
Abstract
Background:
Bevacizumab prolongs progression-free survival (PFS) in patients with metastatic colorectal cancer. We analysed the protein expression levels of vascular endothelial growth factor (VEGF) ligands and receptors to determine their prognostic and predictive effects.
Methods:
We graded expression of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-R1, and VEGF-R2 to assess whether overexpression predicted bevacizumab resistance in samples from 268 of 471 patients randomised to capecitabine (C), capecitabine and bevacizumab (CB), or CB and mitomycin (CBM) in the MAX trial and extended the analysis to the CAIRO-2 population.
Results:
Patients with low expression of VEGF-D (0, 1+) benefited from bevacizumab treatment (PFS hazard ratio (HR) (C vs CB+CBM), 0.21; 95% CI, 0.08–0.55; overall survival (OS) HR, 0.35; 95% CI, 0.13–0.90). Patients with higher VEGF-D expression received less benefit (VEGF-D 2+ PFS HR, 0.67; 95% CI, 0.45–1.00; OS HR, 0.82; 95% CI, 0.52–1.30; VEGF-D 3+ PFS HR, 0.77; 95% CI, 0.50–1.17; OS HR, 1.28; 95% CI, 0.79–2.09) (P interaction <0.05). In CAIRO-2, there was no difference in PFS or OS according to VEGF-D expression.
Conclusions:
The predictive value of VEGF-D expression for bevacizumab may depend on the chemotherapy backbone used. Further evaluation is required before clinical utilisation.
Similar content being viewed by others


Main
Angiogenesis is necessary for tumour proliferation and metastasis and presents an attractive target for drug therapy. Circulating angiogenic factors include the vascular endothelial growth factor (VEGF) family (A–D), placental growth factor (PlGF), platelet-derived growth factors (PDGFs), and fibroblast growth factors (FGFs). They bind to a range of receptors, including VEGF receptors (VEGFRs) 1–3 on endothelial and tumour cells, to regulate angiogenesis (Kerbel, 2008). Bevacizumab, a monoclonal antibody that binds to and inactivates VEGF-A, is an antiangiogenic drug that, when used in combination with chemotherapy, improves progression-free survival (PFS) in patients with metastatic colorectal cancer (Hurwitz et al, 2013). However, the benefit gained is modest (Hurwitz et al, 2005; Saltz et al, 2008; Tebbutt et al, 2010), highlighting the need to identify responsive and resistant subgroups through biomarker studies.
Although several studies have been undertaken, none has identified or validated a clinically applicable predictive biomarker for bevacizumab efficacy (Lambrechts et al, 2013). These studies have included: serum or plasma biomarkers such as VEGF-A levels; tissue biomarkers such as VEGF-A, VEGFR-2, and neuropilin-1; levels of circulating endothelial cells; imaging changes, VEGF or VEGFR single-nucleotide polymorphisms (SNPs); and dynamic biomarkers, such as hypertension (Jubb et al, 2006; Dowlati et al, 2008; Schneider et al, 2008; Foernzler et al, 2010; Jubb and Harris, 2010; Jubb et al, 2011; Van Cutsem et al, 2012; Miles et al, 2013).
It is recognised that there may be redundancy among family members of angiogenic regulators (Achen and Stacker, 1998; Cao et al, 1998; Pan et al, 2007; Zhang et al, 2010). We hypothesised therefore that related VEGF family members such as VEGF-C and VEGF-D may continue to stimulate angiogenesis despite inhibition of VEGF-A by bevacizumab (Jubb et al, 2011).
The AGITG MAX trial was an investigator-initiated study evaluating the effect on PFS of adding bevacizumab to capecitabine chemotherapy as first-line therapy for metastatic colorectal cancer. We evaluated the expression of angiogenesis-related factors (ARFs) VEGF-A to VEGF-D, VEGFR-1, and VEGFR-2 in tumour tissue as predictors of efficacy of bevacizumab by correlating the expression with clinical outcomes in the MAX study and for significant biomarkers attempted to support the result in separate tumour specimens from patients in the CAIRO2 trial (Tol et al, 2009).
Subjects and Methods
Patients and study design
The primary objective of the phase III MAX study (Tebbutt et al, 2010) was to evaluate the effect on PFS of adding bevacizumab with or without mitomycin to capecitabine among patients receiving first-line chemotherapy for metastatic colorectal cancer. Eligible patients were enrolled between July 2005 and June 2007 and randomly assigned to capecitabine (C), capecitabine and bevacizumab (CB), or capecitabine, bevacizumab, and mitomycin (CBM). Patients were evaluated for tumour response or progression every 6 weeks. Treatment was continued until disease progression in the absence of significant toxicity. All patients provided written informed consent for the main study, and most provided additional optional consent for provision of tumour tissue for biomarker analyses. Separate ethics approval for biomarker studies was obtained centrally.
Tumour collection and processing
Formalin-fixed, paraffin-embedded samples of tumour tissue from archival specimens collected at the time of diagnosis were retrieved where possible. Although the majority of specimens (83%) were from the primary tumour, a minority only had specimens from metastatic sites to assess. Fourteen patients had both primary and secondary specimens to compare the expression levels. Tumour blocks were collected and analysed centrally by technicians blinded to trial outcome data.
Guided by a haematoxylin and eosin-stained slide, three representatve 1-mm adjacent tumour cores were extracted from each patient’s tumour section and assembled into a recipient block using a Beecher mark II tissue arrayer (Beecher Instruments, Sun Prairie, WI, USA). Cores from a renal cortex specimen were also inserted on each tissue microarray (TMA) for orientation and to serve as a positive control for staining.
Immunohistochemical analysis
Four 1-μm sections were cut from each TMA and mounted on individual microscope slides. The sections were deparaffinised by baking at 60 °C for 1 h, followed by two washes each with xylene and then with ethanol. Slides were rehydrated with a wash in deionised water. Antigen retrieval was performed for 30 min at 100 °C using Citrate (Labvision, Fremont, CA, USA) or for 60 min with Dako target retrieval solution (Dako, Glostrup, Denmark). No antigen retrieval was required for the anti-VEGF-B antibody. After cooling for 20 min, slides were washed in tap water and then twice in trisethanolamine-buffered saline (Dako) with 0.05% Tween 20 detergent (TBS-T). Endogenous peroxidase was quenched by incubating the slides in 3% hydrogen peroxide for 10 min at room temperature, and the slides were washed twice more with TBS-T.
Slides were incubated with primary antibody at room temperature or at 37 °C at an optimised concentration (Supplementary Table S1). Slides were washed twice more in TBS-T before exposure to the appropriate secondary antibody at room temperature, again washed twice in TBS-T, and incubated with chromogen 3-amino-9-ethylcarbazole (Sigma-Aldrich, St Louis, MO, USA) for 5–15 min until a signal developed. They were then washed in water and counterstained with haematoxylin and Scott’s solution (20 g MgSO4 and 3.5 g NaHCO3 per litre DH20). Following further washes in water, slides were coated with CC/Mount (Sigma-Aldrich) aqueous mounting medium and coverslips were applied. For each antibody, a negative control was prepared by parallel staining of each slide with an appropriate subclass control antibody.
Scoring was performed independently by two investigators, who were blinded to treatment allocation and outcome. Grading of intensity and extent of staining, in tumour cells but not adjacent stroma, was 0=negative; 1=weak widespread/very limited moderate staining (i.e., <25% of cells); 2=moderate widespread; and 3=strong widespread staining (Supplementary Figure S1). Where possible, the patient’s metastatic tissue rather than primary tumour was assessed for expression. For the purpose of this analysis, when there was a discrepancy in the scores for a given patient, it was resolved by consensus on a second review by two scorers.
Statistical analysis
Statistical analyses were in accordance with a protocol for statistical analysis developed with blinding to treatment allocation and patient outcomes. No prior information was available regarding the distribution of these biomarkers. The final choice of cut points for each biomarker (0, 1+; 2+; 3+) was based on a pooled distribution of biomarkers in all the three treatment groups. Tests for each biomarker considered these as ordered categories.
All patients for whom data on biomarker expression were available were included in the analysis. PFS, the primary end point, was defined as the time from randomisation until documented evidence of disease progression according to the Response Evaluation Criteria in Solid Tumours (RECIST, version 1.0), occurrence of new disease, or death from any cause. The secondary end point was OS, defined as the time from randomisation until death from any cause.
Each of the six biomarkers was initially analysed individually. The PFS of patients according to biomarker expression (0, 1+ vs 2+ vs 3+) and treatment group were summarised in Kaplan–Meier curves, and the differences between these groups were compared in a log-rank test. A proportional-hazards model with biomarker expression, a treatment covariate (C vs CB and CBM), and their interaction was used to assess whether increasing biomarker expression predicted resistance to bevacizumab. Each analysis was adjusted for baseline clinicopathological factors, using the same variables identified to be significant in multivariate models of the intention-to-treat MAX population.
Multivariate proportional-hazards analysis with treatment, all six biomarkers, and their individual treatment-by-biomarker interactions assessed the predictive values of these biomarkers simultaneously. Only statistically significant biomarkers and the biomarker interactions (P <.05) were retained in the final multivariate model. A global assessment of the predictive values of all biomarkers combined was tested using the log-likelihood ratio test to compare this multivariate model with another model with treatment and the expression of all six biomarkers only. The reported P-values were adjusted for the effects of all six biomarkers and their interactions with treatment; P<0.05 indicated that overexpression of biomarkers predicts resistance to bevacizumab. These P-values were not adjusted for multiple comparisons. Similar methods were adopted in assessing the predictive values of these biomarkers for OS. All reported P-values were two-sided and not adjusted for multiple comparisons.
Secondary cohort
We attempted to support the findings for VEGF-D expression with an independent patient population from the CAIRO2 trial (Tol et al, 2009). In this study, 251 patients with metastatic colorectal cancer were randomised to capecitabine, oxaliplatin, and bevacizumab (control arm) or the same regimen plus weekly cetuximab (experimental arm). Only tumour samples, in the form of a TMA, from patients in the control arm (all treated with bevacizumab) were analysed, using the same methods as for the MAX samples. We hypothesised that, if there was a prognostic difference in the VEGF-D 0–1+ compared with 2–3+ patients in PFS and OS, this would support but not validate our findings.
Results
Characteristics of the patients
Of the 471 randomised patients, 389 consented to the biomarker analysis study, from whom 268 tumour specimens were available for examination of VEGF and VEGFR expression (57% of the study population) (Figure 1). They were representative of the study population (Supplementary Table S2). The median follow-up time for these patients was 30.6 months (range 1.2–42.4 months). Tumour specimens from the remaining patients could not be retrieved or were unsuitable for TMA construction. The grading of staining intensity and its distribution for each biomarker is illustrated in Supplementary Figure S2. The concordance between the independent scorers was determined by weighted Kappa score and was >0.79 for each biomarker. Fourteen patients had matched primary and secondary specimens allowing comparison of tissue expression between each, with good-to-very-good concordant expression (weighted Kappa scores >0.70).

CONSORT diagram.
Progression-free survival
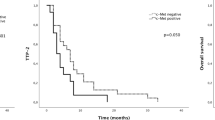
When each of the six biomarkers was examined singly, only VEGF-D predicted the effect of bevacizumab on PFS (Table 1). Among patients with VEGF-D expression scores of 0 or 1+, the median PFS was 5.8 months in the C group and 16.8 months in the CB+CBM group (P=0.0001; Figure 2A, Table 2). Among patients with VEGF-D expression 2+, the median PFS was 6.0 months in the C group and 8.8 months in the CB+CBM group (P=0.05). Among patients with VEGF-D expression 3+, the median PFS was 7.0 months in the C group and 9.0 months in the CB+CBM group (P=0.22; Figure 2A, Table 2). The additional benefit of bevacizumab was significantly greater among the patients with lower expression of VEGF-D than among those with higher expression of VEGF-D (P=0.02 for the interaction between VEGF-D expression and treatment group; Figure 3). The interaction remained significant after multivariate adjustments for baseline factors (performance status, prior resection of the primary tumour, number of organ sites of metastasis, baseline serum alkaline phosphatase, and baseline serum bilirubin). For the other biomarkers, the interactions between biomarker expression and treatment were not significant (Table 1). In the step-down multivariable analysis with a treatment covariate and individual treatment-by-biomarker interactions, only treatment, VEGF-D, and treatment-by-VEGF-D interaction remained significant. In a step-down multivariable model where only significant predictors were retained, treatment, VEGF-D, and treatment-by-VEGF-D interaction were the only predictors. However, the global interaction was not significant for PFS (log-likelihood χ2=8.23 (with six degrees of freedom); P=0.22).

Kaplan–Meier curves for (A) PFS and (B) OS according to VEGF-D expression in unadjusted analyses.

Forest plots for (A) PFS and (B) OS for groups with different levels of VEGF-D expression. *Hazard ratio for all patients in the MAX trial, including those whose tumour samples were not analysed.
Overall survival
Among patients with VEGF-D expression 0 or 1+, the median OS was 18.9 months in the C group and the median OS was not reached in the CB+CBM group (P=0.03; Figure 2B, Table 2). Among patients with VEGF-D expression 2+, the median OS was 20.6 months in the C group and 21.6 months in the CB+CBM group (P=0.40). Among patients with VEGF-D expression 3+, the median OS was 24.5 months in the C group and 19.4 months in the CB+CBM group (P=0.32). Therefore, the additional benefit of bevacizumab for OS was only evident in patients with lower expression of VEGF-D (P=0.01 for the interaction between VEGF-D expression and the assigned treatment; Figure 2B). The interaction remained significant after multivariate adjustments for baseline clinicopathological factors (performance status, prior resection of the primary tumour, prior radiotherapy, baseline serum alkaline phosphatase, and baseline serum neutrophils). The interaction between VEGF-A expression and assigned treatments was not significant (Table 1). For the other biomarkers, the interactions between biomarker expression and assigned treatments were also significant when examined singly for VEGF-B, VEGF-C, VEGFR-1, and VEGFR-2.
In the step-down multivariable model with the treatment and individual treatment-by-biomarker interactions, only treatment, VEGFR-1, and treatment by VEGFR-1 interaction remained significant. Even with adjustment for multiple comparisons, the global assessment of treatment by VEGFR-1 interaction remained significant for OS (Figure 4). The global test of interaction with all six biomarkers combined was significant for OS (log-likelihood χ2=15.12 (with six degrees of freedom); P=0.02).

Kaplan–Meier curves for (A) PFS and (B) OS according to VEGF-R1 expression in analyses adjusted for multiple comparisons.
Sensitivity analysis
C was compared with CB and C with CBM for VEGF-D only (Supplementary Figure S2). Among patients with VEGF-D expression scores of 0 or 1+, the median PFS was 11.5 months in the CB group and was not reached in the CBM group. Among patients with VEGF-D expression 2+, the median PFS was 9.5 months in the CB group and 8.4 months in the CBM group. Among patients with VEGF-D expression 3+, the median PFS was 8.8 months in the CB group and 9.3 months in the CBM group. The additional benefit of bevacizumab was consistently significantly greater among the patients with lower expression of VEGF-D than among those with higher expression of VEGF-D (P=0.03 for the interaction between VEGF-D expression and the C vs CB treatment comparison and P=0.04 for the interaction between VEGF-D expression and the C vs CBM treatment comparison).Additionally, there is no difference in the treatment outcome between CB vs CBM, and no significant interaction between VEGF-D and treatment (CB vs CBM) for PFS (P=0.94) and OS (P=0.62).
Outcomes of CAIRO2 patients according to VEGF-D expression
VEGF-D expression of 0 or 1+, 2+, and 3+ were recorded for 25, 126, and 100 patients, respectively. The median PFS for patients with VEGF-D expression 0 or 1+, 2+, and 3+ was 10.1 (95% CI, 7.3–14.8), 9.7 (95% CI, 7.5–11.4), and 9.8 (95% CI, 6.9–11.8) months, respectively (P=0.35). The median OS for patients with VEGF-D expression 0 or 1+, 2+, and 3+ was 22.0 (95% CI, 12.5–26.9), 21.7 (95% CI, 18.6–23.4), and 21.7 (95% CI,16.9–25.4) months, respectively (P=0.67).
Discussion
In the MAX study, high expression of VEGF-D shown on immunohistochemistry predicted resistance to bevacizumab. The PFS benefit of bevacizumab was greater for patients with lower expression of VEGF-D than those with higher expression.
This study represents a comprehensive evaluation of tumoural expression of VEGF ligands and receptors in colorectal cancer patients treated with bevacizumab. The study used a large cohort of tumour samples (57%). Other randomised phase III colorectal bevacizumab biomarker studies tested not >34% of patient samples (Jubb et al, 2006; Foernzler et al, 2010). Expression levels of biomarkers were scored by two independent reviewers, one of whom was an anatomical pathologist, blinded to trial allocation and treatment outcome. There was good inter-rater agreement, with weighted Kappa >0.79 between the two scorers. The investigators remained blinded to trial allocation and treatment outcome when deciding the appropriate categorisation of ARFs into different expression groups of 0, 1+; 2+; and 3+.
This study also has several limitations. First, the optimal methods for examining these novel biomarker expression levels are not well established. There are conflicting reports on the rate of discordance between primary and metastatic tissue in the expression of VEGF-A levels (Cascinu et al, 2000; Kuramochi et al, 2006; Jubb and Harris, 2010). No study has examined the rate of discordance in the expression of the other ARFs. Most biological samples in this study were from archived primary colorectal tumours (83%) although expression levels between paired primary and secondary tumours (14 samples) had at least good-to-very-good concordant expression. Second, there is currently no consistent and validated scoring system to define the expression levels of ARFs as assessed by immunohistochemistry, which, moreover, is a semiquantitative technique (Jubb et al, 2006; Schneider et al, 2008; Foernzler et al, 2010). Although commercially available antibodies with prior published use with immunohistochemistry were used in the analysis, their specificity for epitopes was not tested independently. In determining the ARF expression in tumours using immunohistochemistry, we used a simplified scoring system aimed at easy future replication and clinical use. Furthermore, in correlation of ARF expression in tumour with response as an outcome, the number of samples in each group was relatively small, which limits its interpretability when using RECIST response criteria (Chun et al, 2009; Shindoh et al, 2012). We acknowledge that our results are also limited, because there might be potential interactions between mitomycin C with bevacizumab on PFS benefit. However, there was no significant interaction between VEGF-D and treatment (CB vs CBM) for PFS (P=0.94) and OS (P=0.62) (Supplementary Figure S2). Finally, this was a post-hoc analysis of multiple biomarkers of the MAX trial, and our findings may be related to a random effect.
Bevacizumab efficacy has no clinically useful predictive biomarker, such as KRAS mutation status, which is a definitive negative predictive biomarker for efficacy of epidermal growth factor receptor antibody therapy in advanced colorectal cancer (Lievre et al, 2006; Amado et al, 2008; De Roock et al, 2008; Van Cutsem et al, 2008; Bardelli and Siena, 2010; De Roock et al, 2010; Douillard et al, 2010; Rizzo et al, 2010; Van Cutsem et al, 2011; Bokemeyer et al, 2012). Various studies have examined the associations of potential biomarkers with bevacizumab efficacy. These markers include levels of baseline circulating endothelial cells, circulating angiogenesis-related cytokines such as interleukin-8 and PlGF 16 (Jayson et al, 2005; Dellapasqua et al, 2008; Willett et al, 2009; Kopetz et al, 2010), and SNPs in VEGF-A, VEGFR-1, and interleukin-8 (Schneider et al, 2008; Schultheis et al, 2008; Zhang et al, 2009; Jubb and Harris, 2010; Loupakis et al, 2011; Lambrechts et al, 2012; Collinson et al, 2013; Maru et al, 2013; Miles et al, 2013). Technical measurement difficulties and lack of validation in large randomised trials have limited their translation into routine clinical use (Furstenberger et al, 2005; Rowand et al, 2007; Mancuso et al, 2009; Maru et al, 2013). SNPs studies have produced conflicting findings and are compromised by multiple analyses that limit interpretation of apparently significant published P-values (Schneider et al, 2008; Schultheis et al, 2008; Zhang et al, 2009; Jubb and Harris, 2010; Lambrechts et al, 2012).
VEGF-A is thought to mediate angiogenesis through interaction with VEGFR-2, leading to an increase in blood vessel formation through changes in endothelial proliferation, cellular permeability, and cell migration (Kerbel, 2008). The VEGF family members VEGF-C and VEGF-D bind to VEGFR-3, leading to lymphangiogenesis, but can also bind to VEGFR-2, causing angiogenesis (Achen and Stacker, 1998; Rissanen et al, 2003). This interaction occurs after the removal of N- and C-terminal propeptides from the central VEGF homology domains and receptor flanking sites, generating mature forms of the protein (Stacker et al, 1999). These interactions led us to hypothesise that overexpression of either VEGF-C or VEGF-D could lead to resistance to bevacizumab.
The association of elevated VEGF-D levels with inferior efficacy of bevacizumab in this study is biologically plausible and is supported by preclinical data demonstrating that VEGF-D has been shown to bind to VEGFR-2, triggering angiogenesis (Achen and Stacker, 1998; Stacker et al, 1999; Rissanen et al, 2003). Other studies have independently suggested a possible role for VEGF-D as a mediator of resistance to bevacizumab. Lieu et al (2013) showed that plasma VEGF-D levels increased upon tumour progression in patients with colorectal cancer receiving chemotherapy plus bevacizumab. Similarly, in the CALGB 80303 study, in patients with pancreatic cancer+, the subgroup with low plasma VEGF-D levels derived benefit from bevacizumab, while the main intention-to-treat population did not (Nixon et al, 2011). Unfortunately blood samples were not available from patients from either MAX or CAIRO-2 to assess the predictive role of circulating VEGF-D levels and validate these earlier studies.
Interpreting our results warrants caution, as only 32 patients with 0–1+ expression significantly benefited from bevacizumab treatment. The global test for interaction to account for multiple comparisons did not show statistical significance (P=0.22) for PFS. In the independent population of patients in the CAIRO2 trial, VEGF-D tumour expression did not discriminate PFS or OS, although the 95% confidence intervals were wide. Unlike the MAX study, CAIRO2 could not adequately assess the predictive value of VEGF-D, as all patients in the control arm were treated with bevacizumab and chemotherapy. Yet if VEGF-D is a predictive biomarker for bevacizumab benefit as suggested by the results of the analysis in the MAX trial population, we would expect to see a clear difference in outcome in the CAIRO-2 population according to VEGF-D tumour expression. However, the patient population and the chemotherapy backbone were also different in the two trials and possibly accounted for the different outcomes.
In the MAX study, VEGFR-1 overexpression was also strongly associated with a lack of OS benefit from bevacizumab. VEGFR-1 overexpression, however, did not demonstrate a similar significant association with PFS. Two separate studies have found no association between VEGFR-1 overexpression and OS benefit from bevacizumab (Foernzler et al, 2010; Van Cutsem et al, 2011, 2012). The significance of the finding is therefore uncertain, and replication will be attempted in an appropriate secondary cohort. Given that angiogenesis is a complex phenomenon, there are several other biomarkers, including neuropilin-1 (Van Cutsem et al, 2012) and PlGF, that are worthy of further investigation using our tissue resource from MAX.
The process of identifying predictive biomarkers for bevacizumab and other targeted therapies is important for maximising benefits to patients while minimising cost and toxicity. Also, comprehensive evaluation of a relevant pathway could provide future therapeutic opportunities, because, if VEGF-D is validated as a mechanism of resistance to bevacizumab, therapeutic approaches that target VEGF-D may assist in overcoming resistance to bevacizumab.
In conclusion, our study demonstrates that VEGF-D tumour expression is a potential predictive biomarker for bevacizumab efficacy on PFS. Despite the biological plausibility associated with VEGF-D, the study is hypothesis-generating and further confirmation of its predictive value is still required.
Change history
30 June 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Achen MG, Stacker SA (1998) The vascular endothelial growth factor family; proteins which guide the development of the vasculature. Int J Exp Pathol 79 (5): 255–265.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26 (10): 1626–1634.
Bardelli A, Siena S (2010) Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28 (7): 1254–1261.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Kohne CH (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 48 (10): 1466–1475.
Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L, Alitalo K (1998) Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci USA 95 (24): 14389–14394.
Cascinu S, Graziano F, Catalano V, Staccioli MP, Barni S, Giordani P, Rossi MC, Baldelli AM, Muretto P, Valenti A, Catalano G (2000) Differences of vascular endothelial growth factor (VEGF) expression between liver and abdominal metastases from colon cancer. Implications for the treatment with VEGF inhibitors. Clin Exp Metastasis 18 (8): 651–655.
Chun Y, Vauthey J, Boonsirikamchai P, Maru D, Kopetz S, Palavecino M, Curley S, Abdalla E, Kaur H, Charnsangavej C, Loyer E (2009) Association of computed tomography morphologic criteria with pathologic response and survival in patients treated with bevacizumab for colorectal liver metastases. JAMA 302 (21): 2338–2344.
Collinson F, Hutchinson M, Craven RA, Cairns DA, Zougman A, Wind TC, Gahir N, Messenger MP, Jackson S, Thompson D, Adusei C, Ledermann JA, Hall G, Jayson GC, Selby PJ, Banks RE (2013) Predicting response to bevacizumab in ovarian cancer: a panel of potential biomarkers informing treatment selection. Clin Cancer Res 19 (18): 5227–5239.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11 (8): 753–762.
De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 19 (3): 508–515.
Dellapasqua S, Bertolini F, Bagnardi V, Campagnoli E, Scarano E, Torrisi R, Shaked Y, Mancuso P, Goldhirsch A, Rocca A, Pietri E, Colleoni M (2008) Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol 26 (30): 4899–4905.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28 (31): 4697–4705.
Dowlati A, Gray R, Sandler AB, Schiller JH, Johnson DH (2008) Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab—an Eastern Cooperative Oncology Group Study. Clin Cancer Res 14 (5): 1407–1412.
Foernzler D, Delmar P, Kockx M, Cassidy J, Saltz L, Scherer S (2010) Tumor tissue based biomarker analysis in NO16966: a randomized phase III study of first-line bevacizumab in combination with oxaliplatin-based chemotherapy in patients with mCRC. 2010 Gastrointestinal Cancers Symposium 22–24 January 2010 American Society of Clinical Oncology: Orlando, FL, USA.
Furstenberger G, von Moos R, Senn HJ, Boneberg EM (2005) Real-time PCR of CD146 mRNA in peripheral blood enables the relative quantification of circulating endothelial cells and is an indicator of angiogenesis. Br J Cancer 93 (7): 793–798.
Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, Hambleton J, Novotny WF, Kabbinavar F (2005) Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first-line metastatic colorectal cancer. J Clin Oncol 23 (15): 3502–3508.
Hurwitz HI, Tebbutt NC, Kabbinavar F, Giantonio BJ, Guan Z-Z, Mitchell L, Waterkamp D, Tabernero J (2013) Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. Oncologist 18 (9): 1004–1012.
Jayson GC, Mulatero C, Ranson M, Zweit J, Jackson A, Broughton L, Wagstaff J, Hakansson L, Groenewegen G, Lawrance J, Tang M, Wauk L, Levitt D, Marreaud S, Lehmann FF, Herold M, Zwierzina H (2005) Phase I investigation of recombinant anti-human vascular endothelial growth factor antibody in patients with advanced cancer. Eur J Cancer 41 (4): 555–563.
Jubb AM, Harris AL (2010) Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol 11 (12): 1172–1183.
Jubb AM, Hurwitz HI, Bai W, Holmgren EB, Tobin P, Guerrero AS, Kabbinavar F, Holden SN, Novotny WF, Frantz GD, Hillan KJ, Koeppen H (2006) Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J Clin Oncol 24 (2): 217–227.
Jubb AM, Miller KD, Rugo HS, Harris AL, Chen D, Reimann JD, Cobleigh MA, Schmidt M, Langmuir VK, Hillan KJ, Chen DS, Koeppen H (2011) Impact of exploratory biomarkers on the treatment effect of bevacizumab in metastatic breast cancer. Clin Cancer Res 17 (2): 372–381.
Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358 (19): 2039–2049.
Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S, Lieu C, Yan S, Tran HT, Ellis LM, Abbruzzese JL, Heymach JV (2010) Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol 28 (3): 453–459.
Kuramochi H, Hayashi K, Uchida K, Miyakura S, Shimizu D, Vallbohmer D, Park S, Danenberg KD, Takasaki K, Danenberg PV (2006) Vascular endothelial growth factor messenger RNA expression level is preserved in liver metastases compared with corresponding primary colorectal cancer. Clin Cancer Res 12 (1): 29–33.
Lambrechts D, Claes B, Delmar P, Reumers J, Mazzone M, Yesilyurt BT, Devlieger R, Verslype C, Tejpar S, Wildiers H, de Haas S, Carmeliet P, Scherer SJ, Van Cutsem E (2012) VEGF pathway genetic variants as biomarkers of treatment outcome with bevacizumab: an analysis of data from the AViTA and AVOREN randomised trials. Lancet Oncol 13 (7): 724–733.
Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ (2013) Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol 31 (9): 1219–1230.
Lieu CH, Tran H, Jiang ZQ, Mao M, Overman MJ, Lin E, Eng C, Morris J, Ellis L, Heymach JV, Kopetz S (2013) The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PLoS One 8 (10): e77117.
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66 (8): 3992–3995.
Loupakis F, Ruzzo A, Salvatore L, Cremolini C, Masi G, Frumento P, Schirripa M, Catalano V, Galluccio N, Canestrari E, Vincenzi B, Santini D, Bencardino K, Ricci V, Manzoni M, Danova M, Tonini G, Magnani M, Falcone A, Graziano F (2011) Retrospective exploratory analysis of VEGF polymorphisms in the prediction of benefit from first-line FOLFIRI plus bevacizumab in metastatic colorectal cancer. BMC Cancer 11: 247.
Mancuso P, Antoniotti P, Quarna J, Calleri A, Rabascio C, Tacchetti C, Braidotti P, Wu HK, Zurita AJ, Saronni L, Cheng JB, Shalinsky DR, Heymach JV, Bertolini F (2009) Validation of a standardized method for enumerating circulating endothelial cells and progenitors: flow cytometry and molecular and ultrastructural analyses. Clin Cancer Res 15 (1): 267–273.
Maru D, Venook AP, Ellis LM (2013) Predictive biomarkers for bevacizumab: are we there yet? Clin Cancer Res 19 (11): 2824–2827.
Miles DW, de Haas SL, Dirix LY, Romieu G, Chan A, Pivot X, Tomczak P, Provencher L, Cortes J, Delmar PR, Scherer SJ (2013) Biomarker results from the AVADO phase 3 trial of first-line bevacizumab plus docetaxel for HER2-negative metastatic breast cancer. Br J Cancer 108 (5): 1052–1060.
Nixon AB, Pang H, Starr M, Hollis D, Friedman PN, Bertagnolli MM, Kindler HL, Goldberg RM, Venook AP, Hurwitz H (2011) Prognostic and predictive blood-based biomarkers in patients with advanced pancreatic cancer: results from CALGB 80303. ASCO Meeting Abstr 29 (15 Suppl): 10508.
Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier-Lavigne M, Watts RJ (2007) Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 11 (1): 53–67.
Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, Kholova I, Kauppinen RA, Achen MG, Stacker SA, Alitalo K, Yla-Herttuala S (2003) VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res 92 (10): 1098–1106.
Rizzo S, Bronte G, Fanale D, Corsini L, Silvestris N, Santini D, Gulotta G, Bazan V, Gebbia N, Fulfaro F, Russo A (2010) Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer Treat Rev 36 (Suppl 3): S56–S61.
Rowand JL, Martin G, Doyle GV, Miller MC, Pierce MS, Connelly MC, Rao C, Terstappen LW (2007) Endothelial cells in peripheral blood of healthy subjects and patients with metastatic carcinomas. Cytometry A 71 (2): 105–113.
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang T-S, Rivera F, Couture F, Sirzen F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26 (12): 2013–2019.
Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, Flockhart DA, Hancock B, Davidson N, Gralow J, Dickler M, Perez EA, Cobleigh M, Shenkier T, Edgerton S, Miller KD (2008) Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol 26 (28): 4672–4678.
Schultheis AM, Lurje G, Rhodes KE, Zhang W, Yang D, Garcia AA, Morgan R, Gandara D, Scudder S, Oza A, Hirte H, Fleming G, Roman L, Lenz HJ (2008) Polymorphisms and clinical outcome in recurrent ovarian cancer treated with cyclophosphamide and bevacizumab. Clin Cancer Res 14 (22): 7554–7563.
Shindoh J, Loyer EM, Kopetz S, Boonsirikamchai P, Maru DM, Chun YS, Zimmitti G, Curley SA, Charnsangavej C, Aloia TA, Vauthey JN (2012) Optimal morphologic response to preoperative chemotherapy: an alternate outcome end point before resection of hepatic colorectal metastases. J Clin Oncol 30 (36): 4566–4572.
Stacker SA, Stenvers K, Caesar C, Vitali A, Domagala T, Nice E, Roufail S, Simpson RJ, Moritz R, Karpanen T, Alitalo K, Achen MG (1999) Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J Biol Chem 274 (45): 32127–32136.
Tebbutt NC, Wilson K, Gebski VJ, Cummins MM, Zannino D, van Hazel GA, Robinson B, Broad A, Ganju V, Ackland SP, Forgeson G, Cunningham D, Saunders MP, Stockler MR, Chua Y, Zalcberg JR, Simes RJ, Price TJ (2010) Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: results of the australasian gastrointestinal trials group randomized phase III MAX study. J Clin Oncol 28 (19): 3191–3198.
Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O, Punt CJ (2009) Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 360 (6): 563–572.
Van Cutsem E, de Haas S, Kang Y-K, Ohtsu A, Tebbutt NC, Ming Xu J, Peng Yong W, Langer B, Delmar P, Scherer SJ, Shah MA (2012) Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol 30 (17): 2119–2127.
Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29 (15): 2011–2019.
Van Cutsem E, Lang I, D'Haens G, Moiseyenko V, Zaluski J, Folprecht G, Tejpar S, Kisker O, Stroh C, Rougier P (2008) KRAS status and efficacy in the first-line treatment of patients with metastatic colorectal cancer treated with FOLFIRI with or without cetuximab: the CRYSTAL experience. J Clin Oncol 26 (15_suppl): 2.
Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, Lahdenranta J, Chung DC, Fischman AJ, Lauwers GY, Shellito P, Czito BG, Wong TZ, Paulson E, Poleski M, Vujaskovic Z, Bentley R, Chen HX, Clark JW, Jain RK (2009) Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol 27 (18): 3020–3026.
Zhang D, Li B, Shi J, Zhao L, Zhang X, Wang C, Hou S, Qian W, Kou G, Wang H, Guo Y (2010) Suppression of tumor growth and metastasis by simultaneously blocking vascular endothelial growth factor (VEGF)-A and VEGF-C with a receptor-immunoglobulin fusion protein. Cancer Res 70 (6): 2495–2503.
Zhang W, Dahlberg SE, Yang D, Sandler AB, Brahmer JR, Schiller JH, Carbone DP, Johnson DH, Lenz H (2009) Genetic variants in angiogenesis pathway associated with clinical outcome in NSCLC patients (pts) treated with bevacizumab in combination with carboplatin and paclitaxel: Subset pharmacogenetic analysis of ECOG 4599. J Clin Oncol (Meeting Abstr) 27 (15S): 8032.
Acknowledgements
The MAX study was supported by an unrestricted educational grant from Roche Products Pty Limited (Australia) and an additional untied educational grant from Roche Products Ltd (United Kingdom). The study was also supported by grants for infrastructure to the National Health and Medical Research Council (NHMRC) Clinical Trials Centre from Cancer Australia, the Cancer Institute New South Wales, and the NHMRC (1037786).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Tebbutt has received honoraria and worked in a consulting role for Roche. Dr Price has been an uncompensated member of Roche advisory boards. The other authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Weickhardt, A., Williams, D., Lee, C. et al. Vascular endothelial growth factor D expression is a potential biomarker of bevacizumab benefit in colorectal cancer. Br J Cancer 113, 37–45 (2015). https://doi.org/10.1038/bjc.2015.209
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.209
Keywords
This article is cited by
-
Ang-1 and VEGF: central regulators of angiogenesis
Molecular and Cellular Biochemistry (2024)
-
Exploratory Biomarker Analysis Using Plasma Angiogenesis-Related Factors and Cell-Free DNA in the TRUSTY Study: A Randomized, Phase II/III Study of Trifluridine/Tipiracil Plus Bevacizumab as Second-Line Treatment for Metastatic Colorectal Cancer
Targeted Oncology (2024)
-
Toward Blood-Based Precision Medicine: Identifying Age-Sex-Specific Vascular Biomarker Quantities on Circulating Vascular Cells
Cellular and Molecular Bioengineering (2023)
-
VEGF-A, VEGFR1 and VEGFR2 single nucleotide polymorphisms and outcomes from the AGITG MAX trial of capecitabine, bevacizumab and mitomycin C in metastatic colorectal cancer
Scientific Reports (2022)
-
Development and validation of a tumor microenvironment-related prognostic signature in lung adenocarcinoma and immune infiltration analysis*
Oncology and Translational Medicine (2021)
