNAs, and these include variants of CLIP, such as m6A-miCLIP, which employ antibodies that recognize m6A-containing RNA208. The success of such approaches critically depends on the quality of the antibodies recognizing the modification209. Therefore, similar to studies of protein–RNA interactions, integration of data from complementary methods will be valuable to gain a full picture of RNA modifications and their roles in RNP assembly202,210.
We expect computational methods for site and motif identification to soon reach maturity, leading to high-quality databases of in vivo RBP binding motifs. As most of the computational methods work with uniquely mapping reads, improvements are foreseen in the quantification of sites located in repeat elements as well as at exon–exon boundaries or in splicing and polyadenylation isoforms. Ultimately, we can start to consider what to do next with information on all of the protein–RNA interaction sites; for example, we could construct whole-cell models to predict RNA fates and their roles in cellular changes during development and disease. The path taken towards this ultimate aim will require integration of complementary data sets to gain understanding of the full RNP assembled on each transcript, its spatial dynamics as the transcript moves through the cell and temporal dynamics in response to post-translational protein modifications, RNA methylation and RNA structural switches. As such, RNPs will surely continue to teach us about the highly interconnected and ever-changing world of living cells.
References
Gebauer, F., Schwarzl, T., Valcárcel, J. & Hentze, M. W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 22, 185–198 (2020).
Lerner, M. R. & Steitz, J. A. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc. Natl Acad. Sci. USA 76, 5495–5499 (1979).
Tenenbaum, S. A., Carson, C. C., Lager, P. J. & Keene, J. D. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl Acad. Sci. USA 97, 14085–14090 (2000).
Niranjanakumari, S., Lasda, E., Brazas, R. & Garcia-Blanco, M. A. Reversible cross-linking combined with immunoprecipitation to study RNA–protein interactions in vivo. Methods 26, 182–190 (2002).
Ule, J. et al. CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215 (2003).
Licatalosi, D. D. et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464 (2008). This study introduces HITS-CLIP and validates the RNA map of splicing regulation by Nova proteins.
Hafner, M. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141 (2010). This study describes the development of PAR-CLIP, which enables identification of cross-link sites from the nucleotide substitutions in the sequenced cDNAs.
König, J. et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 17, 909–915 (2010). This study describes the development of iCLIP, which enables amplification of truncated cDNAs and identification of cross-link sites with analysis of truncations.
Chakrabarti, A. M., Haberman, N., Praznik, A., Luscombe, N. M. & Ule, J. Data science issues in studying protein–RNA interactions with CLIP technologies. Annu. Rev. Biomed. Data Sci. 1, 235–261 (2018). This study reviews computational methods and presents the analysis of RNA splicing maps as a way to assess the sensitivity and specificity of CLIP data.
McMahon, A. C. et al. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753 (2016). This study establishes a method to identify RNA binding sites of RBPs through fusion with ADARcd and analysis of RNA editing.
Benhalevy, D., Anastasakis, D. G. & Hafner, M. Proximity-CLIP provides a snapshot of protein-occupied RNA elements in subcellular compartments. Nat. Methods 15, 1074–1082 (2018). In this study, subcellular compartment-specific proximity labelling is combined with CLIP to monitor RNA–protein interactions at specific locations in the cell.
Lin, C. & Miles, W. O. Beyond CLIP: advances and opportunities to measure RBP–RNA and RNA–RNA interactions. Nucleic Acids Res. 47, 5490–5501 (2019).
Ramanathan, M., Porter, D. F. & Khavari, P. A. Methods to study RNA–protein interactions. Nat. Methods 16, 225–234 (2019).
Lee, F. C. Y. & Ule, J. Advances in CLIP technologies for studies of protein–RNA interactions. Mol. Cell 69, 354–369 (2018).
Ule, J., Jensen, K., Mele, A. & Darnell, R. B. CLIP: a method for identifying protein–RNA interaction sites in living cells. Methods 37, 376–386 (2005). This study gives a detailed description of the CLIP protocol, establishes the CLIP workflow and explains the stages of RNase optimization, SDS-PAGE purification conditions and cDNA library preparation that are used by most later variants.
Jungkamp, A.-C. et al. In vivo and transcriptome-wide identification of RNA binding protein target sites. Mol. Cell 44, 828–840 (2011).
Zhang, Y. et al. Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res. 25, 864–876 (2015).
Meyer, K. et al. Adaptation of iCLIP to plants determines the binding landscape of the clock-regulated RNA-binding protein AtGRP7. Genome Biol. 18, 204 (2017). This is the first plant iCLIP study and identifies RNA-binding partners of an hnRNP-like protein in the reference plant A. thaliana.
Moore, M. J. et al. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc. 9, 263–293 (2014).
Max, K. E. A. et al. Human plasma and serum extracellular small RNA reference profiles and their clinical utility. Proc. Natl Acad. Sci. USA 115, E5334–E5343 (2018).
Hafner, M. et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 44, 3–12 (2008).
Kishore, S. et al. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 8, 559–564 (2011). This study evaluates how differences in cross-linking and ribonuclease digestion affect the sites obtained with HITS-CLIP and PAR-CLIP, both marked by specific cross-linking-induced mutations.
Friedersdorf, M. B. & Keene, J. D. Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biol. 15, R2 (2014).
König, J., Zarnack, K., Luscombe, N. M. & Ule, J. Protein–RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genet. 13, 77–83 (2012).
Castello, A. et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406 (2012).
Patton, R. D. et al. Chemical crosslinking enhances RNA immunoprecipitation for efficient identification of binding sites of proteins that photo-crosslink poorly with RNA. RNA 26, 1216–1233 (2020).
Porter, D. F. & Khavari, P. A. easyCLIP quantifies RNA–protein interactions and characterizes recurrent PCBP1 mutations in cancer. Preprint at bioRxiv https://doi.org/10.1101/635888 (2019).
Feng, H. et al. Modeling RNA-binding protein specificity in vivo by precisely registering protein–RNA crosslink sites. Mol. Cell 74, 1189–1204.e6 (2019). This study performs de novo motif discovery on >100 RBPs using eCLIP data by joint modelling of sequence specificity and cross-link sites, and evaluation of motifs by allele imbalance.
Sugimoto, Y. et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein–RNA interactions. Genome Biol. 13, R67 (2012).
Kramer, K. et al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 11, 1064–1070 (2014).
Lau, N. C., Lim, L. P., Weinstein, E. G. & Bartel, D. P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862 (2001).
Granneman, S., Kudla, G., Petfalski, E. & Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Natl Acad. Sci. USA 106, 9613–9618 (2009).
Zhang, C. & Darnell, R. B. Mapping in vivo protein–RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 29, 607–614 (2011).
Zarnegar, B. J. et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nat. Methods 13, 489–492 (2016). This study presents a non-isotopic method for the detection of protein–RNA complexes using an infrared-labelled adapter, which simplifies their visualization after SDS-PAGE separation.
Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Blondal, T. et al. Isolation and characterization of a thermostable RNA ligase 1 from a Thermus scotoductus bacteriophage TS2126 with good single-stranded DNA ligation properties. Nucleic Acids Res. 33, 135–142 (2005).
Buchbender, A. et al. Improved library preparation with the new iCLIP2 protocol. Methods 178, 33–48 (2020).
Ascano, M., Hafner, M., Cekan, P., Gerstberger, S. & Tuschl, T. Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 3, 159–177 (2012).
Chi, S. W., Zang, J. B., Mele, A. & Darnell, R. B. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460, 479–486 (2009).
Helwak, A., Kudla, G., Dudnakova, T. & Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013).
Grosswendt, S. et al. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol. Cell 54, 1042–1054 (2014).
Sugimoto, Y. et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494 (2015).
Corley, M. et al. Footprinting SHAPE-eCLIP reveals transcriptome-wide hydrogen bonds at RNA–protein interfaces. Mol. Cell 80, 903–914.e8 (2020).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178, 473–490.e26 (2019).
Padrón, A., Iwasaki, S. & Ingolia, N. T. Proximity RNA labeling by APEX-seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol. Cell 75, 875–887.e5 (2019).
Kaewsapsak, P., Shechner, D. M., Mallard, W., Rinn, J. L. & Ting, A. Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein–RNA crosslinking. eLife 6, e29224 (2017).
Hung, V. et al. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 11, 456–475 (2016).
Chen, C.-L. & Perrimon, N. Proximity-dependent labeling methods for proteomic profiling in living cells. Wiley Interdiscip. Rev. Dev. Biol. 6, e272 (2017).
Choder, M. mRNA imprinting: additional level in the regulation of gene expression. Cell. Logist. 1, 37–40 (2011).
Wang, P. et al. Mapping spatial transcriptome with light-activated proximity-dependent RNA labeling. Nat. Chem. Biol. 15, 1110–1119 (2019).
Li, Y., Aggarwal, M. B., Ke, K., Nguyen, K. & Spitale, R. C. Improved analysis of RNA localization by spatially restricted oxidation of RNA–protein complexes. Biochemistry 57, 1577–1581 (2018).
Li, Y., Aggarwal, M. B., Nguyen, K., Ke, K. & Spitale, R. C. Assaying RNA localization in situ with spatially restricted nucleobase oxidation. ACS Chem. Biol. 12, 2709–2714 (2017).
van Steensel, B. & Henikoff, S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat. Biotechnol. 18, 424–428 (2000).
Xu, W., Rahman, R. & Rosbash, M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 24, 173–182 (2018).
Brannan, K. et al. Robust single-cell discovery of RNA targets of RNA binding proteins and ribosomes. Preprint at Research Square https://doi.org/10.21203/rs.3.rs-87224/v1 (2020).
Gräwe, C., Stelloo, S., van Hout, F. A. H. & Vermeulen, M. RNA-centric methods: toward the interactome of specific RNA transcripts. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2020.11.011 (2020).
Gemmill, D., D’souza, S., Meier-Stephenson, V. & Patel, T. R. Current approaches for RNA-labelling to identify RNA-binding proteins. Biochem. Cell Biol. 98, 31–41 (2020).
Slobodin, B. & Gerst, J. E. A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 16, 2277–2290 (2010).
Hogg, J. R. & Collins, K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13, 868–880 (2007).
Leppek, K. & Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 42, e13 (2014).
Lee, H. Y. et al. RNA–protein analysis using a conditional CRISPR nuclease. Proc. Natl Acad. Sci. USA 110, 5416–5421 (2013).
Flather, D. et al. Generation of recombinant polioviruses harboring RNA affinity tags in the 5′ and 3′ noncoding regions of genomic RNAs. Viruses 8, 39 (2016).
Hartmuth, K. et al. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl Acad. Sci. USA 99, 16719–16724 (2002).
Windbichler, N. & Schroeder, R. Isolation of specific RNA-binding proteins using the streptomycin-binding RNA aptamer. Nat. Protoc. 1, 637–640 (2006).
Mili, S. & Steitz, J. A. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10, 1692–1694 (2004).
Simon, M. D. et al. The genomic binding sites of a noncoding RNA. Proc. Natl Acad. Sci. USA 108, 20497–20502 (2011).
McHugh, C. A. et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236 (2015).
Munschauer, M. et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 561, 132–136 (2018). This study uses RAP-MS and CLIP maps in a complementary fashion to map the assembly of NORAD lncRNA into an RNP that links proteins involved in DNA replication or repair.
Chu, C. et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416 (2015).
Theil, K., Imami, K. & Rajewsky, N. Identification of proteins and miRNAs that specifically bind an mRNA in vivo. Nat. Commun. 10, 4205 (2019).
Flynn, R. A. et al. Systematic discovery and functional interrogation of SARS-CoV-2 viral RNA–host protein interactions during infection. Preprint at bioRxiv https://doi.org/10.1101/2020.10.06.327445 (2020).
Schmidt, N. et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol. https://doi.org/10.1038/s41564-020-00846-z (2020).
Mukherjee, J. et al. β-Actin mRNA interactome mapping by proximity biotinylation. Proc. Natl Acad. Sci. USA 116, 12863–12872 (2019).
Yi, W. et al. CRISPR-assisted detection of RNA–protein interactions in living cells. Nat. Methods 17, 685–688 (2020).
Han, Y. et al. Directed evolution of split APEX2 peroxidase. ACS Chem. Biol. 14, 619–635 (2019).
Sharma, D. et al. The kinetic landscape of an RNA-binding protein in cells. Nature https://doi.org/10.1038/s41586-021-03222-x (2021).
Lee, C.-Y. S. et al. Recruitment of mRNAs to P granules by condensation with intrinsically-disordered proteins. eLife 9, e52896 (2020).
Hafner, M. et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17, 1697–1712 (2011).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
De, S. & Gorospe, M. Bioinformatic tools for analysis of CLIP ribonucleoprotein data. Wiley Interdiscip. Rev. RNA 8, e1404 (2017).
Ameur, A. et al. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 18, 1435–1440 (2011).
Sibley, C. R. et al. Recursive splicing in long vertebrate genes. Nature 521, 371–375 (2015).
Rogelj, B. et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2, 603 (2012).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Siddharthan, R., Siggia, E. D. & van Nimwegen, E. PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny. PLoS Comput. Biol. 1, e67 (2005).
Badis, G. et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723 (2009).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015).
Wright, J. E. et al. A quantitative RNA code for mRNA target selection by the germline fate determinant GLD-1. EMBO J. 30, 533–545 (2011).
Zhao, Y. & Stormo, G. D. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat. Biotechnol. 29, 480–483 (2011).
Van Nostrand, E. L. et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020). This study performs eCLIP experiments for 103 RBPs from HepG2 and 120 RBPs from K562 cell lines, each in duplicate and with SMI controls, and carried out comparative analysis; the data are available as part of the ENCODE project.
Mukherjee, N. et al. Deciphering human ribonucleoprotein regulatory networks. Nucleic Acids Res. 47, 570–581 (2019). This study produces 114 PAR-CLIP experiments for 64 RBPs in the HEK cell line, and presents a comparative analysis of these RBPs.
Liu, N. et al. N6-Methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564 (2015).
Brümmer, A., Kishore, S., Subasic, D., Hengartner, M. & Zavolan, M. Modeling the binding specificity of the RNA-binding protein GLD-1 suggests a function of coding region-located sites in translational repression. RNA 19, 1317–1326 (2013).
Ray, D. et al. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 27, 667–670 (2009).
Fukunaga, T. et al. CapR: revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol. 15, R16 (2014).
Maticzka, D., Lange, S. J., Costa, F. & Backofen, R. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 15, R17 (2014). This study presents the first computational framework for modelling sequence-binding and structure-binding preferences of RBPs from CLIP data.
Bahrami-Samani, E., Penalva, L. O. F., Smith, A. D. & Uren, P. J. Leveraging cross-link modification events in CLIP-seq for motif discovery. Nucleic Acids Res. 43, 95–103 (2015).
Pietrosanto, M., Mattei, E., Helmer-Citterich, M. & Ferrè, F. A novel method for the identification of conserved structural patterns in RNA: from small scale to high-throughput applications. Nucleic Acids Res. 44, 8600–8609 (2016).
Paraskevopoulou, M. D., Karagkouni, D., Vlachos, I. S., Tastsoglou, S. & Hatzigeorgiou, A. G. microCLIP super learning framework uncovers functional transcriptome-wide miRNA interactions. Nat. Commun. 9, 3601 (2018).
Livi, C. M., Klus, P., Delli Ponti, R. & Tartaglia, G. G. catRAPID signature: identification of ribonucleoproteins and RNA-binding regions. Bioinformatics 32, 773–775 (2016).
Khorshid, M., Hausser, J., Zavolan, M. & van Nimwegen, E. A biophysical miRNA–mRNA interaction model infers canonical and noncanonical targets. Nat. Methods 10, 253–255 (2013).
Breda, J., Rzepiela, A. J., Gumienny, R., van Nimwegen, E. & Zavolan, M. Quantifying the strength of miRNA–target interactions. Methods 85, 90–99 (2015).
Krakau, S., Richard, H. & Marsico, A. PureCLIP: capturing target-specific protein–RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol. 18, 240 (2017).
Drewe-Boss, P., Wessels, H.-H. & Ohler, U. omniCLIP: probabilistic identification of protein–RNA interactions from CLIP-seq data. Genome Biol. 19, 183 (2018).
Stražar, M., Žitnik, M., Zupan, B., Ule, J. & Curk, T. Orthogonal matrix factorization enables integrative analysis of multiple RNA binding proteins. Bioinformatics 32, 1527–1535 (2016).
Pan, X. & Shen, H.-B. RNA–protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinforma. 18, 136 (2017).
Van Nostrand, E. L. et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol. 21, 90 (2020).
Ule, J. et al. An RNA map predicting Nova-dependent splicing regulation. Nature 444, 580–586 (2006).
Gruber, A. J. et al. Discovery of physiological and cancer-related regulators of 3′ UTR processing with KAPAC. Genome Biol. 19, 44 (2018).
Zarnack, K. et al. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152, 453–466 (2013). This study demonstrates the quantitative capacity of CLIP to compare binding of an RBP between conditions — in this case, to demonstrate the displacement of U2AF2 by hnRNP C at cryptic splice sites within intronic Alu elements.
Wang, S. et al. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes Dev. 33, 1048–1068 (2019).
Haberman, N. et al. Insights into the design and interpretation of iCLIP experiments. Genome Biol. 18, 7 (2017).
Ellington, A. D. & Szostak, J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990).
Tuerk, C. & Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990).
Lambert, N. et al. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell 54, 887–900 (2014).
Ghanbari, M. & Ohler, U. Deep neural networks for interpreting RNA-binding protein target preferences. Genome Res. 30, 214–226 (2020).
Wang, Q. et al. The PSI–U1 snRNP interaction regulates male mating behavior in Drosophila. Proc. Natl Acad. Sci. USA 113, 5269–5274 (2016).
Zisoulis, D. G. et al. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 17, 173–179 (2010).
Licatalosi, D. D. & Darnell, R. B. RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 11, 75–87 (2010).
Gerstberger, S., Hafner, M. & Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 15, 829–845 (2014).
Gerstberger, S., Hafner, M., Ascano, M. & Tuschl, T. Evolutionary conservation and expression of human RNA-binding proteins and their role in human genetic disease. Adv. Exp. Med. Biol. 825, 1–55 (2014).
Yamaji, M. et al. DND1 maintains germline stem cells via recruitment of the CCR4–NOT complex to target mRNAs. Nature 543, 568–572 (2017).
Kim, K. K., Yang, Y., Zhu, J., Adelstein, R. S. & Kawamoto, S. Rbfox3 controls the biogenesis of a subset of microRNAs. Nat. Struct. Mol. Biol. 21, 901–910 (2014).
Xu, Q. et al. Enhanced crosslinking immunoprecipitation (eCLIP) method for efficient identification of protein-bound RNA in mouse testis. J. Vis. Exp. https://doi.org/10.3791/59681 (2019).
Li, W., Jin, Y., Prazak, L., Hammell, M. & Dubnau, J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS ONE 7, e44099 (2012).
Vourekas, A. et al. The RNA helicase MOV10L1 binds piRNA precursors to initiate piRNA processing. Genes Dev. 29, 617–629 (2015).
Vourekas, A. et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat. Struct. Mol. Biol. 19, 773–781 (2012).
Vourekas, A., Alexiou, P., Vrettos, N., Maragkakis, M. & Mourelatos, Z. Sequence-dependent but not sequence-specific piRNA adhesion traps mRNAs to the germ plasm. Nature 531, 390–394 (2016).
Miller, M. R., Robinson, K. J., Cleary, M. D. & Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods 6, 439–441 (2009).
Ule, J., Hwang, H.-W. & Darnell, R. B. The future of cross-linking and immunoprecipitation (CLIP). Cold Spring Harb. Perspect. Biol. 10, a032243 (2018).
Saito, Y. et al. Differential NOVA2-mediated splicing in excitatory and inhibitory neurons regulates cortical development and cerebellar function. Neuron 101, 707–720.e5 (2019).
Hwang, H.-W. et al. cTag-PAPERCLIP reveals alternative polyadenylation promotes cell-type specific protein diversity and shifts araf isoforms with microglia activation. Neuron 95, 1334–1349.e5 (2017). This study describes the development of a knock-in mouse in which a GFP-tagged RBP is conditionally expressed in selected cell populations, enabling cell type-specific CLIP; in this case, GFP-PABP is used to map the 3′ ends of mRNAs in excitatory and inhibitory neurons, astrocytes and microglia.
Sawicka, K. et al. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. eLife 8, e46919 (2019).
Köster, T., Reichel, M. & Staiger, D. CLIP and RNA interactome studies to unravel genome-wide RNA–protein interactions in vivo in Arabidopsis thaliana. Methods 178, 63–71 (2020).
Schmal, C., Reimann, P. & Staiger, D. A circadian clock-regulated toggle switch explains AtGRP7 and AtGRP8 oscillations in Arabidopsis thaliana. PLoS Comput. Biol. 9, e1002986 (2013).
Reichel, M. et al. In planta determination of the mRNA-binding proteome of Arabidopsis etiolated seedlings. Plant Cell 28, 2435–2452 (2016).
Zhang, Z. et al. UV crosslinked mRNA-binding proteins captured from leaf mesophyll protoplasts. Plant Methods 12, 42 (2016).
Marondedze, C., Thomas, L., Serrano, N. L., Lilley, K. S. & Gehring, C. The RNA-binding protein repertoire of Arabidopsis thaliana. Sci. Rep. 6, 29766 (2016).
Bach-Pages, M. et al. Discovering the RNA-binding proteome of plant leaves with an improved RNA interactome capture method. Biomolecules 10, 661 (2020).
Köster, T., Marondedze, C., Meyer, K. & Staiger, D. RNA-binding proteins revisited — the emerging Arabidopsis mRNA interactome. Trends Plant. Sci. 22, 512–526 (2017).
Beckmann, B. M. et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 6, 10127 (2015).
Sibley, C. R., Blazquez, L. & Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 17, 407–421 (2016).
Blazquez, L. et al. Exon junction complex shapes the transcriptome by repressing recursive splicing. Mol. Cell 72, 496–509.e9 (2018).
Tollervey, J. R. et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 (2011).
Yamazaki, T. et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell 70, 1038–1053.e7 (2018).
Modic, M. et al. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency–differentiation transition. Mol. Cell 74, 951–965 (2019).
Horos, R. et al. The small non-coding vault RNA1-1 acts as a riboregulator of autophagy. Cell 176, 1054–1067.e12 (2019).
Holmqvist, E. et al. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 35, 991–1011 (2016).
Gottwein, E. et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10, 515–526 (2011).
Gay, L. A., Sethuraman, S., Thomas, M., Turner, P. C. & Renne, R. Modified cross-linking, ligation, and sequencing of hybrids (qCLASH) identifies Kaposi’s sarcoma-associated herpesvirus microRNA targets in endothelial cells. J. Virol. 92, e02138-17 (2018).
Kutluay, S. B. et al. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159, 1096–1109 (2014).
Apolonia, L. et al. Promiscuous RNA binding ensures effective encapsidation of APOBEC3 proteins by HIV-1. PLoS Pathog. 11, e1004609 (2015).
Flynn, R. A. et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA 21, 135–143 (2015).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339 (2020).
Nabeel-Shah, S. et al. SARS-CoV-2 nucleocapsid protein attenuates stress granule formation and alters gene expression via direct interaction with host mRNAs. Cold Spring Harb. Lab. https://doi.org/10.1101/2020.10.23.342113 (2020).
Pandya-Jones, A. et al. A protein assembly mediates Xist localization and gene silencing. Nature 587, 145–151 (2020).
Tauber, D., Tauber, G. & Parker, R. Mechanisms and regulation of RNA condensation in RNP granule formation. Trends Biochem. Sci. 45, 764–778 (2020).
Lyon, A. S., Peeples, W. B. & Rosen, M. K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-020-00303-z (2020).
Formicola, N., Vijayakumar, J. & Besse, F. Neuronal ribonucleoprotein granules: dynamic sensors of localized signals. Traffic 20, 639–649 (2019).
Uren, P. J. et al. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol. 13, 400–411 (2016).
Blackinton, J. G. & Keene, J. D. Functional coordination and HuR-mediated regulation of mRNA stability during T cell activation. Nucleic Acids Res. 44, 426–436 (2016).
Wheeler, D. L. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 36, D13–D21 (2008).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013).
Blin, K. et al. doRiNA 2.0 — upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 43, D160–D167 (2015).
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H. & Yang, J.-H. starBase v2.0: decoding miRNA–ceRNA, miRNA–ncRNA and protein–RNA interaction networks from large-scale CLIP-seq data. Nucleic Acids Res. 42, D92–D97 (2013).
Zhu, Y. et al. POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic Acids Res. 47, D203–D211 (2019).
Lewinski, M., Bramkamp, Y., Köster, T. & Staiger, D. SEQing: web-based visualization of iCLIP and RNA-seq data in an interactive python framework. BMC Bioinforma. 21, 113 (2020).
Ray, D. et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177 (2013).
Giudice, G., Sánchez-Cabo, F., Torroja, C. & Lara-Pezzi, E. ATtRACT — a database of RNA-binding proteins and associated motifs. Database 2016, baw035 (2016).
Jankowsky, E. & Harris, M. E. Specificity and nonspecificity in RNA–protein interactions. Nat. Rev. Mol. Cell Biol. 16, 533–544 (2015).
Attig, J. et al. Heteromeric RNP assembly at LINEs controls lineage-specific RNA processing. Cell 174, 1067–1081.e17 (2018).
Beltran, M. et al. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res. 26, 896–907 (2016).
Warner, J. R. & McIntosh, K. B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 34, 3–11 (2009).
Briese, M. et al. A systems view of spliceosomal assembly and branchpoints with iCLIP. Nat. Struct. Mol. Biol. 26, 930–940 (2019). This study describes an adaptation of CLIP for simultaneously profiling the RNA interactome of many RBPs that are associated with stable RNPs, in this case determining the RNA interaction profiles of spliceosomal proteins.
Cai, S. et al. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 143, 5317–5338 (2018).
Garcia, J. F. & Parker, R. MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system. RNA 21, 1393–1395 (2015).
McHugh, C. A. & Guttman, M. RAP-MS: a method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol. Biol. 1649, 473–488 (2018).
Zeng, F. et al. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat. Protoc. 1, 920–927 (2006).
Bell, T. J., Eiríksdóttir, E., Langel, U. & Eberwine, J. PAIR technology: exon-specific RNA-binding protein isolation in live cells. Methods Mol. Biol. 683, 473–486 (2011).
Matia-González, A. M., Iadevaia, V. & Gerber, A. P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA–protein complexes. Methods 118–119, 93–100 (2017).
Mellacheruvu, D. et al. The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 10, 730–736 (2013).
Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping [version 1; peer review: 2 approved]. F1000Res. 8, 135 (2019).
Cronan, J. E. Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J. Nutr. Biochem. 16, 416–418 (2005).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Kim, D. I. et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 (2016).
Kido, K. et al. AirID, a novel proximity biotinylation enzyme, for analysis of protein–protein interactions. eLife 9, e54983 (2020).
Witten, J. T. & Ule, J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 27, 89–97 (2011).
Kapusta, A. & Feschotte, C. Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet. 30, 439–452 (2014).
Attig, J. & Ule, J. Genomic accumulation of retrotransposons was facilitated by repressive RNA-binding proteins: a hypothesis. Bioessays 41, e1800132 (2019).
Martí-Gómez, C., Lara-Pezzi, E. & Sánchez-Cabo, F. dSreg: a Bayesian model to integrate changes in splicing and RNA-binding protein activity. Bioinformatics 36, 2134–2141 (2020).
Rot, G. et al. High-resolution RNA maps suggest common principles of splicing and polyadenylation regulation by TDP-43. Cell Rep. 19, 1056–1067 (2017).
Goering, R. et al. FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. eLife 9, e52621 (2020).
Dermit, M. et al. Subcellular mRNA localization regulates ribosome biogenesis in migrating cells. Dev. Cell 55, 298–313.e10 (2020).
del Campo, E. M. Post-transcriptional control of chloroplast gene expression. Gene Regul. Syst. Bio. 3, 31–47 (2009).
Sutandy, F. X. R. et al. In vitro iCLIP-based modeling uncovers how the splicing factor U2AF2 relies on regulation by cofactors. Genome Res. 28, 699–713 (2018). This study describes the development of ‘in vitro iCLIP’ for the study of how protein–RNA interactions are determined by cis-acting sequences and modulated by trans-acting RBPs.
Strittmatter, L. M. et al. PsiCLIP reveals dynamic RNA binding by DEAH-box helicases before and after exon ligation. Preprint at bioRxiv https://doi.org/10.1101/2020.03.15.992701 (2020).
Ule, J. & Blencowe, B. J. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol. Cell 76, 329–345 (2019).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Capitanchik, C. A., Toolan-Kerr, P., Luscombe, N. M. & Ule, J. How do you identify m6A methylation in transcriptomes at high resolution? A comparison of recent datasets. Front. Genet. 11, 398 (2020).
Lu, Z. & Chang, H. Y. Decoding the RNA structurome. Curr. Opin. Struct. Biol. 36, 142–148 (2016).
Cai, Z. et al. RIC-seq for global in situ profiling of RNA–RNA spatial interactions. Nature 582, 432–437 (2020).
Foley, S. W. et al. A global view of RNA–protein interactions identifies post-transcriptional regulators of root hair cell fate. Dev. Cell 41, 204–220.e5 (2017).
Casas-Vila, N., Sayols, S., Pérez-Martínez, L., Scheibe, M. & Butter, F. The RNA fold interactome of evolutionary conserved RNA structures in S. cerevisiae. Nat. Commun. 11, 2789 (2020).
Hussain, S. et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261 (2013).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
Helm, M., Lyko, F. & Motorin, Y. Limited antibody specificity compromises epitranscriptomic analyses. Nat. Commun. 10, 5669 (2019).
Tang, Y. et al. m6A-Atlas: a comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 49, D134–D143 (2020).
Miniard, A. C., Middleton, L. M., Budiman, M. E., Gerber, C. A. & Driscoll, D. M. Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression. Nucleic Acids Res. 38, 4807–4820 (2010).
Choudhury, N. R. et al. Tissue-specific control of brain-enriched miR-7 biogenesis. Genes Dev. 27, 24–38 (2013).
Zielinski, J. et al. In vivo identification of ribonucleoprotein–RNA interactions. Proc. Natl Acad. Sci. USA 103, 1557–1562 (2006).
Rogell, B. et al. Specific RNP capture with antisense LNA/DNA mixmers. RNA 23, 1290–1302 (2017).
Sharma, S. Isolation of a sequence-specific RNA binding protein, polypyrimidine tract binding protein, using RNA affinity chromatography. Methods Mol. Biol. 488, 1–8 (2008).
Tsai, B. P., Wang, X., Huang, L. & Waterman, M. L. Quantitative profiling of in vivo-assembled RNA–protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 10, M110.007385 (2011).
Yoon, J.-H., Srikantan, S. & Gorospe, M. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods 58, 81–87 (2012).
Bardwell, V. J. & Wickens, M. Purification of RNA and RNA–protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 18, 6587–6594 (1990).
Meredith, E. K., Balas, M. M., Sindy, K., Haislop, K. & Johnson, A. M. An RNA matchmaker protein regulates the activity of the long noncoding RNA HOTAIR. RNA 22, 995–1010 (2016).
Carey, J., Cameron, V., de Haseth, P. L. & Uhlenbeck, O. C. Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry 22, 2601–2610 (1983).
Lim, F., Downey, T. P. & Peabody, D. S. Translational repression and specific RNA binding by the coat protein of the Pseudomonas phage PP7. J. Biol. Chem. 276, 22507–22513 (2001).
Deckert, J. et al. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol. 26, 5528–5543 (2006).
Wallace, S. T. & Schroeder, R. In vitro selection and characterization of streptomycin-binding RNAs: recognition discrimination between antibiotics. RNA 4, 112–123 (1998).
Zhang, Z. et al. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res. 48, e52 (2020).
Han, S. et al. RNA–protein interaction mapping via MS2 or Cas13-based APEX targeting. Proc. Natl Acad. Sci. USA 117, 22068–22079 (2020).
Lin, X. & Lawrenson, K. In vivo analysis of RNA proximity proteomes using RiboPro. Preprint at bioRxiv https://doi.org/10.1101/2020.02.28.970442 (2020).
Kucukural, A., Özadam, H., Singh, G., Moore, M. J. & Cenik, C. ASPeak: an abundance sensitive peak detection algorithm for RIP-seq. Bioinformatics 29, 2485–2486 (2013).
Golumbeanu, M., Mohammadi, P. & Beerenwinkel, N. BMix: probabilistic modeling of occurring substitutions in PAR-CLIP data. Bioinformatics 32, 976–983 (2016).
Zhang, Z. & Xing, Y. CLIP-seq analysis of multi-mapped reads discovers novel functional RNA regulatory sites in the human transcriptome. Nucleic Acids Res. 45, 9260–9271 (2017).
Park, S. et al. CLIPick: a sensitive peak caller for expression-based deconvolution of HITS-CLIP signals. Nucleic Acids Res. 46, 11153–11168 (2018).
Lovci, M. T. et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol. 20, 1434–1442 (2013).
Shah, A., Qian, Y., Weyn-Vanhentenryck, S. M. & Zhang, C. CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567 (2017).
Wang, Z. et al. iCLIP predicts the dual splicing effects of TIA–RNA interactions. PLoS Biol. 8, e1000530 (2010).
Chen, B., Yun, J., Kim, M. S., Mendell, J. T. & Xie, Y. PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol. 15, R18 (2014).
Uren, P. J. et al. Site identification in high-throughput RNA–protein interaction data. Bioinformatics 28, 3013–3020 (2012).
Tree, J. J., Granneman, S., McAteer, S. P., Tollervey, D. & Gally, D. L. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell 55, 199–213 (2014).
Comoglio, F., Sievers, C. & Paro, R. Sensitive and highly resolved identification of RNA–protein interaction sites in PAR-CLIP data. BMC Bioinforma. 16, 32 (2015).
Palmer, L. E., Weiss, M. J. & Paralkar, V. R. YODEL: peak calling software for HITS-CLIP data. F1000Res. 6, 1138 (2017).
Lunde, B. M., Moore, C. & Varani, G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 8, 479–490 (2007).
Corley, M., Burns, M. C. & Yeo, G. W. How RNA-binding proteins interact with RNA: molecules and mechanisms. Mol. Cell 78, 9–29 (2020).
Masliah, G., Barraud, P. & Allain, F. H.-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell. Mol. Life Sci. 70, 1875–1895 (2013).
Huppertz, I. et al. iCLIP: protein–RNA interactions at nucleotide resolution. Methods 65, 274–287 (2014).
Zhao, Y. et al. SpyCLIP: an easy-to-use and high-throughput compatible CLIP platform for the characterization of protein–RNA interactions with high accuracy. Nucleic Acids Res. 47, e33–e33 (2019).
Schneider, C., Kudla, G., Wlotzka, W., Tuck, A. & Tollervey, D. Transcriptome-wide analysis of exosome targets. Mol. Cell 48, 422–433 (2012). This study describes the development of split-CRAC, where an RBP undergoes in vitro cleavage during affinity purification and allows separate identification of RNA sites cross-linked to the N-terminal and C-terminal regions of the RBP.
Acknowledgements
The authors thank F. Lee, A. Chakrabarti and R. Abouward for suggestions on the manuscript. This work was supported by the German Research Foundation (DFG) (grants STA653/13-1 and STA653/14-1 to D.S. and KO5364/1-1 to T.K.), the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH) to M.H. and J.Ma., the European Union’s Horizon 2020 research and innovation programme (835300-RNPdynamics) to J.U. and J.Mu., the Swiss National Science Foundation (310030_189063) to M.Z. and the Biozentrum Basel International Ph.D. Program Fellowships for Excellence to M.K. The Francis Crick Institute receives its core funding from Cancer Research UK (FC001110), the UK Medical Research Council (FC001110) and the Wellcome Trust (FC001110).
Author information
Authors and Affiliations
Contributions
Introduction (D.S. and J.U.); Experimentation (M.H., J.Ma., J.Mu. and J.U.); Results (M.Z., M.K. and J.U.); Applications (M.H., J.Ma., D.S., T.K., J.Mu. and J.U.); Reproducibility and data deposition (M.Z., M.K. and J.U.); Limitations and optimizations (J.U. and J.Mu.); Outlook (J.U. and D.S.); Oversight of Primer (J.U.).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Methods Primers thanks U. Ohler, L. Penalva, R. Skalsky, G. Yeo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ASPeak: https://sourceforge.net/projects/as-peak/
Bmix: https://github.com/cbg-ethz/BMix
CLAM: https://github.com/Xinglab/CLAM
CLIPick: https://gitlab.com/CLIPick/CLIPick-package
CLIPper: https://github.com/YeoLab/clipper
CLIP nf-core analysis pipeline: https://nf-co.re/clipseq
CLIP Tool Kit: https://github.com/chaolinzhanglab/ctk
doRiNA: https://dorina.mdc-berlin.de/
ENCODE project: https://www.encodeproject.org/
ENCORI (previously known as starBase): http://starbase.sysu.edu.cn
European Nucleotide Archive: http://www.ebi.ac.uk/ena
Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/
iCount: https://github.com/tomazc/iCount
iMaps: https://imaps.genialis.com/iclip
Integrative Genomics Viewer: http://software.broadinstitute.org/software/igv/
omniCLIP: https://github.com/philippdre/omniCLIP
PIPE-CLIP: https://github.com/QBRC/PIPE-CLIP
Piranha: https://github.com/smithlabcode/piranha
POSTAR2: http://lulab.life.tsinghua.edu.cn/postar
PureCLIP: https://github.com/skrakau/PureCLIP
pyCRAC: https://git.ecdf.ed.ac.uk/sgrannem/pycrac
wavClusteR: https://github.com/FedericoComoglio/wavClusteR
Supplementary information
Glossary
- Watson–Crick face
-
Part of the nucleobases that is involved in hydrogen bonding for canonical base pairing.
- Position-specific weight matrices
-
(PWMs). A commonly used representation of motifs, showing the proportion of the four nucleotides at each position in a set of biological sequences (such as RNA-binding protein binding sites).
- Recursive splicing
-
A process in which an intron is spliced sequentially in two or more distinct steps.
- Biomolecular condensates
-
Membraneless assemblies of proteins and/or nucleic acids that are bound together by multivalent interactions formed by protein domains, intrinsically disordered regions and/or nucleic acids.
- Intrinsically disordered regions
-
(IDRs). Polypeptide regions that do not form a defined three-dimensional structure in solution but tend to contain multivalent, assembly-promoting segments, the functionality of which is heavily modulated by post-translational modifications.
Rights and permissions
About this article

Cite this article
Hafner, M., Katsantoni, M., Köster, T. et al. CLIP and complementary methods. Nat Rev Methods Primers 1, 20 (2021). https://doi.org/10.1038/s43586-021-00018-1
Accepted:
Published:
DOI: https://doi.org/10.1038/s43586-021-00018-1
This article is cited by
-
Profiling of RNA-binding protein binding sites by in situ reverse transcription-based sequencing
Nature Methods (2024)
-
KARR-seq reveals cellular higher-order RNA structures and RNA–RNA interactions
Nature Biotechnology (2024)
-
TREX reveals proteins that bind to specific RNA regions in living cells
Nature Methods (2024)
-
Structure-based prediction and characterization of photo-crosslinking in native protein–RNA complexes
Nature Communications (2024)
-
A ubiquitous GC content signature underlies multimodal mRNA regulation by DDX3X
Molecular Systems Biology (2024)
