
Abstract
Five to seven percent of lung tumours are estimated to occur because of occupational asbestos exposure. Using cDNA microarrays, we have earlier detected asbestos exposure-related genomic regions in lung cancer. The region at 2p was one of those that differed most between asbestos-exposed and non-exposed patients. Now, we evaluated genomic alterations at 2p22.1-p16.1 as a possible marker for asbestos exposure. Lung tumours from 205 patients with pulmonary asbestos fibre counts from 0 to 570 million fibres per gram of dry lung, were studied by fluorescence in situ hybridisation (FISH) for DNA copy number alterations (CNA). The prevalence of loss at 2p16, shown by three different FISH probes, was significantly increased in lung tumours of asbestos-exposed patients compared with non-exposed (P=0.05). In addition, a low copy number loss at 2p16 associated significantly with high-level asbestos exposure (P=0.02). Furthermore, 27 of the tumours were studied for allelic imbalances (AI) at 2p22.1–p16.1 using 14 microsatellite markers and also AI at 2p16 was related to asbestos exposure (P=0.003). Our results suggest that alterations at 2p16 combined with other markers could be useful in diagnosing asbestos-related lung cancer.
Similar content being viewed by others

Main
Lung cancer is the leading cause of cancer-related deaths, causing almost 1.2 million deaths per year worldwide (Parkin et al, 2005). Several risk factors are known to contribute to the outbreak of lung cancer, of which tobacco is by far the most predominant (Peto, 1994). However, exposures to occupational carcinogens, such as asbestos, are also well-established causal factors of lung cancer. Asbestos refers to a group of naturally occurring fibrous silicate minerals, which have been widely exploited in, for example, the building industry. Although banned in many countries, asbestos exposure continues to be a serious risk factor for lung cancer, because of the long latency period between initial exposure and outbreak of the disease (Consensus report, 1997; Henderson et al, 2004). Five to seven percent of all lung cancer cases are estimated to occur because of occupational asbestos exposure (LaDou, 2004). So far, probability based on epidemiological studies of lung cancer risk with a certain asbestos exposure level has been considered the best way to estimate whether, in individual cases, a lung tumour has been caused by the patient's earlier exposure to asbestos.
Several mechanisms have been proposed to be involved in asbestos-induced cytotoxicity and genotoxicity, the formation of reactive species of oxygen or nitrogen being particularly central (Xu et al, 1999; Unfried et al, 2002). Asbestos fibres have been shown to induce damage at both DNA and chromosomal levels, as well as abnormal chromosome segregation (Dopp and Schiffmann, 1998; Okayasu et al, 1999; Unfried et al, 2002). We were recently able to identify specific gene copy number and expression profiles in lung cancer of asbestos-exposed patients (Nymark et al, 2006; Wikman et al, 2007). In the earlier studies, six chromosomal regions were identified to be affected by both DNA copy number alterations (CNA) and gene expression changes, and differed between lung tumours of asbestos-exposed and non-exposed patients. One of the identified regions was 2p21–p16.3 (Nymark et al, 2006; Wikman et al, 2007). Furthermore, treatment with crocidolite asbestos fibres in the lung cell lines, A549 and Beas-2B, has been shown to result in altered gene expression at 2p22 (Nymark et al, 2007). Here we have further explored 2p22–2p16 by fragment analysis for detecting allelic imbalance (AI) and to localise the specific asbestos-targeted core region. Furthermore, CNA was studied in a large number of lung tumours by fluorescence in situ hybridisation (FISH). Particularly, we showed that low copy number loss and AI at 2p16 in lung cancer were significantly associated with patients' past occupational exposure to asbestos.
Material and methods
Lung tumours and normal lung tissue
Lung tumour samples from 205 patients were used in this study. The study population is presented in Table 1. Fresh-frozen lung tumour samples (n=81) have been obtained from patients operated during 1990–1996 at the Department of Thoracic and Cardiovascular Surgery of the Helsinki University Central Hospital. The informed consents of these patients were obtained and they have been personally interviewed for their work history. The Ethical Review Board for Research in Occupational Health and Safety, and the Coordinating Ethical Review Board, Helsinki and Uusimaa Hospital District (75/E2/2001) have approved the study protocol and the collection of fresh-frozen lung tumour samples and personally interviewed data.
Formalin-fixed paraffin-embedded (FFPE) samples of lung tumours (n=124) were received from different Hospitals in Finland. These patients' pulmonary asbestos fibre counts had been analysed at the Finnish Institute of Occupational Health as a part of earlier exposure assessment. The National Agency for Medicolegal Affairs (4476/33/300/05) permitted the use of tissue samples originally obtained for histopathological diagnosis, and the Ministry for Social Affairs and Health (STM/2474/2005) permitted the collection of patient information for this study.
The pulmonary asbestos fibre counts of all the patients were analysed by electron microscopy with energy dispersive spectrometry (Karjalainen et al, 1993). We considered a patient to be asbestos-exposed if the fibre count was ⩾1.0 million fibres per gram of dry weight, which is usually considered as a sign of occupational asbestos exposure and to be associated with increased risk for lung cancer (Karjalainen et al, 1993, 1994). The median fibre count among the exposed patients was 6.6 million g−1 (range 1.0–570 million g−1). Furthermore, asbestos-exposed patients with fibre counts of ⩾5 million fibres per gram were considered as highly exposed. It has been reported that the background level of asbestos in the general population may rise up to 1.0 million fibres per gram (Miserocchi et al, 2008). To highlight the contrast between the exposed and non-exposed, we considered patients with fibre counts of ⩽0.5 million fibres per gram to be non-exposed. Specimens obtained from patients with a fibre count between 0.5 and 1 million fibres per gram were not collected. In addition, morphologically normal lung from 27 of the lung tumour patients were studied for AI. The 27 patients included in the AI study have been studied earlier by cDNA microarrays, and among this group there were non-exposed patients and highly asbestos-exposed patients with pulmonary fibre count ⩾5.0 million fibres per gram (Nymark et al, 2006).
Fluorescence in situ hybridisation
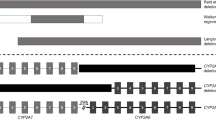
Bacterial artificial chromosomes (BAC) from the clones RP11-1114A19, RP11-347F1, RP11-703K23, RP11-963J22, and RP11-183P21 were used as FISH probes. The probes are shown in Figure 1, together with the corresponding microsatellite markers. The BAC clones were obtained from BACPAC Resources (CHORI, Oakland, CA, USA). DNA was extracted using Qiagen plasmid midi kit (Qiagen, Valencia, CA, USA) and labelled by nick-translation with either biotin-14-dATP (Invitrogen Life Technologies, Carlsbad, CA, USA) (Kettunen et al, 2000) or Spectrum Green dUTP using a Nick Translation kit (Vysis Inc./Abbott Molecular Inc., Downers Grove, IL, USA). For the centromere of chromosome 2, either a Spectrum Orange-labelled CEP2 DNA probe (alpha satellite; Vysis, Inc./Abbott Molecular, Inc.) or a rhodamine-labelled chromosome 2 satellite probe (Qbiogene, KREATECH Biotechnology BV, Amsterdam, The Netherlands) was used. Both are referred to as CEP2 in the following.

P arm of chromosome 2 showing microsatellite markers and bacterial artificial chromosome (BAC) clones used in fragment analyses and in fluorescent in situ hybridisation. Start and end base pairs are presented according to the University of California Santa Cruz (UCSC) genome browser.
A total of 87% of the FFPE tumours were sampled and put onto the tissue microarrays (TMA), and 13% of the FFPE tumours were studied as biopsy section slides. Before fluorescence microscopic analysis, serial sections of TMAs and biopsies were stained with haematoxylin and eosin for identification of tumour cells. After paraffin removal of TMA and biopsy section slides, and hydration of fresh-frozen tissue slides, the samples were treated in 0.01 M citrate, pH 6.0 at 80°C for 2 h (Chin et al, 2003). For digestion, we used DigestAll3 (Zymed Laboratories Invitrogen Immunodetection, San Francisco, CA, USA) at 37°C for 10 min. Denaturation, dehydrations, and hybridisation were carried out as described earlier (Kettunen et al, 2000; Andersen et al, 2001). Post-hybridisation washes for directly labelled BAC probes and for CEP2 were carried out according to manufacturer's instructions. Biotin-conjugated BAC probes were washed as described earlier (Kettunen et al, 2000), using fluorescein–avidin D and biotinylated anti-avidin D (Vector Laboratories Inc. Burlingame, CA, USA) for visualisation. The slides were counterstained with diamidino-2-phenylindole (DAPI) to identify tumour cells by morphology in fluorescence microcopy.
All the BAC probes were hybridised on metaphase chromosomes to ensure correct localisation at 2p (Figure 2A). FISH hybridisations were analysed using a Zeiss Axioplan fluorescence microscope (Zeiss, Jena, Germany). To calculate the locus-specific copy number changes in each case, we divided the copy numbers of each probe by the mean count of centromere 2.

(A) An example of a metaphase showing hybridisation signals of the RP11-703K23 probe in chromosome 2. (B) Lung tumour cells hybridised with the CEN2 centromeric probe (red) showing diploid to tetraploid cells and with the RP11-963J22 probe (green) showing gain of DNA at 2p21. (C) Lung tumour cells hybridised with the CEN2 centromeric probe (red) showing diploid to tetraploid cells and with the RP11-703K23 probe (green) showing loss at 2p16.
For each specimen, at least 100 cells were scored wherever possible. Owing to the large number of overlapping cells, we were able to count only 50–100 cells in 19% of the TMA specimens, and 30–50 cells in 12% of the TMA specimens and in 35% of the biopsy specimens. The mean signal count was calculated for each specimen. The FISH slides were divided into two sets containing specimens from both asbestos-exposed and non-exposed patients, and were analysed by two persons independently. Tissue cores with normal lung or lymph node material were used as negative controls in the TMA slides. In addition, lymphocytes or other non-cancerous cells showing two signals served as internal controls in each specimen.
To obtain locus-specific CNA at 2p in each case, we calculated the ratio between the mean signal count of each BAC probe and the mean signal count of CEP2. It has been reported that approximately half of all lung tumours are polyploid (Mitelman et al, 2009). In a case, in which the genome of the tumour cells is triploid, the fourth copy of locus DNA would show as a 33% increase in DNA copy number compared with the mean of centromeres. Thus, to be able to detect low CNA, we used the thresholds ⩾1.3 for gain and ⩽0.75 for loss of DNA.
Fragment analyses for detection of allelic imbalances
DNA from snap-frozen normal lung tissue was extracted using QIAamp Tissue Kit (Qiagen). Sections (4 μm) from snap-frozen lung tumour tissue specimens were stained with 1% toluidine blue–0.2% methylene blue solution for 20 s and dehydrated. The laser capturing procedure was carried out using Arcturus Veritas microdissecting device (Arcturus, Mountain View, CA, USA). Tumour cells were captured in CapSure Macro LCM caps (Arcturus, Mountain View, CA, USA) and DNA was extracted using Pico Pure DNA extraction kit (Arcturus).
Fragment analysis was carried out at 2p22–p16.1 (16.6 Mbp) using 14 microsatellite markers (Figure 1) to detect AI. Primers were labelled with FAM, HEX, or NED fluorochrome (TIB MOLBIOL Syntheselabor GmbH, Berlin, Germany or Applied Biosystems, Warrington, Cheshire, UK). The target sequences were amplified in tumour and corresponding normal lung tissue. PCR products of different sizes and fluorochromes were further combined and run with electrophoresis using 3100 Avant Genetic Analyzer (Applied Biosystems). The results were analysed with GeneMapper Analysis Software version 3.5 (Applied Biosystems). Allelic ratios were calculated between the two highest peaks with the expected fragment length for heterozygous markers in both tumour and normal DNA. Allelic imbalances were suggested when the ratio of the allelic ratios between the tumour and normal DNA was 2.0 or higher.
Results
Copy number alterations
Using FISH, we obtained the DNA copy numbers at 2p21–p16 in 55–156 lung tumours, depending on the probe and at centromere of chromosome 2 (alpha satellite) in 181 lung tumours. Locus-specific CNA was obtainable in 54–134 cases, depending on the BAC probe. The CEP2 signal count in lung tumour cells was on average 2.7 (range 1.6–5.7), varying between the lung tumour types as follows: adenocarcinoma of the lung (AC): 2.7; squamous cell carcinoma of the lung (SCC): 2.6; large cell lung cancer (LCLC): 3.0; and small cell lung cancer (SCLC): 2.6. Among the asbestos-exposed and non-exposed cases, the CEP2 mean signal counts were 2.7 (range 1.6–5.4) and 2.6 (range 1.6–5.7), respectively.
Locus-specific CNA results are shown in Table 2a. Both gains and losses of DNA were found (Figure 2B and C). In gains, the signals were evenly spread in the nuclei (Figure 2B).
The difference in DNA status between the exposed and non-exposed patients' lung tumours was tested at two stages. At first, we compared the frequencies of cases with losses, gains, and no CNA (Fisher's exact test or χ2-test). If P was <0.10, we continued by comparing the frequency of losses with the frequency of gains as well as the frequency of losses with the frequency of no CNA (Fisher's exact test or χ2-test). The RP11-703K23 probe at 2p16.3 showed association with asbestos exposure (P=0.09, Fisher's exact test) when comparing the difference between frequencies of losses, gains, and no CNA. Pair-wise comparisons, as explained above, of the number of cases with lost DNA and no CNA showed that losses at 2p16.3 were significantly associated with asbestos exposure (P=0.03, Fisher's exact test). Among the highly exposed patients (⩾5 million fibres per gram) the 2p16.3 losses were even more significantly associated with asbestos exposure (P=0.02, Fisher's exact test). An example of the lost RP11-703K23 signals is shown in Figure 2C. Gains and/or losses at 2p16 occurred in all histological types of lung cancer. When the frequencies of losses, gains, and no CNA were compared in different histological types, we found losses at 2p16 in the exposed patients with any histological tumour type, but the case numbers in each group were small and only SCC showed significant P-values. CNA in SCC were significantly associated with asbestos exposure at 2p16.2 (RP11-1114A19/CEN2, P=0.04, Fisher's exact test) and with high-level exposure at 2p16.3 (RP11-703K23/CEN2, P=0.02, Fisher's exact test).
In addition, the prevalence of probes that showed a CNA in each case at 2p16 (probes RP11-703K23, RP11-347F1, and RP11-1114A19) and at 2p21 (probes RP11-183P21 and RP11-963J22) was calculated. At 2p16, the prevalence of at least one probe loss was significantly increased in asbestos-exposed, particularly in highly exposed, in comparison with non-exposed cases (Table 2b). The difference was also significant between the exposed (100%, 2 out of 2 cases) and non-exposed (0%, 0 out of 7 cases) patients with SCLC (P=0.03, Fisher's exact test). No significant association was identified with asbestos when similar calculations were carried out for the gains (data not shown). It can be assumed that among the non-exposed of our study population no asbestos-induced cancers exist, whereas among the exposed patients the proportion of excess lung cancers because of asbestos exposure depends on the risk level in the group, and may be roughly 50% (Karjalainen et al, 1994). Thus, when combining the results of the three probes at 2p16, the specificity of copy number losses at 2p16 in detecting asbestos-related lung tumours would be 93%, but the sensitivity cannot be estimated because it is not known which of the lung cancers in the exposed group are asbestos-related.
Allelic imbalance in 2p22.1–p16.1
A total of 27 lung tumours were microdissected for allele analyses and their microsatellite allelotyping profiles were compared with those in normal lung tissue of the same patients. Allelic imbalances at 2p22.1–p16.1 in the lung tumours of the asbestos-exposed and the non-exposed patients are shown in Table 3. The average proportion of informative microsatellite markers was 67% (range 41–83%). This agrees well with the reported heterozygosity rate of these markers. Microsatellite instability at 2p was observed in three cases: in two ACs, one from an asbestos-exposed and one from a non-exposed patient, and in one non-exposed SCLC. Although AI at 2p was observed to some extent in every lung tumour, we identified more AI in the lung tumours of asbestos-exposed (on average 52%) than in those of non-exposed patients (on average 32%). At 2p16.3 (D2S123), AI occurred in 63% of the asbestos-exposed and in 0% of the non-exposed patients' tumours (P=0.08, Fisher's exact test, Table 3). As the markers, D2S2739 and D2S2251, adjacent to D2S123 at 2p16 showed similar trends of asbestos association, we also looked at the five markers at 2p16 together (Table 3, Figure 3). Tumours having at least two informative markers out of five were included in the analysis. Allelic imbalances at 2p16 were scored for a tumour if at least two markers showed AI. We found significantly more AI in asbestos-exposed patients compared with non-exposed (P=0.003, Fisher's exact test; Figure 3).

Significantly more allelic imbalances (AI) at 2p16 were found in asbestos-exposed patients' lung tumours compared with those of non-exposed (P=0.003, Fisher's exact test). aAC, adenocarcinoma; SCC, squamous cell carcinoma; SCLC, small cell lung cancer; LCLC, large cell lung carcinoma; bmillion g−1 dry lung; cwhite, normal; light grey, no result; dark grey, allelic imbalance; MSI, microsatellite instability; bold when at least two markers showed AI.
Discussion
We analysed 205 lung tumours for CNA at 2p and 27 lung tumours for AI at the same region. We showed that copy number loss at 2p16 is significantly associated with a patients' past occupational asbestos exposure. Particularly, we found that the prevalence of low copy number loss at 2p16 shown with three different FISH probes was significantly associated with asbestos exposure (P=0.04, χ2-test). In addition, AI at 2p16 was detected more frequently in the tumours of asbestos-exposed patients (P=0.003, Figure 3).
The chromosome count and structure in lung cancer cells often show great variation. Some aberrations in chromosome 2 have been detected in lung cancer, for example the amplification of the MYCN locus (2p24) in SCLC (Knuutila, 2004; Kim et al, 2006). Recently the fusion gene, EML4-ALK, resulting from an inversion at 2p21/2p23, was found in 3–7% of non-small cell lung cancer (NSCLC) (Soda et al, 2007; Koivunen et al, 2008). Furthermore, by array comparative genomic hybridisation it has been shown that 52% of NSCLC carried gained sequences at 2p21.1–p14 independent of histological type (Dehan et al, 2007). However, because the asbestos exposure burden of a given lung cancer patient has not usually been determined, the majority of earlier studies understandably lack association analyses between genomic findings and asbestos exposure status. Our earlier array studies on lung cancer showed that genomic alterations at 2p21–p16.3 may be related to asbestos exposure in lung cancer patients (Nymark et al, 2006; Wikman et al, 2007).
In this study, we have further characterised the asbestos-related genetic aberrations at 2p in a large group of lung cancer patients. We detected asbestos-associated loss and AI at 2p16 and localised the core region at 2p16.3–p16.2 (Table 2a).
Our results indicate that asbestos specifically targets regions such as on chromosome 2. Asbestos has been shown to cause hyperdiploidy, loss of chromosomes, and chromosomal breakage (Dopp and Schiffmann, 1998). Although the precise mechanisms and outcomes of asbestos-induced DNA damage have not yet been thoroughly established, the finding of DNA loss and AI at 2p16 in the lung tumours of asbestos-exposed patients is in accordance with the current knowledge on the behaviour of asbestos fibres. Furthermore, common fragile sites may be prone to replicative stress and increased frequency of AI. We have earlier detected an association between fragile sites and some of the asbestos-associated CNA (Nymark et al, 2006). The aphidicolin-sensitive common fragile site, FRA2D, is located at 2p16.2 between the markers, D2S123 and D2S2153, and seems to overlap with the region showing asbestos-associated loss (Schwartz et al, 2006). Some tumours, such as a cancer syndrome called the Carney complex (CNC), have shown both amplifications and deletions at 2p21-p16 (Matyakhina et al, 2003). The CNC syndrome is a genetically heterogeneous disease associated with multiple neoplasms, such as skin, cardiac, and breast myxomas, as well as endocrine tumours. The size of the CNA at 2p has been reported to vary from case to case and from cell to cell, indicating that 2p is indeed prone to variable alterations.
In our earlier work, the affected sequences in which the DNA copy number differed significantly between lung tumours of asbestos-exposed and non-exposed patients, was estimated to situate at 2p21–p16.3 (Nymark et al, 2006). Here we were able to more specifically localise the affected region to 2p16 (RP11-703K23). Alternation of normal and abnormal loci makes it challenging to identify typical asbestos-related alterations, particularly in a complex genome, such as in lung cancer, which often shows polyploid genome (Mitelman et al, 2009). Nevertheless, by comparing locus-specific probes with a centromere probe to obtain locus-specific CNA at 2p, we were able to identify asbestos-associated CNA, although the alterations occurred at low level. We anticipate that genomic alterations occurring at 2p may reflect distinct events in lung tumours, some related to asbestos exposure and some not. Independently of patients' asbestos exposure status, we detected frequent low copy number gains at 2p21, which suggests that 2p21 may harbour genes important to the development of lung cancer, such as the earlier mentioned oncogenic EML4-ALK fusion gene. It seems that, although chromosome 2 may generally be affected by gains in lung tumours, asbestos may cause breaks in the chromosome and thus result in additional alterations, such as losses.
In conclusion, we show that locus-specific copy number loss and AI at 2p16.3-p16.2 are associated with asbestos exposure in lung tumours, whereas low-level copy number gains at 2p21 are common in lung cancer in general. Probability based on epidemiological studies has for a long time been used as a method of indicating that asbestos exposure burden may be associated with an individual's lung cancer. We believe that the findings we describe here may benefit the diagnosis of asbestos-associated lung tumours, when combined with earlier identified asbestos-associated alterations in other chromosomal regions, such as 19p13 and 9q33.1 (Ruosaari et al, 2008; Nymark et al, 2009).
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andersen CL, Hostetter G, Grigoryan A, Sauter G, Kallioniemi A (2001) Improved procedure for fluorescence in situ hybridization on tissue microarrays. Cytometry 45: 83–86
Chin S, Daigo Y, Huang H, Iyer N, Callagy G, Kranjac T, Gonzalez M, Sangan T, Earl H, Caldas C (2003) A simple and reliable pretreatment protocol facilitates fluorescent in situ hybridisation on tissue microarrays of paraffin wax embedded tumour samples. Mol Pathol 56: 275–279
Consensus report (1997) Asbestos, asbestosis, and cancer: the Helsinki criteria for diagnosis and attribution. Scand J Work Environ Health 23: 311–316
Dehan E, Ben-Dor A, Liao W, Lipson D, Frimer H, Rienstein S, Simansky D, Krupsky M, Yaron P, Friedman E, Rechavi G, Perlman M, Aviram-Goldring A, Izraeli S, Bittner M, Yakhini Z, Kaminski N (2007) Chromosomal aberrations and gene expression profiles in non-small cell lung cancer. Lung cancer 56: 175–184
Dopp E, Schiffmann D (1998) Analysis of chromosomal alterations induced by asbestos and ceramic fibers. Toxicol Lett 96–97: 155–162
Henderson DW, Rödelsperger K, Woitowitz HJ, Leigh J (2004) After Helsinki: a multidisciplinary review of the relationship between asbestos exposure and lung cancer, with emphasis on studies published during 1997–2004. Pathology 36: 517–550
Karjalainen A, Anttila S, Heikkilä L, Karhunen P, Vainio H (1993) Asbestos exposure among Finnish lung cancer patients: occupational history and fiber concentration in lung tissue. Am J Ind Med 23: 461–471
Karjalainen A, Anttila S, Vanhala E, Vainio H (1994) Asbestos exposure and the risk of lung cancer in a general urban population. Scand J Work Environ Health 20: 243–250
Kettunen E, El-Rifai W, Björkqvist AM, Wolff H, Karjalainen A, Anttila S, Mattson K, Husgafvel-Pursiainen K, Knuutila S (2000) A broad amplification pattern at 3q in squamous cell lung cancer – a fluorescence in situ hybridization study. Cancer Genet Cytogenet 117: 66–70
Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, Pollack JR (2006) Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 25: 130–138
Knuutila S (2004) Amplicons: Respiratory tract tumors. http://www.infobiogen.fr/services/chromcancer/Ampl/Resp.html. Atlas Genet Cytogenet Oncol Haematol
Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, Lee J, Richards WG, Sugarbaker DJ, Ducko C, Lindeman N, Marcoux JP, Engelman JA, Gray NS, Lee C, Meyerson M, Jänne PA (2008) EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 14: 4275–4283
LaDou J (2004) The asbestos cancer epidemic. Environ Health Perspect 112: 285–290
Matyakhina L, Pack S, Kirschner LS, Pak E, Mannan P, Jaikumar J, Taymans SE, Sandrini F, Carney JA, Stratakis CA (2003) Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet 40: 268–277
Miserocchi G, Sancini G, Mantegazza F, Chiappino G (2008) Translocation pathways for inhaled asbestos fibers. Environ Health 7: 4
Mitelman F, Johansson B, Mertens F (2009) Mitelman Database of Chromosome Aberrations in Cancer, http://cgap.nci.nih.gov/Chromosomes/Mitelman
Nymark P, Kettunen E, Aavikko M, Ruosaari S, Kuosma E, Vanhala E, Salmenkivi K, Pirinen R, Karjalainen A, Knuutila S, Wikman H, Anttila S (2009) Molecular alterations at 9q33.1 and polyploidy in asbestos-related lung cancer. Clin Cancer Res 15: 468–475
Nymark P, Lindholm P, Korpela M, Lahti L, Ruosaari S, Kaski S, Hollmen J, Anttila S, Kinnula V, Knuutila S (2007) Gene expression profiles in asbestos-exposed epithelial and mesothelial lung cell lines. BMC Genomics 8: 62
Nymark P, Wikman H, Ruosaari S, Hollmén J, Vanhala E, Karjalainen A, Anttila S, Knuutila S (2006) Identification of specific gene copy number changes in asbestos-related lung cancer. Cancer Res 16: 5737–5743
Okayasu R, Takahashi S, Yamada S, Hei TK, Ullrich RL (1999) Asbestos and DNA double strand breaks. Cancer Res 59: 298–300
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108
Peto R (1994) Smoking and death: the past 40 years and the next 40. BMJ 309: 937–939
Ruosaari ST, Nymark PEH, Aavikko MM, Kettunen E, Knuutila S, Hollmen J, Norppa H, Anttila SL (2008) Aberrations of chromosome 19 in asbestos-associated lung cancer and in asbestos-induced micronuclei of bronchial epithelial cells in vitro. Carcinogenesis 29: 913–917
Schwartz M, Zlotorynski E, Kerem B (2006) The molecular basis of common and rare fragile sites. Cancer Lett 232: 13–26
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S-i, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448: 561–566
Unfried K, Schurkes C, Abel J (2002) Distinct spectrum of mutations induced by crocidolite asbestos: clue for 8-hydroxydeoxyguanosine-dependent mutagenesis in vivo. Cancer Res 62: 99–104
Wikman H, Ruosaari S, Nymark P, Sarhadi VK, Saharinen J, Vanhala E, Karjalainen A, Hollmén J, Knuutila S, Anttila S (2007) Gene expression and copy number profiling suggests the importance of allelic imbalance in 19p in asbestos-associated lung cancer. Oncogene 26: 4730–4737
Xu A, Wu L-J, Santella RM, Hei TK (1999) Role of oxyradicals in mutagenicity and DNA damage induced by crocidolite asbestos in mammalian cells. Cancer Res 59: 5922–5926
Acknowledgements
We are grateful to Päivi Tuominen, Tuula Stjernvall, and Helinä Hämäläinen for excellent technical assistance, and to Hanna Vauhkonen for valuable advice in fragment analyses. This work has been financially supported by the Foundation for the Finnish Cancer Institute (EKe), the K Albin Johansson Foundation (EKe), the Academy of Finland Grant no. 115372 (EKe), the Finnish Funding Agency for Technology and Innovation (TEKES) (SA), and the Graduate School in Computational Biology, Bioinformatics and Biometry (SR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kettunen, E., Aavikko, M., Nymark, P. et al. DNA copy number loss and allelic imbalance at 2p16 in lung cancer associated with asbestos exposure. Br J Cancer 100, 1336–1342 (2009). https://doi.org/10.1038/sj.bjc.6605012
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605012
Keywords
This article is cited by
-
CDKN2A copy number and p16 expression in malignant pleural mesothelioma in relation to asbestos exposure
BMC Cancer (2019)
-
Arsenic, asbestos and radon: emerging players in lung tumorigenesis
Environmental Health (2012)
-
Genetic ecotoxicology of asbestos pollution in the house mouse Mus musculus domesticus
Environmental Science and Pollution Research (2011)
-
Arsenic-related DNA copy-number alterations in lung squamous cell carcinomas
British Journal of Cancer (2010)
