
Abstract
The pseudokinase, MLKL (mixed-lineage kinase domain-like), is the most terminal obligatory component of the necroptosis cell death pathway known. Phosphorylation of the MLKL pseudokinase domain by the protein kinase, receptor interacting protein kinase-3 (RIPK3), is known to be the key step in MLKL activation. This phosphorylation event is believed to trigger a molecular switch, leading to exposure of the N-terminal four-helix bundle (4HB) domain of MLKL, its oligomerization, membrane translocation and ultimately cell death. To examine how well this process is evolutionarily conserved, we analysed the function of MLKL orthologues. Surprisingly, and unlike their mouse, horse and frog counterparts, human, chicken and stickleback 4HB domains were unable to induce cell death when expressed in murine fibroblasts. Forced dimerization of the human MLKL 4HB domain overcame this defect and triggered cell death in human and mouse cell lines. Furthermore, recombinant proteins from mouse, frog, human and chicken MLKL, all of which contained a 4HB domain, permeabilized liposomes, and were most effective on those designed to mimic plasma membrane composition. These studies demonstrate that the membrane-permeabilization function of the 4HB domain is evolutionarily conserved, but reveal that execution of necroptotic death by it relies on additional factors that are poorly conserved even among closely related species.
Similar content being viewed by others

Main
Necroptosis is a form of programmed cell death that can be induced following ligation of death ligand and Toll-like receptors (TLRs). Most experimental work has focused on necroptosis induced by tumour necrosis factor (TNF). The key effectors in the pathway are the protein kinases, receptor interacting protein kinase (RIPK)-1 and RIPK3,1, 2, 3, 4 and the mixed-lineage kinase domain-like (MLKL) pseudokinase.5, 6, 7, 8 RIPK3 phosphorylates the pseudokinase domain of MLKL, the most terminal known essential component of the pathway,5, 6 which is believed to induce a conformational change and unleash the N-terminal four-helix bundle (4HB) domain of MLKL: an executioner domain.5, 9, 10 Several models have been proposed for how this 4HB domain might induce cell death, including activation of downstream effectors, such as ion channels,11, 12 direct permeabilization of membranes and/or formation of a transmembrane pore,13, 14 all of which remain the subject of debate. The consensus from these and other studies is that in order to kill, MLKL must translocate to membranes and assemble into high molecular weight signalling complexes, which are likely to be MLKL oligomers, although the stoichiometry of these MLKL oligomers remains an open question.10, 11, 12, 13, 14 Nonetheless, phosphorylation appears to be a key cue for MLKL activation15 and, as the most terminal known post-translational modification in the pathway, could potentially be utilized as a biomarker in pathologies arising from necroptotic cell death.14, 16
A model whereby RIPK3-mediated phosphorylation of the MLKL pseudokinase domain activation loop (S345 in mouse; T357/S358 in human) leads to unleashing of the executioner 4HB domain is supported by several lines of evidence. First, expression of the isolated mouse MLKL 4HB or the complete N-terminal domain (NTD), which encompasses the 4HB domain and two "brace" helices, killed mouse dermal fibroblasts (MDFs).10 Second, cell death occurred to a similar extent when full-length mouse MLKL harbouring the S345D mutation, that mimics activation loop phosphorylation by RIPK3, was expressed in murine fibroblasts.5, 15, 17 In addition, three studies have attributed a direct membrane-permeabilization function to recombinant human MLKL 4HB domain in in vitro liposome dye-release assays.13, 14, 18 However, a number of questions remain unanswered. It is unclear why S345D mouse MLKL is a potent killer of murine fibroblasts, while the human counterpart, T357D/S358E, induced death of HT29 cells in one study,19 yet did not induce pronounced death of human U2OS cells unless dimerized via a fused domain.14 Similarly, it is unclear why forced dimerization of mouse MLKL 4HB domain was required for cell death in L929, CHO and HeLa cells,12 but not in MDFs.10 It is also unclear why recombinant human MLKL 4HB domain exhibits a preference for substrate liposomes containing cardiolipin, high concentrations of which are considered to be confined to mitochondrial inner membranes,14, 18 even though mitochondria are dispensable for necroptotic death.20 In addition, the human MLKL 4HB domain potently and rapidly induced membrane permeabilization in liposome assays,13, 14, 18 yet a substantial delay in cell death is observed following MLKL membrane translocation.15 These observations led us to investigate the differential susceptibility of different cell types to MLKL-induced necroptotic death and whether reduced susceptibility could be overcome by inducible dimerization of either full-length MLKL or the 4HB domain. Finally we determined the extent to which the necroptosis inducing properties are conserved between MLKL orthologues.
We found that the human MLKL NTD, and 4HB domain encoded within, did not cause death of the commonly studied human cell lines, U937, HT29 and HeLa. However, inducible dimerization of the human MLKL 4HB domain via a fused gyrase domain led to robust killing of these cell lines as well as wild-type, but not Mlkl−/− MDFs. Analogously, dimerization of full-length wild-type mouse MLKL via a fused gyrase domain led to the death of wild-type and Mlkl−/− MDFs in the absence of necroptotic stimuli. Interestingly, NTDs from mouse, horse and frog MLKL, but not human, chicken and stickleback MLKL, induced death of Mlkl−/− MDFs. Nonetheless, using liposome permeabilization assays, we demonstrated that like the mouse and frog MLKL NTDs, human and chicken MLKL NTDs compromised membrane integrity, and were more effective on liposomes whose composition resembled that of plasma membranes than on those mimicking mitochondrial membranes. Collectively, these studies demonstrate that although the MLKL 4HB domain encodes an evolutionarily conserved membrane-permeabilization function, execution of necroptotic death relies on the presence or absence of endogenous factors that are not universally expressed in U937, HT29, HeLa and MDF cells to either mediate MLKL oligomerization, membrane translocation and/or downstream signalling.
Results
Cell death induction by the NTD of human MLKL requires dimerization
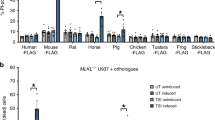
Our earlier work demonstrated that expression of the mouse MLKL (mMLKL) N-terminal domain (NTD; residues 1–180) or the mMLKL four-helix bundle (4HB) domain (residues 1–125) killed mouse fibroblasts in the absence of a conventional necroptotic stimulus such as TSQ: TNF (T), Smac-mimetic (S) and Q-VD-OPh (Q).10 In contrast, in the present work, we observed that expression of the analogous human MLKL (hMLKL) NTD (residues 1–180) in U937, HT29 and HeLa cells did not induce stimulus-independent cell death (Figures 1a–c).

Human MLKL N-terminal domain and 4HB domain constructs require forced dimerization to induce cell death in human cell lines. (a–c) U937, HT29 and HeLa cells were stably infected with doxycycline-inducible constructs encoding human MLKL (1–180). Expression was induced for 4 h with 10 ng/ml doxycycline followed by the induction of apoptosis (TS) or necroptosis (TSQ) or no treatment (untreated; UT) for 48 h. (d and e) Schematic representing coumermycin-induced dimerization of MLKL (1–180) (d) or MLKL 4HB domain (e) fused to gyrase. (f–k) U937 (f and g), HT29 (h and i) and HeLa (j and k) cells were stably infected with doxycycline-inducible constructs encoding human MLKL (1–180)-gyrase or human MLKL 4HB domain (1–125)-gyrase. Expression and dimerization were induced for 4 h with 10 ng/ml doxycycline and 700 nM coumermycin, followed by the induction of apoptosis with TS or necroptosis with TSQ or no further treatment (UT) for 48 h. Cell death was quantified by measuring PI-permeable cells using flow cytometry throughout. A statistical comparison of untreated, dox-induced conditions in the absence versus presence of coumermycin using a paired t-test yielded P-values of 0.081 and 0.0012 in (f and g), respectively. All the data are plotted as the mean±S.E.M. of at least three independent experiments
Our earlier studies demonstrated that mMLKL (1–180) spontaneously assembled into a high molecular weight complex in membranes.10 The lack of killing by hMLKL (1–180) led us to determine whether the human domain lacked an intrinsic capacity to oligomerize. We tested this hypothesis by fusing E. coli DNA gyrase (Figures 1d and e), a domain that can be dimerized by the divalent antibiotic coumermycin, to the C termini of hMLKL (1–180; NTD) and hMLKL (1–125; 4HB) domains.21 In the absence of coumermycin, the fusion proteins behaved the same as the unfused domains in the absence of apoptotic (TS) or necroptotic (TSQ) stimuli in U937 cells (Figures 1a, f and g). Similarly, a C-terminally StrepII-tagged version of hMLKL (1–125) did not induce stimulus-independent cell death (Supplementary Figures 1A and C). However, addition of coumermycin to cells expressing either of these domains led to their death without requiring other stimuli (Figures 1f and g), suggesting that the hMLKL NTD was simply less effective at oligomerizing than its murine counterpart. Notably, the observed cell death confirms that fusion to gyrase did not compromise NTD folding or stability, nor impose a dimer configuration that is incompatible with induction of necroptosis. To test this more rigorously, we expressed the constructs in HT29 cells (Figures 1h and i). Unexpectedly, even though hMLKL (1–180) gyrase was well expressed (Supplementary Figure 1F), coumermycin-induced dimerization did not induce stimulus-independent cell death in HT29 cells (Figure 1h). It is possible that the lack of HT29 death at 48 h is reflective of the generally slower necroptosis kinetics in HT29s, as observed upon TSQ stimulation. However, expression of the hMLKL 4HB domain (1–125) in HT29 cells induced cell death in the absence of coumermycin, and addition of coumermycin increased cell death (Figure 1i), at 48 h. In HeLa cells, coumermycin-mediated dimerization of hMLKL (1–180) and (1–125) gyrase fusions was required to induce death (Figures 1j and k), with more death observed for hMLKL (1–125) than hMLKL (1–180). These findings are consistent with the idea that oligomerization is not an intrinsic property of the hNTD but requires additional factors that are not present in all the cells. Furthermore, these data support the notion that the amino acids from 125–180 can inhibit the killing function of the hMLKL 4HB domain.
Human and mouse MLKL NTDs and activated mutant constructs are poor killers of cells of the opposite species
Although expression of mMLKL (1–180) or the 4HB domain (1–125) potently kills wild-type and Mlkl-deficient MDFs,10 the capacity of human constructs to kill these murine cells has not yet been examined. We therefore expressed the hMLKL (1–180) gyrase fusion in wild-type and Mlkl−/− MDFs, and observed no cell death either in the absence or presence of dimerization by coumermycin (Figures 2a and b). In contrast, forced dimerization of the hMLKL (1–125) gyrase fusion caused wild-type, but not Mlkl−/−, MDFs to die (Figures 2c and d). These data suggest that the hMLKL 4HB domain cannot induce death in MDFs independently, but can augment the activity of endogenous mouse MLKL when dimerized.

Human and mouse N-terminal domain constructs or full-length phosphomimetic MLKL mutants rarely induce cell death in cells of the opposite species. (a–d) Wild-type and Mlkl−/− mouse dermal fibroblasts (MDFs) were stably infected with doxycycline-inducible constructs encoding human NTD (1–180)-gyrase (a and b) or human 4HB domain (1–125)-gyrase (c and d). Expression and dimerization were induced for 4 h with 10 ng/ml doxycycline and 700 nM coumermycin, before induction of apoptosis (TS) or necroptosis (TSQ) or no treatment (UT) for 24 h. (e) Mlkl−/− MDFs were stably infected with doxycycline-inducible constructs encoding human MLKL (1–471). After 4 h of doxycycline (10 ng/ml) treatment to induce expression, cells were either stimulated for 24 h with TS to induce apoptosis or TSQ to induce necroptosis or left untreated (UT). Cell death was quantified by measuring PI-permeable cells using flow cytometry and data are plotted as the mean±S.E.M. of three biological replicates assayed in three independent experiments (n=9). (f–k) Wild-type and Mlkl−/− MDFs, U937 and HT29 were stably infected with doxycycline-inducible constructs encoding human full-length MLKL mutant (T357E/S358E), mouse full-length MLKL mutant (S345D) and mouse MLKL (1–180), as indicated. After 4 h of doxycycline (10 ng/ml) treatment to induce expression, the cells were stimulated for 48 h (U937, HT29) or 24 h (MDFs) with TS to induce apoptosis or TSQ to induce necroptosis or left untreated (UT). Data are plotted as the mean±S.E.M. of at least three independent experiments for U937 and HT29 and of at least three biological replicates each assayed in a minimum of two independent experiments for MDFs. Cell death was quantified by measuring PI-permeable cells using flow cytometry throughout. (l) Membrane complex (complex II) formation monitored by Blue-Native PAGE after a 14-h induction of mouse full-length MLKL S345D and human MLKL NTD (1–180) in U937 cells using 10 ng/ml doxycycline. Membrane fractionation purity and protein abundance was assessed by immunoblotting for GAPDH and VDAC. Data are representative of two independent repeats
To preclude the possibility that the absence of the pseudokinase domain from these expression constructs might compromise their killing functions, we attempted to reconstitute necroptosis signalling in Mlkl−/− MDFs with constructs encoding full-length (FL) mouse and human MLKL (Supplementary Figure 2A and Figure 2e, respectively). As observed previously,5 mMLKL reconstituted the necroptosis pathway (Supplementary Figure 2A), however, consistent with an earlier report,22 human MLKL did not (Figure 2e), despite evident expression (Supplementary Figure 2B). Taken together, these data demonstrate evolutionary divergence in the intrinsic capabilities of MLKL 4HB domains to induce cell death, and the evolution of species-specific necroptosis mechanisms in which MLKL orthologues do not readily complement one another.
Full-length mouse MLKL (residues 1–464) bearing the S345D mutation that mimics activation by RIPK3-mediated phosphorylation, is a potent killer of mouse fibroblasts in the absence of stimuli.5, 15, 17 To determine whether a deficit in endogenous mouse RIPK3-mediated activation of hMLKL underpins the inability of hMLKL to reconstitute mouse fibroblasts, we tested whether full-length hMLKL bearing the phosphomimetic mutations, T357E/S358E (TSEE), was lethal to wild-type or Mlkl−/− MDFs (Figure 2f). Unlike the S345D mMLKL mutant,5, 15, 17 the hMLKL TSEE mutant did not induce death of either wild-type or Mlkl−/− MDFs (Figure 2f). Similarly, expression of the hMLKL TSEE mutant in the human cell lines, U937 and HT29, did not induce cell death (Figures 2g and h). More surprisingly, expression of this mutant construct inhibited TSQ-induced necroptosis in U937 cells (Figure 2g), suggesting a dominant negative activity. Because mMLKL (1–464) bearing the S345D mutation is a potent death effector in mouse fibroblasts,5, 15, 17 we next asked whether it induced cell death in human cell lines. However, neither S345D mMLKL (1–464) nor mMLKL (1–180) were able to induce death of HT29 cells (Figures 2i and j), despite both being potent killers of mouse fibroblasts.5, 10, 15, 17 In contrast, expression of S345D mMLKL (1–464) in U937 cells led to a modest, but reproducible, amount of death (Figure 2k). The killing activity of S345D mMLKL (1–464) corresponded with its capacity to translocate from the cytoplasmic (C) fraction to membranes (M) and assemble into a high molecular weight species (Figure 2l). In contrast, hMLKL (1–180) did not undergo oligomerization or membrane translocation in parallel experiments (Figure 2l), in keeping with its inability to induce death of U937 cells. Collectively, these data support the idea that cell-specific factors determine whether mouse or human MLKL NTD or activated mutant constructs are able to kill, and whether dimerization mediated by a fused domain can, at least in part, overcome a requirement for these factors.
Dimerization of full-length MLKL drives cell death
Consistent with the hypothesis that MLKL must be activated by RIPK3 phosphorylation, ectopic expression of mMLKL alone does not induce necroptosis.5, 10, 15 Although expression of activating mutants can overcome the requirement for RIPK3 phosphorylation,5, 15 it has not been established whether forced dimerization of mMLKL can similarly cause death in the absence of stimuli. We examined this by fusing full-length mMLKL to a gyrase domain (Figure 3a) and expressing in wild-type and Mlkl−/− MDFs (Figures 3c and d). Stimulus-independent death was only observed upon coumermycin-mediated dimerization in wild-type and Mlkl−/− MDFs (Figures 3c and d), confirming the importance of oligomerization in MLKL activation as a likely prerequisite for membrane translocation. We explored this hypothesis by characterizing two loss-of-function mouse MLKL 4HB domain point mutants, R105A/D106A and E109A/E110A, that we have shown fail to translocate to membranes and assemble into high molecular weight complexes10 (Figure 3b). As expected from our earlier studies with the untagged versions,10 these mMLKL-gyrase mutants failed to reconstitute TSQ-mediated necroptosis when expressed in Mlkl−/− MDFs10 (Figures 3f and h). Interestingly, however, they did not induce death even when dimerization was forced by coumermycin, either in wild-type (Figures 3e and g) or Mlkl−/− MDFs (Figures 3f and h). The inability of dimerization to rescue the killer function of these mutants suggests that the Ala substitutions compromise higher order oligomerization or interactions with other proteins that are crucial for MLKL-mediated cell death.

Gyrase-mediated dimerization of full-length mouse MLKL causes cell death, but is unable to overcome loss-of-function mutations in the 4HB domain. (a) Schematic representing coumermycin-induced dimerization of full-length mouse MLKL fused to gyrase at the C terminus. (b) Cartoon of the 4HB domain showing the positions of loss-of-function mutations as sticks in blue (R105/D106) and red (E109/E110). Figure drawn using PyMol (www.pymol.org) from the co-ordinates of the full-length mouse MLKL structure (PDB accession, 4BTF5). (c, e and g) Wild-type and (d, f and h) Mlkl−/− mouse dermal fibroblasts (MDFs) were stably infected with doxycycline-inducible constructs encoding for wild-type, E109A/E110A, R105A/D106A full-length mouse MLKL (1–464), as indicated. Expression and dimerization were induced for 4 h with 10 ng/ml doxycycline and 700 nM coumermycin, before induction of apoptosis (TS) or necroptosis (TSQ) or no treatment (UT) for 24 h. Cell death was quantified by measuring PI-permeable cells using flow cytometry, and data are plotted as the mean±S.E.M. of at least three biological replicates each assayed in a minimum of two independent experiments (n⩾6)
We used an analogous approach to examine whether forced dimerization of wild-type or the phosphomimetic T357E/S358E (TSEE) human MLKL could induce death of wild-type or Mlkl−/− MDFs, U937, HT29 and HeLa cells (Figure 4). Interestingly, death of wild-type MDFs, HT29 and HeLa cells only occurred following forced dimerization via the fused gyrase domain and, surprisingly, to the same extent for wild-type and phosphomimetic mutant. As observed for TSEE hMLKL expression in U937 (Figure 2g), dimerized TSEE hMLKL suppressed endogenous necroptosis signalling upon TSQ stimulation (Figure 4f), whereas wild-type hMLKL did not (Figure 4e).

Gyrase-mediated dimerization of full-length wild-type or T357E/S358E hMLKL is required for cell death. Wild-type (a and b) and Mlkl−/− (c and d) mouse dermal fibroblasts (MDFs), U937 (e and f), HT29 (g and h) and HeLa (i and j) were stably infected with doxycycline-inducible constructs encoding C-terminal gyrase fusions of wild-type (left panels) or T357E/S358E (TSEE; right panels) human MLKL (1–471). Expression and dimerization were induced for 4 h with 10 ng/ml doxycycline and 700 nM coumermycin, before induction of apoptosis (TS) or necroptosis (TSQ) or no treatment (UT) for 24 h for MDFs or 48 h for human cell lines. Cell death was quantified by measuring PI-permeable cells using flow cytometry. Data are plotted as the mean±S.E.M. of at least three independent experiments for U937 and HT29 and of at least three biological replicates each assayed in a minimum of two independent experiments for MDFs
The NTDs of mouse, horse and frog, but not human, chicken and fish, MLKL orthologues kill mouse cells
Our observations that the human MLKL NTD did not kill mouse fibroblasts, and mMLKL (1–180) did not kill human HT29 cells (Figure 2), led us to test the killing capacities of MLKL 4HB domains from other species (Figure 5a). We therefore expressed the NTD of mouse, human, horse, frog, chicken and stickleback MLKL, all bearing C-terminal StrepII tags to allow expression to be monitored by western blot (Supplementary Figures 2C and H), in Mlkl−/− MDFs, and evaluated their intrinsic cell-killing capacities (Figures 5b–g). Expression of mouse, horse and frog MLKL NTD induced death of Mlkl−/− MDFs (Figures 5d and e), while the human, chicken and stickleback counterparts did not (Figures 5c, f and g). Using the capacity of the 3H1 anti-MLKL antibody to detect mouse and horse MLKL NTD, we observed that horse MLKL (1–189) translocated to membranes and assembled into high molecular weight complexes by Blue-Native PAGE, two hallmarks of MLKL activation, as observed for mMLKL (1–180; Figure 5h).10 These data suggest a common mechanism of action between mouse and horse MLKL NTDs in inducing cell death.

Mouse, horse and frog MLKL N-terminal domains kill mouse dermal fibroblasts (MDFs), but chicken and stickleback NTDs do not. (a) Alignment of the 4HB domain amino-acid sequences of MLKL orthologues. Numbering and schematic depiction of secondary structure shown above sequences correspond to that of the mouse orthologue. Green shaded sequences are orthologous to R105 and E109 in mouse MLKL. (b–g) Three biologically independent MDF cell lines derived from Mlkl−/− mice were stably infected with the indicated doxycycline-inducible NTD construct from different MLKL orthologues. Expression was induced for 4 h with 10 ng/ml doxycycline before inducing apoptosis (TS) or necroptosis (TSQ) or no treatment (UT) for 24 h. Cell death was analysed in at least two independent experiments by detecting PI-permeable cells using flow cytometry. Data are plotted as the mean±S.E.M (n⩾6). Expression of these constructs was confirmed by western blot in Supplementary Figure 2. In e, a statistical comparison of uninduced versus dox-induced untreated cells using a paired t-test yielded a P-value of 0.0081. (h) Separation of cytoplasmic and membrane fractions on Blue-Native PAGE after a 16 h induction using 50 ng/ml doxycycline of mouse MLKL NTD (1–180) and horse MLKL NTD (1–189) C-terminally tagged with StrepII. Membrane fractionation purity and protein abundance was assessed by immunoblotting for GAPDH and VDAC. Data are representative of three independent repeats
Recombinant MLKL NTDs permeabilize liposomes with compositions resembling those of plasma membranes
Although the foregoing results are consistent with the hypothesis that there are cell-specific factors required for the NTDs to kill cells (summarized in Figure 6a and Supplementary Figure 3), an alternative explanation is that deficits in cell death induction among MLKL orthologues arise from an intrinsic inability to permeabilize membranes. To test this, we prepared recombinant mouse, human, chicken and frog MLKL proteins (Figure 6b), and tested their ability to directly permeabilize liposomes mimicking plasma or mitochondrial membrane compositions in vitro (Figures 6c–k). Unfortunately, we were unable to express and purify recombinant frog NTD. Each of the NTD and full-length MLKL proteins were more effective in permeabilizing liposomes with plasma membrane-like composition than those of resembling mitochondrial membranes (Figures 6c–k). This preference was most apparent among the NTDs of mouse and chicken MLKL (Figures 6c and d). Among the NTD constructs, hMLKL (2–154; Figure 6e) was a poorer mediator of membrane permeabilization than either mMLKL (1–169) or chicken MLKL (2–156; Figures 6c and d). Full-length MLKL proteins were more potent membrane disruptors than their NTDs, raising the possibility that the pseudokinase domain may facilitate either stabilization of the NTD or organization of MLKL monomers into higher order, membrane permeabilizing assemblies. Importantly, we detected only negligible liposome permeabilization in control experiments using recombinant pseudokinase domains (Figures 6i–k), in keeping with a role for the NTDs within the full-length MLKL proteins in mediating membrane perforation. These results indicate that the NTD of MLKL encodes an intrinsic, evolutionarily conserved capacity to permeabilize plasma membranes.

Plasma membrane permeabilization is an intrinsic property of MLKL NTDs. (a) Schematic representation of the steps preceding membrane permeabilization downstream of full-length MLKL activation following RIPK3-mediated phosphorylation (left panel) or expression of NTD constructs (right). The cellular data presented in Figures 1–5 suggest that other putative factors (X1, X2, X3 and X4) regulate MLKL activation (A), oligomerization (B), membrane translocation (C) and permeabilization (D), and that the brace helices can dampen 4HB domain-induced death in some cell lines. Recent work has implicated the co-chaperone system, Cdc37-HSP90, as one such factor that operates as a putative X1 on full-length MLKL.23 RIPK1 and RIPK3 are shown in brown and yellow, respectively; MLKL is depicted in orange; the putative regulators, X1–X4, are shown in blue. (b) Left panel, recombinant mouse MLKL pseudokinase domain (179–464) and N-terminal domains of mouse (residues 1–169), human (2–154) and chicken (2–156) MLKL (~1 μg) were resolved by reducing SDS-PAGE and stained with Simply Blue Safe Stain (Life Technologies). Right panel, recombinant human (190–471) and frog (195–498) MLKL pseudokinase domains and full-length human (2–471), chicken (2–486) and frog (2–498) MLKL (~1 μg) were similarly resolved and stained. (c–k) Liposomes containing the self-quenching dye 5(6)-carboxyfluorescein were exposed to 1 μM recombinant protein as indicated and dye release was monitored spectrophotometrically over 3.25 h. Data are plotted as the mean±S.D. of three to five independent experiments, except for frog MLKL (2–498), which corresponds to mean±S.D. of two independent experiments. Red data and curves represent plasma membrane composition liposomes, while those in black signify mitochondrial-like membranes
Discussion
MLKL contains a C-terminal pseudokinase domain that, until phosphorylated by RIPK3 in its activation loop, suppresses the executioner function of the N-terminal four-helix bundle (4HB) domain. Several studies have implicated the N-terminal domain (NTD), which encompasses the 4HB domain, as the mediator of cell death.10, 11, 12, 13 These studies suggest the simple hypothesis that RIPK3 phosphorylates MLKL leading to a conformational change, exposure of the NTD, resulting in MLKL membrane localization, oligomerization and membrane permeabilization. However, a more complicated mechanism is suggested by studies in L929, CHO and HeLa cells, where dimerization via a fused domain was necessary for the NTD of mouse MLKL to induce cell death.12
Taken together, these studies led us to hypothesize that cell type-specific factors, such as proteins that modulate MLKL activation and 4HB domain exposure, oligomerization, membrane translocation and permeabilization are necessary for MLKL 4HB domain-induced cell death (summarized in Figure 6a). The identities of these proteins (termed X1–X4 in Figure 6a) are of enormous interest to the field and we have recently identified one such factor, the co-chaperone system Cdc37-HSP90, as an important mediator of MLKL activation,23 which can be considered as putative X1 proteins. In addition, we have shown that phosphorylation of the MLKL activation loop coincides with MLKL oligomerization and membrane translocation but that these events precede membrane permeabilization by several hours,15 again strongly implying that this is a regulated, rate-limiting stage in MLKL killing and imply the existence of X4 factors. We also recently described sites of MLKL phosphorylation outside of the activation loop that can positively and negatively regulate necroptosis signalling.15 These findings suggest that putative X1 proteins may post-translationally modify MLKL and thereby determine the threshold for MLKL-induced cell death.15, 24, 25
A key step in MLKL activation is its oligomerization (Figure 6a; Supplementary Figure 3). Although this is readily accomplished by mMLKL NTD or 4HB domains expressed in MDFs, forced dimerization of their human counterparts was required to kill human cells. Notably, hNTD dimerization could only measurably kill U937 cells, whereas h4HB domain dimerization killed wild-type MDF, U937, HT29 and HeLa cells, suggesting that in some cell types the brace helix encodes an inhibitory function, possibly preventing oligomerization, consistent with previous findings.13, 18 Furthermore, these data imply that there are cell-specific factors that are required to enable hMLKL to translocate to membranes, oligomerize and permeabilize membranes.
Recently, we established that hMLKL phosphorylation led to immediate membrane translocation and assembly of phospho-MLKL into higher order complexes, with cell death following some hours later.15 To further test the simple hypothesis that phosphorylation by RIPK3 induces a conformational change in MLKL leading to exposure of the NTD, we examined whether full-length MLKL constructs bearing mutations that mimicked RIPK3-mediated phosphorylation, T357E/S358E (TSEE) hMLKL and S345D mMLKL, could induce death in MDFs, U937 and HT29 cells. Consistent with our hNTD results, TSEE hMLKL did not induce death in wild-type or Mlkl−/− MDFs, HT29 or U937 cells, whereas the murine S345D MLKL mutant caused death of U937 cells. Thus the phosphorylation equals activation hypothesis is too simplistic. Expression of a related T357E/S358D hMLKL construct in U2OS or HT29 cells was previously reported to induce ~30% cell death,14, 19 which was enhanced to ~50% in U2OS cells via dimerization of a fused domain.14 In contrast, using our expression system, we only observed measurable death of wild-type MDF, HT29 or HeLa cells following forced dimerization of wild-type or T357E/S358E hMLKL via a fused gyrase domain, and the extent of death was comparable for wild-type and phosphomimetic mutant constructs. Taken together, these studies imply that killing by hMLKL requires activating signals or interactions in addition to RIPK3 phosphorylation, possibly by promoting MLKL oligomerization.
To further understand the unexpected species-specific difference in the ability of human and mouse MLKL NTDs to kill cells, we examined whether the NTDs of MLKL orthologues could induce cell death. In contrast to the human, chicken and stickleback MLKL NTDs, the mouse, horse and frog MLKL orthologues induced cell death. By comparing amino-acid sequences, we identified the α4-helix residues that align with mouse R105 and E109 as conserved basic and acidic residues, respectively, among mouse, horse and frog MLKL NTDs, but divergent among the non-killing human, chicken and stickleback MLKL. The importance of these residues for cell death was previously inferred from our earlier alanine scanning mutagenesis studies of the mMLKL 4HB domain, where R105A/D106A and E109A/E110A were loss-of-function mutants that had lost the capacity to translocate to membranes and assemble into high molecular complexes.10 Here, we showed that wild-type mMLKL dimerized via a fused gyrase domain can induce cell death in wild-type and Mlkl−/− MDFs, whereas forced dimerization of constructs harbouring the R105A/D106A or E109A/E110A mutations could not. These data indicate that the earlier reported defective membrane translocation by these mutants10 cannot be rescued by forced oligomerization, consistent with an essential role for these residues in either assembly of higher order MLKL oligomers, membrane translocation or recruitment of downstream factors required for the induction of necroptosis rather than MLKL activation per se.
Previous studies have demonstrated that the hMLKL 4HB domain possesses an intrinsic capacity to permeabilize membranes in in vitro liposome assays,13, 14, 18 however, it remained unclear whether this is a broadly conserved function among orthologues and, if so, why all MLKL 4HB domains do not induce death upon expression in cells. Earlier work established a preference of hMLKL 4HB domain for permeabilizing liposomes with 15% cardiolipin or high PIP2 compositions.13, 14 The relevance of the former is unclear because cardiolipin is believed to be exclusively mitochondrially localized, yet mitochondria and the PGAM5-Drp1 mitochondrial fragmentation pathway are not universally required for necroptosis.5, 20, 26, 27, 28 Here, we observed that the 4HB domains of mouse and frog MLKL, which kill MDFs, and human and chicken 4HB domains, which do not, could permeabilize membranes and exhibited a clear preference for plasma membrane over mitochondrial composition liposomes. Unexpectedly, full-length human, frog and chicken MLKL were more potent and rapid inducers of membrane permeabilization than the recombinant NTDs alone, suggesting a function for the pseudokinase domain in augmenting 4HB domain-mediated membrane permeabilization. Although an intrinsic membrane-permeabilization function is clearly conserved between orthologous 4HB domains, no RIPK3 orthologues have been identified in frog, chicken and stickleback, suggesting that other kinases are likely to have evolved compensatory roles as modulators of MLKL activation. Considered together with the cellular studies, our data argue for the existence of additional factors in cells that either suppress MLKL-mediated killing (which might be absent in MDFs) or are required for MLKL to induce necroptosis, for example, by promoting its oligomerization. How the conserved membrane-permeabilization function of the MLKL 4HB domain is regulated within cells remains of enormous interest and highlights the importance of identifying additional effectors within these cells, which either serve to augment MLKL-driven necroptosis or to suppress the membrane-permeabilization function of the 4HB domain.
Materials and Methods
Expression constructs
Synthetic cDNAs encoding mouse (Mus musculus; residues 1–464) and human (Homo sapiens; residues 1–471) MLKL were synthesized by DNA2.0 (CA, USA) cDNAs encoding chicken (Gallus gallus; residues 1–486), frog (Xenopus laevis; residues 1–498), horse (Equus caballus; residues 1–189, N-terminal domain only) MLKL were synthesized by Bioneer (Daejeon, South Korea) and stickleback MLKL (Gasterosteus aculeatus; residues 1–471) was synthesized by GeneArt (Regensberg, Germany). All oligonucleotides for PCR amplifications and oligonucleotide-directed mutagenesis were synthesized by IDT (Singapore or Coralville, IA, USA). For expression in mammalian cell lines, cDNAs were ligated into the doxycycline-inducible puromycin-resistant lentiviral vector, pFTRE3G PGK Puro,5, 10 or cDNAs lacking a stop codon were ligated in-frame as BamHI-NheI fragments into derivative vectors encoding either a C-terminal StrepII tag for detection of expression15 or E. coli DNA GyraseB domain for oligomerization studies.21 For expression of recombinant proteins, cDNAs encoding human MLKL (2–154) was ligated in-frame C-terminal to a TEV protease-cleavable GST tag in the vector pGEX-2T-TEV, and chicken MLKL (2–156) was ligated in-frame C-terminal to a NusA-His6 tag in pETNusH Htb, as described for mouse MLKL (1–169) previously.10 Full-length human (2–471), chicken (2–486) and frog (2–498) MLKL and frog (195–498) pseudokinase domains were expressed from insect cells as described previously for mouse MLKL5, 10 using the Bac-to-Bac system (Life Technologies, Carlsbad, CA, USA), but with a TEV-cleavable N-terminal GST tag for expression of human (2–471) and frog (2–498) MLKL rather than an N-terminal His6 tag. Human MLKL pseudokinase domain (190–471) was expressed and purified as before.29 The sequences of all DNA inserts were verified by Sanger sequencing (Micromon Facility, Melbourne, Victoria, Australia), and are available on request.
Cell lines and cell death assays
U937 and HT29 cells were cultured in human tonicity RPMI medium and HeLa cells in Dulbecco’s modified Eagle’s medium (DMEM) both supplemented with 8–10% v/v fetal calf serum (FCS) and puromycin (5 μg/ml) for lines stably transduced with MLKL expression constructs. Cell death assays were carried out in 48-well plates, seeded at 1.5 × 104 for HT29 and 3 × 104 for U937 and HeLa cells per well, as described previously.15 The cells were left for 48 h and treated for 4 h with 10 ng/ml doxycycline and coumermycin (700 nM) when gyrase fusions were expressed. The cells were either left untreated or treated with TNF (100 ng/ml) and the Smac-mimetic, Compound A (500 nM), or TNF, Compound A and the pan-Caspase inhibitor, Q-VD-OPh (10 μM), after 24 or 48 h collected as described in the figure legends, stained with propidium iodide (PI; 1 μg/ml) and quantified by flow cytometry.
Three independent lines of Mlkl−/− and wild-type MDFs were generated as described previously.5 MDFs and HEK293T were maintained in DMEM supplemented with 8–10% v/v FCS, and puromycin (2 μg/ml) for lines stably transduced with MLKL expression constructs. Cell death assays were carried out in 24-well plates, seeded at 5 × 104 cells per well, as before.15 The cells were attached overnight and then induced for 4 h with 10 ng/ml doxycycline and then left either untreated or treated with TNF and Smac-mimetic, or the TNF, Smac-mimetic and Q-VD-OPh cocktail, as above. Quantification of cell death by flow cytometry was performed 24 h post stimulation.
Statistical analyses
Error bars represent mean±S.E.M. of specified number of independent and/or biological repeats of cell death assays, not technical replicates. Liposome permeabilization assays are presented as the mean±S.D. of independent experiments.
Fractionation and blue native PAGE
Fractionation of cells into cytoplasmic and membrane fractions was performed as described previously.10, 15 Briefly, U937 or MDF cells stably transduced with mutant MLKL constructs were induced as indicated in the figure legends, before cells were collected and permeabilized in buffer (20 mM HEPES pH 7.5, 100 mM KCl, 2.5 mM MgCl2 and 100 mM sucrose) containing 0.025% digitonin (BIOSYNTH, Staad, Switzerland), 2 μM N-ethyl maleimide, phosphatase and protease inhibitors. Crude membrane and cytoplasmic fractions were separated by centrifugation (5 min at 11 000 × g), and the respective fractions prepared in buffers to a final concentration of 1% w/v digitonin. The samples were resolved on a 4–16% Bis-Tris Native PAGE gel, transferred to PVDF for western blot analyses.
Western blot antibodies
Antibodies used for western blotting were: the rat anti-MLKL 3H1 monoclonal (produced in-house;5 available as MABC604, EMD Millipore, Billerica, MA, USA), which detected mouse, human and horse MLKL; anti-Strep-tag II (ab76949, Abcam, Cambridge, UK); StrepTactin-HRP (2-1502-001, IBA, Life Sciences, Gottingen, Germany); rabbit anti-VDAC1 (AB10527, EMD Millipore); rabbit anti-GAPDH (2118, Cell Signalling Technology, Danvers, MA, USA); mouse anti-Actin (A-1987, Sigma-Aldrich, St Louis, MO, USA).
Recombinant protein production
Pseudokinase domains from mouse (179–464) and frog (195–498) MLKL, full-length frog (2–498) and chicken (2–486) MLKL were expressed and purified from Sf21 insect cells, as described for full-length mouse MLKL and its pseudokinase domain previously,5, 10, 15 albeit with a TEV protease-cleavable N-terminal GST tag for frog MLKL (2–498) instead of the N-terminal His6 used otherwise. Mouse MLKL N-terminal domain (1–169) was expressed fused to an N-terminal NusA-His6 tag and purified from E. coli BL21 Codon Plus as previously.10 Chicken MLKL (2–156) was prepared analogously. The N-terminal domain of human MLKL (2–154) was expressed in E. coli BL21 Codon Plus fused to an N-terminal GST tag encoded by the vector, pGEX-2T-TEV. Purification was performed using standard protocols.21, 30 Briefly, 0.6 L Super broth cultures containing 100 μg/ml ampicillin were inoculated with transformed E. coli and cultured at 37 °C with shaking to OD600 of 0.6–0.8. Cultures were then cooled to 18 °C, protein expression induced by the addition of 1 mM IPTG with continued shaking and incubation at 18 °C overnight. The cell pellets were resuspended in lysis buffer (200 mM NaCl, 20 mM HEPES pH 7.5, 0.5 mM TCEP) supplemented with 2 mM PMSF, before lysis by sonication, elimination of debris by centrifugation at 45 000 × g, 0.45 μm filtration of the lysate and incubation with glutathione agarose (UBP Bio, Aurora, CO, USA) at 4 °C with agitation for 1–2 h. The beads were collected and washed extensively with lysis buffer before incubation with 200 μg TEV protease at 20 °C for 2 h. Supernatant containing cleaved MLKL N-terminal domain was concentrated by centrifugal ultrafiltration and loaded on to Superdex S200 gel filtration column pre-equilibrated with gel filtration buffer (200 mM NaCl, 20 mM HEPES pH 7.5). Fractions containing purified MLKL N-terminal domain as assessed by SDS-PAGE were pooled, concentrated by centrifugal ultrafiltration to 5–10 mg/ml, snap frozen as 50 μl aliquots in liquid nitrogen and stored at −80 °C until required. All recombinant proteins used in liposome studies were eluted from Superdex-200 in gel filtration buffer.
Liposome permeabilization assays
Liposomes of plasma membrane (20% POPE, 40% POPC, 10% phosphoinositol, 20% DOPS, 10% POPG; based on refs 31, 32) and mitochondrial (30% POPE, 45% POPC, 10% phosphoinositol, 15% cardiolipin; based on ref. 18) mimetic compositions were assembled from lipids purchased from Avanti Polar Lipids (Alabaster, AL, USA) and resuspended in chloroform at 20 mg/ml. Lipids were mixed to a final volume of 100 μl, solvent was evaporated under N2 gas followed by vacuum. 18.8 mg of 5(6)-carboxyfluorescein was solubilized in 100 μl of 1 M NaOH, spun at 14 000 × g for 1 min, before 25 μl of 1 M HEPES pH 7.5 added and the volume made up to 1 ml with milliQ water to a final concentration of 50 mM. Dried lipids were resuspended in 1 ml 50 mM 5(6)-carboxyfluorescein solution, vortexed and sonicated, then liposomes extruded through a 100 nm pore membrane. Liposomes were purified from excess 5(6)-carboxyfluorescein by gel filtration using PD10 columns (GE Healthcare, Sydney, NSW, Australia) equilibrated in SUV buffer (10 mM HEPES pH 7.5, 135 mM KCl, 1 mM MgCl2) and eluted in 2.5 ml SUV buffer to yield liposomes at 30 μM concentration. Liposomes (10 μM) were incubated with 1 μM recombinant protein at 20 °C with dye release monitored spectrophotometrically (excitation 485 nm, emission 535 nm) starting 15 min after components were assembled, and collecting every subsequent 30 min for a further 3 h. A 100% dye release was determined in parallel by incubation of liposomes with 1% Triton-X100.
Abbreviations
- 4HB:
-
4-helix bundle
- Cdc37:
-
cell division cycle37
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- DOPS:
-
1,2-dioleoyl-sn-glycero-3-phosphoserine
- Dox:
-
doxycycline
- FCS:
-
fetal calf serum
- GST:
-
glutathione-S-transferase
- HSP90:
-
heat shock protein90
- MDF:
-
mouse dermal fibroblasts
- MLKL:
-
mixed-lineage kinase domain-like
- mMLKL:
-
mouse MLKL
- hMLKL:
-
human MLKL
- NTD:
-
N-terminal domain
- PIP:
-
phosphatidylinositol-phosphate
- POPE:
-
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPC:
-
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG:
-
2-oleoyl-1-pamlitoyl-sn-glyecro-3-glycerol
- Puro:
-
puromycin
- RIPK:
-
receptor interacting protein kinase
- TLR:
-
Toll-like receptor
- TNF:
-
tumour necrosis factor
- TSQ:
-
TNF (T), Smac-mimetic Compound A (S) and QVD-OPh (Q)
References
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489–495.
Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313–321.
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013; 39: 443–453.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–227.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 2012; 109: 5322–5327.
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 2013; 23: 994–1006.
Czabotar PE, Murphy JM . A tale of two domains—a structural perspective of the pseudokinase, MLKL. FEBS J 2015; 282: 4268–4278.
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 2014; 111: 15072–15077.
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16: 55–65.
Chen X, Li W, Ren J, Huang D, He WT, Song Y et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014; 24: 105–121.
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014; 7: 971–981.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–146.
Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C et al. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J 2015; 471: 255–265.
Linkermann A, Green DR . Necroptosis. N Engl J Med 2014; 370: 455–465.
Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 2016; 23: 76–88.
Su L, Quade B, Wang H, Sun L, Wang X, Rizo J . A plug release mechanism for membrane permeation by MLKL. Structure 2014; 22: 1489–1500.
Yoon S, Bogdanov K, Kovalenko A, Wallach D . Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ (e-pub ahead of print 17 July 2015 doi:doi:10.1038/cdd.2015.92).
Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 2013; 5: 878–885.
Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ 2014; 21: 1600–1612.
Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem 2013; 288: 16247–16261.
Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis 2016; 7: e2051.
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 2015; 6: 6282.
Murphy JM, Vince JE . Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Res 2015; 4: 1297.
Moujalled DM, Cook WD, Murphy JM, Vaux DL . Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis 2014; 5: e1086.
Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 2014; 5: e1004.
Schenk B, Fulda S . Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 2015; 34: 5796–5806.
Murphy JM, Lucet IS, Hildebrand JM, Tanzer MC, Young SN, Sharma P et al. Insights into the evolution of divergent nucleotide-binding mechanisms among pseudokinases revealed by crystal structures of human and mouse MLKL. Biochem J 2014; 457: 369–377.
Murphy JM, Zhang Q, Young SN, Reese ML, Bailey FP, Eyers PA et al. A robust methodology to subclassify pseudokinases based on their nucleotide binding properties. Biochem J 2014; 457: 323–334.
Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B, McLafferty FW . Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry 1999; 38: 8056–8063.
Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 2008; 135: 702–713.
Acknowledgements
We thank Professor Mark Hampton and Dr. Andreas Konigstorfer for kindly providing HT29 cells and Dr. Toru Okamoto for development of the inducible lentiviral vector. This work was supported by National Health and Medical Research Council of Australia (NHMRC) grants (461221, 1046010, 1051210, 1057888, 1057905, 1067289) and fellowships to JMH (541951), DLV (1020136), JEV (1052598), PEC (1079700) and JS (541901, 1058190); an Australian Research Council Future Fellowship to JMM (FT100100100); a Victorian International Research Scholarship to MCT; with additional support from the Victorian State Government Operational Infrastructure Support and NHMRC IRIISS grant (9000220).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
JS is a member of the Scientific Advisory Board of TetraLogic Pharmaceuticals Corporation. JMM co-leads a programme funded by Catalyst Therapeutics Pty Ltd and the Walter and Eliza Hall Institute to develop necroptosis inhibitors. The remaining authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Tanzer, M., Matti, I., Hildebrand, J. et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ 23, 1185–1197 (2016). https://doi.org/10.1038/cdd.2015.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.169
This article is cited by
-
LUBAC-mediated M1 Ub regulates necroptosis by segregating the cellular distribution of active MLKL
Cell Death & Disease (2024)
-
MLKL post-translational modifications: road signs to infection, inflammation and unknown destinations
Cell Death & Differentiation (2023)
-
Phosphorylation-dependent pseudokinase domain dimerization drives full-length MLKL oligomerization
Nature Communications (2023)
-
A common human MLKL polymorphism confers resistance to negative regulation by phosphorylation
Nature Communications (2023)
-
Membrane permeabilization is mediated by distinct epitopes in mouse and human orthologs of the necroptosis effector, MLKL
Cell Death & Differentiation (2022)
