
Abstract
The antipsychotic effect of neuroleptics cannot be attributed entirely to acute blockade of postsynaptic D2-like dopamine (DA) receptors, but may arise in conjunction with the delayed depolarization block of the presynaptic neurons and reduced DA synthesis capacity. Whereas the phenomenon of depolarization block is well established in animals, it is unknown if a similar phenomenon occurs in humans treated with neuroleptics. We hypothesized that haloperidol treatment should result in decreased DA synthesis capacity. We used 6-[18F]fluoro-L-dopa (FDOPA) and positron emission tomography (PET) in conjunction with compartmental modeling to measure the relative activity of DOPA decarboxylase (DDC) (kD3, min−1) in the brain of nine unmedicated patients with schizophrenia, first in the untreated condition and again after treatment with haloperidol. Patients were administered psychometric rating scales at baseline and after treatment. Consistent with our hypothesis, there was a 25% decrease in the magnitude of kD3 in both caudate and putamen following 5 weeks of haloperidol therapy. In addition, the magnitudes of kD3 in cerebral cortex and thalamus were also decreased. Psychopathology as measured with standard rating scales improved significantly in all patients. The decrease of kD3 in the thalamus was highly significantly correlated with the improvement of negative symptoms. Subchronic treatment with haloperidol decreased the activity of DDC in the brain of patients with schizophrenia. This observation is consistent with the hypothesis that the antipsychotic effect of chronic neuroleptic treatment is associated with a decrease in DA synthesis, reflecting a depolarization block of presynaptic DA neurons. We link an alteration in cerebral catecholamine metabolism in human brain with the therapeutic action of neuroleptic medication.
Similar content being viewed by others

INTRODUCTION
The dopamine (DA) hypothesis of schizophrenia, which attributes the positive schizophrenic symptoms to an excess of dopaminergic neurotransmission, was formulated on the basis of observations that typical antipsychotic drugs block D2-like DA receptors in vitro (Carlsson, 1988) with affinities that correlate with clinical potency (Seeman and Lee, 1975). PET studies show that the treatment of schizophrenic patients with neuroleptics at clinically relevant doses leads to occupancy of D2-like DA receptors in the range of 65–90% in the case of haloperidol and other typical neuroleptics and 20–40% in the case of clozapine, an atypical neuroleptic (Farde et al, 1992).
For several reasons, the antipsychotic effect of neuroleptics cannot be entirely attributed to the acute pharmacological blockade of postsynaptic D2-like DA receptors. First, DA receptor blockade occurs shortly after the administration of these compounds (Nordström et al, 1992), while the onset of antipsychotic effects is delayed by a period ranging from days to weeks (Johnstone et al, 1978). Second, similar striatal DA receptor occupancies by neuroleptics have been observed in responders to neuroleptic drug treatment and in nonresponders (Wolkin et al, 1989). Thus, the blockade of DA D2 receptors may be necessary, but cannot be sufficient to account fully for antipsychotic drug action. This discrepancy led to the suggestion that the antipsychotic effect of neuroleptics may arise in conjunction with the delayed depolarization block of the presynaptic neuron and reduced DA synthesis capacity, a phenomenon which occurs in experimental animals treated chronically with neuroleptics (Grace and Bunney, 1986). Several positron emission tomography (PET) studies with radiolabeled L-DOPA analogues have reported elevated activity of the enzyme DOPA decarboxylase (DDC) in the striatum of untreated patients with schizophrenia (Hietala et al, 1995; Lindström et al, 1999; Reith et al, 1994), indicating the presence of elevated capacity for DA synthesis within the terminals of DA neurons (Cumming and Gjedde, 1998). However, the transition of DA neurons to a partially inactivated state of depolarization block has not been demonstrated in schizophrenic patients under treatment with neuroleptics. Therefore, we used the PET tracer [18F]fluoro-L-DOPA (FDOPA) in conjunction with compartmental modeling to test the hypothesis that subchronic treatment with a typical neuroleptic decreases the capacity for DA synthesis in the brain of patients with schizophrenia.
METHODS
The study was approved by the local ethics committee.
Subjects
Nine male patients meeting DSM-IV criteria for schizophrenia, who were acutely admitted to the hospital, gave written, informed consent to participate in the study. The age of the subjects ranged from 27 to 47 years (mean±SD: 37.6±6.0 years). Except for their psychiatric disorder, they were healthy with respect to medical history, physical examination, 12-lead electrocardiogram (ECG), electroencephalogram (EEG), fasting clinical laboratory tests, and magnetic resonance imaging (MRI) of the brain. Among the inclusion criteria was the requirement that patients were free from any psychoactive medication for at least 6 months prior to the PET study. Three patients were entirely drug naïve, the remainder had been treated with various doses of oral neuroleptics. Two PET scans with FDOPA were performed, one at baseline and one after treatment with haloperidol for a period of 20–45 days (mean±SD: 35.3±7.6 days). To ensure that truly subchronic, adaptive effects of haloperidol were measured, the second PET scan was scheduled no earlier than 30 days after initiation of treatment in all but one patient. In this single patient, the second scan had to be performed earlier for logistical reasons. All patients improved during this course of treatment; haloperidol dose was selected so as to yield the optimal clinical benefit while minimizing extrapyramidal symptoms. The mean daily dose on the day of the second PET scan was 8.9±4.5 mg (range 1–15 mg).
Psychometric Rating Scales
Psychopathological symptoms and EPS were derived by trained raters on the day of the first PET scan, prior to initiation of haloperidol, and again on the day of the second PET scan by means of the following standard instruments: Positive and Negative Syndrome Scale (PANSS, Kay et al, 1987), Clinical Global Impression (CGI, Guy, 1976), Extrapyramidal Symptoms Rating Scale (ESRS; akinesia score, Chouinard et al, 1980) and the Barnes Akathisia Rating Scale (BARS, Barnes, 1989).
PET Data Acquisition
For 24 h prior to the PET scans, patients were maintained on a diet low in protein in order to minimize the variance in the competition between dietary large neutral amino acids and the tracer FDOPA for facilitated diffusion across the blood–brain barrier (Stout et al, 1998). Decarboxylation of FDOPA in the peripheral tissue was blocked by oral administration of the peripheral DDC inhibitor carbidopa (Merck Sharp & Dome, 2 mg/kg body weight) 1 h prior to injection of the FDOPA (Cumming et al, 1993). Emission recordings from the first two patients were obtained with a GE 4096+ whole-body scanner with 15 planes, an in-plane resolution of 5–6 mm, and an axial resolution of 6–7 mm. All other studies were performed with a Siemens ECAT EXACT whole-body PET scanner with a field of view of 16.2 cm in 47 planes, in interplane spacing of 3.375 mm and an axial resolution of 5.4 mm FWHM. After a brief attenuation scan, a sequence of 28 emission frames lasting a total of 128 min was recorded. Frame length increased progressively from 1 to 10 min.
A mean of 153 MBq FDOPA (SD: 52 MBq, range: 95–300 MBq) was injected intravenously as a bolus into a cubital vein. A series of 23 blood samples were collected from a radial artery at the following intervals after FDOPA injection: 8 × 30 s, 1 × 1 min, 2 × 2.5 min, 2 × 5 min, and 10 × 10 min. Blood samples were immediately centrifuged and the total plasma radioactivity counted by gamma spectroscopy. The fractions of untransformed FDOPA and its major plasma metabolite O-methyl-FDOPA were measured in nine plasma samples by HPLC (Cumming et al, 1993). The fractions at each of the 23 sample times were estimated by fitting multiexponential functions to the nine fractions measured by HPLC and the continuous plasma input functions were calculated.
After the first PET scan, a T1-weighted 3D gradient echo magnetic resonance scan with 1.5 mm slice thickness and 128 slices was acquired from each patient The MRI was resliced according to the ac-pc-line, which was identified in a midsagittal plane. The summed PET images were then coregistered with the resliced MRI to permit anatomical positioning of volumes of interest (VOIs) using the automatic image registration (AIR) algorithm (Woods et al, 1992). For caudate, putamen, and thalamus, VOIs were generated from polygonal regions of interest (ROIs) on all MRI planes containing the respective tissue types. VOIs for the cortical areas were created from ten consecutive MRI slices.
PET Data Analysis
A constrained two-compartment model assuming irreversible trapping in the brain of decarboxylated metabolites was fitted to the timeradioactivity curves (TACs) recorded in brain (Gjedde et al, 1991; Huang et al, 1991). The temporal changes in the plasma concentrations of FDOPA and its principal metabolite O-methyl-FDOPA were used to calculate the apparent first-order rate constant for the formation of O-methyl-FDOPA (kD0, min−1), corresponding to the whole-body activity of catechol-O-methyltransferase, and the fractional rate constant for the elimination of O-methyl-FDOPA from circulation , corresponding to the renal elimination rate (Cumming et al, 1993). In order to test for possible treatment effects on the peripheral pharmacokinetics of FDOPA (see Cumming et al, 1999), the magnitudes of these rate constants were calculated for each subject in the baseline condition and after haloperidol treatment.
In brain regions such as the cerebellum or the occipital cortex that are nearly devoid of DDC activity, the concentration of FDOPA in the brain tissue at any time after intravenous injection is determined by the convolution of the arterial input with the unidirectional blood–brain clearance of FDOPA (KD1, ml g−1 min−1) and the fractional rate constant for the diffusion of FDOPA from brain back to plasma (kD2, min−1). The ratio of these two terms, KD1/kD2, corresponds to the tracer blood–brain distribution volume at equilibrium (VeD, ml g−1). In brain regions containing DDC, FDOPA is decarboxylated at rate constant kD3 (min−1), corresponding to the local activity of DDC (Cumming et al, 1993). The product [18F]fluorodopamine is retained in synaptic vesicles, resulting in the progressive accumulation of radioactivity in the caudate and putamen, which are richly endowed with DDC. Here, the solution of the two-compartment model is facilitated by constraining the magnitude of VeD to the individual observation obtained in the cortical reference region. This approach allows stable estimates of the term of interest, the local relative activity of DOPA decarboxylase kD3 (Cumming and Gjedde, 1998; Gjedde et al, 1991; Huang et al, 1991). Thus, the parameters to be estimated in the ROIs were the apparent plasma volume occupied by FDOPA Vo (ml g−1), the local magnitude of KD1, and the magnitude of kD3.
The quantitative analysis of dynamic emission sequences obtained with FDOPA is based upon the compartmental model established for the quantification of cerebral glucose metabolism, in which the rate constant k3 there corresponds to the phosphorylation of glucose analogues by the hexokinase reaction (Sokoloff et al, 1977). An additional complication of FDOPA modeling is presented by the peripherally formed metabolite O-methyl-FDOPA, which becomes the predominant radiolabeled compound in the blood and brain during prolonged experiments (Cumming et al, 1993). The entry of O-methyl-FDOPA into brain requires additional physiological constraints in the compartmental model for estimating the DDC activity. In the present model, the two amino acids O-methyl-FDOPA and FDOPA are assumed to have a single common distribution volume, and are assumed to have a fixed permeability ratio, q, of 1.5, based upon the mean of several estimates (Cumming and Gjedde, 1998). The sensitivity of the estimation of kD3 to the constrained values of q and VeD was investigated in the monkey brain (Léger et al, 1998).
In our study, individual estimates of VeD in occipital cortex were used to constrain fitting of the same model to time radioactivity recorded in caudate and putamen during 45 min of tracer circulation. This procedure has been shown to optimize the precision and accuracy of the estimation of kD3 when the eventual elimination of decarboxylated metabolites from the brain is neglected (see Figure 3 in Cumming and Gjedde, 1998). For thalamus and cortical VOIs, 60 min emission recordings were used in order to improve the estimation of kD3 in brain regions of low DDC activity. However, this claim has not been completely examined in the literature.
Statistical Analyses
Means and standard deviations were calculated for psychopathological and kD3 values. Shifts from baseline to end point were analyzed by means of Wilcoxon matched-pairs signed-ranks tests. Spearman rank correlations were calculated for treatment-related changes in kD3 and psychopathology or EPS side effects, respectively. To account for baseline values, proportional change scores ((end point−baseline)/baseline × 100) were computed for kD3, CGI, and PANSS, while absolute changes in BARS and EPS scores (end point−baseline) were used. In all analyses, the two-tailed level of statistical significance was set at α=0.05. Owing to the exploratory character of the study, no adjustment for multiple testing was performed.
RESULTS
The mean magnitude of the FDOPA distribution volume VeD in occipital cortex was 0.60±0.11 ml g−1 in the baseline condition and 0.70±0.22 ml g−1 after haloperidol treatment. This increase in VeD failed to reach statistical significance (p=0.051). Also, the change in VeD was not significantly correlated to change in kD3. The unidirectional blood–brain clearance of FDOPA (KD1, ml g−1 min−1) ranged from 0.028 ml g−1 min−1 in the left caudate nucleus to 0.038 ml g−1 min−1 in the left putamen in the baseline condition; haloperidol was without effect on the magnitude of KD1 in any region. Also, haloperidol did not alter kD0, the apparent rate constant for the formation of the O-methylated FDOPA metabolite in the plasma (pretreatment, mean±SD: 0.011±0.002 min−1; post-treatment: 0.011±0.002 min−1) or .
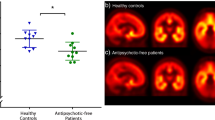
In the baseline PET study, no significant differences in the magnitude of kD3 between hemispheres were detectable, although estimates tended to be higher in the left striatum (Table 1). kD3 decreased significantly with treatment in all brain regions investigated except for the right caudate (Table 1, Figure 1). Baseline kD3 in the cerebral cortex VOIs was low, but also decreased significantly with neuroleptic treatment (Table 1). Thalamic kD3, which was significantly higher than in cortex, was also significantly reduced. The reduction in kD3 was most pronounced in the left caudate (Table 1, Figure 1). There was no statistically significant correlation between treatment duration and the magnitude of change in kD3. However, the single subject who was scanned after only 20 days treatment, had very small decreases (eg lowest decrease in left caudate) or even increases in kD3.

Scatter plots of individual changes (in % from baseline) in kD3 (min−1) in striatal and thalamic regions. Subjects scanned on the GE 4096+ camera are indicated with a black circle: —, sample median.
Mean baseline and post-treatment psychopathological and extrapyramidal side effect (EPS) rating scores are presented in Table 2. There were significant improvements in all psychopathological rating scores after haloperidol treatment. EPS scores were significantly increased by haloperidol (Table 2). At baseline, we could not detect any statistically significant correlation between kD3 in any region and psychopathological or side effect rating scales, respectively. The same was true, when the follow-up condition was investigated separately or when both conditions were analyzed together. However, when the change between pre- and post-treatment kD3 was correlated with change in psychopathology or side effects, respectively, we could indeed detect statistically significant relations. The change in thalamus kD3 was highly significantly correlated with percent change in the PANSS negative subscore (RS=0.93; p<0.0005), but not with change in the PANSS positive subscore (RS=−0.18; p=0.651). Moreover, there were positive, although less robust, correlations between thalamic kD3 and CGI (RS=0.74; p=0.023) as well as BARS rating scores (RS=0.71; p=0.032).
DISCUSSION
Soon after the introduction of chlorpromazine, the first neuroleptic drug, Carlsson and Lindqvist discovered that acute treatment of rats with antipsychotics increases DA metabolite concentrations in the brain and stimulated the activity of tyrosine hydroxylase in the rat striatum suggesting that pharmacological blockade of presynaptic DA ‘autoreceptors’ increased the rate of DA synthesis and release in the brain (Carlsson and Lindqvist, 1963). This claim was substantiated by observations that neuroleptics increase the basal firing rate of DA neurons and block the amphetamine-induced inhibition of the depolarization of DA neurons (Grace, 1992; Grace and Bunney, 1986). However, these effects of neuroleptics are obtained acutely, and therefore seem insufficient to account for the onset of antipsychotic effects, and might indeed be considered paradoxical. Furthermore, the clinical response can be delayed by several weeks, whereas potentiation of DA transmission by neuroleptics occurs immediately. Subsequent electrophysiological studies showed that chronic administration of neuroleptics decreased the number of spontaneously active DA neurons in the rat substantia nigra, a condition known as depolarization block. Grace hypothesized that the antipsychotic effect of neuroleptic drugs occurs in conjunction with the development of this depolarization block (Grace, 1992). Others have stressed the importance of block in mesolimbic, but not striatal neurons for obtaining therapeutic effects (Chiodo and Bunney, 1983; White and Wang, 1983), whereas depolarization block of nigrostriatal neurons may underlie the extrapyramidal side effects (White and Wang, 1983). The present documentation of a haloperidol-induced decrease of DA synthesis capacity is in good agreement with observations of amphetamine-evoked changes in DA D2 receptor occupancy. Amphetamine-induced changes in [123I]iodobenzamide binding in the striatum suggest the presence of a supernormal amphetamine-releasable DA pool in acutely psychotic patients, which was normalized with medication (Laruelle et al, 1999).
In the present study, we find that a course of treatment with haloperidol producing significant clinical improvement was associated with a decline in kD3, the DDC activity, calculated by fitting of a constrained compartment model to the recorded TACs, in several brain regions. This finding was robust, occurring in eight out of nine patients (Figure 1). Although we found no correlation between kD3 and treatment duration, it is remarkable that the single subject who for logistical reasons had to be scanned 20 days after initiation of haloperidol, demonstrated the mildest decrease in kD3 or even increases in some regions. In this patient, 20 days treatment may have been too short to observe a decrease in kD3.
In our study, the unidirectional blood–brain clearance of FDOPA (KD1, ml g−1 min−1) was not influenced by haloperidol treatment. Thus, the estimation of kD3 is isolated from the possible effects of drug treatment on the delivery of tracer to the brain. Also, haloperidol did not alter the plasma metabolism of FDOPA. However, we have documented a mild, but almost significant increase in distribution volume VeD, which is the ratio KD1/kD2. Since KD1 does not change at all in our study, the increase in VeD could possibly be attributed to a decrease in kD2, which is the fractional rate constant for the diffusion of FDOPA from the brain back to the plasma. Blood flow changes induced by haloperidol have been reported, but the literature is inconsistent in this regard. Most studies found blood flow changes in frontal cortex (Miller et al, 2001), basal ganglia (Miller et al, 1997), or hippocampus (Medoff et al, 2001), respectively, but not in occipital cortex, which served as reference region in our study, from which the equilibrium distribution volume VeD was determined. Thus, although we cannot totally exclude the possibility that the documented changes in kD3 are influenced by brain perfusion or tracer distribution, our study has the advantage that it actually measures these effects. Owing to numerous methodological factors, our present baseline estimates of kD3 (Reith et al, 1994), which represents the rate constant for the decarboxylation of FDOPA, cannot be compared directly with previously reported normative values obtained by other laboratories, and cannot readily be compared with estimates of the net clearance of FDOPA (Hietala et al, 1995) or [β-11C]DOPA (Lindström et al, 1999) to striatum KiD (ml g−1 min−1), which reflect the composite of perfusion, diffusion back to blood, and decarboxylation in nigrostriatal terminals. Consequently, estimates of the magnitude of Ki are weighted by the unknown equilibrium volume (VeD) of FDOPA in the brain tissue. Therefore, the effects of treatment or condition on the magnitude of Ki in the human brain cannot be unambiguously attributed to the altered rate of trapping of FDOPA in the brain (Hietala et al, 1995; Meyer-Lindenberg et al, 2002; Yatham et al, 2002). The present compartmental model explicitly evaluates the magnitude of VeD, and therefore isolates the parameter of interest, the activity of DDC, from the effects of perfusion and distribution.
FDOPA and other exogenous DDC substrates are tracers for the ultimate step in DA synthesis. Since concentrations of L-DOPA occurring in living brain are far below the saturating concentration for DDC, it is usually concluded that this enzyme occurs in vast excess and consequently cannot contribute to the regulation of DA synthesis. On the other hand, tyrosine hydroxylase, the penultimate enzyme in catecholamine synthesis, is nearly saturated with its main substrate tyrosine, and is consequently generally considered to be the sole rate-limiting step in DA synthesis (for a review, see Roth and Elsworth, 1995). However, in the living brain the DDC activity influences the fraction of substrate, which is directed to catecholamine synthesis rather than being exported from the brain. The presence of alternate fates for L-DOPA in vivo requires that regulation of DDC must also influence the rate of DA synthesis (Cumming and Gjedde, 1998; Gjedde et al, 1993). In support of this claim, the DDC activity in the striatum of living rats was potentiated by acute neuroleptic treatment and decreased by the DA presynaptic autoreceptor agonist apomorphine (Cumming et al, 1997). In an FDOPA/PET study, acute haloperidol markedly stimulated DDC activity in pig striatum (Danielsen et al, 2001). Whereas the functional state of DA autoreceptors may account for the acute effects of neuroleptics on DDC activity, the present finding of decreased DDC activity in human brain suggests that additional factors must influence DDC activity during chronic neuroleptic treatment.
It could be expected that decreased decarboxylation of FDOPA should predict decreased synthesis and turnover of endogenous DA in the brain. Concentrations of the DA metabolite homovanillic acid (HVA) measured in biological fluids have often served as an index of cerebral DA catabolism in humans. Thus, the baseline plasma HVA concentration and the initial increase occurring upon initiation of neuroleptic treatment both correlate with treatment response (Chang et al, 1988; Davila et al, 1988). Plasma HVA concentrations decrease with treatment in responders, whereas they are essentially unchanged in nonresponders (Bowers et al, 1984). Patients with predominantly negative schizophrenic symptoms show a lower initial increase in HVA concentrations and a flat subsequent decrease (Davila et al, 1988). However, much of the plasma HVA is actually derived from the synthesis in catecholamines in sympathetic organs, and so is an imperfect index of central dopaminergic transmission (Amin et al, 1992).
Although left–right asymmetry in the baseline magnitude of kD3 did not reach significance, the decline in the magnitude of kD3 after haloperidol was significantly greater in the left caudate ( Table 1). Similarly, Hietala et al (1999) reported the presence of asymmetry in the FDOPA influx to caudate in normals, with the greater influx to the right side; this asymmetry was absent in untreated schizophrenic patients. Together, these findings suggest the presence of considerable asymmetry in the uptake of FDOPA is characteristic of caudate in normal subjects, and that haloperidol treatment preferentially reduced the FDOPA consumption on the abnormal left side of patients with schizophrenia ( Table 1). In a meta-analysis of numerous [123I]iodobenzamide/SPECT studies, higher D2 binding in the right striatum of healthy subjects is consistently reported (Larisch et al, 1998). Altered asymmetry in pre- and postsynaptic markers of DA might underlie the left-turning bias reported in chronic schizophrenic patients (Bracha et al, 1993; Lyon and Satz, 1991). While healthy control subjects consistently turn right to collect coin reinforcers, schizophrenic patients tend to turn left. However, this phenomenon has been attributed to a dopaminergic overactivity in the right hemisphere striatal or mesoprefrontal areas (Bracha et al, 1993). A correlation between excess [123I]IBZM binding in the left basal ganglia of schizophrenic patients was also demonstrated to correlate with stereotyped behavior (Pedro et al, 1994). However, with regard to the structure of the basal ganglia in untreated patients with schizophrenia, there are several reports showing either no change or decreases in volume, but studies demonstrating structural asymmetry in these structures are lacking (Corson et al, 1999; Gunduz et al, 2002; Keshavan et al, 1998; Shihabuddin et al, 1998,2001). Interestingly, Scheepers et al found a reduction in left caudate nucleus volume in patients who responded to clozapine treatment, but not in nonresponders (Scheepers et al, 2001).
We find that chronic haloperidol also decreased the magnitude of kD3 in the thalamus. The thalamus is the only structure in which the change of kD3 is related to the change in psychopathology. We find a very marked positive correlation between change in kD3 and improvement in negative symptoms; that is, patients with the strongest decrease in thalamic kD3 demonstrated the most pronounced decrease in the PANSS negative symptoms subscore. The thalamus seems to play a crucial role in the pathophysiology of schizophrenic symptomatology and is probably also an important site through which the clinical effects of antipsychotics are mediated (Carlsson et al, 1999b). It is an essential structure for controlling arousal and the transmission of sensory input—both from the outside world as well as from internal states of the body—to the cerebral cortex (Carlsson et al, 1999a). Thalamic volume has been reported to be reduced in most but not all of the available studies of schizophrenia, but an asymmetry in structure has not been shown (Andreasen et al, 1994; Arciniegas et al, 1999; Hazlett et al, 1999; Wolkin et al, 1998). The thalamus has a low, but significant dopaminergic innervation (Bouthenet et al, 1987; Farde et al, 1997). DA transmission in the ventral thalamus has been implicated in the mechanism for prepulse inhibition of the startle reflex (Young et al, 1995), which is abnormal in schizophrenia (Kumari et al, 2000). Moreover, the concentration of DA was elevated in subregions of the thalamus from patients dying with schizophrenia (Oke and Adams, 1987). However, little is known about the origin and function of dopaminergic projections to the thalamus. The present observations are suggestive that inhibition of catecholaminergic transmission in parts of the thalamus may mediate aspects of therapeutic response to neuroleptics. However, at present it can only be a smaller of speculation about the functional relation between thalamic DA metabolism and the negative symptoms of schizophrenia. Studies in larger patient samples are necessary to relate presynaptic dopaminergic function to psychopathology.
In an earlier PET/FDOPA study, we have reported that the NMDA antagonist amantadine potentiates DA synthesis capacity in the basal ganglia of normal humans (Deep et al, 1999). Our present findings could represent a linkage of therapeutic response to neuroleptics with reduced DA synthesis, consistent with the development of a drug-induced depolarization block of presynaptic DA neurons in human patients with schizophrenia. This lends support to Grace's hypothesis that an antipsychotic drug-induced blockade of DA synthesis and release need not necessarily lead to decreased extracellular DA levels, but instead serves to suppress the abnormal phasic DA release in response to external excitatory stimuli (Grace, 1992). Taken together, these results show that psychotic symptoms reflect increased responsiveness to DA, due in part to an elevated rate of DA synthesis that can be rectified by neuroleptic-induced depolarization block of DA neurons.
References
Amin F, Davidson M, Davis KL (1992). Homovanillic acid measurement in clinical research: a review of methodology. Schizophr Bull 18: 123–148.
Andreasen NC, Arndt S, Swayze II V, Cizadlo T, Flaum M, O'Leary D et al (1994). Thalamic abnormalities in schizophrenia visualized through magnetic resonance image averaging. Science 266: 294–298.
Arciniegas D, Rojas DC, Teale P, Sheeder J, Sandberg E, Reite M (1999). The thalamus and the schizophrenia phenotype: failure to replicate reduced volume. Biol Psychiatry 45: 1329–1335.
Barnes TR (1989). A rating scale for drug-induced akathisia. Br J Psychiatry 154: 672–676.
Bouthenet ML, Martres MP, Sales N, Schwartz JC (1987). A detailed mapping of dopamine D-2 receptors in rat central nervous system by autoradiography with [125I]iodosulpride. Neuroscience 20: 117–155.
Bowers Jr MB, Swigar ME, Jatlow PI, Goicoechea N (1984). Plasma catecholamine metabolites and early response to haloperidol. J Clin Psychiatry 45: 248–251.
Bracha HS, Livingston RL, Clothier J, Linington BB, Karson CN (1993). Correlation of severity of psychiatric patients' delusions with right hemispatial inattention (left-turning behavior). Am J Psychiatry 150: 330–332.
Carlsson A (1988). The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1: 179–186.
Carlsson A, Hansson LO, Waters N, Carlsson ML (1999a). A gluatamatergic deficiency model of schizophrenia. Br J Psychiatry 174 (Suppl 37): 2–6.
Carlsson A, Lindqvist M (1963). Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol 20: 140–144.
Carlsson A, Waters N, Carlsson ML (1999b). Neurotransmitter interactions in schizophrenia—therapeutic implications. Biol Psychiatry 46: 1388–1395.
Chang WH, Chen TY, Lee CF, Hung JC, Hu WH, Yeh EK (1988). Plasma homovanillic acid levels and subtyping of schizophrenia. Psychiatry Res 23: 239–244.
Chiodo LA, Bunney BS (1983). Typical and atypical neuroleptics: differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J Neurosci 3: 1607–1619.
Chouinard G, Ross-Chouinard A, Annable L, Jones BD (1980). The extrapyramidal symptom rating scale. Can J Neurol Sci 7: 233.
Corson PW, Nopoulos P, Andreasen NC, Heckel D, Arndt S (1999). Caudate size in first-episode neuroleptic-naive schizophrenic patients measured using an artificial neural network. Biol Psychiatry 46: 712–720.
Cumming P, Ase A, Laliberte C, Kuwabara H, Gjedde A (1997). In vivo regulation of DOPA decarboxylase by dopamine receptors in rat brain. J Cereb Blood Flow Metab 17: 1254–1260.
Cumming P, Gjedde A (1998). Compartmental analysis of dopa decarboxylation in living brain from dynamic positron emission tomograms. Synapse 29: 37–61.
Cumming P, Léger GC, Kuwabara H, Gjedde A (1993). Pharmacokinetics of plasma 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine ([18F]Fdopa) in humans. J Cereb Blood Flow Metab 13: 668–675.
Cumming P, Yokoi F, Chen A, Deep P, Dagher A, Reutens D et al (1999). Pharmacokinetics of radiotracers in human plasma during positron emission tomography. Synapse 34: 124–134.
Danielsen EH, Smith D, Hermansen F, Gjedde A, Cumming P (2001). Acute neuroleptic stimulates DOPA decarboxylase in porcine brain in vivo. Synapse 41: 172–175.
Davila R, Manero E, Zumarraga M, Andia I, Schweitzer JW, Friedhoff AJ (1988). Plasma homovanillic acid as a predictor of response to neuroleptics. Arch Gen Psychiatry 45: 564–567.
Deep P, Dagher A, Sadikot A, Gjedde A, Cumming P (1999). Stimulation of dopa decarboxylase activity in striatum of healthy human brain secondary to NMDA receptor antagonism with a low dose of amantadine. Synapse 34: 313–318.
Farde L, Nordström AL, Wiesel FA, Pauli S, Halldin C, Sedvall G (1992). Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry 49: 538–544.
Farde L, Suhara T, Nyberg S, Karlsson P, Nakashima Y, Hietala J et al (1997). A PET-study of [11C]FLB 457 binding to extrastriatal D2-dopamine receptors in healthy subjects and antipsychotic drug-treated patients. Psychopharmacology (Berl) 133: 396–404.
Gjedde A, Léger GC, Cumming P, Yasuhara Y, Evans AC, Guttman M et al (1993). Striatal L-dopa decarboxylase activity in Parkinson's disease in vivo: implications for the regulation of dopamine synthesis. J Neurochem 61: 1538–1541.
Gjedde A, Reith J, Dyve S, Léger G, Guttman M, Diksic M et al (1991). Dopa decarboxylase activity of the living human brain. Proc Natl Acad Sci USA 88: 2721–2725.
Grace AA (1992). The depolarization block hypothesis of neuroleptic action: implications for the etiology and treatment of schizophrenia. J Neural Transm 36 (Suppl): 91–131.
Grace AA, Bunney BS (1986). Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. J Pharmacol Exp Ther 238: 1092–1100.
Gunduz H, Wu H, Ashtari M, Bogerts B, Crandall D, Robinson DG et al (2002). Basal ganglia volumes in first-episode schizophrenia and healthy comparison subjects. Biol Psychiatry 51: 801–808.
Guy W (1976). Clinical global impressions CGI. In: Guy W (ed). ECDEU Assessment Manual for Psychopharmacology, Revised. DHHS Publication: Rockville, MD.
Hazlett EA, Buchsbaum MS, Byne W, Wei TC, Spiegel-Cohen J, Geneve C et al (1999). Three-dimensional analysis with MRI and PET of the size, shape, and function of the thalamus in the schizophrenia spectrum. Am J Psychiatry 156: 1190–1199.
Hietala J, Syvalahti E, Vilkman H, Vuorio K, Rakkolainen V, Bergman J et al (1999). Depressive symptoms and presynaptic dopamine function in neuroleptic-naive schizophrenia. Schizophr Res 35: 41–50.
Hietala J, Syvalahti E, Vuorio K, Rakkolainen V, Bergman J, Haaparanta M et al (1995). Presynaptic dopamine function in striatum of neuroleptic-naive schizophrenic patients. Lancet 346: 1130–1131.
Huang SC, Yu DC, Barrio JR, Grafton S, Melega WP, Hoffman JM et al (1991). Kinetics and modeling of L-6-[18F]fluoro-dopa in human positron emission tomographic studies. J Cereb Blood Flow Metab 11: 898–913.
Johnstone EC, Crow TJ, Frith CD, Carney MW, Price JS (1978). Mechanism of the antipsychotic effect in the treatment of acute schizophrenia. Lancet 1: 848–851.
Kay SR, Fiszbein A, Opler LA (1987). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13: 261–276.
Keshavan MS, Rosenberg D, Sweeney JA, Pettegrew JW (1998). Decreased caudate volume in neuroleptic-naive psychotic patients. Am J Psychiatry 155: 774–778.
Kumari V, Soni W, Mathew VM, Sharma T (2000). Prepulse inhibition of the startle response in men with schizophrenia: effects of age of onset of illness, symptoms, and medication. Arch Gen Psychiatry 57: 609–614.
Larisch R, Meyer W, Klimke A, Kehren F, Vosberg H, Müller-Gartner HW (1998). Left–right asymmetry of striatal dopamine D2 receptors. Nucl Med Commun 19: 781–787.
Laruelle M, Abi Dargham A, Gil R, Kegeles L, Innis R (1999). Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry 46: 56–72.
Léger G, Gjedde A, Kuwabara H, Guttman M, Cumming P (1998). Effect of catechol–methyltransferase inhibition on brain uptake of [18F]fluorodopa: implications for compartmental modelling and clinical usefulness. Synapse 30: 351–361.
Lindström LH, Gefvert O, Hagberg G, Lundberg T, Bergström M, Hartvig P et al (1999). Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(β-11C) DOPA and PET. Biol Psychiatry 46: 681–688.
Lyon N, Satz P (1991). Left turning (swivel) in medicated chronic schizophrenic patients. Schizophr Res 4: 53–58.
Medoff DR, Holcomb HH, Lahti AC, Tamminga CA (2001). Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus 11: 543–550.
Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M et al (2002). Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci 5: 267–271.
Miller DD, Andreasen NC, O'Leary DS, Watkins GL, BolesPonto LL, Hichwa RD (2001). Comparison of the effects of risperidone and haloperidol on regional cerebral blood flow in schizophrenia. Biol Psychiatry 49: 704–715.
Miller DD, Rezai K, Alliger R, Andreasen NC (1997). The effect of antipsychotic medication on relative cerebral blood perfusion in schizophrenia: assessment with technetium-99m hexamethyl-propyleneamine oxime single photon emission computed tomography. Biol Psychiatry 41: 550–559.
Nordström AL, Farde L, Halldin C (1992). Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology (Berl) 106: 433–438.
Oke AF, Adams RN (1987). Elevated thalamic dopamine: possible link to sensory dysfunctions in schizophrenia. Schizophr Bull 13: 589–604.
Pedro BM, Pilowsky LS, Costa DC, Hemsley DR, Ell PJ, Verhoeff NP et al (1994). Stereotypy, schizophrenia and dopamine D2 receptor binding in the basal ganglia. Psychol Med 24: 423–429.
Reith J, Benkelfat C, Sherwin A, Yasuhara Y, Kuwabara H, Andermann F et al (1994). Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci USA 91: 11651–11654.
Roth RH, Elsworth JD (1995). Biochemical pharmacology of midbrain dopamine neurons. In: Bloom FE and Kupfer DJ (eds). Psychopharmacology: The Fourth Generation of Progress. Raven: New York. pp 227–243.
Scheepers FE, Gispen de Wied CC, Hulshoff Pol HE, Kahn RS (2001). Effect of clozapine on caudate nucleus volume in relation to symptoms of schizophrenia. Am J Psychiatry 158: 644–646.
Seeman P, Lee T (1975). Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188: 1217–1219.
Shihabuddin L, Buchsbaum MS, Hazlett EA, Silverman J, New A, Brickman AM et al (2001). Striatal size and relative glucose metabolic rate in schizotypal personality disorder and schizophrenia. Arch Gen Psychiatry 58: 877–884.
Shihabuddin I, Shihabuddin L, Buchsbaum MS, Hazlett EA, Haznedar MM, Harvey PD et al (1998). Dorsal striatal size, shape, and metabolic rate in never-medicated and previously medicated schizophrenics performing a verbal learning task. Arch Gen Psychiatry 55: 235–243.
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD et al (1977). The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28: 897–916.
Stout DB, Huang SC, Melega WP, Raleigh MJ, Phelps ME, Barrio JR (1998). Effects of large neutral amino acid concentrations on 6-[F-18]Fluoro-L-DOPA kinetics. J Cereb Blood Flow Metab 18: 43–51.
White FJ, Wang RY (1983). Differential effects of classical and atypical antipsychotic drugs on A9 and A10 dopamine neurons. Science 221: 1054–1057.
Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue CY et al (1989). Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry 146: 905–908.
Wolkin A, Rusinek H, Vaid G, Arena L, Lafargue T, Sanfilipo M et al (1998). Structural magnetic resonance image averaging in schizophrenia. Am J Psychiatry 155: 1064–1073.
Woods RP, Cherry SR, Mazziotta JC (1992). Rapid automated algorithm for aligning and reslicing PET images. J Comput Assist Tomogr 16: 620–633.
Yatham LN, Liddle PF, Shiah IS, Lam RW, Ngan E, Scarrow G et al (2002). PET study of [18F]6-Fluoro-L-Dopa uptake in neuroleptic- and mood-stabilizer-naive first-episode nonpsychotic mania: effects of treatment with divalproex sodium. Am J Psychiatry 159: 768–774.
Young KA, Randall PK, Wilcox RE (1995). Startle and sensorimotor correlates of ventral thalamic dopamine and GABA in rodents. Neuroreport 15: 2495–2499.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gründer, G., Vernaleken, I., Müller, M. et al. Subchronic Haloperidol Downregulates Dopamine Synthesis Capacity in the Brain of Schizophrenic Patients In Vivo. Neuropsychopharmacol 28, 787–794 (2003). https://doi.org/10.1038/sj.npp.1300103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300103
Keywords
This article is cited by
-
The relationship between striatal dopamine and anterior cingulate glutamate in first episode psychosis changes with antipsychotic treatment
Translational Psychiatry (2023)
-
Striatal dopaminergic alterations in individuals with copy number variants at the 22q11.2 genetic locus and their implications for psychosis risk: a [18F]-DOPA PET study
Molecular Psychiatry (2023)
-
Prospective F-18 FDOPA PET Imaging Study in Human PD
Nuclear Medicine and Molecular Imaging (2022)
-
The relationship between grey matter volume and striatal dopamine function in psychosis: a multimodal 18F-DOPA PET and voxel-based morphometry study
Molecular Psychiatry (2021)
-
Dopamine dysregulation in psychotic relapse after antipsychotic discontinuation: an [18F]DOPA and [11C]raclopride PET study in first-episode psychosis
Molecular Psychiatry (2021)
