
Abstract
The common variants in lysyl oxidase-like 1 gene (LOXL1) are associated with exfoliation glaucoma (XFG) patients developed through exfoliation syndrome (XFS). However, the risk allele of a variant in LOXL1 has been found to be inverted between Asian and Caucasian populations. Therefore, we newly performed a genome-wide association study using 201 XFS/XFG and 697 controls in Japanese and identified 34 genome-wide significant single-nucleotide polymorphisms (SNPs) distributing in not only LOXL1 but also TBC1D21 and PML at the 15q24.1 locus. These SNPs were confirmed by an independent population consisted of 121 XFS/XFG and 263 controls in Japanese. Moreover, further analyses revealed a unique haplotype structure only from the combination of TBC1D21 and LOXL1 variants showing a high XFS/XFG susceptibility specific for the Asian population. Although there still should be other gene(s) in the other region(s) contributing to the disease process, these results suggested that the combination of newly discovered variants in these genes might be useful for precise XFG risk assessment, as well as for elucidating the molecular mechanism of XFG pathogenesis through XFS.
Similar content being viewed by others


Introduction
Glaucoma is one of the leading causes of irreversible blindness worldwide1. It is a complex age-related disorder consisting of heterogeneous subtypes that share common features for the development of the disease, i.e., progressive loss of retinal ganglion cells and optic nerve axons that result in visual field defects2. Therefore, it is important to investigate each distinct type of glaucoma in order to elucidate the different molecular pathways that lead to the common optic-nerve degeneration.
Exfoliation glaucoma (XFG) is categorized as secondary open-angle glaucoma and reportedly develops from exfoliation syndrome (XFS), as manifested by an observation of abnormal fibrillar deposits (known as pseudoexfoliation (PEX)) on the lens and iris epithelium3. In general, XFG has a worse prognosis than primary open-angle glaucoma (POAG), the major type of glaucoma, as it is often observed in patients as elevated intraocular pressure (IOP) combined with severe damage of visual function at the time of initial presentation due to the asymptomatic nature of glaucoma. Moreover, according to the findings reported in the Tajima Study4, a robust epidemiology study conducted in Japan, XFG accounts for 0.8% of the 5.0% of all types of glaucoma observed in Japanese people over 40 years of age and the prevalence of XFG increases with advancing age. Consequently, it would be of enormous benefit if the predisposing genetic factors of XFG could be used to predict the risk of developing the disease prior to onset in order to stop, or at least hinder, the irreversible progression toward blindness by initiating early medical intervention.
In 2007, Thorleifsson et al. reported a genome-wide association study (GWAS) using the populations from Iceland and Sweden and described that one intronic variant and two nonsynonymous coding variants of lysyl oxidase-like 1 gene (LOXL1) confer the risk of developing XFG, possibly through XFS5. From the functional point of view, the involvement of LOXL1 in XFG pathogenesis is quite rational, because LOXL1 plays an indispensable role in elastic fiber homeostasis and maintaining the structures of the trabecular meshwork, the tissue that determines the IOP level in the eye6. However, LOXL1, in and of itself, fails to sufficiently explain the complex mechanism of the disease progression of XFG through XFS. This observation is also supported from the point of view of genetics, as many replication studies using populations derived from different genetic backgrounds (Caucasians from the US7,8,9 and Europe10,11, Asians from Japan12,13,14,15,16 and China17,18 and Africans from South Africa19) have succeeded in replicating the association of the LOXL1 variants, although the allele frequency of the variants varied among the ethnicities. In fact, the risk allele of one of the exonic variants (rs1048661) has been found to be inverted between Japanese12 and Nordic populations5. Consequently, it is wise to investigate other modifying or causative variants in genes other than LOXL1 by performing a GWAS using a non-Caucasian population in order to identify variants that are determining the different etiology of XFS onset and its progression to XFG among the ethnicities.
In this present study, we conducted a GWAS and replication study using two independent populations of Japanese case-control subjects in an attempt to identify new variants associated with XFS/XFG. As a result, we identified a cluster of genome-wide significant SNPs only from the chromosome 15q24.1, which is well known as a locus including LOXL15. However, the genome-wide significant SNPs were distributed in not only LOXL1, but also in the two adjacent genes, TBC1D21 and PML, without being in significant linkage disequilibrium (LD). We also identified a suggestive association only between TBC1D21 and LOXL1 variants by a conditional analysis and revealed a unique haplotype structure of the variants derived from these two genes in our study population. Together with the accumulating knowledge of LOXL1-XFS/XFG relationships, the variants and genes newly discovered in this study would help to provide a better prediction of XFG risk assessment, as well as to elucidate the molecular mechanism of XFG pathogenesis through XFS.
Results
GWAS for XFS/XFG
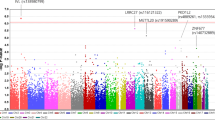
The samples used in this study are summarized in Table 1. In order to perform GWAS, we first selected the XFS/XFG subjects based on our strict diagnosis criteria and then obtained the genotype data. We performed a GWAS using a population consisting of 201 XFS/XFG patients and 697 healthy controls (Table 1). In total, 652,792 SNPs that passed the quality controls (QC) were used for the association study (Figure 1A). According to the quantile-quantile (Q-Q) plot (Supplementary Material, Figure S1), the genomic inflation factor (λ) showed 1.020, suggesting that the population substructure should not have any substantial effects on the association analysis. Under those conditions, we observed a strong association signal from a locus on chromosome 15 (Figure 2A). In fact, we obtained 34 genome-wide significant SNPs (Supplementary Material, Table S1) that passed the Bonferroni correction threshold (0.05/652,792 = 7.66 × 10−8) on chromosome 15q24.1 (Figure 2A). Surprisingly, these significant SNPs were found to be broadly distributed in not only LOXL1, but also the adjacent two genes, TBC1D21 and PML. The significance for this region was also confirmed by an imputation analysis (Figure 2B). According to the LD plots (Supplementary Material, Figure S2A), the SNPs in each gene seemed to be derived from 3 distinct LD blocks, which was also confirmed by the LD values using 34 genome-wide significant SNPs (Supplementary Material, Table S2).

Study design.
(A) First, a GWAS was performed and 652,792 SNPs were analyzed for 201 XFS/XFG patients and 697 controls. As a result, 34 genome-wide significant SNPs were obtained from the genes of TBC1D21, LOXL1 and PML at15q24.1 locus. (B) Next, the GWAS results were replicated by analyzing 43 SNPs from the 15q24.1 locus by using an independent population of 121 XFS/XFG patients and 263 controls. (C) Finally, a meta-analysis was performed by combining the two data sets by use of the Mantel-Haenszel test. In addition, the pairs of significant SNPs were assessed by conditional and haplotype analyses. The result suggested the susceptible haplotype might exist for XFS/XFG from TBC1D21 and LOXL1 variants.

Association results.
(A) The SNPs with a strong association signal from the GWAS result appeared to exist as a cluster on chromosome 15 (red arrow). Horizontal dotted line represents Bonferroni correction threshold (P < 7.66 × 10−8). (B) Because the genome-wide significant SNPs were intensively identified on the 15q24.1 locus, we also imputed the SNPs across the locus and plotted against the positions based on the NCBI Build 36 coordinates. The LD value of r2 is referred to the most significant SNP (rs893818) in the GWAS. The cSNP (rs16958445) and two genes (TBC1D21 and LOXL1) used in the conditional and haplotype association analyses are highlighted by red. The genetic recombination rates (cM/Mb) estimated by HapMap Project31 in Release 22 are indicated by the thin blue line.
Replication study
Next, we attempted to replicate the GWAS results by analyzing an independent Japanese population consisting of 121 XFS/XFG patients and 263 controls (Table 1). We assessed a total of 43 SNPs at 15q24.1 locus, including 34 genome-wide significant SNPs from our GWAS results and 2 nonsynonymous coding SNPs (cSNPs) (rs1048661 and rs3825942) in exon 1 of LOXL1 that reportedly are strongly associated with XFS/XFG5. As a result, almost every genome-wide significant SNP, except 1 SNP (rs723434), passed the Bonferroni correction threshold in the replication study (0.05/43 = 1.16 × 10−3) (Supplementary Material, Figure S2B and Table S3), confirming the association results obtained by GWAS. We also confirmed the association result of the reported 2 cSNPs in LOXL1 using our Japanese population, of which the risk allele of rs1048661 was indeed inverted from the reported result with Caucasian. When the combined P values of GWAS and the replication study were calculated by use of the Mantel-Haenszel test20, the level of significance of almost every SNP increased. In fact, rs893818, which was the most significant SNP in intron 1 of LOXL1, reached to P = 8.21 × 10−84 (Supplementary Material, Figures S2C and S2D, Table S4). The rs893818 SNP was clearly independent from the SNPs in TBC1D21 and PML as in reference to the LD values calculated by applying the whole samples used in this study (Supplementary Material, Table S2). Moreover, the LD blocks based on the whole samples demonstrated that LOXL1, TBC1D21 and PML were in 3 distinct LD blocks, suggesting that the variants from different genes were independently contributing to the disease (Supplementary Material, Figures S2E and S2F).
Meta-analysis of two studies
We then performed a conditional analysis for 34 genome-wide significant SNPs on the basis of rs893818 according to our GWAS result. Overall, it turned out that the effect of rs893818 was very strong, while the variants in TBC1D21 and PML were generally weak, although we found a suggestive signal (P = 0.027) from a nonsynonymous variant in exon 4 of TBC1D21 (rs16958445) (Supplementary Material, Table S5). These two SNPs were considered to be truly associated with XFS/XFG because there were no confounding factors such as age and gender (Supplementary Material, Table S6). Consequently, we obtained 3 susceptible cSNPs, including 2 reported cSNPs in LOXL1, showing a weak correlation (r2 < 0.2) to each other (Supplementary Material, Figures S3). Therefore, we then evaluated the combinational effect of 3 cSNPs (rs16958445 from TBC1D21 and rs1048661 and rs3825942 from LOXL1) in order to analyze the susceptibility to XFS/XFG by performing a haplotypic association analysis with the samples used in the replication study (Table 2). As a result, haplotype H6 (rs16958445: G, rs1048661: T and rs3825942: G) was most significant for the association with XFS/XFG. Interestingly, haplotype H6 was more significant than haplotypes H3 and H4, including the significant haplotype (rs1048661: G and rs3825942: G) in the LOXL1 for the Caucasian population5. In addition, according to the dbSNP database, rs16958445 in TBC1D21 was found to be monomorphic for the G allele in Caucasian and African populations. Consequently, we obtained a unique haplotype structure in this present study showing a high XFS/XFG susceptibility specific for the Asian population.
Taken together, the newly identified genes in 15q24.1 locus, together with the unique haplotype of TBC1D21 and LOXL1 variants, could add some information to explain the molecular mechanism of XFS/XFG pathogenesis, which has been considered difficult to explain by LOXL1 alone.
Discussion
In this study, we successfully obtained the genetic markers strongly associated with XFS/XFG patients by analyzing two independent Japanese populations totaling 1,282 subjects from GWAS and its replication study. As a result, we discovered the genome-wide significant variants in not only LOXL1, but also 2 other genes, TBC1D21 and PML, both of which are located on chromosome 15q24.1, which is well known as the “LOXL1 locus”5. We also found a suggestive combinational effect of the variants related to the XFS/XFG susceptibility only between LOXL1 and TBC1D21, which seemed to be specific to Asian population.
In the first reported GWAS for XFS/XFG, Thorleifsson et al. detected a strong association signal only in LOXL1 at the 15q24.1 locus from a Nordic population5. Their GWAS was carried out based on the genotype data generated by using the Infinium HumanHap300 BeadChip (Illumina, Inc., San Diego, California, USA) array, whereas we used the Genome-Wide Human SNP Array 6.0 (SNP 6.0; Affymetrix, Inc., Santa Clara, California, USA), in which the SNP coverage is more than three-fold. We precisely compared the probe disposition derived from each microarray as well as the association results from the two studies across the locus (Supplementary Material, Figure S4A and B). Although the number of QC-passed SNPs in the locus was different in the two studies depending on which microarrays and sample ethnicities were used, we were able to directly compare the results for some SNPs (Supplementary Material, Figure S4A; connected with green lines). As a result, a genome-wide significant SNP in LOXL1 was reproducible in both studies. However, the genome-wide significant SNPs in other genes at the 15q24.1 locus, especially in TBC1D21 (Supplementary Figure S4B), obtained in our study using Japanese samples were not significant in the first reported GWAS using the Caucasian samples. Moreover, we found that 1 of the significant SNPs in TBC1D21, rs16958445, which together with 2 cSNPs in LOXL1 seemed to be contributing to the disease according to the results of the conditional analysis, was a monomorphic SNP in all of the HapMap populations other than Asian (see dbSNP in NCBI).
With regard to rs16958445, the non-risk allele “A” was a minor allele, whose frequency was about 0.144–0.157 or 0.166 in Japanese, according to the dbSNP or our control samples, respectively (Supplementary Material, Table S4). This meant that the contribution of rs16958445 to the XFS/XFG susceptibility might be limited for explaining the different susceptibility among the ethnicities. However, the results of the haplotype association analysis by 3 cSNPs (H6: GTG in Table 2) demonstrated that the significance was higher than that by use of only 2 cSNPs in LOXL1 (Supplementary Material, H4: TG in Table S7). In addition, the non-risk allele of rs16958445 decreased the significance of the major susceptible haplotype in Caucasians (Supplementary Material, H3: GG in Table S7). Taken all together, we concluded that the discrepancy of the results from the two studies was due to the different genetic backgrounds of the samples and not due to practical reasons such as the microarray density.
The association results of LOXL1 variants found in the Nordic population has been extensively characterized in many other populations with different ethnic backgrounds. In particular, out of the three variants reported, two coding variants of rs1048661 (Arg141Leu) and rs3825942 (Gly153Asp) in exon 1 were mostly analyzed by the replication analysis21 (Supplementary Material, Figure S5). Although both of the variants were found to be associated with the disease broadly beyond the ethnicities, the risk alleles of rs1048661 and rs3825942 were, intriguingly, inverted in Japanese12/Chinese18 and South African19 populations, respectively. The inverted genotypes of the risk alleles among different populations may be reflecting the different patterns of historical recombination between these SNPs and the unidentified causative variants. However, the one thing that is apparent is that the particular amino-acid changes in LOXL1 protein were not functionally sufficient enough to be the causative factor for the disease pathogenesis. Consequently, it would have been wise to investigate other modifying or causative variants in genes other than LOXL1. Thus, we decided to perform a GWAS using our Japanese population in order to identify unique variants contributing to the onset of XFS and its progression to XFG.
To date, only a limited number of disease-associated variants identified by GWAS were applicable to the actual diagnosis in the clinic setting. Thus far, a case in point is the diagnosis that has been applied to predict the risk of age-related macular degeneration (AMD)22,23. In order to predict the prevalence and incidence of AMD, multiple risk models have been developed by incorporating both the genetic and environmental factors, resulting in a marked increase of the accuracy to discriminate risk and non-risk individuals24. However, in the case of XFS/XFG, the situation is completely different from that of AMD, due to the limited number of identified genetic factors as well as their characteristics. Although the prevalence of LOXL1 variants in affected patients is strikingly high (>80%), which means that the sensitivity is high, the prevalence of unaffected controls is also high enough (up to 88%) to render the specificity being low, indicating that the simple allelic testing of LOXL1 variants themselves cannot exclude the individuals who would not develop XFS/XFG21. Although the variants identified in this study seemed to possess a weaker effect on the disease than the LOXL1 variants have, we also discovered a unique haplotype specific for the Japanese population (Table 2). Combining the information of each allele and the haplotypes would help in developing a better diagnosis to predict XFS/XFG risk by the genetic testing of these variants.
Since XFS is characterized by clinically visible abnormal fibrillar deposits (i.e., PEX), progressive accumulation of those deposits have been considered to be a trigger of developing glaucoma by increasing the outflow resistance of the trabecular meshwork, resulting in the continuous elevation of IOP3. The LOXL1 protein, which catalyzes the formation of elastin fibers and contributes to the trabecular meshwork structures, is thus probably involved in the XFS/XFG pathogenesis6. However, the exonic variants in LOXL1 fail to sufficiently explain the complex mechanism of the disease (e.g., as if they were affecting the quality (amino-acid changes) and/or quantity (expression level) of LOXL1 protein) due to the differences in allelic distribution among different genetic backgrounds and the high prevalence of the variants in the healthy population21.
According to our GWAS result, the significant variants for XFS/XFG other than the LOXL1 variants were newly discovered from TBC1D21 and PML genes (Figure 2B, Supplementary Material, Figure S2). In fact, the TBC1D21 gene, which belongs to the TBC1 domain family, was suggested to be related to LOXL1 by haplotype association analysis (Table 2). The TBC (Tre-2/Bub2/Cdc16) domain consists of approximately 200 amino acids and is considered to function as a GTPase activator of Rab proteins. However, the functional details of more than 40 members of the protein families in humans and mice, including TBC1D21, have to date not been well investigated25. Although there are few functional analyses of the gene, our RT-PCR analysis revealed that TBC1D21 is expressed in the human retina (Supplementary Material, Figure S6). When we compared the sequence conservation of the variant (rs16958445) among several species, we found that the variant was conserved within primates (Supplementary Material, Figure S7). We also performed in silico prediction in order to evaluate the influence of amino acid change in rs16958445 by means of SIFT26 and PolyPhen-227 and programs predicted as “TOLERATED” and “benign”, respectively. The results suggested that the amino acid change in rs16958445 does not affect the function of TBC1D21 protein. Consequently, further functional investigation is necessary to elucidate whether or not TBC1D21 involves in the disease etiology of XFS/XFG.
On the other hand, PML is functionally well characterized and would be a good target for analyzing the relationship to XFS/XFG etiology, although the contribution to XFS/XFG susceptibility was considered to be marginal than TBC1D21 and LOXL1 based on our analysis. PML is a member of the tripartite motif (TRIM) family and famous for being involved in generating fusion protein as a result of chromosomal translocation with the retinoic acid receptor alpha gene, which is often associated with acute promyelocytic leukemia28. The original function of PML protein is to form a nuclear body, which carries several components, such as pRB, p53, CBP, elf-4, Daxx, SUMO-1, etc29. Therefore, if the variants in PML cause inappropriate formation of the nuclear body, broad biological functions would be affected, including transcriptional regulation, initiation of translation and sumoylation, which could associate with the elastic microfibrillopathy. In fact, our RT-PCR analysis shows that PML is expressed in the human retina (Supplementary Material, Figure S6). Consequently, we consider that it would be interesting to further investigate the involvement of PML in the XFS/XFG pathogenesis.
Although we succeeded in obtaining some new variants and genes associated with XFS/XFG, it should be noted that there must be other gene(s) in other region(s) contributing to the disease in order to explain the complex mechanism of XFS/XFG pathogenesis. Moreover, under the current GWAS condition, we are unable to verify whether or not the variants were specifically related to the mechanism for the progression to glaucoma from XFS. Since there are patients who possess PEX, but never develop glaucoma, other factors must be involved in determining whether or not XFS patients develop glaucoma. Together with the variants/genes identified in this study and the accumulated knowledge of LOXL1, the identification of the remaining variants as well as the variants specific for XFS patients without glaucoma could provide a complete set of genetic factors associated with XFS and XFG, which should be useful for their diagnostic tools as well as for revealing the molecular mechanism of their etiology.
Methods
Subjects
All of the procedures in this study were performed in accordance with the tenets set forth in the Declaration of Helsinki and were approved by the Institutional Review Board of Kyoto Prefectural University of Medicine, Kyoto, Japan. Written informed consent was obtained from all XFS/XFG participants after receiving a detailed and thorough explanation of the study. Those participants were interviewed to determine their familial history of glaucoma and the medical histories of other ocular or general diseases. The Japanese volunteers were recruited between March 2005 and August 2012 at the University Hospital of Kyoto Prefectural University of Medicine in order to provide peripheral blood samples. The genotyping data of this current study for XFS/XFG patients and healthy volunteers was simultaneously obtained with the data of our previous study for POAG30. From the complete data set, XFS/XFG cases and controls suitable for this study were selectively chosen from the complete data set based on the following strict diagnosis: 1) all XFG patients were diagnosed by slit-lamp examination for the existence of exfoliation materials on the pupil and surface anterior lens capsule with open angle, 2) IOP higher than 21 mmHg and 3) visual field defect according to the optic nerve appearance. The category “XFG with other glaucoma” group includes normal tension glaucoma or primary angle closure glaucoma with XFS. All of the patients and healthy volunteers were diagnosed by 3 ophthalmologists (Y.I., M.U. and K.M.) from a single institution. The demographics (i.e., subject age and male-to-female ratio) of all of the subjects used in this study are shown in Table 1. To examine the possible confounding effects of the age and gender of the subjects, correlations between the case and control samples were assessed by use of the Student's t test or chi-square test (Table 1).
SNP genotyping and quality control for the GWAS
As described previously30, 906,600 SNPs were genotyped for 2,126 Japanese subjects including POAG patients, XFS/XFG patients and healthy volunteers by Genome-Wide Human SNP Array 6.0 (Affymetrix) and strict QC was applied. For the GWAS, 203 XFS/XFG patients and 718 controls were initially selected from the total number of subjects based on the latest diagnosis. In order to exclude subjects within the population who were genetically related, identity-by-descent estimates were performed for all possible combinations by PLINK v1.07 (see URLs). As a result, 2 XFS/XFG patients and 21 controls who were assumed to be genetically related were excluded from the GWAS population. Ultimately, the population ended up with 201 XFS/XFG patients and 697 controls (Figure 1A and Table 1). Population stratification was examined by principal component analysis using EIGENSTRAT software v3.0 (see URLs). The four HapMap populations (CEU, YRI, JPT and CHB) were simultaneously applied into EIGENSTRAT as the reference. The generated cluster plots indicated that the XFG and control population were genetically clustered within the JPT population and there was no outlier sample (Supplementary Material, Figure S8). In order to extract high-quality autosomal SNP genotype data, the following QC filters were applied: (i) call rate per SNPs in each case and control samples ≥95%, respectively, (ii) minor allele frequency (MAF) in case and control samples ≥1% and (iii) Hardy-Weinberg equilibrium (HWE) in control samples with a P-value ≥0.001. Consequently, the remaining 652,792 SNPs were analyzed for the XFS/XFG GWAS (Figure 1A).
Replication study
For the replication study, an independent population of 384 samples consisting of 121 XFS/XFG patients and 263 controls was prepared (Table 1). Those samples were extensively checked in order to remove any familial relationships. In total, 41 SNPs were applied to the replication by referencing the GWAS results as follows: 34 SNPs that passed the Bonferroni's correction in our GWAS and 7 SNPs which were selected to be useful for confirming the LD region of 15q24.1 locus. In addition, 2 cSNPs (rs1048661 and rs3825942) were assessed that have been reported by Thorleifsson et al.5, yet are not designed in SNP 6.0. Furthermore, 4 SNPs associated with POAG in our previous study30 were included in this replication assay. In total, 47 SNPs were divided into each of the two groups of 30 and 17 SNPs per assay plate, respectively, according to the manufacturer's recommendation and genotyped simultaneously for 384 samples in each group by use of the MassARRAY iPLEX Genotyping system (Sequenom, Inc., San Diego, California, USA). All of the resulting call rates of these SNPs were over 90%.
Meta-analysis
A meta-analysis was performed for 41 SNPs, which were genotyped in both the GWAS and replication study, by use of the Mantel-Haenszel test20. Among these SNPs, rs2165241 and rs4243042 were found to be monomorphic for the XFS/XFG subjects used in the replication study (i.e., the risk allele frequency was 100%). Therefore, those 2 SNPs were excluded from the Mantel-Haenszel test. Genotype imputation for GWAS was performed using MACH ver. 1.0.16 software (see URLs). The genotype data of JPT and CHB in the HapMap Phase 231 (release #24) were applied as the reference for haplotyping. After the QC was applied for the imputation results (MAF ≥0.01 and Rsq ≥0.7), association analysis of the SNPs for XFS/XFG was performed by use of mach2dat ver. 1.1.9 software (Windows version). Conditional analysis was assessed for 34 genome-wide significant SNPs using the combined samples from the GWAS and the replication study (i.e., 322 XFS/XFG patients and 960 controls). Haplotype analysis was examined with the samples from the replication study in order to assess the reported cSNPs in LOXL1. Both conditional and haplotype analyses were performed by use of the PLINK v1.07 whole genome association analysis toolset.
Data management and statistical analysis
In order to manage and analyze all of the genotype data, our in-house Genoika Server System30,32,33 was used. The Genoika Server System comes with PLINK, R program with some packages, EIGENSTRAT and Haploview (see URLs for the details of each software) built in and all of the analyses were performed by use of this system. In addition, Microsoft Office Excel 2003 (Microsoft Corporation, Redmond, Washington, USA) was used for preparing the data sets and statistical analysis. The frequency of alleles in the case and control samples was compared by use of the basic allele test. The odds ratio (OR) and the upper and lower limit of the 95% confidence interval (CI) of each SNP were calculated for the allele possessing a higher frequency in the case samples than in the control samples. The HWE was evaluated by use of the chi-square test. A regional plot in Figure 2 was drawn by use of LocusZoom ver. 1.1 (see URLs) genetic analysis software. Q-Q plots were generated by ranking the observed values from minimum to maximum and plotting them against their expected chi-square values using the “snpStats” package ver. 1.10.0 in the R program (see URLs).
URLs
PLINK software v1.07, http://pngu.mgh.harvard.edu/~purcell/plink/; EIGENSTRAT software v3.0, http://genetics.med.harvard.edu/reich/Reich_Lab/Welcome.html; MACH ver. 1.0.16 and mach2dat ver. 1.1.9 softwares, http://www.sph.umich.edu/csg/abecasis/MACH/index.html; Haploview v4.2, http://www.broadinstitute.org/scientific-community/science/programs/medical-and-population-genetics/haploview/haploview; LocusZoom version 1.1, http://csg.sph.umich.edu/locuszoom/. R programs, http://www.r-project.org/; SIFT dbSNP DB (build 132), http://sift.jcvi.org/; PolyPhen-2 version 2.2.2 r398, http://genetics.bwh.harvard.edu/pph2/.
References
Quigley, H. A. & Broman, A. T. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90, 262–7 (2006).
Kwon, Y. H., Fingert, J. H., Kuehn, M. H. & Alward, W. L. Primary open-angle glaucoma. N Engl J Med 360, 1113–24 (2009).
European Glaucoma Society. Terminology and guidelines for glaucoma, 3rd Edition. (Dogma, Savona, Italy, 2008).
Iwase, A. et al. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology 111, 1641–8 (2004).
Thorleifsson, G. et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 317, 1397–400 (2007).
Schlotzer-Schrehardt, U. Molecular pathology of pseudoexfoliation syndrome/glaucoma--new insights from LOXL1 gene associations. Exp Eye Res 88, 776–85 (2009).
Fingert, J. H. et al. LOXL1 mutations are associated with exfoliation syndrome in patients from the midwestern United States. Am J Ophthalmol 144, 974–975 (2007).
Challa, P. et al. Analysis of LOXL1 polymorphisms in a United States population with pseudoexfoliation glaucoma. Mol Vis 14, 146–9 (2008).
Fan, B. J. et al. DNA sequence variants in the LOXL1 gene are associated with pseudoexfoliation glaucoma in a U.S. clinic-based population with broad ethnic diversity. BMC Med Genet 9, 5 (2008).
Pasutto, F. et al. Association of LOXL1 common sequence variants in German and Italian patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci 49, 1459–63 (2008).
Mossbock, G. et al. Lysyl oxidase-like protein 1 (LOXL1) gene polymorphisms and exfoliation glaucoma in a Central European population. Mol Vis 14, 857–61 (2008).
Mori, K. et al. LOXL1 genetic polymorphisms are associated with exfoliation glaucoma in the Japanese population. Mol Vis 14, 1037–40 (2008).
Hayashi, H., Gotoh, N., Ueda, Y., Nakanishi, H. & Yoshimura, N. Lysyl oxidase-like 1 polymorphisms and exfoliation syndrome in the Japanese population. Am J Ophthalmol 145, 582–585 (2008).
Ozaki, M. et al. Association of LOXL1 gene polymorphisms with pseudoexfoliation in the Japanese. Invest Ophthalmol Vis Sci 49, 3976–80 (2008).
Mabuchi, F. et al. Lysyl oxidase-like 1 gene polymorphisms in Japanese patients with primary open angle glaucoma and exfoliation syndrome. Mol Vis 14, 1303–8 (2008).
Fuse, N. et al. Evaluation of LOXL1 polymorphisms in eyes with exfoliation glaucoma in Japanese. Mol Vis 14, 1338–43 (2008).
Lee, K. Y. et al. Association of LOXL1 polymorphisms with pseudoexfoliation in the Chinese. Mol Vis 15, 1120–6 (2009).
Chen, L. et al. Evaluation of LOXL1 polymorphisms in exfoliation syndrome in a Chinese population. Mol Vis 15, 2349–57 (2009).
Williams, S. E. et al. Major LOXL1 risk allele is reversed in exfoliation glaucoma in a black South African population. Mol Vis 16, 705–12 (2010).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22, 719–48 (1959).
Challa, P. Genetics of pseudoexfoliation syndrome. Curr Opin Ophthalmol 20, 88–91 (2009).
Klein, R. J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–9 (2005).
Maller, J. et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet 38, 1055–9 (2006).
Chen, Y., Bedell, M. & Zhang, K. Age-related macular degeneration: genetic and environmental factors of disease. Mol Interv 10, 271–81 (2010).
Fukuda, M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci Rep 31, 159–68 (2011).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–81 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–9 (2010).
de The, H. & Chen, Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer 10, 775–83 (2010).
Zhong, S., Salomoni, P. & Pandolfi, P. P. The transcriptional role of PML and the nuclear body. Nat Cell Biol 2, E85–90 (2000).
Nakano, M. et al. Common variants in CDKN2B-AS1 associated with optic-nerve vulnerability of glaucoma identified by genome-wide association studies in Japanese. PLoS One 7, e33389 (2012).
International HapMap, C. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–61 (2007).
Nakano, M. et al. Three susceptible loci associated with primary open-angle glaucoma identified by genome-wide association study in a Japanese population. Proc Natl Acad Sci U S A 106, 12838–42 (2009).
Ueta, M. et al. Association between prostaglandin E receptor 3 polymorphisms and Stevens-Johnson syndrome identified by means of a genome-wide association study. J Allergy Clin Immunol 126, 1218-25 e10 (2010).
Acknowledgements
The authors wish to thank all the patients and volunteers who enrolled in the study. We also thank Professor T. Miki for managing the anonymous code-assignment process; S. Ohashi, M. Yamashita and N. Saito for processing blood samples and performing genotyping; H. Yamada for the assistance in clinical information analysis; F. Sato for the management of genotype data and building the analyzing system; T. Ichikawa for excellent secretarial assistance; and J. Bush for reviewing the manuscript. This work was supported by the grants from the Takeda Science Foundation to MN, the Collaborative Development of Innovative Seeds of Japan Science and Technology Agency (JST) to MK and KT, the Ministry of Health, Labor and Welfare of Japan to MN, KM, SK and KT and Santen Pharmaceutical Co. Ltd. to SK and KT.
Author information
Authors and Affiliations
Contributions
Designed the research: M.K., S.K. and K.T. Performed the research: M.N., Y.I., M.U., K.I., M.F., N.O., H.A. and K.M. Analyzed the data: M.N., Y.T., R.S. and M.F. Wrote the paper: M.N., Y.I., Y.T., K.M., S.K. and K.T. All authors reviewed the manuscript.
Ethics declarations
Competing interests
This study has been completed under the Collaborative Research Agreement executed by Kyoto Prefectural University of Medicine and Santen Pharmaceutical Co., Ltd. All materials and information produced throughout this study are parts of co-owned intellectual properties. The authors MF and MK are employees of Santen Pharmaceutical Co., Ltd. All the other authors declare that no conflict of interest exists.
Electronic supplementary material
Supplementary Information
Supplementary Material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Nakano, M., Ikeda, Y., Tokuda, Y. et al. Novel common variants and susceptible haplotype for exfoliation glaucoma specific to Asian population. Sci Rep 4, 5340 (2014). https://doi.org/10.1038/srep05340
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05340
This article is cited by
-
Analysis of genetically determined gene expression suggests role of inflammatory processes in exfoliation syndrome
BMC Genomics (2023)
-
Efficient and reliable establishment of lymphoblastoid cell lines by Epstein-Barr virus transformation from a limited amount of peripheral blood
Scientific Reports (2017)
-
A common variant mapping to CACNA1A is associated with susceptibility to exfoliation syndrome
Nature Genetics (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
