
Abstract
Background:
Menopausal hormone therapy (MHT) use has been consistently associated with a decreased risk of colorectal cancer (CRC) in women. Our aim was to use a genome-wide gene–environment interaction analysis to identify genetic modifiers of CRC risk associated with use of MHT.
Methods:
We included 10 835 postmenopausal women (5419 cases and 5416 controls) from 10 studies. We evaluated use of any MHT, oestrogen-only (E-only) and combined oestrogen–progestogen (E+P) hormone preparations. To test for multiplicative interactions, we applied the empirical Bayes (EB) test as well as the Wald test in conventional case–control logistic regression as primary tests. The Cocktail test was used as secondary test.
Results:
The EB test identified a significant interaction between rs964293 at 20q13.2/CYP24A1 and E+P (interaction OR (95% CIs)=0.61 (0.52–0.72), P=4.8 × 10−9). The secondary analysis also identified this interaction (Cocktail test OR=0.64 (0.52–0.78), P=1.2 × 10−5 (alpha threshold=3.1 × 10−4). The ORs for association between E+P and CRC risk by rs964293 genotype were as follows: C/C, 0.96 (0.61–1.50); A/C, 0.61 (0.39–0.95) and A/A, 0.40 (0.22–0.73), respectively.
Conclusions:
Our results indicate that rs964293 modifies the association between E+P and CRC risk. The variant is located near CYP24A1, which encodes an enzyme involved in vitamin D metabolism. This novel finding offers additional insight into downstream pathways of CRC etiopathogenesis.
Similar content being viewed by others


Main
The use of menopausal hormone therapy (MHT) has been consistently associated with a reduced risk of developing colorectal cancer (CRC; Calle et al, 1995; Fernandez et al, 1998; Grodstein et al, 1998, 1999; Anderson et al, 2004; Campbell et al, 2007; Johnson et al, 2009; Rennert et al, 2009; Green et al, 2012; Lin et al, 2012). According to a recently published meta-analysis, the relative risk (RR) of CRC was 0.74 (95% confidence interval (CI) 0.68–0.81) for ever use of oestrogen plus progestogen (E+P) therapy and 0.79 (95% CI 0.69–0.91) for ever use of oestrogen-only (E-only; Lin et al, 2012). Compared to placebo, menopausal women randomised to combined E+P hormone therapy in the Women’s Health Initiative (WHI) also had a lower risk of CRC (Chlebowski et al, 2004), although their cancers tended to be of higher stage with poorer prognosis (Simon et al, 2012). However, randomisation to conjugated E-only in the WHI trial was not associated with risk of CRC (Anderson et al, 2004).
The underlying mechanisms of how MHT use influences colon carcinogenesis are largely unknown. Insight into potential biological pathways could be gained by investigating genetic modifiers of CRC risk associated with use of MHT. Furthermore, certain loci associated with susceptibility for CRC may only be evident in presence or absence of a specific environmental factor such as use of MHT. Thus, studies that specifically examine the association of MHT use with CRC risk in the context of varying genetic backgrounds are needed. Previous studies that have investigated gene–environment (G × E) interactions between single nucleotide polymorphisms (SNPs), MHT use and CRC risk have been largely based on candidate genes, which encompass only limited genetic variance (Lin et al, 2010, 2011; Rudolph et al, 2011; Slattery et al, 2011). A genome-wide scan to examine G × E interactions is a crucial next step to comprehensively examine additional variants and identify novel interactions with MHT. We therefore carried out a genome-wide association analysis to assess G × E interactions with use of MHT, using recently developed statistical methods and data from several studies comprising in total 5419 CRC cases and 5416 controls.
Materials and Methods
Study participants
Our overall genome-wide association study (GWAS) design has been described previously (Hutter et al, 2012; Peters et al, 2013). In brief, this analysis is based on 10 studies (a case–control study from the Colon Cancer Family Registry (CCFR), and nine studies from the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO)). Study-specific details are described in Supplementary Methods. All cases were defined as colorectal adenocarcinoma, and confirmed by medical records, pathology reports or death certificate. All participants provided written informed consent, and studies were approved by their respective institutional review boards.
Harmonisation of environmental data
Information on basic demographics and environmental risk factors was collected by using in-person interviews and/or structured questionnaires, as detailed previously (The Women’s Health Initiative Study Group, 1998; Colditz and Hankinson, 2005; Newcomb et al, 2007; Hoffmeister et al, 2009). Postmenopausal status was defined by using either (i) study-derived menopausal status, if available; (ii) self-reported menopausal status, if study derived was not available; or (iii) age ⩾55, if study derived and self-report were not available. MHT use was considered either as any MHT use, E-only use or E+P use at reference time. Non-users (of any MHT) at reference time were used as reference group. The reference time for nested case–control studies was time of enrolment into the cohort (VITAL, WHI and PLCO) or blood draw (NHS). For case–control studies, the reference time was at diagnosis and 2 years prior to diagnosis for CCFR and DALS. The harmonisation procedure is described in more detail in the Supplementary Methods.
Genotyping, quality assurance/quality control and imputation
All analyses were based on genotyped data generated from genome-wide association scans and imputation to HapMap II. Genotyped SNPs were excluded if they were triallelic, not assigned an rs number, or were reported or observed as not performing consistently across platforms. Furthermore, genotyped SNPs were excluded based on call rate (<98%), lack of Hardy-Weinberg equilibrium in controls (HWE, P<1 × 10−4) and minor allele frequency (MAF<5%). Further details on DNA extraction, genotyping and quality assurance/quality control for each of the involved studies can be found in the Supplementary Methods and in Peters et al (2012).
All autosomal SNPs of all studies were imputed to the CEU population in HapMap II release 24, with the exception of OFCCR, which was imputed to HapMap II release 22. CCFR set 1 was imputed using IMPUTE (Marchini et al, 2007), OFCCR was imputed using BEAGLE (Browning and Browning, 2007), and all other studies were imputed using MACH (Li et al, 2010). Imputed data were merged with genotype data such that genotype data were used if a SNP had both types of data, unless there was a difference in terms of reference allele frequency (>0.1) or position (>100 base pairs), in which case imputed data were used. SNPs were restricted based on per study MAF ⩾5% of samples and per study imputation accuracy (r2>0.3). After imputation and QC analyses, a total of about 2.7 M SNPs were used in the analysis. The inflation factor λ (a measurement of the overdispersion of the test statistics from the marginal association tests) obtained from the fixed-effect meta-analysis of GWAS scans was 1.029, 1.027 and 1.027 for the samples used for any MHT use, E+P and E-only analyses, respectively, indicating little evidence of population substructure (Quantile–quantile (Q–Q) plots in Supplementary Figure 1).
Statistical Methods
Models were adjusted for age at reference time, centre and the first three principal components from EIGENSTRAT (Price et al, 2006) to account for population substructure. Each directly genotyped SNP was coded as 0, 1 or 2 copies of the variant allele. For imputed SNPs, we used the expected number of copies of the variant allele (the ‘dosage’), which has been shown to give unbiased test statistics (Jiao et al, 2011). SNPs are treated as continuous variables (i.e., assuming log-additive effects). Each study was analysed separately using logistic regression, and study-specific results were combined using fixed-effect meta-analysis to obtain summary odds ratios (ORs) and 95% CIs across studies. We calculated the heterogeneity P-values by Woolf’s test (Woolf, 1955). Q–Q plots were assessed to determine whether the distribution of the P-values was consistent with the null distribution (except for the extreme tail). To test for multiplicative interactions between SNPs and environmental risk factors, we primarily used the empirical Bayes (EB) test (Mukherjee and Chatterjee, 2008; Mukherjee et al, 2008, 2010) given that this test can be more powerful than the conventional case–control logistic regression analysis while maintaining the desired type I error. It calculates the interaction log OR that corresponds to a weighted average of the case-only and case–control estimators. Thus, the method makes use of the greater precision of the case-only estimator by simultaneously reducing the chance of generating biased estimates due to violations of the assumption of gene–environment independence in controls (Mukherjee and Chatterjee, 2008). We modelled the SNP by environment (G × E) interaction by the product of the SNP and the dichotomised environmental variable. A two-sided P-value <5 × 10−8 was considered genome-wide significant, yielding a genome-wide significance level of 0.05 (Risch and Merikangas, 1996; International HapMap Consortium, 2005; Wellcome Trust Case Control Consortium, 2007; Dudbridge and Gusnanto, 2008; Hoggart et al, 2008; Pe’er et al, 2008). We secondarily used the multiplicative Cocktail method (Hsu et al, 2012), to evaluate how robust the findings were under the use of an alternative test to evaluate a multiplicative interaction (Supplementary Methods).
To estimate the effects of the environment variable stratified by genotype, we fit the following model: logit(d)=b0+b1e+c1p1+c2p2+β1p1e+β2p2e+covariates, where p1 and p2 are the imputation posterior probabilities for genotype A/B, B/B. Then the stratified effects of environment variable can be estimated as  ,
,  and
and  for genotypes A/A, A/B and B/B, respectively. The s.e. are estimated by using the standard formula for a linear combination of two parameters based on the covariance matrix of these parameters.
for genotypes A/A, A/B and B/B, respectively. The s.e. are estimated by using the standard formula for a linear combination of two parameters based on the covariance matrix of these parameters.
We estimated the CRC incidence rates associated with E+P use among individuals with each genotype of SNPs, which would provide more direct interpretation for G × E interaction effects on public health. We based the estimation of the incidence rate on the Surveillance, Epidemiology, and End Results (SEER) age-adjusted CRC incidence rate (denoted by ‘I’) 1992–2010 among the White population, which is 59.5 per 100 000 women per year. By using I, we estimated the reference incidence rate of CRC (denoted by ‘Iref’) using the population attributable risk (PAR), which is estimated by one minus the average of the inverse of estimated risk score exp(−X β) in cases (Bruzzi et al, 1985). Specifically, the formula for computing the PAR estimator is:

where Yj=1 for case and 0 for control; Xj=covariates; β=estimated regression coefficients in the logistic regression analysis. We can then estimate the reference incidence rate of CRC for X=0 by Iref =(1−PAR) × I (Gail et al, 1989). On the basis of this reference incidence rate of CRC (i.e., Iref), we further calculated the CRC incidence rate for each subgroup defined by genotypes of the SNP according to E+P use or non-use by multiplying the Iref with each corresponding OR estimate. We calculated the 95% CIs using a resampling technique with 1000 weighted bootstraps. Since SEER incidence rates are based on a large number of individuals, the uncertainty of ‘I’ is negligible compared to the uncertainty from the PAR estimate, and hence was not considered in the calculation of 95% CIs.
Methods used for functional follow-up on promising loci are described in the Supplementary Methods.
Results
Our study population comprised 10 835 menopausal women: 5419 cases and 5416 controls. For all 10 835 women information on use of any MHT was available and information on the type of MHT preparation was available for 9004 participants. At reference time, 3384 women used any MHT (31.2%), 1283 (11.8%) used E+P and 1606 (14.8%) used E-only (Supplementary Table 1). MHT use was inversely associated with the risk of CRC. Compared to non-users of any MHT at reference time, the OR for CRC was 0.70 (95% CI 0.62–0.79, P=1.9 × 10−9, P for heterogeneity (phet)=0.14) for women using any MHT preparation, 0.76 (95% CI 0.64–0.90, P=0.0015, phet=0.049) for women who used E+P and 0.71 (95% CI 0.61–0.84, P=7.1 × 10−5, phet=0.017) for women who used E-only.
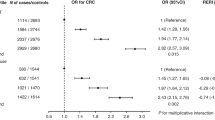
The EB test identified a significant interaction of the variant rs964293 with E+P, with OR=0.61 (95% CI 0.52–0.72, P=4.8 × 10−9). This interaction showed borderline evidence of heterogeneity across studies (phet=0.044; Figure 1A). The same variant (rs964293) showed an interaction with E+P use on CRC risk in our secondary analysis, using the Cocktail test (OR=0.64, 95% CI 0.52–0.78, P=1.2 × 10−5, alpha threshold for significance in the group where the variant is assigned according to its rank=3.1 × 10−4). Table 1 summarises the results of the interaction analyses of rs964293 with MHT use. When testing for interaction between rs964293 and use of any MHT or E-only, no significant results were observed (Table 1).

Forest plot for meta-analysis of the interaction between SNP and oestrogen plus progestogen use, using the empirical Bayes (A and C) and case–control logistic regression method (B and D) for rs964293 (A and B) and rs6023015 (C and D). DALS and PLCO studies are not plotted because they do not have information on the type of hormone compound.
Table 2 presents the association of the different strata of MHT by rs964293 with CRC risk. The OR of CRC for women taking E+P compared with women not using MHT is 0.96 (95% CI 0.61–1.50, P=0.84), 0.61 (95% CI 0.39–0.95, P=0.03) and 0.40 (95% CI 0.22–0.73, P=0.0026) for women with C/C, A/C and A/A genotype of rs964293, respectively (Table 2). The per study ORs of E+P on CRC stratified by rs964293 genotype are shown in Figure 2A–C. No significant heterogeneity between study wise estimates was observed (phet=0.3, 0.66 and 0.23, respectively).

Forest plot for meta-analysis of the marginal association of oestrogen plus progestogen with colorectal cancer risk in strata defined by zero, one or two minor alleles of rs964293 (A, B and C, respectively) and rs6023015 (D, E and F, respectively).
The variant rs964293 is located in an intergenic region 28 kb upstream of CYP24A1 on chromosome 20q13.2. Supplementary Figure 2 shows the interaction P-value (EB test) of rs964293 as well as SNPs surrounding rs964293 in region 20q13.2. The variant rs964293 was directly genotyped in five of the study sets included in the analysis and imputation accuracy was high in the remaining six study sets (r2 ranging from 0.82 to 0.99). The MAF ranged from 0.34 to 0.38 in the 11 study sets.
The SNP rs964293 is in moderate LD (r2=0.61 in 1000 Genomes pilot CEU) with a strong functional candidate, rs6023015. Our in silico functional analysis demonstrates that this SNP is located in a region with strong DNase hypersensitivity and histone methylation patterns consistent with enhancer activity. Supplementary Figure 3 shows how these two patterns of DNase hypersensitivity and histone methylation are stronger in some cancerous cell lines (including the CRC cell lines CACO2 and HCT-116) relative to non-cancerous cell lines. ChIP-seq experiments indicate that several transcription factors bind to this region (Supplementary Figure 4). In data from 169 tissue samples of the transverse colon available through the GTEx portal (Ardlie et al, 2015), both variants, rs964293 and rs6023015, are expression quantitative trait loci (eQTL) for CYP24A1 expression (P=0.037 and 0.018, respectively). Expression of CYP24A1 varied slightly stronger by genotypes of another SNP rs2256649 in LD with rs964293 (r2=0.60 in 1000 Genomes pilot CEU), with P=0.0069. Compared to homozygous carriers of the wild-type allele, expression of CYP24A1 was higher in homozygous carriers of the minor allele for all three SNPs. Supplementary Figure 5 displays the distribution of the rank normalised gene expression of CYP24A1 by genotypes of rs964293, rs6023015 and rs2256649.
We estimated absolute risks for CRC according to the use of E+P and genotypes of rs964293 and rs6023015 (Table 3). Compared to not-using E+P and carrying the wild-type CC genotype of rs964293, using E+P was associated with 30.6 fewer cases of CRC within a year among carriers of the AA genotype. Likewise, carrying the CC genotype of rs6023015 and using E+P was associated with 32.4 fewer cases of CRC within a year when compared to carrying the GG genotype of rs6023015 and not-using E+P.
Aside from results reported above, we did not observe any additional genome-wide significant G × E interactions with use of E+P and none for any MHT and E-only use.
Discussion
In this study, including over 10 000 women, we evaluated genome-wide G × E interactions with the use of any MHT preparation as well as separately with use of E+P and of E-only. Employing the powerful yet robust EB method, we found evidence of an interaction between the variant rs964293 at 20q13.2/CYP24A1 and use of E+P.
Although most prior studies of G × E interactions in CRC have utilised a candidate gene approach (Slattery et al, 2010; Zhong et al, 2013, Figueiredo et al (2011), examined G × E interactions using a genome-wide scan and 14 environmental variables, including MHT use, and did not observe any genome-wide significant associations within the CCFR and OFCCR study. However, that study had limited power to detect any significant G × MHT interactions since it included only 572 highly selected CRC cases of menopausal women (age <50 years or with a family history) and did not assess the possible differential interaction with use of MHT according to type of preparation.
Our finding of a genome-wide significant interaction between rs964293 and use of E+P has convincing biologic plausibility. This SNP is located in an intergenic region 28 kb upstream of CYP24A1. As it is not in strong LD with any coding variants, we hypothesised that the underlying causal variant(s) exerts its effect through a regulatory mechanism. We found that rs964293 is in moderate LD with rs6023015 (r2>0.61 in CEU), which lies in a putative enhancer region for CYP24A1. The rs6023015 SNP was imputed in all 11 studies with high accuracy (mean r2=0.95, range 0.87–1.00), and interaction and association of E+P across strata defined by number of minor alleles of rs6023015 paralleled that of rs964293 (Supplementary Table 4 and Figure 2D–F). Also the estimated ORs for interaction of rs6023015 with use of E+P were comparable to those found for rs964293 (Figure 1C and D), but P-values observed with the EB test were less extreme (P=2.8 × 10−6). Both SNPs are eQTL for CYP24A1 expression in 169 normal tissue samples of the transverse colon. CYP24A1 expression was increased in homozygous carriers of the minor allele compared to homozygous carriers of the wild-type allele (Supplementary Figure 5). Details of the in silico functional analyses of rs6023015 are provided in the Supplementary Methods.
CYP24A1 codes for a protein that belongs to the cytochrome P450 family. These mitochondrial proteins are monooxygenases that catalyse several reactions involved in drug metabolism and synthesis of cholesterol, steroids, sex hormones and other lipids. Specifically, CYP24A1 plays a key role in the metabolism of the steroid hormone vitamin D by degrading its active form. CYP24A1 is highly expressed in malignant colon tumours as compared with healthy colonic epithelium at both the mRNA (Bareis et al, 2001) and protein level (Matusiak and Benya, 2007), and some variants in CYP24A1 have been associated with CRC risk in candidate gene studies (Dong et al, 2009). Vitamin D refers to a group of lipid soluble cholesterol-based compounds that, in contrast with other vitamins, can be synthesised endogenously. The active form of vitamin D exerts several functions relevant to regulation of tumour pathogenesis and progression, such as the activation of apoptotic pathways, anti-proliferative effects and angiogenesis inhibition (Deeb et al, 2007). Substantial experimental and epidemiological data support an inverse association with risk of CRC (Chan and Giovannucci, 2010). Moreover, there is evidence linking the vitamin D pathway and sex hormones in CRC aetiology. A secondary analysis of data from the WHI suggested that the association between low-dose vitamin D plus calcium supplementation and CRC risk is modified by MHT (E-only and E+P; Ding et al, 2008). Women randomised to receive calcium and 400 international units (IU) vitamin D3 supplementation in the placebo arms of the factorial oestrogen therapy trials were at suggestively decreased risk of developing CRC (HR=0.71, 95% CI 0.46–1.09) compared to women randomised to receive calcium, vitamin D3 and concurrent MHT (HR=1.30 (95% CI 0.83–2.03), P for interaction=0.05; Ding and Giovannucci, 2009). Furthermore, the difference in the association of calcium and vitamin D supplementation with CRC between users and non-users of MHT appeared to be more evident for E+P than for E-only.
The results observed here suggest that CYP24A1 may be regulated by sex hormones exposure and that the modifying effect observed with rs964293 may be caused by a disruption of the increased CYP24A1 expression induced by sex hormones exposure. Alternatively, given that we found an interaction only with E+P exposure and not with E-only intake, CYP24A1 may be a metabolising enzyme for progestogens but not oestrogen. Taken together, this evidence provides a strong rationale for additional functional studies to determine the role of rs964293/rs6023015 in CYP24A1, particularly in relation to exposure to exogenous E+P in colorectal carcinogenesis.
Our study has several strengths. First, our large sample size facilitated the detection of genome-wide G × E interactions even accounting for the stringent threshold for significance necessary for control of type I error and the generally small magnitude of effect modification that can be reasonably expected. We followed a ‘whole set’ approach rather than a ‘discovery-validation’ strategy to maximise efficiency (Skol et al, 2006). Second, as previously described (Hutter et al, 2012), we have carefully harmonised data on a range of environmental variables, including MHT use across 10 studies. Third, two different methods (EB and Cocktail) to evaluate G × E interaction identified the same variant as having a significant G × E interaction, providing greater confidence that this association is not a false positive finding (although the EB test can be part of the Cocktail test itself, this was not the case for rs964293). The magnitude of the interaction between rs964293 and the use of E+P yielded by the traditional case–control logistic regression analysis was similar to the one found by the EB test (OR=0.64 and 0.61, respectively; Figure 1 and Table 1), though it did not reach the threshold of genome-wide significance (P=1.2 × 10−5). We observed some degree of heterogeneity among studies for the interaction of rs964293 with E+P (phet=0.044). Figure 1 shows that the OR for interaction is ⩽1 for all the studies but the VITAL, which constitutes about 2.5% of the total sample size. We do not consider this heterogeneity as a strong limitation to the results of the study.
The use of E+P for the purpose of chemoprevention is not routinely recommended for the general population due to concerns about the potential adverse consequences of long-term exposure. However, our results suggest the possibility that the benefit of E+P may be enhanced in women carrying genetic variants at rs964293 (Table 3). In conjunction with other strategies for risk-stratification and after its evaluation across other relevant clinical outcomes, this finding could be exploited to more specifically identify individuals for whom the benefits of chemoprevention may outweigh potential risks (Collins, 2015).
In summary, we have identified a CYP24A1-related variant as effect modifier of CRC risk associated with use of E+P using a genome-wide approach. This finding offers important insight into the role of E+P and its downstream pathways, including its potential interaction with vitamin D in the etiopathogenesis of CRC and supports the need for further studies to confirm the involvement of CYP24A1 in modulating CRC risk.
Change history
19 January 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O’Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S (2004) Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 291: 1701–1712.
Ardlie KG, Deluca DS, Segre AV, Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, Lek M, Ward LD, Kheradpour P, Iriarte B, Meng Y, Palmer CD, Esko T, Winckler W, Hirschhorn JN, Kellis M, MacArthur DG, Getz G, Shabalin AA, Li G, Zhou Y-H, Nobel AB, Rusyn I, Wright FA, Lappalainen T, Ferreira PG, Ongen H, Rivas MA, Battle A, Mostafavi S, Monlong J, Sammeth M, Mele M, Reverter F, Goldmann JM, Koller D, Guigo R, McCarthy MI, Dermitzakis ET, Gamazon ER, Im HK, Konkashbaev A, Nicolae DL, Cox NJ, Flutre T, Wen X, Stephens M, Pritchard JK, Tu Z, Zhang B, Huang T, Long Q, Lin L, Yang J, Zhu J, Liu J, Brown A, Mestichelli B, Tidwell D, Lo E, Salvatore M, Shad S, Thomas JA, Lonsdale JT, Moser MT, Gillard BM, Karasik E, Ramsey K, Choi C, Foster BA, Syron J, Fleming J, Magazine H, Hasz R, Walters GD, Bridge JP, Miklos M, Sullivan S, Barker LK, Traino HM, Mosavel M, Siminoff LA, Valley DR, Rohrer DC, Jewell SD, Branton PA, Sobin LH, Barcus M, Qi L, McLean J, Hariharan P, Um KS, Wu S, Tabor D, Shive C, Smith AM, Buia SA, Undale AH, Robinson KL, Roche N, Valentino KM, Britton A, Burges R, Bradbury D, Hambright KW, Seleski J, Korzeniewski GE, Erickson K, Marcus Y, Tejada J, Taherian M, Lu C, Basile M, Mash DC, Volpi S, Struewing JP, Temple GF, Boyer J, Colantuoni D, Little R, Koester S, Carithers LJ, Moore HM, Guan P, Compton C, Sawyer SJ, Demchok JP, Vaught JB, Rabiner CA, Lockhart NC (2015) The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660.
Bareis P, Bises G, Bischof MG, Cross HS, Peterlik M (2001) 25-Hydroxy-vitamin D metabolism in human colon cancer cells during tumor progression. Biochem Biophys Res Commun 285: 1012–1017.
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097.
Bruzzi P, Green SB, Byar DP, Brinton LA, Schairer C (1985) Estimating the population attributable risk for multiple risk factors using case-control data. Am J Epidemiol 122: 904–914.
Calle EE, Miracle-McMahill HL, Thun MJ, Heath CW Jr (1995) Estrogen replacement therapy and risk of fatal colon cancer in a prospective cohort of postmenopausal women. J Natl Cancer Inst 87: 517–523.
Campbell PT, Newcomb P, Gallinger S, Cotterchio M, McLaughlin JR (2007) Exogenous hormones and colorectal cancer risk in Canada: associations stratified by clinically defined familial risk of cancer. Cancer Causes Control 18: 723–733.
Chan AT, Giovannucci EL (2010) Primary prevention of colorectal cancer. Gastroenterology 138: 2029–2043.e10.
Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, Rosenberg Ca, Taylor VM, Harris R, Chen C, Adams-Campbell LL, White E (2004) Estrogen plus progestin and colorectal cancer in postmenopausal women. N Engl J Med 350: 991–1004.
Colditz Ga, Hankinson SE (2005) The Nurses’ Health Study: lifestyle and health among women. Nat Rev Cancer 5: 388–396.
Collins FS (2015) Precision medicine: who benefits from aspirin to prevent colorectal cancer? Available at: http://directorsblog.nih.gov/2015/03/24/precision-medicine-who-benefits-from-aspirin-to-prevent-colorectal-cancer/ (accessed on 07 August 2015).
Deeb KK, Trump DL, Johnson CS (2007) Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 7: 684–700.
Ding EL, Giovannucci EL (2009) Reply to comment on: interaction of hormone replacement therapy with calcium and Vitamin D supplementation on colorectal cancer risk. Int J Cancer 124: 1737–1738.
Ding EL, Mehta S, Fawzi WW, Giovannucci EL (2008) Interaction of estrogen therapy with calcium and vitamin D supplementation on colorectal cancer risk: reanalysis of Women’s Health Initiative randomized trial. Int J Cancer 122: 1690–1694.
Dong LM, Ulrich CM, Hsu L, Duggan DJ, Benitez DS, White E, Slattery ML, Farin FM, Makar KW, Carlson CS, Caan BJ, Potter JD, Peters U (2009) Vitamin D related genes, CYP24A1 and CYP27B1, and colon cancer risk. Cancer Epidemiol Biomarkers Prev 18: 2540–2548.
Dudbridge F, Gusnanto A (2008) Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 32: 227–234.
Fernandez E, La Vecchia C, Braga C, Talamini R, Negri E, Parazzini F, Franceschi S (1998) Hormone replacement therapy and risk of colon and rectal cancer. Cancer Epidemiol Biomarkers Prev 7: 329–333.
Figueiredo JC, Lewinger JP, Song C, Campbell PT, Conti DV, Edlund CK, Duggan DJ, Rangrej J, Lemire M, Hudson T, Zanke B, Cotterchio M, Gallinger S, Jenkins M, Hopper J, Haile R, Newcomb P, Potter J, Baron JA, Le Marchand L, Casey G (2011) Genotype-environment interactions in microsatellite stable/microsatellite instability-low colorectal cancer: results from a genome-wide association study. Cancer Epidemiol Biomarkers Prev 20: 758–766.
Gail MH, Brinton La, Byar DP, Corle DK, Green SB, Schairer C, Mulvihill JJ (1989) Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 81: 1879–1886.
Green J, Czanner G, Reeves G, Watson J, Wise L, Roddam A, Beral V (2012) Menopausal hormone therapy and risk of gastrointestinal cancer: Nested case-control study within a prospective cohort, and meta-analysis. Int J Cancer 130: 2387–2396.
Grodstein F, Martinez ME, Platz EA, Giovannucci E, Colditz GA, Kautzky M, Fuchs C, Stampfer MJ (1998) Postmenopausal hormone use and risk for colorectal cancer and adenoma. Ann Intern Med 128: 705–712.
Grodstein F, Newcomb PA, Stampfer MJ (1999) Postmenopausal hormone therapy and the risk of colorectal cancer: a review and meta-analysis. Am J Med 106: 574–582.
Hoffmeister M, Raum E, Krtschil A, Chang-Claude J, Brenner H (2009) No evidence for variation in colorectal cancer risk associated with different types of postmenopausal hormone therapy. Clin Pharmacol Ther 86: 416–424.
Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ (2008) Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol 32: 179–185.
Hsu L, Jiao S, Dai JY, Hutter C, Peters U, Kooperberg C (2012) Powerful cocktail methods for detecting genome-wide gene-environment interaction. Genet Epidemiol 36: 183–194.
Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, Nan H, Lemire M, Rangrej J, Figueiredo JC, Jiao S, Harrison TA, Liu Y, Chen LS, Stelling DL, Warnick GS, Hoffmeister M, Küry S, Fuchs CS, Giovannucci E, Hazra A, Kraft P, Hunter DJ, Gallinger S, Zanke BW, Brenner H, Frank B, Ma J, Ulrich CM, White E, Newcomb PA, Kooperberg C, LaCroix AZ, Prentice RL, Jackson RD, Schoen RE, Chanock SJ, Berndt SI, Hayes RB, Caan BJ, Potter JD, Hsu L, Bézieau S, Chan AT, Hudson TJ, Peters U (2012) Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res 72: 2036–2044.
International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437: 1299–1320.
Jiao S, Hsu L, Hutter CM, Peters U (2011) The use of imputed values in the meta-analysis of genome-wide association studies. Genet Epidemiol 35: 597–605.
Johnson JR, Lacey JV, Lazovich D, Geller Ma, Schairer C, Schatzkin A, Flood A (2009) Menopausal hormone therapy and risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev 18: 196–203.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010) MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834.
Lin J, Zee RYL, Liu KY, Zhang SM, Lee IM, Manson JE, Giovannucci E, Buring JE, Cook NR (2010) Genetic variation in sex-steroid receptors and synthesizing enzymes and colorectal cancer risk in women. Cancer Causes Control 21: 897–908.
Lin JH, Manson JE, Kraft P, Cochrane BB, Gunter MJ, Chlebowski RT, Zhang SM (2011) Estrogen and progesterone-related gene variants and colorectal cancer risk in women. BMC Med Genet 12: 78.
Lin KJ, Cheung WY, Lai JYC, Giovannucci EL (2012) The effect of estrogen vs. combined estrogen-progestogen therapy on the risk of colorectal cancer. Int J Cancer 130: 419–430.
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39: 906–913.
Matusiak D, Benya RV (2007) CYP27A1 and CYP24 expression as a function of malignant transformation in the colon. J Histochem Cytochem 55: 1257–1264.
Mukherjee B, Ahn J, Gruber SB, Ghosh M, Chatterjee N (2010) Case-control studies of gene-environment interaction: Bayesian design and analysis. Biometrics 66: 934–948.
Mukherjee B, Ahn J, Gruber SB, Rennert G, Moreno V, Chatterjee N (2008) Tests for gene-environment interaction from case-control data: a novel study of type I error, power and designs. Genet Epidemiol 32: 615–626.
Mukherjee B, Chatterjee N (2008) Exploiting gene-environment independence for analysis of case-control studies: an empirical Bayes-type shrinkage estimator to trade-off between bias and efficiency. Biometrics 64: 685–694.
Newcomb PA, Baron J, Cotterchio M, Gallinger S, Grove J, Haile R, Hall D, Hopper JL, Jass J, Le Marchand L, Limburg P, Lindor N, Potter JD, Templeton AS, Thibodeau S, Seminara D (2007) Colon cancer family registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev 16: 2331–2343.
Pe’er I, Yelensky R, Altshuler D, Daly MJ (2008) Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 32: 381–385.
Peters U, Hutter CM, Hsu L, Schumacher FR, Conti DV, Carlson CS, Edlund CK, Haile RW, Gallinger S, Zanke BW, Lemire M, Rangrej J, Vijayaraghavan R, Chan AT, Hazra A, Hunter DJ, Ma J, Fuchs CS, Giovannucci EL, Kraft P, Liu Y, Chen L, Jiao S, Makar KW, Taverna D, Gruber SB, Rennert G, Moreno V, Ulrich CM, Woods MO, Green RC, Parfrey PS, Prentice RL, Kooperberg C, Jackson RD, Lacroix AZ, Caan BJ, Hayes RB, Berndt SI, Chanock SJ, Schoen RE, Chang-Claude J, Hoffmeister M, Brenner H, Frank B, Bézieau S, Küry S, Slattery ML, Hopper JL, Jenkins Ma, Le Marchand L, Lindor NM, Newcomb Pa, Seminara D, Hudson TJ, Duggan DJ, Potter JD, Casey G (2012) Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet 131: 217–234.
Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron Ja, Berndt SI, Bézieau S, Brenner H, Butterbach K, Caan BJ, Campbell PT, Carlson CS, Casey G, Chan AT, Chang-Claude J, Chanock SJ, Chen LS, Coetzee Ga, Coetzee SG, Conti DV, Curtis KR, Duggan D, Edwards T, Fuchs CS, Gallinger S, Giovannucci EL, Gogarten SM, Gruber SB, Haile RW, Harrison Ta, Hayes RB, Henderson BE, Hoffmeister M, Hopper JL, Hudson TJ, Hunter DJ, Jackson RD, Jee SH, Jenkins Ma, Jia WH, Kolonel LN, Kooperberg C, Küry S, Lacroix AZ, Laurie CC, Laurie Ca, Le Marchand L, Lemire M, Levine D, Lindor NM, Liu Y, Ma J, Makar KW, Matsuo K, Newcomb Pa, Potter JD, Prentice RL, Qu C, Rohan T, Rosse Sa, Schoen RE, Seminara D, Shrubsole M, Shu XO, Slattery ML, Taverna D, Thibodeau SN, Ulrich CM, White E, Xiang Y, Zanke BW, Zeng YX, Zhang B, Zheng W, Hsu L (2013) Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 144: 799–807.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick Na, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909.
Rennert G, Rennert HS, Pinchev M, Lavie O, Gruber SB (2009) Use of hormone replacement therapy and the risk of colorectal cancer. J Clin Oncol 27: 4542–4547.
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273: 1516–1517.
Rudolph A, Sainz J, Hein R, Hoffmeister M, Frank B, Försti A, Brenner H, Hemminki K, Chang-Claude J (2011) Modification of menopausal hormone therapy-associated colorectal cancer risk by polymorphisms in sex steroid signaling, metabolism and transport related genes. Endocr Relat Cancer 18: 371–384.
Simon MS, Chlebowski RT, Wactawski-Wende J, Johnson KC, Muskovitz A, Kato I, Young A, Hubbell FA, Prentice RL (2012) Estrogen plus progestin and colorectal cancer incidence and mortality. J Clin Oncol 30: 3983–3990.
Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38: 209–213.
Slattery ML, Herrick J, Curtin K, Samowitz W, Wolff RK, Caan BJ, Duggan D, Potter JD, Peters U (2010) Increased risk of colon cancer associated with a genetic polymorphism of SMAD7. Cancer Res 70: 1479–1485.
Slattery ML, Lundgreen A, Herrick JS, Kadlubar S, Caan BJ, Potter JD, Wolff RK (2011) Variation in the CYP19A1 gene and risk of colon and rectal cancer. Cancer Causes Control 22: 955–963.
The Women’s Health Initiative Study Group (1998) Design of the Women’s Health Initiative clinical trial and observational study. The Women’s Health Initiative Study Group. Control Clin Trials 19: 61–109.
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678.
Woolf B (1955) On estimating the relation between blood group and disease. Ann Hum Genet 19: 251–253.
Zhong R, Liu L, Zou L, Sheng W, Zhu B, Xiang H, Chen W, Chen J, Rui R, Zheng X, Yin J, Duan S, Yang B, Sun J, Lou J, Liu L, Xie D, Xu Y, Nie S, Miao X (2013) Genetic variations in the TGFβ signaling pathway, smoking and risk of colorectal cancer in a chinese population. Carcinogenesis 34: 936–942.
Acknowledgements
DACHS: we thank all participants and cooperating clinicians, and Ute Handte-Daub, Renate Hettler-Jensen, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance. GECCO: we would like to thank all those at the GECCO Coordinating Center for helping bring together the data and people that made this project possible. NHS: we would like to acknowledge Patrice Soule and Hardeep Ranu of the Dana Farber Harvard Cancer Center High-Throughput Polymorphism Core who assisted in the genotyping for NHS under the supervision of Dr Immaculata Devivo and Dr David Hunter, Qin (Carolyn) Guo and Lixue Zhu who assisted in programming for NHS. We would like to thank the participants and staff of the Nurses’ Health Study for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA and WY. PLCO: we thank Drs Christine Berg and Philip Prorok, Division of Cancer Prevention, National Cancer Institute; the Screening Center investigators and staff or the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial; Mr Tom Riley and staff, Information Management Services, Inc.; Ms Barbara O’Brien and staff, Westat, Inc.; and Drs Bill Kopp, Wen Shao and staff, SAIC-Frederick. Most importantly, we acknowledge the study participants for their contributions to making this study possible. PMH: we would like to thank the study participants and staff of the Hormones and Colon Cancer study. WHI: we thank the WHI investigators and staff for their dedication, and the study participants for making the programme possible. A full listing of WHI investigators can be found at: https://cleo.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf. Funding details are as follows: GECCO: National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088; R01 CA059045). Mengmeng Du is supported by grants R25 CA094880 and P30 CA008748 from NCI. CCFR: National Institutes of Health (RFA # CA-95–011) and through cooperative agreements with members of the Colon Cancer Family Registry and P.I.s. This genome-wide scan was supported by the National Cancer Institute, National Institutes of Health by U01 CA122839. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the CFRs, nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government or the CFR. The following Colon CFR centres contributed data to this manuscript and were supported by National Institutes of Health: Australasian Colorectal Cancer Family Registry (U01 CA097735), Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783) and Seattle Colorectal Cancer Family Registry (U01 CA074794). DACHS: German Research Council (Deutsche Forschungsgemeinschaft, BR 1704/6–1, BR 1704/6–3, BR 1704/6–4 and CH 117/1–1), and the German Federal Ministry of Education and Research (01KH0404 and 01ER0814). DALS: National Institutes of Health (R01 CA48998 to MLS); NHS is supported by the National Institutes of Health (R01 CA137178, P01 CA 087969 and P50 CA 127003). MEC: National Institutes of Health (R37 CA54281, P01 CA033619 and R01 CA63464). OFCCR: National Institutes of Health, through funding allocated to the Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783); see CCFR section above. As subset of ARCTIC, OFCCR is supported by a GL2 grant from the Ontario Research Fund, the Canadian Institutes of Health Research, and the Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society Research Institute. TJH and BZ are recipients of Senior Investigator Awards from the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation. PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. In addition, a subset of control samples were genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) Prostate Cancer GWAS, Colon CGEMS pancreatic cancer scan (PanScan) and the Lung Cancer and Smoking study. The prostate and PanScan study data sets were accessed with appropriate approval through the dbGaP online resource (http://cgems.cancer.gov/data/) accession numbers phs000207v.1p1 and phs000206.v3.p2, respectively, and the lung data sets were accessed from the dbGaP website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093 v2.p2. Funding for the Lung Cancer and Smoking study was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446 and NIH GEI U01 HG 004438. For the lung study, the GENEVA Coordinating Center provided assistance with genotype cleaning and general study coordination, and the Johns Hopkins University Center for Inherited Disease Research conducted genotyping. PMH: National Institutes of Health (R01 CA076366 to PAN). VITAL: National Institutes of Health (K05 CA154337). WHI: The WHI programme is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C and HHSN271201100004C. XG-A is a recipient of an ASISA Fellowship and SEOM (Sociedad Española de Oncología Médica) grant. ATC is a Damon Runyon Clinical Investigator and is also supported by NIDDK K24DK098311. WJG is supported by grant #HL115606. SO is supported by grant R35 CA197735.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Garcia-Albeniz, X., Rudolph, A., Hutter, C. et al. CYP24A1 variant modifies the association between use of oestrogen plus progestogen therapy and colorectal cancer risk. Br J Cancer 114, 221–229 (2016). https://doi.org/10.1038/bjc.2015.443
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.443
Keywords
This article is cited by
-
Genetic risk impacts the association of menopausal hormone therapy with colorectal cancer risk
British Journal of Cancer (2024)
-
The microbiome, genetics, and gastrointestinal neoplasms: the evolving field of molecular pathological epidemiology to analyze the tumor–immune–microbiome interaction
Human Genetics (2021)
-
Association study of genetic variants in estrogen metabolic pathway genes and colorectal cancer risk and survival
Archives of Toxicology (2018)
