
Abstract
Purpose:
To assess whether the combination of letrozole, metronomic cyclophosphamide and sorafenib (LCS) is well tolerated and shows activity in primary breast cancer (BC).
Methods:
Thirteen oestrogen receptor-positive, postmenopausal, T2-4, N0-1 BC patients received the LCS combination for 6 months. In these patients we examined the pharmacokinetics of sorafenib and cyclophosphamide, toxicity of the regimen, the clinical response to therapy and changes in the levels of biologically relevant biomarkers.
Results:
Adequate plasma concentrations of sorafenib were achieved in patients when it was dosed in combination with L+C. The mean plasma concentrations of C were consistently lower following administration of LCS, compared with administration of L+C only. The most common drug-related grade 3/4 adverse events were skin rash (69.3%), hand-foot skin reaction (69.3%) and diarrhoea (46.1%). According to RECIST Criteria, a clinical complete response was observed in 6 of 13 patients. A significant reduction in tumour size, evaluated with MRI, was also observed between baseline and 14 days of treatment in all 13 patients (P=0.005). A significant reduction in SUV uptake, measured by 18FDG-PET/CT, was observed in all patients between baseline and 30 days of treatment (P=0.015) and between baseline and definitive surgery (P=0.0002). Using modified CT Criteria, a response was demonstrated in 8 out of 10 evaluable patients at 30 days and in 11 out of 13 evaluable patients at the definitive surgery. A significant reduction in Ki67 expression was observed in all patients at day 14 compared with baseline (P<0.00001) and in 9 out of 13 patients at the definitive surgery compared with baseline (P<0.03). There was also a significant suppression of CD31 and VEGF-A expression in response to treatment (P=0.01 and P=0.007, respectively).
Conclusions:
The LCS combination is feasible and tolerable. The tumour response and target biomarker modulation indicate that the combination is clinically and biologically active.
Similar content being viewed by others

Main
Treatment with aromatase inhibitors (AIs), such as letrozole, has demonstrated increased response rate compared with tamoxifen in patients with breast cancer (BC) (Smith and Dowsett, 2003). However, the development of resistance to endocrine therapy is a significant problem involving the activation of other signalling pathways such as EGFR, HER2, IGFR, MAPK, PI3K-AKT or PKC (Nicholson et al, 2004, 2005; Osborne et al, 2005; Generali et al, 2009). This provides a strong rationale for combining endocrine therapies with signal-transduction inhibitors to bypass endocrine therapy resistance and achieve a more potent anti-tumour effect (Gligorov et al, 2007; Johnston et al, 2007).
Sorafenib (Nexavar, BAY43-9006) is an oral multi-targeted kinase inhibitor, which is capable of inhibiting several kinases involved in tumour progression and angiogenesis (Iyer et al, 2010). It was also recently shown that sorafenib can inhibit both mTOR and (Kumar et al, 2007; Huynh et al, 2009) and MAPK signalling (Keswani et al, 2008). Moreover, our preclinical data demonstrate that the combination of letrozole with sorafenib in aromatase-expressing BC cells shows a synergistic inhibition of cell proliferation in vitro (Bonelli et al, 2010). We showed that sorafenib inhibited BC-cell proliferation in a concentration-dependent manner through a mechanism involving downregulation of mTORC and its downstream targets p70S6K and 4E-binding protein 1 (4E-BP1) (Bonelli et al, 2010). In BC cells treated with letrozole and sorafenib, suppression of cell proliferation was associated with enhanced accumulation of cells in the G0/G1 phase of the cell cycle due to a downregulation of c-myc, cyclin D1 and phospho-Rb expression (Bonelli et al, 2010). Moreover, experiments involving long-term exposure to sorafenib showed that it prevented the acquisition of resistance towards letrozole in BC cells in vitro (Bonelli et al, 2010). These preclinical data provide a strong rational for testing letrozole combined with sorafenib in patients with BC.
Metronomic chemotherapy is the frequent administration of cytotoxic drugs at a low dose that is designed to avoid dose-limiting adverse effects that would require rest periods. In addition, metronomic chemotherapy is proposed to target tumour growth indirectly by inhibiting angiogenesis, rather than directly targeting tumour cells (Pasquier et al, 2010). Our group has previously shown that the combination of letrozole with a metronomic regimen of cyclophosphamide is active in elderly patients with primary BC (Bottini et al, 2006). These data do contrast with studies that combined endocrine therapy with conventional high-dose chemotherapy regimens, where the combination was shown to be antagonistic (Osborne et al, 1989; Albain et al, 2009). However, the favourable results we reported with metronomic cyclophosphamide probably stem from the lower-toxicity profile observed with this regimen and the fact that this therapy targets the vascular compartment instead of targeting tumour cells directly.
On the basis of these findings, we have planned a randomised phase II trial to explore the activity of the letrozole, metronomic chemotherapy and sorafenib combination. This is a neoadjuvant trial in postmenopausal breast cancer patients with ER positive, HER2 negative primary breast cancer and is currently ongoing (Eudract Number 2007-006208-39). In this paper, we present pharmacokinetic, toxicity, response and biomarker data for this regimen in the first thirteen consecutive patients enrolled in the study before randomisation.
Materials and methods
Patients and treatment
This study involved 13 postmenopausal women with ER positive, HER2 negative, T2-4, N0-1 BC enrolled in a single institution. Inflammatory breast cancer was excluded from the study. Eligibility criteria for inclusion were the same as those described for our previous study, which examined the combination of letrozole with metronomic cyclophosphamide in BC (Bottini et al, 2006). Patients received one tablet (2.5 mg) of letrozole per day (Femara; Novartis), one tablet (50 mg) of cyclophosphamide per day (Endoxan; Baxter) and every 5th day they received two tablets (400 mg bid) of sorafenib (Nexavar, Bayer). These drugs were given continuously for 6 months. At the end of treatment the patients underwent definitive surgery. All adverse events (AE) were graded by the National Cancer Institute Common Toxicity Criteria (NCI-CTC), version 3.0.
With regard to the pharmacokinetic profile, during the first 5 days the patients received only letrozole and cyclophosphamide. The administration of sorafenib started from day 6 onwards. The pharmacokinetic profile of sorafenib was assessed on day 29. The pharmacokinetic profile of cyclophosphamide and 4-hydroxy-cyclophosphamide was assessed on days 5 and 29. This approach permitted us to test the safety and tolerability of this regimen and the pharmacokinetic interaction between sorafenib and cyclophosphamide. The decision to continue the study or not depended on evaluations of tolerability, safety and pharmacokinetic profile of cyclophosphamide after 6 weeks. A dose escalation was not planned. If <33% of these patients experienced no significant toxicity on the treatment, the study continued until the conclusion of the pre-planned enrolment. If 33% or more of the patients experienced a significant toxicity during the treatment, the study would have been closed due to unacceptable toxicity. The dose-limiting unacceptable toxicity was defined by the following adverse events during the first cycle that could be correlated to the drug combination:
-
1
Haematological toxicity:
-
Neutropenia <0.5 × 109 l−1 not associated with fever lasting longer than 10 days
-
Neutropenia <0.5 × 109 l−1 associated to fever >38.5 °C
-
Piastrinopenia <25 × 109 l−1 or grade 3 piastrinopenia associated with bleeding
-
-
2
Non-haematological toxicity:
-
grade 3–4 non-haematological toxicity with the exclusion of:
-
a
allergic reactions controlled by an adequate therapy
-
b
grade 4 hypertension crises related to sorafenib
-
c
grade 3 hypertension
-
a
-
Any toxicity correlated to the study drug was unacceptable according to the opinion of the investigators.
Pharmacokinetic analysis
The pharmacokinetic analysis was performed in the Laboratory of Dr Frank-Thorsten Hafner (Bayer HealthCare AG, Preclinical Pharmacokinetics—Bioanalytics, Pharma Research Center, Building 468 D-42096 Wuppertal, Germany). For measurement of sorafenib, plasma samples were collected on day 1 of Cycle 2 (Day 29) following multiple dosing of both 400 mg bid sorafenib and 50 mg of cyclophosphamide in combination with 2.5 mg of letrozole (see Figure 1A). Plasma samples from 13 patients were collected before dosing and at 0.5, 1, 2, 4, 8, 10 and 12 h after dosing. For measurement of cyclophosphamide and its metabolite 4-hydroxy-cyclophosphamide, plasma samples were collected on Day 5 of Cycle 1 (before sorafenib treatment) and on Day 1 of Cycle 2 (Day 29) (after concomitant treatment with multiple oral doses of both drugs) (see Figure 1A). This allowed us to evaluate the effect of multiple oral doses of sorafenib on the pharmacokinetics of cyclophosphamide. Plasma samples from 13 patients were collected before dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h after dosing. The concentration of the drugs was assessed by using a one-way analysis of variance model for dose-normalised and log-transformed maximum observed serum concentration considering the concentration-time curve (AUC). Statistical significance of the factor of dose was set at P=0.05.

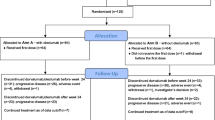
(A) Schematics illustrating the design of the study. (B) Pharmacodynamic profile of the study.
Response assessment
The size of the primary tumour and the axillary lymph nodes was measured monthly with callipers by the same clinician. Tumour size measurements obtained by breast MRI, using protocols described elsewhere (Bonelli et al, 2010), were made at baseline and at the 14th day of treatment and were independently reviewed by two experienced radiologists. Any discrepant readings were resolved by consensus. Response evaluated by either clinical palpation or breast MRI was assessed by the measurement of the changes in the product of the two largest diameters recorded at baseline, at the 14th day and at the end of therapy before surgery, according to the RECIST Criteria.
The 18FDG-PET/CT scans (CT 8 slice) were carried out using a Discovery ST PET/CT scanner (GE Healthcare, Milwaukee, WI, USA) and performed at baseline, after 30 days of treatment and before definitive surgery. The 18FDG-PET/CT images were read independently by two experienced radiologists. Standardised uptake values (SUVs) were calculated. Lesions were scored for response by Modified CT Criteria using 18FDG-PET/CT scans obtained at baseline, after 30 days of treatment and just before definitive surgery. Lesions were scored as follows: CR (disappearance of lesion), PR (decrease in tumour size of ⩾10% or decrease in tumour density of ⩾15%), SD (does not meet the criteria for CR, PR or PD) and PD (an increase in tumour size of ⩾10% and does not meet criteria of PR by tumour density on CT) (Choi et al, 2007).
The observers were blinded for any clinical data including the site of the primary tumour. Discrepancies were resolved by consensus. During the study, clinicians and pathologists were blinded to the 18FDG-PET/CT related data.
Collection and processing of tissue samples
Tissue was obtained from patients at three time points: tissue from incision biopsy performed at presentation, tissue from tru-cut biopsy performed after 14 days of treatment and tissue obtained at definitive surgery. Surgery (quadrantectomy or mastectomy) was planned after full clinical reassessment. All patients subjected to quadrantectomy underwent irradiation of the residual breast (60 Gys delivered in 6 weeks). Immunohistochemistry was performed on paraffin-embedded tumour samples obtained at all three time points. HER-2, ER, PgR, Ki67, CD31 and VEGF-A staining was performed using standard protocols as described elsewhere (Koukourakis et al, 2005; Bottini et al, 2006).
Statistical analysis
Ki67, CD31, VEGF-A, MRI and SUVmax were analysed as continuous variables. Non-parametric statistical methods (Mann–Whitney test for unpaired data, Wilcoxon’s matched-pairs signed-rank test for paired data, Spearman Rho for simple correlation analysis) were used in the primary analyses of the data. The PK analysis was carried out using ANOVA test. All tests were two-sided; P<0.05 was considered as statistically significant. Statistical analysis was performed on an IBM-compatible personal computer using Statistica software (Statsoft, Tulsa, OK, USA) for Windows (Microsoft, Redmond, WA, USA) software.
Ethical approval
The Local Ethical Committee of A.O. Istituti Ospitalieri di Cremona approved this prospective study and signed informed consent was obtained from each patient.
Results
Details of the study population
Thirteen postmenopausal women (median 67 years old, range 53–79) with T2-4, N0-1 (11 patients with T2, and 2 with T4, 6 with node negative and 7 with node positive, 5 with grade 2 and 8 with grade 3) and ER+ve (13/13) and HER2-ve (0/13) BC were prospectively enrolled between April 2009 and May 2010 into the trial. See Table 1A and 1B for details of patient characteristics.
Pharmacokinetic analysis of sorafenib
We examined whether the pharmacokinetics of sorafenib were affected when sorafenib was dosed in combination with letrozole and metronomic cyclophosphamide. Plasma samples were collected on day 1 of Cycle 2 (Day 29) at which time point patients had already received multiple doses of sorafenib, cyclophosphamide and letrozole (Figure 1A). Plasma samples from 13 patients were collected before dosing and at 0.5, 1, 2, 4, 8, 10 and 12 h after dosing. A time course of circulating sorafenib concentrations is shown in Figure 2A. The results for AUC (0–12) and Cmax of sorafenib for Day 1 of Cycle 2 are given as individual and geometric mean values (see Table 1A). The geometric means for AUC (0–12) and Cmax calculated were within the range of values observed after multiple dosing with 400 mg per b.i.d. sorafenib in previous studies (Strumberg et al, 2005, 2007). These findings show that circulating concentrations of sorafenib were not significantly affected by dosing in combination with cyclophosphamide and letrozole.

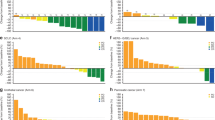
Pharmaockinetics of sorafenib and cyclophosphamide. (A) Plasma concentrations of sorafenib following multiple doses of 400 mg per b.i.d. sorafenib in combination with 50 mg of cyclophosphamide and 2.5 mg of letrozole on Day 1 of Cycle 2 (geometric means, geometric s.d.; n=13). (B) Plasma concentrations of cyclophosphamide (*) following multiple doses of 50 mg of cyclophosphamide in combination with 2.5 mg of letrozole without (Day 5, Cycle 1) and after concomitant treatment (Day 1, Cycle 2) with multiple oral doses of 400 mg per b.i.d. sorafenib (G.M.=geometric means, G.s.d.= geometric standard deviation; n=13) *More than one-third of individual plasma concentrations were below LLOQ at 24 h after dosing on both profile days. No corresponding geometric mean concentrations was calculated.
Pharmacokinetic analysis of cyclophosphamide
We then examined whether dosing of sorafenib affects the pharmacokinetics of cyclophosphamide. Plasma samples were collected on Day 5 of Cycle 1 (before sorafenib treatment) and on Day 1 of Cycle 2 (Day 29) (after concomitant treatment with multiple oral doses of both sorafenib and cyclophosphamide) (see Figure 1A). This allowed us to evaluate the effect of multiple oral doses of sorafenib on the pharmacokinetics of cyclophosphamide. Although cyclophosphamide was measurable in all samples up to 12 h, only a few isolated plasma samples obtained at 24 h contained concentrations above the lower limit of quantification (LLOQ), so we limited our pharmacokinetic analysis to the time points up to 12 h. Geometric mean plasma concentration time courses of cyclophosphamide obtained in patients before and after exposure to sorafenib are shown in Figure 2A. The results for AUC (0–24), Cmax and t1/2 of cyclophosphamide for Day 5 of Cycle 1 and for Day 1 of Cycle 2 are given as individual and geometric mean values in Table 1B. Mean plasma concentrations of cyclophosphamide were consistently lower following concomitant administrations of sorafenib and letrozole compared with concomitant administration of letrozole alone (Figure 2B). This trend was confirmed by the comparison of corresponding AUC (0–24) and Cmax values from Day 5, Cycle 1 and Day 1 of Cycle 2. AUC (0–24) of cyclophosphamide was consistently lower in all subjects when both drugs were co-administered, on average by 29% (Table 1B). Cmax was also reduced in most subjects on Day 1 of Cycle 2 but to a lesser degree, on average by 12%. The decrease in AUC (0–24) was accompanied by a corresponding decrease in t1/2, on average by 27%. These data indicate that concomitant administration of sorafenib moderately decreases the exposure to cyclophosphamide.
Toxicity assessment
We did not observe particular haematological toxicity; only neutropenia grade 1–2 (38.5% of the cases) and grade 3 (7.7% of the cases). The non-haematological toxicity of letrozole and cyclophospamide in combination with sorafenib was mainly grade 1 or 2 as reported in Table 2. The most common drug-related grade 3–4 adverse events were skin rash (69.3%), hand-foot skin reaction (69.3%), diarrhoea (46.1%). Examples of skin rash and hand-foot skin reaction are depicted in Figure 3A and B, respectively. Dosing interruptions or reductions due to an AE did not occur.

Common drug-related adverse events. (A) Grade 2 skin rash. (B) Grade 2 hand-foot syndrome. Both of these reactions are adverse events related to the treatment with sorafenib.
Response assessment by palpation, mammography and pathology
Complete response by clinical palpation, evaluated at the end of treatment before definitive surgery, was observed in 6 out of 13 patients (46%), whereas partial response was observed in the remaining 7 patients (54%). Complete response by mammography was observed in 5 out of 13 patients (38.4%), whereas partial response was observed in the remaining 8 patients (61.6%) before definitive surgery. At the definitive surgery, we observed two patients with pT1b tumour, three patients with pT1c tumour, five patients with pT2 tumour, two patients with pT4b tumour, with 11 out of 13 patients having lymph node involvement. No change of tumour grading between the baseline sample and post-treatment samples was noted. None of the enrolled patients showed a post-treatment pathologic complete response.
Imaging assessment by MRI, FDG-PET and Modified CT criteria
Tumour response was assessed by an MRI scan at baseline and at 14 days of treatment (Figure 1B). Median tumour size evaluated by an MRI was 31 mm (range 17–90 mm) at baseline, 27 mm (range 11–90 mm) at day 14 with a small but statistically significant reduction in tumour size being observed in all 13 patients at day 14 (P=0.005).
The metabolic activity of the tumours was evaluated by a 18FDG -PET/CT scan at baseline (PET1), after 1 month of treatment (PET2) and at the end of the treatment before definitive surgery (PET3) for all 13 patients (Figures 1B, 4A and 4B). At PET1 the mean average SUVmax was 5.01 with a median of 4.5 (range 1.8–12). At PET 2 the mean average SUVmax was 2.39 with a median of 1.9 (range 0–4.5). At PET3 the mean average SUVmax was 0.71 with a median of 0 (range 0–3.9). In this series of patients we identified a significant reduction between PET1 and PET2 (P=0.015), between PET2 and PET3 (P=0.006) and between PET1 and PET3 (P=0.0002) (Figure 4B).

Measurment of responses with 18FDG PET/CT imaging. (A) Example images of 18FDG PET/CT scan obtained from the same patient before (PET1) and after treatment (PET3). A change in the metabolic activity of the tumour between baseline and end of the treatment is clearly observed. (B) Tumour SUVmax values by the 18FDG PET/CT scan. The trend of the tumour metabolic activity during treatment for each patient. A significant reduction in SUVmax has been quantified in all thirteen patients from baseline to the definitive surgery (P-value: between PET1 and PET2 (P=0.015), between PET2 and PET3, (P=0.006) and between PET1 and PET3 (P=0.0002)).
We also monitored the SUVmax in the lymph nodes. At baseline, 10 out of 13 patients presented a positive uptake suggesting tumour involvement of lymph nodes. After 1 month of treatment, seven patients showed a reduction in SUVmax values in the lymph nodes. By the end of treatment, all of the patients had a complete metabolic response in the lymph nodes and in the remaining patients the lymph nodes still showed lower metabolic activity compared with baseline. The average of the SUVmax at PET1 was 2.89 (median=2.50, range 1.1–7.1); at PET2 it was 1.52 (median=1.30, range 0–3.3) and at PET3 it was 0.17 (median=0, range 0–1.7). A significant reduction of SUVmax in the lymph nodes was therefore observed between PET1 and PET2 (P=0.045), PET2 and PET3 (P=0.0008), and PET1 and PET3 (P=0.0002). Using the Modified CT Criteria (Choi et al, 2007), to evaluate response at 30 days and response just before surgery, our study showed response (CR or PR) in 8 patients and 2 patients with stable disease (n=10 evaluable patients) at 30 days and a response in 11 patients (n=11 evaluable patients) at definitive surgery (Table 3).
Assessment of biological markers
Biological markers were assessed in samples obtained at baseline, at 14 days and at definitive surgery (Figure 1B). Ki67 labelling index was assessable in 11 of the 13 patients, but there was insufficient tissue available for the remaining two patients. At baseline the mean average Ki67 index was 20.69% with median of 20% (range 8–36), at day 14 the mean was 10.64% with median of 10% (range 1–20) and at definitive surgery the mean was 11.38% with median of 7% (range 0–34) (Figure 5A). We observed a marked suppression of the Ki67 proliferation index after 14 days in the 11 patients that could be evaluated (P<0.00001), and there was a significant reduction in Ki67 index at definitive surgery compared with baseline (P=0.03). However, 4 patients out of the 11 evaluated showed an increase in Ki67 index at definitive surgery. At the end of treatment, these four patients showed a clinical partial response, which corresponded to a pT2 and pT4 at the pathology report. It is possible that these four patients experienced early-onset resistance to the therapeutic regimen.

Analysis of molecular markers. (A) Ki67 labelling index at baseline, day 14 and at the definitive surgery. Proliferation trend is reported for each patient. A significant reduction in mean average proliferation index is observed between baseline and day 14 (P=0.00001) and between baseline and end of treatment (P=0.031). (B) Immunohistochemistry demonstrating the expression of angiogenesis-related markers (CD31 and VEGF-A) in pre-treatment and post-treatment samples.
Change in expression of the angiogenesis-related markers CD31 and VEGF-A was also evaluated (Figure 5B). CD31 expression could be evaluated in 10 of the 13 patients at baseline (median expression=3, range 1–5), in 8 out of 13 at day 14 (median expression=1, range 1–3) and in 8 out of 13 at definitive surgery (median expression=1, range 1–3). The CD31 expression showed a significant reduction at day 14 (P=0.006) and at definitive surgery (P=0.01) compared with baseline. VEGF-A expression could be evaluated in 9 of the 13 patients at baseline (median expression=2, range 1–2), in 8 out of 13 at day 14 (median expression=1, range 1–3) and in 9 out of 13 at definitive surgery (median expression=1, range 1–3). No significant difference in VEGF-A expression was found between baseline and day 14, but a significant reduction at the definitive surgery compared with baseline was observed (P=0.007) (Figure 5B). A correlation was observed between the decrease in CD31 and the decrease in VEGF-A expression, both at day 14 (P<0.0001) and at the definitive surgery (P<0.0001).
Discussion
Endocrine resistance presents a major challenge in the management of ER+ve breast cancer and is an area under intense investigation. Non-genomic cross-talk between ER and growth factor receptors can activate intracellular signalling cascades involved in oestrogen-independent growth, such as the Ras-Raf-mitogen-activated protein kinase (MAPK) (Massarweh et al, 2008) and the phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR pathways (Johnston et al, 2005). Moreover, both MAPK and AKT can directly enhance the classic genomic transcriptional activity of ER in the absence of oestrogens (Le Goff et al, 1994) further contributing to endocrine resistance. It has been shown that letrozole-based treatment is able to significantly reduce PI3K and phospho-mTOR expression (Generali et al, 2008) and that HIF-1alpha and p44/42 MAPK expression is independent factor for endocrine resistance in neoadjuvant setting (Generali et al, 2009). These data suggest that targeting these pathways could potentiate the aromatase inhibitor effect, preventing or postponing the onset of endocrine resistance. Therefore, combined inhibition of the oestroge pathway and growth factor signalling represents an attractive target for pharmacological intervention. Sorafenib is an oral multikinase inhibitor, capable of inhibiting mTOR signalling and HIF-1alpha expression (Kumar et al, 2007; Huynh et al, 2009) or MAPK expression and related proliferation proteins (Keswani et al, 2008). Our previous in vitro experiments with the combination of letrozole and sorafenib demonstrated a potential role for this combination in the treatment of hormone-dependent BC cells (Bonelli et al, 2010). However, to date, the combination of sorafenib with letrozole and metronomic administration of cyclophosphamide has never been tested in a clinical setting. We have therefore investigated the pharmacology, toxicity and efficacy of this regimen in primary breast cancer patients.
Pharmacokinetic analysis showed there was no impact of the combination regimen on the circulating levels of sorafenib in patients. Moreover, the analysis of the pharmacokinetic data showed that there was no relevant impact of sorafenib (400 mg BID) on the pharmacokinetics of cyclophosphamide when these compounds were co-administered. However, the data do indicate that concomitant sorafenib moderately decreased the exposure (Cmax and AUC0–24) to cyclophosphamide, without being influenced by the sorafenib total dose. Indeed it is recognised that cyclophosphamide is a potent inducer of microsomal enzymes and is able to increase levels of CYP2C8, 2C9 and 3A4 inducing its own metabolism (Chang et al, 1997). As previously shown, a similar induction by cyclophosphamide of human CYP2B6 and 3A4 enzyme expression occurs (Gervot et al, 1999; Lindley et al, 2002). This auto-induction in cyclophosphamide metabolism is detectable within 24 h after the start of the treatment and might explain the reduction of cyclophosphamide plasma concentrations. However, further investigation in a larger population of cancer patients and a more accurate evaluation of all cyclophosphamide-derived metabolites is warranted.
The metronomic administration of chemotherapy compounds is a good treatment option for breast cancer patients, has a low-toxicity profile and high efficacy in most patients (Montagna et al, 2014). We previously demonstrated that co-administration of endocrine therapy with the metronomic administration of cyclophosphamide is well tolerated without any particular adverse events (Bottini et al, 2006).
In the current study, we show that sorafenib administered with cyclophosphamide and letrozole had a safety profile consistent with that of the individual agents in patients with breast cancer. The most common toxicities were skin rash, hand-foot skin reaction and diarrhea Although all are reported to be associated with sorafenib (Gomez & Lacouture, 2011), it is difficult to determine whether sorafenib was the sole agent responsible for the relative high incidence of these events among the enrolled patients. Across three phase 2 randomised studies where advanced breast cancer patients were treated with sorafenib in combination with chemotherapy (Baselga et al, 2012; Gradishar et al, 2013; Schwartzberg et al, 2013), the incidence of hand-foot syndrome, rash and diarrhea was 31–44, 2–4 and 3–6%, respectively. By comparison, the incidence of hand-foot syndrome (69.3%), rash (69.3%) and diarrhea (46.1%) was elevated in our study. On the contrary, the incidence of other grade 3/4 toxicities reported in our study, such as fatigue and neutropenia, was similar to those reported in these previous studies that combined chemotherapy with sorafenib. However, despite this incidence of toxicity, in our experience the timely detection of adverse events in patients helps to mitigate their clinical consequences.
It is also of interest to compare the toxicity profile of LCS with a previous study that combined endocrine therapy with sorafenib in breast cancer. Isaacs et al (2011) reported a phase I/II study that employed a combination of sorafenib and anastrazole in patients with metastatic breast cancer. In that study, 77% of patients required dose reductions of sorafenib and 31% came off study due to toxicity. The most common grade 3/4 toxicities were hand-foot syndrome (34%), fatigue (17%), rash (11%), emesis (11%) and hypertension (11%). Among the 35 women enrolled into that phase I/II trial, 46% had an ECOG ⩾1, 23% had visceral metastasis, 43% had bone metastasis and all had received one or two lines of previous therapy. In contrast, the women enrolled in our study were naive of any treatment and without any visceral/bone involvement. These differences in the patient populations may explain the different toxicity profile reported by our group in comparison with the study by Isaacs et al (2011).
Clinical anti-cancer activity (complete and partial responses) was observed in all patients. This was objectively confirmed by the 18FDG-PET/CT scan, with decreased SUVmax values observed in all 13 patients after treatment. However, FDG PET/CT failed to detect 11 instances of isolated tumour cells, micro-embolic or plural-embolic in axillary nodes, which were reported in the definitive pathological report. In some studies the role of FDG PET/CT in the axillary staging of breast cancer has been questioned with FDG PET/CT proving to have poor sensitivity compared with immunohistochemistry on lymph nodes biopsy in which multistep sectioning is commonly used to identify axillary micrometastases (that is, clusters of malignant cells >0.2 mm to ⩽2.0 mm in diameter) (Guller et al, 2002; Kelemen et al, 2002; van der Hoeven et al, 2002). Thus, the limited spatial resolution of the current FDG PET/CT scanners in comparison with the more accurate pathological examination techniques is a reason for these modest results of PET in the detection of micro-metastases.
The Ki67 labelling index after 2 weeks of therapy and at definitive surgery was significantly lower than the labelling index at the baseline for eight patients. In particular, we noticed that the reduction of Ki67 expression was greater between the beginning and the end of the treatment than after 15 days of treatment. Taken together, these data suggest that the tried and tested letrozole-cyclophosphamide combination (Bottini et al, 2006) with sorafenib is promising in terms of metabolic activity and suppression proliferation index. Of note, however, are four patients that did not show a reduction in tumour proliferation activity at the end of the treatment. As previously described by Dowsett et al (2005), this might be an indicator of endocrine resistance. However, Ki67 may not be the most reliable indicator of tumour cell proliferation. As Dowsett et al (2005) have proposed, to better investigate the anti-proliferative effect of therapy, it might be more appropriate to analyse other indices of cell proliferation, such as S phase markers or MCM2.
Previous reports showed the metronomic schedule is able to modulate several factors, such as VEGF-A, circulating endothelial cells and fibroblast growth factor 2, which are all involved in remodelling of cancer vessels (Mancuso et al, 2006; Calleri et al, 2009; Isaacs et al, 2011). Our group has shown that the combination of letrozole and metronomic cyclophosphamide has an anti-angiogenic effect with a significant reduction of VEGF-A after 6 months of neoadjuvant treatment (Bottini et al, 2006). Here we investigated the expression of two robust markers, CD31 and VEGF-A, supported also by published data on their modulation and their correlation with treatment response to bevacizumab (Yang et al, 2008). The expression of CD31 and VEGF-A was significantly decreased after treatment, supporting the contention that the administered regimen has an anti-angiogenic effect in primary BC.
In this study, we also used the breast MRI technique as a more accurate method than breast ultrasound and mammography or clinical palpation (Bodini et al, 2004) in evaluating the disease response to therapy and because of its ability to delineate anatomic detail of breast cancers. In our study, there was only a modest evidence of significant changes in tumour dimensions after 14 days of treatment, maybe due to the short interval between the two evaluations. In the future, it would be preferable to perform the breast MRI at time points further into treatment to monitor properly any differences in clinical response induced by the treatment (Bodini et al, 2004). Moreover, improvements in an MRI technology for the monitoring of blood-flow as a marker of tumour angiogenesis (Mehta et al, 2011) are warranted in future trials containing anti-angiogenic drugs such as sorafenib and or metronomic cyclophosphamide.
In conclusion, the addition of sorafenib to the already tested letrozole-cyclophosphamide combination was associated with a clinical response, a significant decrease in tumour metabolic activity, in proliferation index and in angiogenesis in patients with hormone receptor positive primary breast cancer. Conceivably, this benefit may be due to the ability of sorafenib to overcome resistance to endocrine therapy and/or the increased anti-angiogenic activity when sorafenib is combined with the metronomic administration of cyclophosphamide.
Change history
06 January 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Albain KS, Barlow WE, Ravdin PM, Farrar WB, Burton GV, Ketchel SJ, Cobau CD, Levine EG, Ingle JN, Pritchard KI, Lichter AS, Schneider DJ, Abeloff MD, Henderson IC, Muss HB, Green SJ, Lew D, Livingston RB, Martino S, Osborne CK Breast Cancer Intergroup of North A (2009) Adjuvant chemotherapy and timing of tamoxifen in postmenopausal patients with endocrine-responsive, node-positive breast cancer: a phase 3, open-label, randomised controlled trial. Lancet 374 (9707): 2055–2063.
Baselga J, Segalla JG, Roche H, Del Giglio A, Pinczowski H, Ciruelos EM, Filho SC, Gomez P, Van Eyll B, Bermejo B, Llombart A, Garicochea B, Duran MA, Hoff PM, Espie M, de Moraes AA, Ribeiro RA, Mathias C, Gil Gil M, Ojeda B, Morales J, Kwon Ro S, Li S, Costa F (2012) Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol 30 (13): 1484–1491.
Bodini M, Berruti A, Bottini A, Allevi G, Fiorentino C, Brizzi MP, Bersiga A, Generali D, Volpi D, Marini U, Aguggini S, Tampellini M, Alquati P, Olivetti L, Dogliotti L (2004) Magnetic resonance imaging in comparison to clinical palpation in assessing the response of breast cancer to epirubicin primary chemotherapy. Breast Cancer Res Treatment 85 (3): 211–218.
Bonelli MA, Fumarola C, Alfieri RR, La Monica S, Cavazzoni A, Galetti M, Gatti R, Belletti S, Harris AL, Fox SB, Evans DB, Dowsett M, Martin LA, Bottini A, Generali D, Petronini PG (2010) Synergistic activity of letrozole and sorafenib on breast cancer cells. Breast Cancer Res Treatment 124 (1): 79–88.
Bottini A, Generali D, Brizzi MP, Fox SB, Bersiga A, Bonardi S, Allevi G, Aguggini S, Bodini G, Milani M, Dionisio R, Bernardi C, Montruccoli A, Bruzzi P, Harris AL, Dogliotti L, Berruti A (2006) Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J Clin Oncol 24 (22): 3623–3628.
Calleri A, Bono A, Bagnardi V, Quarna J, Mancuso P, Rabascio C, Dellapasqua S, Campagnoli E, Shaked Y, Goldhirsch A, Colleoni M, Bertolini F (2009) Predictive potential of angiogenic growth factors and circulating endothelial cells in breast cancer patients receiving metronomic chemotherapy plus bevacizumab. Clin Cancer Res 15 (24): 7652–7657.
Chang TK, Yu L, Maurel P, Waxman DJ (1997) Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res 57 (10): 1946–1954.
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS (2007) Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 25 (13): 1753–1759.
Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, Griffith C, Boeddinghaus I, Salter J, Detre S, Hills M, Ashley S, Francis S, Walsh G, Trialists I (2005) Short-term changes in Ki-67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence-free survival. Clin Cancer Res 11 (2 Pt 2): 951s–958ss.
Generali D, Buffa FM, Berruti A, Brizzi MP, Campo L, Bonardi S, Bersiga A, Allevi G, Milani M, Aguggini S, Papotti M, Dogliotti L, Bottini A, Harris AL, Fox SB (2009) Phosphorylated ERalpha, HIF-1alpha, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J Clin Oncol 27 (2): 227–234.
Generali D, Fox SB, Brizzi MP, Allevi G, Bonardi S, Aguggini S, Milani M, Bersiga A, Campo L, Dionisio R, Vergoni F, Giardini R, Dogliotti L, Bottini A, Harris AL, Berruti A (2008) Down-regulation of phosphatidylinositol 3'-kinase/AKT/molecular target of rapamycin metabolic pathway by primary letrozole-based therapy in human breast cancer. Clin Cancer Res 14 (9): 2673–2680.
Gervot L, Rochat B, Gautier JC, Bohnenstengel F, Kroemer H, de Berardinis V, Martin H, Beaune P, de Waziers I (1999) Human CYP2B6: expression, inducibility and catalytic activities. Pharmacogenetics 9 (3): 295–306.
Gligorov J, Azria D, Namer M, Khayat D, Spano JP (2007) Novel therapeutic strategies combining antihormonal and biological targeted therapies in breast cancer: focus on clinical trials and perspectives. Crit Rev Oncol Hematol 64 (2): 115–128.
Gomez P, Lacouture ME (2011) Clinical presentation and management of hand-foot skin reaction associated with sorafenib in combination with cytotoxic chemotherapy: experience in breast cancer. Oncologist 16 (11): 1508–1519.
Gradishar WJ, Kaklamani V, Sahoo TP, Lokanatha D, Raina V, Bondarde S, Jain M, Ro SK, Lokker NA, Schwartzberg L (2013) A double-blind, randomised, placebo-controlled, phase 2b study evaluating sorafenib in combination with paclitaxel as a first-line therapy in patients with HER2-negative advanced breast cancer. Eur J Cancer 49 (2): 312–322.
Guller U, Nitzsche EU, Schirp U, Viehl CT, Torhorst J, Moch H, Langer I, Marti WR, Oertli D, Harder F, Zuber M (2002) Selective axillary surgery in breast cancer patients based on positron emission tomography with 18F-fluoro-2-deoxy-D-glucose: not yet!. Breast Cancer Res Treatment 71 (2): 171–173.
Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Thng CH, Chow P, Ong HS, Chung A, Soo KC (2009) Sorafenib and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J Cell Mol Med 13 (8B): 2673–2683.
Isaacs C, Herbolsheimer P, Liu MC, Wilkinson M, Ottaviano Y, Chung GG, Warren R, Eng-Wong J, Cohen P, Smith KL, Creswell K, Novielli A, Slack R (2011) Phase I/II study of sorafenib with anastrozole in patients with hormone receptor positive aromatase inhibitor resistant metastatic breast cancer. Breast Cancer Res Treatment 125 (1): 137–143.
Iyer R, Fetterly G, Lugade A, Thanavala Y (2010) Sorafenib: a clinical and pharmacologic review. Expert Opin Pharmacother 11 (11): 1943–1955.
Johnston SR, Martin LA, Head J, Smith I, Dowsett M (2005) Aromatase inhibitors: combinations with fulvestrant or signal transduction inhibitors as a strategy to overcome endocrine resistance. J Steroid Biochem Mol Biol 95 (1-5): 173–181.
Johnston SR, Martin LA, Leary A, Head J, Dowsett M (2007) Clinical strategies for rationale combinations of aromatase inhibitors with novel therapies for breast cancer. J Steroid Biochem Mol Biol 106 (1-5): 180–186.
Kelemen PR, Lowe V, Phillips N (2002) Positron emission tomography and sentinel lymph node dissection in breast cancer. Clin Breast Cancer 3 (1): 73–77.
Keswani RN, Chumsangsri A, Mustafi R, Delgado J, Cohen EE, Bissonnette M (2008) Sorafenib inhibits MAPK-mediated proliferation in a Barrett's esophageal adenocarcinoma cell line. Dis Esophagus 21 (6): 514–521.
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL Tumour, Angiogenesis Research G (2005) Inclusion of vasculature-related variables in the Dukes staging system of colon cancer. Clin Cancer Res 11 (24 Pt 1): 8653–8660.
Kumar SM, Yu H, Edwards R, Chen L, Kazianis S, Brafford P, Acs G, Herlyn M, Xu X (2007) Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res 67 (7): 3177–3184.
Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269 (6): 4458–4466.
Lindley C, Hamilton G, McCune JS, Faucette S, Shord SS, Hawke RL, Wang H, Gilbert D, Jolley S, Yan B, LeCluyse EL (2002) The effect of cyclophosphamide with and without dexamethasone on cytochrome P450 3A4 and 2B6 in human hepatocytes. Drug Metab Dispos 30 (7): 814–822.
Mancuso P, Colleoni M, Calleri A, Orlando L, Maisonneuve P, Pruneri G, Agliano A, Goldhirsch A, Shaked Y, Kerbel RS, Bertolini F (2006) Circulating endothelial-cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood 108 (2): 452–459.
Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R (2008) Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 68 (3): 826–833.
Mehta S, Hughes NP, Buffa FM, Li SP, Adams RF, Adwani A, Taylor NJ, Levitt NC, Padhani AR, Makris A, Harris AL (2011) Assessing early therapeutic response to bevacizumab in primary breast cancer using magnetic resonance imaging and gene expression profiles. J Natl Cancer Inst Monogr 2011 (43): 71–74.
Montagna E, Cancello G, Dellapasqua S, Munzone E, Colleoni M (2014) Metronomic therapy and breast cancer: A systematic review. Cancer Treatment Rev 40 (8): 942–950.
Nicholson RI, Hutcheson IR, Hiscox SE, Knowlden JM, Giles M, Barrow D, Gee JM (2005) Growth factor signalling and resistance to selective oestrogen receptor modulators and pure anti-oestrogens: the use of anti-growth factor therapies to treat or delay endocrine resistance in breast cancer. Endocr Relat Cancer 12 (Suppl 1): S29–S36.
Nicholson RI, Hutcheson IR, Knowlden JM, Jones HE, Harper ME, Jordan N, Hiscox SE, Barrow D, Gee JM (2004) Nonendocrine pathways and endocrine resistance: observations with antiestrogens and signal transduction inhibitors in combination. Clin Cancer Res 10 (1 Pt 2): 346S–354SS.
Osborne CK, Kitten L, Arteaga CL (1989) Antagonism of chemotherapy-induced cytotoxicity for human breast cancer cells by antiestrogens. J Clin Oncol 7 (6): 710–717.
Osborne CK, Shou J, Massarweh S, Schiff R (2005) Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11 (2 Pt 2): 865s–870ss.
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7 (8): 455–465.
Schwartzberg LS, Tauer KW, Hermann RC, Makari-Judson G, Isaacs C, Beck JT, Kaklamani V, Stepanski EJ, Rugo HS, Wang W, Bell-McGuinn K, Kirshner JJ, Eisenberg P, Emanuelson R, Keaton M, Levine E, Medgyesy DC, Qamar R, Starr A, Ro SK, Lokker NA, Hudis CA (2013) Sorafenib or placebo with either gemcitabine or capecitabine in patients with HER-2-negative advanced breast cancer that progressed during or after bevacizumab. Clin Cancer Res 19 (10): 2745–2754.
Smith IE, Dowsett M (2003) Aromatase inhibitors in breast cancer. N Engl J Med 348 (24): 2431–2442.
Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B (2007) Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12 (4): 426–437.
Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, Schwartz B, Awada A, Voigtmann R, Scheulen ME, Seeber S (2005) Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol 23 (5): 965–972.
van der Hoeven JJ, Hoekstra OS, Comans EF, Pijpers R, Boom RP, van Geldere D, Meijer S, Lammertsma AA, Teule GJ (2002) Determinants of diagnostic performance of [F-18]fluorodeoxyglucose positron emission tomography for axillary staging in breast cancer. Ann Surg 236 (5): 619–624.
Yang SX, Steinberg SM, Nguyen D, Wu TD, Modrusan Z, Swain SM (2008) Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin Cancer Res 14 (18): 5893–5899.
Acknowledgements
This study was partly funded by the FIRM (Fondazione Internazionale per la Ricerca In Medicina), Cremona, Italy. Dr Reynolds acknowledges support from Breakthrough Breast Cancer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have declared no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Bazzola, L., Foroni, C., Andreis, D. et al. Combination of letrozole, metronomic cyclophosphamide and sorafenib is well-tolerated and shows activity in patients with primary breast cancer. Br J Cancer 112, 52–60 (2015). https://doi.org/10.1038/bjc.2014.563
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.563
Keywords
This article is cited by
-
PET/CT-Based Response Evaluation in Cancer—a Systematic Review of Design Issues
Molecular Imaging and Biology (2020)
-
Tumor endothelial cells as a potential target of metronomic chemotherapy
Archives of Pharmacal Research (2019)
-
Current achievements and future perspectives of metronomic chemotherapy
Investigational New Drugs (2017)
-
Pharmacokinetics of metronomic chemotherapy: a neglected but crucial aspect
Nature Reviews Clinical Oncology (2016)
