
Abstract
Cold-inducible RNA-binding protein (CIRP) is a damage-associated molecular pattern (DAMP) molecule which stimulates proinflammatory cytokine release in hemorrhage and sepsis. Under these medical conditions, disruption of endothelial homeostasis and barrier integrity, typically induced by proinflammatory cytokines, is an important factor contributing to morbidity and mortality. However, the role of CIRP in causing endothelial dysfunction has not been investigated. In this study, we show that intravenous injection of recombinant murine CIRP (rmCIRP) in C57BL/6 mice causes lung injury, evidenced by vascular leakage, edema, increased leukocyte infiltration and cytokine production in the lung tissue. The CIRP-induced lung damage is accompanied with endothelial cell (EC) activation marked by upregulation of cell-surface adhesion molecules E-selectin and ICAM-1. Using in vitro primary mouse lung vascular ECs (MLVECs), we demonstrate that rmCIRP treatment directly increases the ICAM-1 protein expression and activates NAD(P)H oxidase in MLVECs. Importantly, CIRP stimulates the assembly and activation of Nlrp3 inflammasome in MLVECs accompanied with caspase-1 activation, IL-1β release and induction of proinflammatory cell death pyroptosis. Finally, our study demonstrates CIRP-induced EC pyroptosis in the lungs of C57BL/6 mice for the first time. Taken together, the released CIRP in shock can directly activate ECs and induce EC pyroptosis to cause lung injury.
Similar content being viewed by others


Introduction
In sepsis, trauma and hemorrhagic shock, proinflammatory cytokines and damage-associated molecular pattern molecules (DAMPs) released from cells of injured tissue have been shown to activate polymorphonuclear cells (PMNs) and endothelial cells (ECs), leading to systemic inflammation and multiple-organ dysfunction1,2,3,4,5. The activated EC layer becomes permeable and loses the integrity of EC barrier, which allows fluids, pathogens and excessive leukocytes entering into tissues to cause damage6. The formation of inflammasome is another cellular response to pathogens or DAMPs for proinflammatory cytokine release7. The inflammasome is a multimolecular platform which assembles as a complex of nucleotide-binding oligomerization domain-like receptor (Nlr) proteins, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) and caspase-1 (or caspase-5) to release proinflammatory cytokines IL-1β and IL-187. The Nlr family, pyrin domain containing 3 (Nlrp3) inflammasome can assemble in response to a wide variety of stimuli and has been implicated in triggering inflammation in a wide variety of disease pathologies, making it the most versatile and clinically implicated inflammasome7. The Nlrp3 inflammasome activation typically occurs in phagocytes7 but recent studies have also shown it in ECs8,9,10,11,12. Moreover, activation of caspase-1 via inflammasome can trigger pyroptosis, an inflammatory form of programmed cell death subroutine that mostly occurs in macrophages13, but has not yet demonstrated in ECs.
Cold-inducible RNA-binding protein (CIRP, official gene name Cirbp) is a highly conserved 172-amino acid nuclear protein that belongs to the family of cold shock proteins14. CIRP contains one amino-terminal consensus sequence RNA-binding domain and one carboxy-terminal glycine-rich domain15. CIRP functions as an RNA chaperone facilitating mRNA translation16,17 and is ubiquitously expressed in various tissues at low levels15,18,19. CIRP is induced upon mild hypothermia14,19,20, by exposure to ultraviolet irradiation21,22 and hypoxia23. It has also been described as a new generation proto-oncogene24. Recently, we discovered that CIRP is a new inflammatory mediator or DAMP25. During shock, CIRP is released into the circulation and stimulates cytokine release in macrophages25,26,27,28. However, whether the released CIRP has any other role in contributing to the endothelial damage has not been well explored.
In this study, we hypothesized that the released CIRP directly contributes to the vascular EC injury through the Nlrp3 inflammasome pathway. We first conducted an in vivo study by injecting mice with recombinant murine CIRP (rmCIRP) and examined for vascular leakage, tissue edema, neutrophil infiltration and expression of proinflammatory cytokines TNF-α and IL-1β as well as cell-surface adhesion molecules E-selectin and ICAM-1 as markers for EC activation in the lungs. To determine whether the EC activation in the lung tissue is a direct effect in response to rmCIRP administration, we treated isolated primary mouse lung vascular ECs (MLVECs) with rmCIRP and examined the expression of EC activation marker. We further determined the mechanism for CIRP-induced EC activation by examining NAD(P)H oxidase activation and formation of Nlrp3 inflammasome in rmCIRP-treated MLVECs. Finally, we identified that rmCIRP induced EC pyroptosis in vitro and in vivo.
Results
Extracellular CIRP causes vascular leakage and endothelial cell activation in the lungs
The lungs are the most susceptible organ for injury in hemorrhagic or septic shock29. CIRP acts as an inflammatory mediator in hemorrhagic and septic shock25. In our previous study, we have demonstrated that intravenous (i.v.) administration of 5 mg/kg body weight rmCIRP to healthy mice increases the levels of serum proinflammatory cytokines (TNF-α, IL-6 and HMGB1) and liver enzymes (AST and ALT)25. To examine the role of CIRP in contributing to the lung injury, we i.v. injected healthy mice with the same dose of rmCIRP to study its effects on the vascular integrity in the lungs. Evans blue dye (EBD) assay is commonly used to assess blood vessel permeability in vivo. When ECs become dysfunctional and lose their close contacts, the i.v. injected EBD leaks into the tissues30. At 5 h after rmCIRP injection, the lungs of mice had increased permeability shown as more blue coloration than the lungs of vehicle-injected mice (Fig. 1A). The amount of the EBD extravasated in the lungs of rmCIRP-injected mice was 2-fold higher than that in the vehicle-injected mice (Fig. 1B).

Extracellular CIRP induces vascular leakage and causes lung injury.
The C57BL/6 mice were intravenously injected with vehicle (normal saline) or recombinant murine CIRP (rmCIRP, 5 mg/kg body weight) and lungs were collected for various analysis 5 h later. (A) Representative images of the lungs stained with Evans blue dye (EBD) post-vehicle or rmCIRP injections. (B) Colorimetric analysis of EBD extracted from stained lungs. Bars represent mean ± SEM (n = 5 per group). *P < 0.05; Student’s t-test. Representative images of H&E stained lung sections from (C) vehicle or (D) rmCIRP injected mice at original magnification ×400. The qPCR analysis of (E) cytokines and (F) endothelial cell adhesion molecules in the lungs of vehicle or rmCIRP injected mice. Bars represent mean ± SEM (n = 3 per group). *P < 0.05; Student’s t-test.
The pulmonary parenchyma of mice injected with rmCIRP also had significant edema and neutrophil infiltration in comparison with that of vehicle group mice (Fig. 1C,D), as judged by H&E staining. In addition, rmCIRP increased the lung expression of proinflammatory cytokines TNF-α and IL-1β mRNA by 16.3- and 3.0- fold, respectively, compared to the vehicle at 5 h after injection (Fig. 1E). These results indicate that CIRP causes an increase in vascular permeability, neutrophil infiltration and inflammation for the lung jury. We next examined the markers for activated status of ECs in the lungs at 5 h after rmCIRP injection by measuring gene expression of the EC adhesion molecules using qPCR. The mRNA levels of E-selectin and intercellular adhesion molecule (ICAM)-1 in the lungs of mice injected with rmCIRP was 3.1- and 2.6- fold higher than those injected with vehicle, respectively (Fig. 1F). Upregulation of EC adhesion molecules after rmCIRP injection indicate that extracellular CIRP can cause activation of ECs in the lung tissue.
CIRP directly activates lung endothelial cells
To determine whether CIRP can have a direct effect on ECs, we isolated primary mouse lung vascular endothelial cells (MLVECs) and then treated them with rmCIRP. The induction of ICAM-1 is a well-known indicator of EC activation. The addition of rmCIRP induced the expression of ICAM-1 in MLVECs in a dose-dependent manner, increasing it by 1.7-fold at 200 ng/ml and by 2.3-fold at 1,500 ng/ml, compared to the absence of rmCIRP at 4 h (Fig. 2A). We then used 200 ng/ml of rmCIRP to conduct a time-course study of the EC activation by rmCIRP. The ICAM-1 levels in MLVECs increased by 2.3-fold at 1 h and further increased by 8.5-fold after 8 h of rmCIRP treatment (Fig. 2B). Taken together, these results indicate that CIRP specifically activates ECs in dose- and time-dependent manners.

CIRP activates vascular endothelial cells.
(A) Western blot analysis of ICAM-1 in the mouse lung vascular endothelial cells (MLVECs) treated with an indicated concentration of rmCIRP for 4 h. The image shown is representative of three independent experiments. Bars represent mean ± SEM (n = 3) from densitometric analysis of blots. *P < 0.05 compared to no rmCIRP; one-way ANOVA, Student-Newman-Keuls test. (B) Western blot analysis of ICAM-1 in MLVECs treated with 200 ng/ml rmCIRP for upto 8 h as indicated. The image is representative of three independent experiments. Bars represent mean ± SEM (n = 3). *P < 0.05 compared to time 0; one-way ANOVA, Student-Newman-Keuls test.
CIRP stimulates NAD(P)H oxidase in lung endothelial cells
Reactive oxygen species (ROS) generated from NAD(P)H oxidase play an important role in activating ECs during lung inflammation, ischemia/reperfusion injury, sepsis, hyperoxia and ventilator-associated lung injury31. NAD(P)H oxidase is a complex of multimeric enzymes consisting of 5 subunits, gp91phox (phox for phagocytic oxidase), p22phox, p47phox, p67phox and small GTPase Rac31. Upon stimulation, the p47phox subunit becomes phosphorylated and binds to gp91phox, generating a conformational change of the entire complex and resulting in its activation31. After treatment of MLVECs with rmCIRP, we observed an increase of the binding of gp91phox to phosphorylated p47phox in a dose-dependent manner by immunoprecipitation with anti-p47phox antibody (Fig. 3A). We also observed that the binding of gp91phox to phosphorylatedp47phox was increased from 1 h to 8 h in the MLVECs treated with 200 ng/ml of rmCIRP (Fig. 3B). These data show that CIRP activates NAD(P)H oxidase and suggest a critical role of endogenous NAD(P)H oxidase in mediating CIRP-induced EC activation.

CIRP induces NAD(P)H oxidase activation.
Western blot analysis of gp91phox, phosphorylated p47phox (phospho-p47phox) and total p47phox from mouse lung vascular endothelial cells (MLVECs) immunoprecipitated with anti-p47phox antibody after treatment with (A) an indicated concentration of rmCIRP for 4 h or (B) 200 ng/ml rmCIRP for upto 8 h as indicated. The images are representative of three independent experiments.
CIRP induces the formation of Nlrp3 inflammasome in lung endothelial cells
The NAD(P)H oxidase-derived ROS has been shown to be a contributing factor for the formation of Nlrp3 inflammasome in ECs8. We then assessed CIRP’s ability to assemble and activate Nlrp3 inflammasome to release IL-1β. A major component of the inflammasome is ASC, an adaptor protein that mediates the interaction between Nlrp3 and caspase-17. To test whether CIRP induced the inflammasome in ECs, we treated MLVECs with rmCIRP at different doses and for different times. We observed an increase of the interaction between Nlrp3 and ASC in a dose-dependent manner by immunoprecipitation with anti-ASC antibody (Fig. 4A). We also observed that the association of Nlrp3 and ASC in MLVECs was induced at 1 h after incubation with 200 ng/ml rmCIRP and was further increased by 8 h (Fig. 4A). Another characteristic of inflammasome formation is the activation of caspase-1, which can be detected by its cleaved P-10 subunit7. In the same setting as for examining the formation of Nlrp3-ASC complex, we also detected an increase of P-10 levels in MLVECs treated with rmCIRP in dose- and time- dependent manners (Fig. 4A). Accordingly, rmCIRP also induced release of IL-1β by MLVECs in a dose-dependent manner, with 32.4-fold increase at 200 ng/ml and 69.5-fold increase at 1,500 ng/ml (Fig. 4B). Furthermore, IL-1β levels were elevated by 11.8-fold at 1 h and by 42.4-fold at 8 h after rmCIRP addition (Fig. 4B). Altogether, these data demonstrate a NLRP3 inflammasome-mediated mechanism of IL-1β release and EC activation by CIRP.

CIRP induces Nlrp3 inflammasome activation and pyroptosis in vitro.
(A,B) Mouse lung vascular endothelial cells (MLVECs) were treated with an indicated concentration of rmCIRP for an indicated period of time. (A) The cells from the treatments were subjected to immunoprecipitation with anti-ASC antibody. Immunoblot analysis of Nlrp3, ASC and cleaved caspase-1 was performed. The images are representative of three independent experiments. (B) IL-1β levels in the cell culture media from above treatments, measured by ELISA. Bars represent mean ± SEM (n = 3). *P < 0.05 compared to no rmCIRP or 0 h; #P < 0.05 compared to 200 ng/ml rmCIRP or 1 h; one-way ANOVA, Student-Newman-Keuls test. (C,D) MLVECs were treated with no rmCIRP or 200 ng/ml rmCIRP for upto 24 h as indicated. Cells were then stained with TMR-red-Cell Death reagent and Alexa Fluor 488-FLICA-activated caspase-1 fluorescent reagent (AF488-caspase-1), followed by flow cytometry. (C) Representative dot blots showing percentages of double-stained pyroptotic MLVECs as indicated by numbers in the top right quadrant. (D) Graph showing the percentages of pyroptotic cells from flow cytometric analysis. Bars represent mean ± SEM (n = 3). *P < 0.05 compared to no rmCIRP; one-way ANOVA, Student-Newman-Keuls test.
CIRP induces lung endothelial cell pyroptosis in vitro and in vivo
We next examined whether the activation of EC inflammasome by CIRP could lead to pyroptosis. By staining the cells with Cell Death and caspase-1 activity reagents and analyzing with flow cytometry, we detected significant increase in the percentage of pyroptotic MLVECs after rmCIRP (200 ng/ml) treatment, which progressed in a time-dependent manner (Fig. 4C,D). The percentage of pyroptotic MLVECs was negligible without rmCIRP treatment (Fig. 4C,D), whereas 8 h after addition of rmCIRP 0.8% MLVECs were pyroptotic which increased by 12-fold (9.8%) after 16 h and by 35-fold (28%) after 24 h of rmCIRP treatment (Fig. 4D). To validate the observation in vivo, we i.v. injected healthy mice with rmCIRP and 24 h later harvested and subjected the lung tissues to immunofluorescent analysis. Lung ECs stained with E-selectin (white fluorescence) exhibited positive staining for activated caspase-1 (green fluorescence) and DNA fragmentation (red fluorescence as assessed by TUNEL) in the rmCIRP-treated mouse, while it was not observed in the vehicle-treated control mouse (Fig. 5). Taken together, our in vitro and in vivo studies clearly indicate that CIRP can directly induce pyroptosis in ECs.

CIRP induces endothelial cell pyroptosis in vivo.
C57BL/6 mice were intravenously injected with vehicle (normal saline) or rmCIRP (5 mg/kg body weight). After 24 h, lung tissues were harvested and subjected to fluorescent immunohistochemistry analysis. Representative images of lung tissues viewed under confocal microscopy (original magnification x600) after staining with Alexa Fluor 488- FLICA-activated caspase-1 (green), TUNEL (red), APC-E-selectin-1 (white) and Hoechst (blue). Images are representative of three independent experiments.
Discussion
We report here that extracellular CIRP, which is an endogenous DAMP, causes endothelial dysfunction in a direct manner and induces lung damage. We show that administration of CIRP to mice results in vascular leakage, edema, increased leukocyte infiltration and proinflammatory cytokines TNF-α and IL-1β in the lung tissues along with upregulation of surface adhesion molecules E-selectin and ICAM-1, reflecting EC activation. CIRP induces EC activation in a direct manner by demonstrating in cultured MLVECs. The NAD(P)H oxidase in ECs is also activated in the presence of CIRP. Importantly, our findings demonstrate that CIRP activates the Nlrp3 inflammasome in ECs, resulting in IL-1β release and caspase-1 mediated induction of EC pyroptosis.
Pulmonary vascular dysfunction and injury are characterized by disruption of the normal microvascular alveolo-capillary permeability barrier, resulting in extra-vascular leak of protein-rich edema into the pulmonary interstitial and alveolar compartments as well as increased PMN sequestration/adhesion in the lungs32,33. Our results clearly show that intravenous administration of rmCIRP to healthy mice resulted in the development of lung edema, increased leukocyte accumulation and increased production of TNF-α and IL-1β in the lungs. In this study, we have further shown that rmCIRP administration increases the expression of E-selectin and ICAM-1 as well as induces vascular leakage in the lungs of mice. Endothelial dysfunction has been linked to EC activation which is defined by the endothelial expression of cell-surface adhesion molecules, such as E-selectin and ICAM-134. Other DAMPs such as HMGB1, mitochondrial DNA, S100 family proteins and Shiga toxin have also been shown to cause endothelial dysfunction and change endothelial permeability35,36,37,38.
The ICAM-1 expressed in ECs is a counter receptor for the leukocyte β2-integrins LFA-1 and Mac-1 and plays an important role in the regulation of PMN sequestration39. The interaction of ICAM-1 with these integrins enables the PMN to adhere firmly to the vascular endothelium and migrate across the microvascular barrier39. EC activation is known to be typically induced by proinflammatory cytokines such as TNF-α, IFN-γ and IL-640. We have previously shown that CIRP treatment results in increased TNF-α release from cultured macrophages and CIRP administration in vivo results in increased serum levels of TNF-α and IL-625. Through induction of cytokines, CIRP can indirectly induce EC activation in vivo. We have further examined the possibility of direct EC activation by CIRP and shown that treatment with rmCIRP increases the ICAM-1 expression and the IL1-β release in cultured MLVECs in a dose- and time- dependent manner. Thus, the released CIRP in hemorrhagic and septic shock can both directly and indirectly cause endothelial activation and dysfunction, leading to the lung damage.
Reactive oxygen species (ROS) function as important signaling molecules to mediate various biological responses. Excessive production of ROS results into oxidative stress and contributes to various pathophysiological responses including lung injury31. Phagocytes such as neutrophils and monocytes are best known to generate large quantities of ROS using the enzyme NAD(P)H oxidase41. The nonphagocytic ECs also contain NAD(P)H oxidase, which is the major enzymatic mechanism to generate ROS within ECs42. The neutrophil NAD(P)H oxidase-derived ROS have been shown to be an important regulator of EC activation43 for the inflammatory responses after trauma and hemorrhage. In this study, we demonstrate that CIRP is directly capable of inducing the NAD(P)H oxidase activation in ECs. NAD(P)H oxidases are a group of multimeric enzymes, which consist of cytosol complex including p40phox, p47phox and p67phox and membrane components p22phox and gp91phox. Upon stimulation, p47phox is phosphorylated and the entire cytosolic complex migrates to the membrane where it associates with gp91phox and cytochrome b558 to assemble the active oxidase31. Since CIRP did not alter total p47phox expression, as shown in Fig. 3, CIRP seems to be a potent stimulator of NAD(P)H oxidase activation by promoting enzymes assembly, rather than by upregulating the expression of the enzyme molecules. Recently, NAD(P)H oxidase has been shown to be required for Nlrp3 inflammasome activation and IL-1β release in ECs in response to hemorrhage8, in macrophages in pulmonary fibrosis44 and in podocytes in response to hyperhomocyteinemia45 and diabetic nephropathy46. In the same token, we also observe that NAD(P)H oxidase activation by CIRP is associated with the formation of Nlrp3 inflammasome in ECs.
Dysregulated Nlrp3 inflammasome activation is associated with both heritable inflammatory diseases such as Muckle-Wells syndrome, familial cold autoinflammatory syndrome and neonatal onset multi-system inflammatory disease as well as acquired inflammatory diseases such as Alzheimer’s disease, gout, pseudogout, obesity, atherosclerosis, metabolic syndrome, type 2 diabetes mellitus and age-related macular degeneration47. A growing list of many endogenous DAMPs, such as extracellular ATP, uric acid crystals, cholesterol crystals, hyaluronan, heparan sulfate, calcium pyrophosphate dihydrate, β-amyloid plaques and islet amyloid polypeptide, also induce Nlrp3 inflammasome activation after their accumulation or alteration under damage or disease conditions47. Although Nlrp3 inflammasome activation was originally thought to be immune cell specific, it has recently been studied in non-immune cells such as ECs during hemorrhage8,12, atherosclerosis10, coronary arteritis9 and obesity11. Our results show that CIRP induced the assembly of Nlrp3 inflammasome in ECs resulting in the activation of caspase-1 and IL-1β production, providing another proinflammatory activity of CIRP besides activating macrophages for cytokine release25. Inflammasome activation is initiated through the assembly of the existing, but not de novo synthesis of, components. This is also supported by our observation that CIRP did not change the protein expression of ASC and pro-caspase-1, as shown in Fig. 4A.
Cell death critically influences the inflammatory process48,49. The death of ECs under pathologic conditions can be through a variety of different cell death mechanisms such as apoptosis, necrosis, necroptosis and autophagy. Proinflammatory cytokines such as IFN-γ have been shown to exacerbate endothelial injury by triggering EC death via caspase-8-dependent apoptosis50. In another study, pulmonary microvascular dysfunction has been shown to be due to MLVEC death via caspase-dependent apoptosis mediated by NAD(P)H oxidase signaling in a murine sepsis model51. Pyroptosis is a newly characterized inflammasome-mediated caspase-1-dependent cell death subroutine that mostly occurs in macrophages13,52. Pyroptosis is morphologically and mechanistically distinct from apoptosis and necrosis as it features plasma membrane rupture and release of inflammatory factors52. It occurs in response to a number of bacterial and viral pathogens but can also be triggered by stroke, heart attack, hemorrhage, or cancer13. Recently, HMGB1 was reported to induce inflammasome activation and pyroptosis in macrophages53. HMGB1 has been shown to trigger Nrlp3 inflammasome-mediated caspase-1 activation in ECs in hemorrhagic shock and uraemia8,54, but EC pyroptosis has not been demonstrated in these studies. We show that extracellular CIRP causes EC pryoptosis both in vitro and in vivo. Our study reports a novel mechanism by which the CIRP, an endogeneous DAMP, can mediate activation of Nlrp3 inflammasome and caspase-1 as well as induces EC pyroptosis, further enhancing the EC dysfunction. Whether like HMGB1, CIRP can also induce Nlrp3 inflammasome activation and pyroptosis in macrophages as well as other cell types needs further investigation.
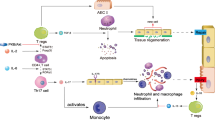
The pathogenesis of vascular EC dysfunction is complex55. Based on this study, we propose a mechanistic model for CIRP-induced vascular EC dysfunction leading to organ injury (Fig. 6). During hemorrhagic and septic shock, CIRP is released in circulation and in turn activates ECs which is defined by the upregulation of adhesion molecules and production of proinflammatory cytokines to increase PMN infiltration. CIRP also induces NAD(P)H oxidase-mediated generation of ROS and Nlrp3 inflammasome activation in ECs leading to caspase-1-mediated EC pyroptosis. This study suggests that targeting CIRP as an adjunct therapy for acute inflammatory diseases via attenuation of EC injury could be an important translational approach.

Model of CIRP-induced vascular endothelial cell dysfunction.
CIRP released during hemorrhagic or septic shock activates endothelial cells (EC) by upregulating adhesion molecules on them to increase infiltration of polymorphonuclear leukocytes (PMN) and produce proinflammatory cytokines and reactive oxygen species (ROS). CIRP also induces activation of Nlrp3 inflammasome and caspase-1-mediated EC pyroptosis. This series of events serve as a mechanism underlying the CIRP-induced EC dysfunction which then leads to organ injury.
Methods
Mice
Male C57BL/6 mice (20 to 25 g) purchased from the Jackson Laboratory (Bar Harbor, ME, USA) were used in all experiments. These mice were housed in a temperature-controlled room on a 12 hour light/dark cycle in the animal facility within the Feinstein Institute for Medical Research or University of Pittsburgh and fed a standard laboratory diet. All experiments were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD, USA). All the animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) of the Feinstein Institute for Medical Research, University of Pittsburgh and VA Pittsburgh Healthcare System. All efforts were made to minimize suffering.
Administration of rmCIRP
rmCIRP was produced as described previously25. Mice were allocated to two groups: vehicle and rmCIRP. A small incision on the neck was made and the internal jugular vein was exposed. Normal saline (vehicle) or rmCIRP at a dose of 5 mg/kg body weight (BW) in 200 μl volume was delivered by injection using 29G × 1/2″ U-100 insulin syringe (Terumo Medical Corporation, Elkton, MD, USA) through the jugular vein. At 5 h after rmCIRP injection, mice were anesthetized and lungs were collected. A section of lung tissue was preserved in 10% formalin for histopathological analysis and rest was frozen in liquid nitrogen and stored at −80 °C for qPCR analysis. An additional set of experiments were performed with Evans blue injected at 5 h after rmCIRP injection and the lungs were harvested for evaluation.
Lung permeability assay
The changes in vascular permeability after rmCIRP administration were measured by Evans blue dye (EBD) leakage from blood into lung airways30. EBD (20 mg/kg; Sigma-Aldrich, St. Louis, MO) in 100 μl volume was administered intravenous (i.v.) by tail vein injection 1 h before the end of the 5 h experiments. One hour later, lungs were perfused with normal saline through the spontaneously beating right ventricle to remove intravascular dye. Lungs were removed, documented by photographs and dried at 60 °C for 48 h. Evans blue dye was extracted from lungs by incubating in formamide (Sigma-Aldrich) at 37 °C for 24 h and centrifuged at 5,000g for 30 min. The EBD in the supernatant was quantitated by a dual wavelength (620 and 740 nm) spectrophotometric method, correcting for contaminating heme pigments by using the formula OD620 (EBD) = OD620 − (1.426 × OD740 + 0.030)56. The extravasated EBD concentration in lung homogenate was calculated against a standard curve and expressed as the dye incorporated per mg of tissue.
Histologic examination
The lung tissues were fixed in 10% formalin followed by paraffin embedding. The paraffin tissue blocks were cut into 5 μm sections, which were transferred to glass slides and stained with hematoxylin and eosin (H&E). Morphologic changes in the lung tissues were examined by light microscopy, documented by photographs and evaluated by two investigators in a blinded manner. Lung injury was assessed based on pulmonary edema judged by thickened alveolar walls with vascular congestion and interstitial and alveolar leukocyte infiltration.
RNA extraction and quantitative real-time PCR
Total RNA was isolated from lung tissue by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction and was reverse-transcribed into cDNA using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA, USA). A PCR reaction was carried out in a 24 μl final volume containing 0.08 μM of each forward and reverse primer, cDNA and 12 μl SYBR Green PCR Master Mix (Life Technologies, Grand Island, NY). The following gene-specific primers were used for amplifying genes: E-selectin forward, 5′-TGACATCGTCCTCATTGCTC-3′ and reverse, 5′-TTTCCTGCTGTCTTCAGCTTATC-3′; ICAM-1 forward, 5′-GGGCTGGCATTGTTCTCTAA-3′ and reverse, 5′-CTTCAGAGGCAGGAAACAGG-3′; TNF-α forward, 5′-AGACCCTCACACTCAGATCATCTTC-3′ and reverse, 5′-TTG CTACGACGTGGGCTACA-3′; IL-1β forward, 5′-CAGGATGAGGACATGAGCACC -3′ and reverse, 5′-CTCTGCAGACTCAAACTCCAC -3′; and β-actin forward, 5′-CGTGAAAAGATGACCCAGATCA -3′ and reverse, 5′-TGGTACGACCAGAGGCATACAG -3′. Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine (Applied Biosystems) under the thermal profile of 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The data was analyzed by the 2−ΔΔCt method for relative quantization and normalized to mouse β-actin mRNA. Relative expression of mRNA was expressed as the fold change in comparison with the vehicle injected lung tissue.
Mouse lung vascular EC isolation and culture
Mouse lung vascular ECs (MLVEC) were isolated using a previously described method57 with modifications8. Briefly, mice were anesthetized by intraperitoneal administration of 50 mg/kg ketamine and 5 mg/kg xylazine and the chest cavity was opened. The right ventricle was cannulated to infuse PBS for removing blood from lungs. Peripheral lung tissue was diced to ∼1 mm3 pieces and cultured in a 60-mm culture dish in growth medium (MEM D-Val medium [Invitrogen Gibco] containing 2 mM glutamine, 10% FBS, 5% human serum, 50 μg/ml penicillin/streptomycin, 5 μg/ml heparin, 1 μg/ml hydrocortisone, 80 μg/ml endothelial cell growth supplement from bovine brain, 5 μg/ml amphotericin and 5 μg/ml mycoplasma removal agent) at 37 °C with 5% CO2 for 60 h. The tissue dices were removed and the adherent cells were continued in culture for 3 d. The MLVECs were purified from these cultured cells using biotin-conjugated rat anti-mouse CD31 (PECAM-1) mAb and BD IMag streptavidin particles plus-DM and the immunomagnetic separation system (BD Biosciences, San Jose, CA, USA) following the manufacturer’s instructions. The purified MLVECs were allowed to grow for 3 to 4 d and then characterized by their cobblestone morphology, uptake of Dil-Ac-LDL (Biomedical Technologies, Stoughton, MA, USA) and staining for factor VIII-related Ag (Sigma-Aldrich). MLVECs from 3 to 5 passages were used in experiments in which cells were treated with increasing concentrations of rmCIRP (200 and 1500 ng/ml) for 4 h or treated with 200 ng/ml rmCIRP for various time points, washed with HBSS three times and harvested for further analysis.
Immunoblot analysis and coimmunoprecipitation
MLVECs were lysed (∼1 × 106 cells/ml) in lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 20 mM PMSF). Protein concentrations were determined by Bio-Rad Laboratories (Hercules, CA, USA) protein assay reagent. 30 μg total protein for each sample was fractionated on a 10% SDS-PAGE gel and then transfered onto polyvinylidene difluoride (PVDF) membrane. The membranes were then blocked for 1 h at room temperature with blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) and probed with anti-ICAM-1 or anti-β-actin antibodies (Santa Cruz Biotechnologies, Santa Cruz, CA, USA). The membranes were then washed with PBS for three times and incubated with the appropriate secondary antibody (LI-COR Biosciences). Protein bands were detected using LI-COR Odyssey Fc Imager (LI-COR Biosciences). Blots were quantitated using NIH Image J densitometric software and normalized to β-actin. For coimmunoprecipitation studies, 600 μg total protein for each sample was immunoprecipitated with anti-p47phox or anti-ASC antibodies (Santa Cruz Biotechnologies). The immunoprecipitated proteins were then fractionated on 10% SDS-PAGE gel and immunoblotted as described above with anti-phosphoserine antibody (Invitrogen) or anti-gp91phox or anti-NLRP3 antibodies (Santa Cruz Biotechnologies) and detected with Clean-Blot IP Detection Reagent (Thermo Scientific, Rockford, IL, USA) following the manufacturer’s instructions. Blots were then stripped and reprobed with anti-p47phox or anti-ASC or anti-mouse caspase-1 p10 antibodies (Santa Cruz Biotechnologies).
Flow cytometry analysis of cell pyroptosis
MLVECs were stimulated with 200 ng/ml rmCIRP or left untreated for 8–24 h followed by staining of the cells with Alexa Fluor 488-labeled Fluorochrome Inhibitor of Caspase-1(FLICA) (ImmunoChemistry Technologies, Bloomington, MN, USA), which binds to activated caspase-1, at 37 °C for 1 h. After fixing with 4% paraformaldehyde, cells were stained with tetramethylrhodamine (TMR) red-labeled In-Situ Cell Death Detection reagent (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer’s instructions. The cells were then analyzed by flow cytometry. Background and auto-fluorescence were determined by a control antibody with the same isotype staining. Acquisition was performed on 10,000 events using a FACScalibur cytometer (BD Biosciences) or BD LSR II (BD Biosciences) and the data was analyzed by CellQuestPro (BD Biosciences) and FlowJo-V10 software (Tree Star, Ashland, OR, USA). The percentage of double-stained cells was taken as percentage of pyroptotic cells.
Measurement of IL-1β
IL-1β in cell-culture media was measured using the ELISA Ready-Set-Go kit for mouse IL-1β (eBioscience, San Diego, CA, USA) following the manufacturer’s instructions.
Immunofluorescence confocal microscopy
Lung tissues were harvested from vehicle (normal saline) and rmCIRP (5 mg/kg body weight) injected mice after 24 h of i.v. injections and subjected to multi-fluorescent immunohistochemistry analysis. Briefly, lung sections were fixed and stained with 1× cell permeable Alexa Fluor 488-labeled FLICA (ImmunoChemistry), followed by TUNEL staining (Roche Applied Science) for dead cells, nuclear staining with 1 μg/ml Hoechst 33258 (Sigma-Aldrich) and allophycocyanin (APC)-labeled E-selectin-1 (R&D Systems, Minneapolis, MN) staining for ECs. The stained lung section was imaged by confocal microscope with excitation at 480 nm for green fluorescence of caspase-1–positive cells, at 540 nm for red fluorescence of TUNEL-stained cells, at 365 nm for blue fluorescence of Hoechst-stained nuclei and at 650 nm for white fluorescence of E-selectin positive ECs.
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) and analyzed using SigmaPlot11 graphing and statistical analysis software (Systat Software Inc., San Jose, CA, USA). Student’s t test was used for comparing two groups. Multiple groups were compared by one-way analysis of variance (ANOVA) with Student-Newman-Keuls’ (SNK) test. Differences were considered significant if P < 0.05.
Additional Information
How to cite this article: Yang, W.-L. et al. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 6, 26571; doi: 10.1038/srep26571 (2016).
References
Schouten, M., Wiersinga, W. J., Levi, M. & van der Poll, T. Inflammation, endothelium and coagulation in sepsis. J Leukoc Biol. 83, 536–545 (2008).
Diebel, L. N., Liberati, D. M., Ledgerwood, A. M. & Lucas, C. E. Changes in lymph proteome induced by hemorrhagic shock: the appearance of damage-associated molecular patterns. J Trauma Acute Care Surg. 73, 41–51 (2012).
Medzhitov, R. Origin and physiological roles of inflammation. Nature. 454, 428–435 (2008).
Kaczorowski, D. J., Mollen, K. P., Edmonds, R. & Billiar, T. R. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 83, 546–552 (2008).
Hutchins, N. A., Wang, F., Wang, Y., Chung, C. S. & Ayala, A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol. 94, 963–970 (2013).
Vandenbroucke, E., Mehta, D., Minshall, R. & Malik, A. B. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 1123, 134–145 (2008).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 13, 397–411 (2013).
Xiang, M. et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol. 187, 4809–4817 (2011).
Chen, Y. et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 1853, 396–408 (2015).
Xiao, H. et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 128, 632–642 (2013).
Chen, Y. et al. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med. 19, 2715–2727 (2015).
Xu, P. et al. Hemorrhagic shock augments Nlrp3 inflammasome activation in the lung through impaired pyrin induction. J Immunol. 190, 5247–5255 (2013).
Bergsbaken, T., Fink, S. L. & Cookson, B. T. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 7, 99–109 (2009).
Nishiyama, H. et al. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol. 137, 899–908 (1997).
Nishiyama, H. et al. Cloning and characterization of human CIRP (cold-inducible RNA-binding protein) cDNA and chromosomal assignment of the gene. Gene. 204, 115–120 (1997).
Dresios, J. et al. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels and enhances global protein synthesis. Proc Natl Acad Sci USA. 102, 1865–1870 (2005).
Al-Fageeh, M. B. & Smales, C. M. Cold-inducible RNA binding protein (CIRP) expression is modulated by alternative mRNAs. Rna. 15, 1164–1176 (2009).
Nishiyama, H. et al. Decreased expression of cold-inducible RNA-binding protein (CIRP) in male germ cells at elevated temperature. Am J Pathol. 152, 289–296 (1998).
Xue, J. H. et al. Effects of ischemia and H2O2 on the cold stress protein CIRP expression in rat neuronal cells. Free Radic Biol Med. 27, 1238–1244 (1999).
Liu, A., Zhang, Z., Li, A. & Xue, J. Effects of hypothermia and cerebral ischemia on cold-inducible RNA-binding protein mRNA expression in rat brain. Brain Res. 1347, 104–110 (2010).
Fornace, A. J., Jr., Alamo, I., Jr. & Hollander, M. C. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci USA. 85, 8800–8804 (1988).
Sheikh, M. S. et al. Identification of several human homologs of hamster DNA damage-inducible transcripts. Cloning and characterization of a novel UV-inducible cDNA that codes for a putative RNA-binding protein. J Biol Chem. 272, 26720–26726 (1997).
Wellmann, S. et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 117, 1785–1794 (2004).
Artero-Castro, A. et al. Cold-inducible RNA-binding protein bypasses replicative senescence in primary cells through extracellular signal-regulated kinase 1 and 2 activation. Mol Cell Biol. 29, 1855–1868 (2009).
Qiang, X. et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 19, 1489–1495 (2013).
Zhou, M., Yang, W. L., Ji, Y., Qiang, X. & Wang, P. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim Biophys Acta. 1840, 2253–2261 (2014).
Rajayer, S. R. et al. Cold-inducible RNA-binding protein is an important mediator of alcohol-induced brain inflammation. PLos One. 8, e79430 (2013).
Godwin, A. et al. Blocking cold-inducible RNA-binding protein protects liver from ischemia-reperfusion injury. Shock. 43, 24–30 (2015).
Bernard, G. R. et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes and clinical trial coordination. Am J Respir Crit Care Med. 149, 818–824 (1994).
Radu, M. & Chernoff, J. An in vivo assay to test blood vessel permeability. J Vis Exp. e50062 (2013).
Frey, R. S., Ushio-Fukai, M. & Malik, A. B. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal. 11, 791–810 (2009).
Schmidt, E. P., Lee, W. L., Zemans, R. L., Yamashita, C. & Downey, G. P. On, around and through: neutrophil-endothelial interactions in innate immunity. Physiology (Bethesda). 26, 334–347 (2011).
Ware, L. B. & Matthay, M. A. The acute respiratory distress syndrome. N Engl J Med. 342, 1334–1349 (2000).
Liao, J. K. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 123, 540–541 (2013).
Wolfson, R. K., Chiang, E. T. & Garcia, J. G. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 81, 189–197 (2011).
Sun, S. et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLos One. 8, e59989 (2013).
Wang, L., Luo, H., Chen, X., Jiang, Y. & Huang, Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLos One. 9, e90472 (2014).
Mayer, C. L. et al. Pro-Coagulant Endothelial Dysfunction Results from EHEC Shiga Toxins and Host Damage-Associated Molecular Patterns. Front Immunol. 6, 155 (2015).
Smith, C. W., Marlin, S. D., Rothlein, R., Toman, C. & Anderson, D. C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 83, 2008–2017 (1989).
Pober, J. S. & Cotran, R. S. Cytokines and endothelial cell biology. Physiol Rev. 70, 427–451 (1990).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 87, 245–313 (2007).
Babior, B. M. The NADPH oxidase of endothelial cells. IUBMB Life. 50, 267–269 (2000).
Fan, J., Frey, R. S., Rahman, A. & Malik, A. B. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-alpha -induced NF-kappa B activation and intercellular adhesion molecule-1 expression in endothelial cells. J Biol Chem. 277, 3404–3411 (2002).
Sun, B. et al. NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Lung Fibrosis by Multiwalled Carbon Nanotubes. Small. 11, 2087–2097 (2015).
Abais, J. M. et al. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal. 18, 1537–1548 (2013).
Gao, P. et al. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J Diabetes Res. 2015, 504761 (2015).
Leemans, J. C., Cassel, S. L. & Sutterwala, F. S. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 243, 152–162 (2011).
Cybulsky, M. I., Chan, M. K. & Movat, H. Z. Acute inflammation and microthrombosis induced by endotoxin, interleukin-1 and tumor necrosis factor and their implication in gram-negative infection. Lab Invest. 58, 365–378 (1988).
Hotchkiss, R. S. et al. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 25, 1298–1307 (1997).
Li, J. H. et al. Interferon-gamma augments CD95(APO-1/Fas) and pro-caspase-8 expression and sensitizes human vascular endothelial cells to CD95-mediated apoptosis. Am J Pathol. 161, 1485–1495 (2002).
Gill, S. E., Rohan, M. & Mehta, S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res. 16, 109 (2015).
Galluzzi, L. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 (2012).
Xu, J. et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 21, 1229–1239 (2014).
Martin-Rodriguez, S. et al. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur J Clin Invest. 45, 160–169 (2015).
Pober, J. S., Min, W. & Bradley, J. R. Mechanisms of endothelial dysfunction, injury and death. Annu Rev Pathol. 4, 71–95 (2009).
Standiford, T. J. et al. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak and early mortality in murine endotoxemia. J Immunol. 155, 1515–1524 (1995).
Tiruppathi, C. et al. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 91, 70–76 (2002).
Acknowledgements
This study was supported by the National Institutes of Health (NIH) grants HL076179 (PW), GM053008 (PW), HL079669 (JF); HL123882 (JF) and VA Merit Award 1I01BX002729 (JF). We thank Xiaoling Qiang and Jian Hua Li (The Feinstein Institute for Medical Research, Manhasset, NY) for technical assistance on providing recombinant murine CIRP for this study. We also thank Max Brenner (The Feinstein Institute for Medical Research) for assistance on depiction of the model figure for the study.
Author information
Authors and Affiliations
Contributions
W.-L.Y., J.F. and P.W. conceived and designed the experiments; Z.W. and Z.L. performed the experiments; W-L.Y., Z.W., Z.L. and A.S. analyzed the data; A.S. and W.-L.Y. wrote the manuscript; and J.F. and P.W. reviewed the manuscript and supervised the research. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yang, WL., Sharma, A., Wang, Z. et al. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep 6, 26571 (2016). https://doi.org/10.1038/srep26571
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26571
This article is cited by
-
Evaluation of serum and tissue levels of cold-inducible RNA-binding protein in non-segmental Vitiligo
Archives of Dermatological Research (2023)
-
Neutrophil trogocytosis during their trans-endothelial migration: role of extracellular CIRP
Molecular Medicine (2022)
-
Extracellular CIRP decreases Siglec-G expression on B-1a cells skewing them towards a pro-inflammatory phenotype in sepsis
Molecular Medicine (2021)
-
Targeting the eCIRP/TREM-1 interaction with a small molecule inhibitor improves cardiac dysfunction in neonatal sepsis
Molecular Medicine (2020)
-
Cold-inducible RNA-binding protein might determine the severity and the presences of major/minor criteria for severe community-acquired pneumonia and best predicted mortality
Respiratory Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
