
Abstract
Recent cases of successful control of human immunodeficiency virus (HIV) by bone marrow transplant in combination with suppressive antiretroviral therapy (ART) and very early initiation of ART have provided proof of concept that HIV infection might now be cured. Current efforts focusing on gene therapy, boosting HIV-specific immunity, reducing inflammation and activation of latency have all been the subject of recent excellent reviews. We now propose an additional avenue of research towards a cure for HIV: targeting HIV apoptosis regulatory pathways. The central enigma of HIV disease is that HIV infection kills most of the CD4 T cells that it infects, but those cells that are spared subsequently become a latent reservoir for HIV against which current medications are ineffective. We propose that if strategies could be devised which would favor the death of all cells which HIV infects, or if all latently infected cells that release HIV would succumb to viral-induced cytotoxicity, then these approaches combined with effective ART to prevent spreading infection, would together result in a cure for HIV. This premise is supported by observations in other viral systems where the relationship between productive infection, apoptosis resistance, and the development of latency or persistence has been established. Therefore we propose that research focused at understanding the mechanisms by which HIV induces apoptosis of infected cells, and ways that some cells escape the pro-apoptotic effects of productive HIV infection are critical to devising novel and rational approaches to cure HIV infection.
Similar content being viewed by others

Facts
-
Human immunodeficiency virus (HIV) has been cured in one and possibly more patients.
-
Efforts are underway to recapitulate a cure for HIV in a generalizable way.
-
Cells containing latent HIV that are induced to reactivate virus do not die due to viral replication.
-
Chronically HIV-infected cells are resistant to apoptosis.
-
Understanding the regulation of apoptosis during HIV infection and latency may be the key to develop a cure for HIV.
Open Questions
-
Why do all cells that are HIV infected not die as a consequence of productive HIV replication?
-
Why do latently HIV-infected cells that are induced to reactivate virus not die as a result of productive HIV replication?
-
Can therapeutic strategies be designed that will both reactivate HIV from latency and induce the death of cells that replicate HIV?
Timothy Ray Brown, ‘the Berlin Patient,’ is now >6 years post bone marrow transplant (BMT) from a donor with the Δ32 mutation in CCR5, and he has no detectable HIV in his blood or tissues while off combination antiretroviral therapy (cART).1, 2, 3 His story has fostered hopes that HIV might be cured, within the foreseeable future. In this article, we will review the recent advances in our understanding of HIV latency, review the approaches that are being tested as a means to cure HIV and discuss the challenges associated with targeting latently HIV-infected CD4 T cells for death, with a focus on ways to alter apoptosis regulation.
Relationship Between Viral Infection, Apoptosis and Viral Persistence
Broadly speaking, viruses that cause human disease cause either acute self-limited infections or chronic persistent infections, each with differing effects on host cell apoptosis and viral persistence.
Acute self-limited viral infections include influenza, Ebola, Hantavirus and dengue, among others. In the best studied of these, influenza virus, hemaglutinin expressed on the surface of an infectious virion binds sialic acid sugars on the surfaces of epithelial cells, typically in the nose, throat, and lungs of the susceptible host.4, 5 Following viral replication, progeny virions bud are released from the infected cell and the cell undergoes apoptosis.6 Apoptosis is associated with an interferon response and decreasing the magnitude of this response increases cell survival following challenge with influenza.7 The influenza nonstructural (NS) protein-1, also induces caspase-dependent apoptosis,8 but to variable degrees according to sequence differences in influenza subtypes,9 likely involving interactions with microtubules.10 As influenza is an acute self-limited infection there are no mechanisms that allow for influenza persistence, and there are no influenza-associated pathways that inhibit apoptosis.
Chronic persistent or chronic latent infections occur with the herpes virus family (Epstein Barr Virus (EBV), cytomegalovirus (CMV) and herpes simplex virus (HSV)), HIV, Hepatitis B and C, and human papilloma virus (HPV), among others. What distinguishes these infections is their ability to persist long after the initial symptoms of infection have resolved and that each virus has evolved strategies to evade apoptosis. The various strategies used by these viruses to evade cell death have been extensively reviewed elsewhere.11
A causal relationship between evasion of apoptosis and the establishment of latency has been demonstrated in the case of EBV. EBV encodes two viral homologs of the cellular anti-apoptosis protein Bcl2, called BALF1 and BHRF1. EBV mutant viruses missing both BALF1 and BHRF1 resulted in robust viral replication but chronic persistent infection as measured by lymphoblastic transformation did not occur.12 Thus in the case of EBV, chronicity and transformation depends upon the presence of apoptosis inhibitors, and so opens the possibility that apoptosis inhibition is required for latency in other viral systems.
HIV is a virus that establishes latency, and has developed multiple strategies to inhibit apoptosis of infected cells under a variety of circumstances. We have previously reviewed ways that HIV infection and individual HIV proteins inhibit apoptosis.13 Although no HIV proteins are in themselves apoptosis inhibitors, the expression of select HIV proteins, such as Vpr, Nef and Tat, alter the transcriptional profile of some cell types to produce more endogenous apoptosis inhibitory proteins. For example, in vitro, Tat induces cellular FLICE inhibitory protein (cFLIP) expression in T cells14 including primary T cells and thereby confers resistance to apoptosis.15 Vpr increases Bcl2 and decreases Bax in the Jurkat T cell line,16 and there is increased X-linked inhibitor of apoptosis (XIAP) expression in latently infected cell line models.17 Both Tat-treated monocytes18 and primary CD4 T cells from HIV-infected patients19 develop tumor necrosis factor (TNF)-related apoptosis inhibitory protein (TRAIL) resistance, potentially through the production of a novel TRAIL splice variant (TRAILshort), which preferentially binds TRAIL receptor (R)2 and prevents pro-apoptotic TRAIL from signaling.20 TRAILshort can be found in the plasma and cells of HIV-infected patients.20 Another possible mechanism is Tat-mediated upregulation of a variety of nuclear factor kappa-B (NFκB)-dependent apoptosis inhibitors, including Bcl2, cFLIP, XIAP and CIAP.21
HIV Latency in CD4+ T-cells
One of the major barriers to HIV cure is the establishment of latency in resting CD4+ T cells. This may arise from cells that are productively infected with HIV and revert to a resting memory T-cell phenotype with integrated pro-virus (post-activation latency) or via direct infection of resting CD4+ T cells (pre-activation latency). The relative contribution of each pathway in vivo is unknown. Once latency is established, latently infected resting memory T cells have a prolonged half-life estimated to be 44 months (reviewed in Finzi et al.22 and Pierson et al.23). Latently infected cells are detected at increased frequency in tissue, such as lymphoid tissue and the gastrointestinal (GI) tract.24 Virus can also persist in other long-lived cells such as infected macrophages,25 naïve T cells,26 follicular dendritic cells27 and the cells in the central nervous system (CNS) including astrocytes28 and microglia.29 The relative contribution of these non-memory T-cell reservoirs is less clear.
The molecular basis of latency—defined as HIV DNA integrated in the host genome but remaining transcriptionally inactive—is complex and may involve multiple mechanisms contributing to transcriptional repression simultaneously. This topic has been extensively reviewed elsewhere30 and include the following mechanisms: (i) insufficient levels of host transcription factor expression, and/or expression of transcription factor complexes with negative regulatory activity;31 (ii) epigenetic silencing and chromatin remodeling that prevent access of transcription factors to transcription initiation sites within the HIV long terminal repeat (LTR);32 (iii) differences in the efficiency of HIV transcription between different insertion sites and different insertion orientations;33 and (iv) the absence of the transactivator of transcription encoded by HIV, Tat.
Models of Latency
There are multiple models of HIV latency, which have been used to understand both the establishment and maintenance of latency and to identify molecules that can activate virus production from latently infected cells.34 Currently available models include latently infected cell lines, latently infected primary CD4+ T cells and resting CD4+ T cells from patients on suppressive cART (generally defined as plasma HIV RNA <50 copies/ml).
Latently infected cell lines
The most commonly used latently infected cell lines include ACH2,35 U1,36 J-lat clones37 and J89.38 Latently infected cell lines have several limitations. First, they represent clonal populations in which a single integration site is present,36, 37 whereas latently infected CD4+ T cells from patients have integration sites randomly distributed throughout the host genome. Second, cell lines are rapidly dividing cells, while latently infected CD4+ T cells found in vivo are resting.39 This impacts reactivation strategies such as histone deacetylase inhibitor (HDACi), which are 10-fold more active in transformed cells compared with non-transformed cells.40 Finally, in latently infected cell lines, integration usually occurs at sites of heterochromatin37 while latently infected primary cells CD4+ T HIV integrates into sites of active gene expression.41
Latently infected primary CD4+ T cells
Several primary CD4+ T-cell models of latency exist where activated cells are infected and subsequently allowed to return to a quiescent latently infected state.42 One model has used naïve CD4+ T cells that are polarized and then infected with a single round virus (which is envelope deficient). Another uses naïve CD4+ T cells co-cultured with antigen-presenting cells and infected with a wild-type HIV (capable of multiple rounds of infection)42 or stimulated with anti-CD3/CD28 before infection. These models are technically demanding as they require a long time in culture ranging from 21 days43 to >60 days.42 Other models have used direct infection of resting CD4+ T cells either via spinoculation44 or in ex vivo tonsil tissue blocks or following incubation with chemokines such as CCL19 or CCL21 (ligands for CCR7), which allows for efficient viral nuclear localization and integration without activation of the cell.45, 46 Finally, CD4+ T cells can also be transduced with Bcl2 to allow for long-term culture, infected with HIV and permitted to return to a resting state.47 The frequency of latently infected cells in these models ranges from 0.1 to 1.0%42, 46, 47 to as high as 20–30%.
Resting CD4+ T cells from HIV-infected patients on cART
The gold standard model of latently infected cells is resting CD4+ T cells from HIV-infected patients on suppressive cART.48 The frequency of latently infected cells can be quantified by activation with a mitogen or anti-CD3/CD28 and then co-culturing with uninfected cells to amplify viral production (also called limiting dilution micro-coculture or infectious units per million (IUPM) cells). While this represents the most accurate assessment of latently infected cells ex vivo, this technique is time consuming; requires the collection of large volumes of blood usually via leukapheresis from patients; and is only semi-quantitative.48 Recent evidence also suggests that IUPM may underestimate the true number of latently infected cells that carry infectious virus and may only represent 10–30% of ‘activatable’ virus.49
Viral Latency and the Role of Apoptosis Pathways
If HIV killed all of the cells that it infects, then logically, HIV persistence would not occur. If persistence is the goal, why then does HIV kill any cells that it infects? Simply, the machinery that is activated during apoptosis (caspase activation) also acts through the BCL10/MALT1/CARMA pathway to activate NFκB, which is the most potent activator of HIV transcription, via the HIV LTR.35 Evidence supporting this includes: (i) HIV replication is increased in T leukemia cells and peripheral blood mononuclear cells treated with the pan-caspase inhibitor z-VAD-fmk;50 (ii) HIV replication in immortalized T-cell lines induced to express the pro-apoptotic proteins FasL, Fas-associated death domain (FADD) protein and p53; (iii) HIV replication is decreased in cells overexpressing the anti-apoptotic proteins Bcl2, FLIP51 or with knockdown of the pro-apoptotic proteins Bax or FADD;52 and (iv) expression of Casp8p41 (a unique cleavage fragment of procaspase 8 generated by HIV protease) in infected cells directly activates NFκB-dependent HIV LTR transcription.53 Given this, one would predict that in settings where the goal of HIV infection is to establish a latent reservoir, it would not be in the interest of HIV to induce cell death.
Apoptosis Susceptibility in Memory T cells in Vivo
Resting memory T cells are metabolically inactive; function to archive historical immune responses; and therefore, need to be long lived and resist stimuli which under normal circumstances favor T-cell death. To understand apoptosis resistance of latently HIV-infected T cells, it is important to first understand the mechanisms of apoptosis resistance in memory CD4 T cells.
T-cell activation and reversion to memory has been the subject of a recent excellent review.54 CD4 T cells undergo activation and proliferation after exposure to a neoantigen that is recognized by the T-cell receptor (TCR) and presented in the context of appropriate major histocompatibility complex (MHC) Class II and co-stimulatory help. Resting CD4 T cells that express CCR7 and CD62L are relatively metabolically inactive and apoptosis resistant before they encounter antigen. However, after antigen exposure with appropriate co-stimulation, T cells proliferate, produce interleukin-2, upregulate Fas and other pro-apoptotic molecules, and change from an apoptosis-resistant state to a post-activation apoptosis-prone state. Forty eight hours after antigen exposure, the CD4 T cell is maximally activated and most sensitive to apoptosis. Thus, in settings such as prolonged TCR engagement, absence of co-stimulatory molecules, or withdrawal of cytokines required for survival, these cells undergo activation-induced cell death (AICD).
The principal molecular mediators of AICD are Fas and Fas ligand, but the susceptibility of the cell to undergo Fas-mediated death is governed by the cumulative expression of apoptosis regulatory proteins, including Bcl2 family members, Inhibitor of Apoptosis proteins (IAPs) and cFLIP. Because productive HIV infection pervasively alters the expression of apoptosis regulatory molecules, the fraction of cells that undergo AICD in an HIV-infected patient is greater than that of an HIV-negative patient and this contributes to the depletion of uninfected CD4 T cells.55
CD4 T-cell memory occurs when antigen concentrations become low but co-stimulation with CD28 and/or interleukin-2 persists. In such settings, anti-apoptotic molecules such as Bcl2 or cIAPs are upregulated to prevent AICD. This process is coincident with the downregulation of CD45RA and low-level expression of CD45RO. There are two types of CD4 memory T cells: central memory T cells (TCM), which express lymph node-homing molecules CCR7 and CD62L;56 and effector memory T cells (TEM), which express receptors such as CCR5.57 HIV preferentially infects HIV-specific activated memory CD4+ T cells, as opposed to memory cells of other specificities.58 Alternately HIV may infect activated cells, which then adopt a central memory phenotype. We have recently shown that stromal-derived factor (SDF-1α) treatment of activated CD4+ T cells resulted in degradation of Bim, resulting in apoptosis resistance and cells adopting a memory phenotype.59 Thus, HIV-infected CD4+ T cells with a memory phenotype, including latently infected cells, are apoptosis resistant, making these cells particularly difficult to eradicate.
Approaches to Cure HIV
Recent advances in our understanding of the biology of HIV have now allowed for proposals to recapitulate either a sterilizing cure (complete elimination of all HIV-infected cells from an individual) or functional cure (long-term control of HIV replication with HIV RNA <50 copies/ml in the absence of cART).
Very early initiation of cART has been associated with a reduced number of latently infected cells and recently there have been reports of successful control of virus replication in individuals who have initiated treatment early and stopped cART.60 The frequency of ‘post-treatment control’ is extremely rare—estimated at <2% of all patients who initiate ART during acute infection.61 Very early initiation of cART following delivery of a baby to an HIV-infected mother not on ART in Mississippi was also recently reported to result in a functional cure.62
Three classes of approaches have been proposed as curative strategies for HIV: reactivation approaches, gene therapy-based approaches, and immune-based therapies (Table 1).
HIV Reactivation
The underlying premise of this approach is that HIV reactivation from a latently infected CD4 T cell will lead to cytotoxicity which, it is hoped, will cause all infected cells to die. A wide variety of approaches have been proposed to reactivate HIV (Table 2). Great interest has focused on epigenetic silencing mechanisms such as histone acetylation, and ways to reverse these processes.
A small proof of concept study of a single dose of the HDACi, vorinostat in HIV-infected patients on suppressive cART resulted in an increase in both histone acetylation and cell-associated HIV RNA in resting memory CD4+ T cells.63 We recently completed a multidose study of 14 days of daily vorinostat in HIV-infected patients (n=20) and demonstrated an increase in cell-associated HIV RNA in 90% of participants, although disappointingly we observed no decline in HIV DNA.64 A similar study is being performed in Denmark with the potent HDACi panobinostat [clinicaltrials.gov] and a single dose study of the HDACi rhomedepsin is underway. Disulfiram, which acts via a totally different mechanism, most likely via depletion of the phosphatase and tensin homolog (PTEN) and activation of AKT phosphorylation has also recently been shown to have some activity in increasing detection of HIV RNA in plasma shortly after dosing in a subset of patients.65
The main mode of action of HDACi in the treatment of cancer is the induction of apoptosis and cell-cycle arrest in rapidly dividing cells. Using the J-Lat cell line, a Jurkat cell line stably infected with HIV-1 that contains a deletion of the Env and Nef genes and encodes for expressed green fluorescent protein (EGFP), under the control of the HIV LTR37 we observed that following treatment with the potent HDACi, MCT1, MCT3 and oxamflatin, EGFP+ cells (i.e., cells induced to express virus) were also enriched for cells expressing activated caspase 3, annexin V and propidium iodide.66 However, in primary cell models HIV reactivation by vorinostat did not appear to induce death.67 In a recent report of elegant studies using latently infected primary T cells that overexpress BCL2, and infected with HIV-1 that contains a deletion of the Nef and pol genes and encodes for EGFP (NL4.3ΔNefΔPol-EGFP), following reactivation of HIV with vorinostat, cell did not die during 18 days of observation. Moreover, vorinostat-reactivated cells only died when co-incubated with autologous CD8 T cells from an elite controller, indicating that immune clearance is possible but requires an effective HIV-specific cytotoxic T-lymphocyte (CTL) response which is often absent in HIV-infected patients treated during chronic infection.67 Consistent with these findings, another study using an in vitro model of latently infected central memory CD4 T cells (TCM) cells, reactivation of latent virus with interleukin-2 and interleukin-7 did not cause cell death, whereas reactivation with CD3–CD28 co-stimulation did kill cells.68
Together, these studies indicate that viral reactivation alone may not be sufficient to induce cell death, and that other interventions may be required for TCM cells to die following viral reactivation. Other pathways that may be important for viral activation include the protein kinase C (PKC) pathway (activated by prostratin and bryostatin), the STAT5 pathway (activated by IL-7), the AKT pathway (via depletion of PTEN with disulfiram) and methylation inhibition (5-azacytadine). The effects of activation of each of these pathways and cell death need to be further explored.
Gene Therapy Approaches
There are three general approaches that use gene therapy to attempt to cure HIV. The first is a gene knockdown to reduce the expression of a host protein that HIV requires to complete its life cycle. An example of such an approach is to use zinc finger nucleases (ZFN) to degrade the message for either CCR569 or CXCR470 and render these cells resistant to either R5 or X4 HIV, respectively. Several early phase trials of autologous CD4 T cells that have been modified ex vivo with ZFN that knockdown CCR5 have now been reported and have demonstrated that this approach was feasible in humans, was safe and well tolerated, reduced HIV RNA rebound levels during cART interruption and induced an increase in total CD4 T-cell numbers.71
A second approach to gene therapy for HIV involves overexpressing proteins, which limit HIV replication and/or pathogenesis, and include chimeric TRIM5α molecules representing human/rhesus fusions that inhibit HIV replication in vitro.72 Another approach involves overexpressing broadly neutralizing anti-HIV antibodies. The antibody b12 has been overexpressed in humanized bone marrow/liver/thymus (hu-BLT) mice resulting in durable production of human b12, and significant improvements in CD4 T-cell numbers following HIV challenge.73, 74
A third gene therapy approach involves expression of chimeric antigen receptors (CARs). These were first proposed more than two decades ago and the first iteration was fusion proteins consisting of the variable immunoglobulin light chain and variable heavy chain region specific for an antigen of interest, fused to the CD3ζ and transmembrane domains (reviewed in Sadelain et al.75). When this chimeric receptor was expressed in T cells, upon exposure to cognate antigen and ligand binding to the variable heavy and light immunoglobulin domains, CD3ζ signaling occurred, causing T-cell activation. Second- and third-generation CARs also include the intercellular signaling domains of CD28, 41BB, or OX40, to provide appropriate co-stimulatory signals. Recently, T cells modified with a third-generation CAR have been reported to successfully treat patients with adult acute lymphoblastic leukemia.75, 76 Trials of first-generation CAR-modified T cells in HIV have now been reported with >11 years follow-up and demonstrated persistence of the CAR-modified T cells and chimeric T-cell function, albeit with minimal anti-HIV effect.77 Ongoing attempts to maximize antiviral activity include different receptor targets, advanced generation CARs, and application of CAR technology for use in natural killer (NK) cells.
Perhaps instructed by early experience with ART, combination-based gene therapy approaches have also been suggested. Indeed, one such approach evaluated T cells transduced with a combination of a CCR5 ribosome, silencing (si)RNA to Tat/Rev and an HIV decoy RNA, transfused post-chemotherapy conditioning to HIV-infected patients with lymphoma.78 In this proof of concept trial, persistence of the transgene declined precipitously within the first few weeks. Other delivery backbones and other conditioning regimens are therefore being considered.79 These include a variety of combination approaches (Clinical Trials # NCT 01734850, NCT 01769911).
Immune-Based Therapies
There are two broad classes of immune-based approaches that have been proposed for therapy of HIV infection—including boosting an effective immune response or reducing immune activation. The underlying premise of boosting an effective immune response is to recapitulate HIV-infected patients who can spontaneously control viral replication (elite controllers). Spurred by multi-disciplinary collaborative groups, a variety of genes and immune functions have been associated with elite control. These associations include the Δ32 allele of CCR5,80, 81, 82 HLA-B5701, and HLA-B27 alleles,83 the NK inhibitory receptors KIR3DS1 and KIR3DL1;83, 84 increased expression of proteins necessary for granule exocytosis-mediated cytotoxicity,85 such as granzyme A, granzyme B, and perforin; and higher numbers of both plasmacytoid dendritic cells and polyfunctional T cells in elite controllers compared with non-controllers or cART-suppressed patients. While it remains unknown which of these associations are required for control of HIV, the existence of these associations has spurred attempts to recapitulate the immune phenotype, to achieve immune control of HIV.
Increased immune activation is associated with morbidity and mortality from both AIDS defining and non-AIDS defining causes.86 Putative causes include translocation of bacterial products across the GI tract, persistent HIV and co-infection with other pathogens, such as CMV and HCV.86 There is a significant correlation between markers of T-cell activation (including expression of HLA-DR, CD38, and PD-1) and markers of viral persistence (including cell-associated HIV DNA and RNA) in T cells in blood and the GI tract.87 However, there is no association between low level plasma viremia and markers of either T-cell or innate immune activation.88
It is possible that targeting inflammation may reduce virus persistence or alternatively targeting virus persistence may reduce inflammation. Although multiple studies of intensification of antiretrovirals have shown no change in HIV DNA or low level plasma viremia, two studies with intensification of raltegravir have shown a reduction in immune activation.89, 90 In addition, both these studies demonstrated that residual virus replication persists in ∼30% of patients on cART. Several approaches that reduce inflammation are currently being evaluated, including anti-inflammatory agents, statins, chloroquine derivatives, leflunomide, pre- and pro-biotics, growth hormone, immunotoxins, and combination approaches.
Another proposed avenue for potential immune-based therapy involves the administration of recombinant cytokines, most commonly members of the IL-2 receptor α subunit family: IL-2, -7, -15, and -21. In the large multi-national SILCAAT and ESPRIT studies, IL-2 therapy increased CD4 T-cell number, but neither improve CD4 T-cell function nor improve health. Of relevance to the HIV cure agenda, IL-2 therapy has also been assessed as a means of decreasing HIV burden; while detectable replication competent HIV was decreased in some patients receiving cART plus IL-2 compared with cART alone,91 all patients had a rapid rebound in virus following treatment cessation indicating that HIV burden was not meaningfully altered by therapy.91
IL-7 therapy has been tested in smaller studies, and shown to increase CD4 T-cell number and function, including increasing anti-HIV-specific CD4 T-cell function.92, 93 IL-7 administration caused modest increases in total intracellular HIV DNA, in proportion to the increases in CD4 T-cell number, suggesting that reservoir size was increased by homeostatic proliferation,94 consistent with the effects of IL-7 in in vitro models of HIV latency.68 Of interest, the increases in reservoir size were associated with increased expression of the anti-apoptotic protein Bcl2,94 consistent with a model of apoptosis resistance favoring HIV persistence.
Both IL-15 and IL-21 enhance innate and adaptive anti-HIV responses. IL-15 augmented both NK cell and HIV- and SIV-specific CD8 T-cell function in vitro, suggesting a potential role for IL-15 as an immunotherapeutic to increase anti-HIV/SIV responses.95 IL-15, but neither IL-7- nor IL-2-treated NK cells increased expression of TRAIL, and killing of autologous CD4 T cells, and reduced the frequency of HIV containing CD4 T cells, ex vivo.96 In vivo, SIV-infected macaques treated with IL-15 had increased numbers of SIV-specific CD8 T cells and increased NK cell numbers with reduced numbers of SIV-infected cells in lymph nodes consistent with an antiviral effect; however, plasma viremia was increased by 2–3 logs.97 Therefore, any therapeutic role of IL-15 may be limited by potentially inducing viral replication.
IL-21 has been shown to enhance antiviral NK cell and CD8 T-cell immunity in animal models of non-HIV viral infections, and has been used successfully in safety trials of patients with advanced malignancy, thereby making it an attractive candidate for anti-HIV therapy.98 In SIV-infected macaques, IL-21 administration increased perforin and granzyme B expression in CD8, effector CD4, and NK cell subsets; however, it remains unknown whether these favorable quantitative changes will translate into improved antiviral function and elimination of latently infected cells.
Induction of Apoptosis of Latently Infected Cells as an Approach to Cure HIV Infection
The case of the Berlin patient is instructive and it teaches many lessons that may be applicable to cure HIV infection in a more generalizable way. First, systemic myeloablative chemotherapy and radiotherapy was used, followed by stem-cell transplant, demonstrating that induction of apoptosis by interventions such as non-selective systemic chemotherapy and radiotherapy with graft versus host disease (GVHD) may be a key to eradicate latently infected cells, when administered with maximally suppressive cART to prevent repopulating the reservoir.1, 2, 3, 99 Second, some degree of toxicity to uninfected cells might be necessary so that once HIV has been eradicated, cells and tissues killed unintentionally (bystander killing) can be repopulated, including lymphoid cells such as uninfected CD4 T cells. Third, multiple treatments may be required. A central principal of cancer chemotherapy is that treatments kill a high fraction of the cancerous cells, and therefore multiple courses of cancer chemotherapy are required so that the number of cancerous cells asymptotically approaches zero. This was the case in the treatment regimen for Timothy Ray Brown, and may likely be the case with future therapies that require induction of cell death.
Chemosensitization
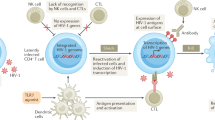
It may also be instructive to consider other similarities with cancer chemotherapy to cure HIV. Cancers that are difficult to treat are often managed with chemosensitization followed by cytotoxic chemotherapy. In the case of HIV, there is ample evidence that productive HIV replication can be cytotoxic (for example, in the case of acute T-cell infection), but also there is emerging evidence that HIV production does not always kill those cells that produce virus100, 101 (i.e., in the case of viral reactivation from latency). Specific reasons why remain unknown, but it is likely that some of the counter apoptotic mechanisms that occur during the development of T-cell memory or the process of chronic HIV infection are at least partially responsible. Thus, we propose to prime cells toward an apoptosis-prone phenotype, reactivate HIV pharmacologically, to induce the death of the reactivating cell—or Prime, Shock, and Kill. Possible approaches to sensitize cells to the cytotoxic effects of HIV productive replication are outlined in Figure 1. There are a multitude of chemosensitization agents used in cancer therapy, many of which are appealing to use in HIV. Some possible agents would include agents that act upon the mitochondrial permeability transition core complex (e.g., adenine nucleotide translocator (ANT) ligands, or voltage-dependent anion channel (VDAC) ligands), Bcl2 inhibitors,102 IAP inhibitors,103 proteasome inhibitors,104 survivin inhibitors,105 PI3K/AKT inhibitors,106 and TRAIL along with TRAIL sensitizers107 (Figure 2). Also, agents known to enhance the susceptibility of immune cells to apoptosis induction such as Toll-like receptor (TLR) agonists, co-stimulatory agents (e.g., anti-CD28), and agents that induce p53 warrant consideration.

Prime, Shock, and Kill hypothesis to eradicate HIV from latently infected cells

Schematic representation of the interaction of HIV proteins with different elements of the apoptosis regulatory network, and possible strategies to promote cell death following HIV reactivation
Caspase 8
One goal for the HIV cure initiative is therefore to design strategies that cause viral reactivation that then results in death of the cells which reactivate virus. We have been studying the involvement of Caspase 8 in the induction of cell death during productive HIV infection, and have observed that during acute infection of CD4 T cells, HIV protease cleaves Caspase 8108 to create a unique protein fragment, Casp8p41. Casp8p41 translocates to mitochondria and independently induces mitochondrial permeability leading to apoptosis, as well as to NFκB activation.53
If latently infected CD4 T cells that reactivate HIV do not die, then it is possible that the Casp8p41 pathway of death is not operational in such cells. Insight into why this pathway is not activated following viral reactivation may lie in observations that short-term activation of CD4 T cells with antigen results in upregulation of procaspase 8 whereas long-term activated cells that acquire a memory phenotype downregulate procaspase 8, and become apoptosis resistant.109 Thus, the low levels of procaspase 8 and intrinsic apoptosis resistance of memory CD4 T cells might explain why such cells do not die following HIV reactivation. Developing strategies to increase Caspase 8 expression in cells that harbor latent HIV and then inducing viral reactivation with agents such as HDACi should lead to testable ‘sensitizing’ strategies designed to enhance death of latently infected cells that are induced to reactivate virus.
TLR stimulation
The rationale for this approach is that TLR-ligand stimulation of T cells will lead to NFκB activation and cytokine production,110 which may upregulate pro-apoptotic regulatory proteins and downregulate anti-apoptosis regulatory proteins, and render TCM cells sensitive to the cytotoxic effects of productive HIV replication. This model is particularly appealing, given recent data that mRNA for TLR1, TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9 has been identified in primary CD4 T cells, and stimulation of resting CD4 T cells with agonists of TLR2, TLR4, and TLR5 triggers interferon-γ production.111 Also, TLR5 stimulation of TCM cells isolated from HIV-infected individuals leads to HIV reactivation, although the viability of these cells after stimulation with the TLR5 ligand flagellin was not specifically examined.112 Likewise, as long ago as 1987, double-stranded RNA (a TLR3 ligand) was shown to have significant anti-HIV activity and caused >90% of productively infected cells to die because of HIV-related cytopathic effects while having no effect on the viability of uninfected cells.113
Auranofin
Another intriguing possibility concerns auranofin, a gold-based compound successfully used for years to treat rheumatoid arthritis. Although the mechanism of action of auranofin is incompletely understood, some reports link it to altered regulation of p53 pathways.114 Therefore, one might predict that auranofin could alter the cellular millieu to favoring apoptosis. Auranofin, in addition to ART (tenofovir, emtricitabine, and raltegravir), in an SIVmac251-infected macaque model induced activation and death of resting memory cells and reduced the amount of cell-associated SIVmac251 DNA in auranofin-treated monkeys compared with those who received ART alone.115 Despite these promising findings, the mechanism by which these latently infected cells were dying was not investigated.
Conclusion
HIV is a disease characterized by altered cell death, wherein the majority of CD4 T cells and other cell types die at an accelerated rate, leading to a significant immune dysregulation. On the other hand, HIV infection fails to cause the death of all of the cells that it infects. This allows for the development of long-lived latently infected HIV reservoirs and the long-term persistence of HIV in the presence of ART. Enhanced understanding of the molecular mechanisms that allow HIV to survive in latently infected cells will allow for interventions that are designed to reverse virus persistence. These interventions could effectively reactivate latent HIV and simultaneously induce cell death—ultimately leading to reduction in the number of latently infected cells and potentially a cure for HIV infection.
Abbreviations
- HIV:
-
human immunodeficiency virus
- ART:
-
antiretroviral therapy
- EBV:
-
Epstein Barr Virus
- CMV:
-
cytomegalovirus
- HSV:
-
herpes simplex virus
- HPV:
-
human papilloma virus
- BMT:
-
bone marrow transplant
- NS:
-
nonstructural
- cFLIP:
-
cellular FLICE inhibitory protein
- XAIP:
-
x-linked apoptosis inhibitory protein
- TRAIL:
-
TNF-related apoptosis inhibitory protein
- TNF:
-
tumor necrosis factor
- GI:
-
gastrointestinal
- CNS:
-
central nervous system
- IUPM:
-
infectious units per million
- FADD:
-
Fas-associated death domain
- TCR:
-
T-cell receptor
- MHC:
-
major histocompatibility complex
- AICD:
-
activation-induced cell death
- TEM:
-
effector memory T cell
- TCM:
-
central memory T cell
- SDF-1α:
-
stromal-derived factor-1α
- HDACi:
-
histone deacetylase inhibitor
- PTEN:
-
phosphatase and tensin homolog
- CTL:
-
cytotoxic T-lymphocyte
- EGFP:
-
expressed green fluorescent protein
- ZFN:
-
zinc finger nucleases
- hu-BLT:
-
humanized bone marrow/liver/thymus mouse
- NK:
-
natural killer
- ANT:
-
adenine nucleotide translocator
- VDAC:
-
voltage-dependent anion channel
- TLR:
-
Toll-like receptor
- cART:
-
combination antiretroviral therapy
References
Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E et al. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 2010; 117: 2791–2799.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K et al. Long-term control of HIV by CCR5 Δ32/Δ32 stem-cell transplantation. N Engl J Med 2009; 360: 692–698.
Hutter G, Thiel E . Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: an update after 3 years and the search for patient no. 2. AIDS 2011; 25: 273–274.
Suzuki Y, Nagao Y, Kato H, Matsumoto M, Nerome K, Nakajima K et al. Human influenza A virus hemagglutinin distinguishes sialyloligosaccharides in membrane-associated gangliosides as its receptor which mediates the adsorption and fusion processes of virus infection. Specificity for oligosaccharides and sialic acids and the sequence to which sialic acid is attached. J Biol Chem 1986; 261: 17057–17061.
Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC . Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988; 333: 426–431.
Ivan FX, Tan KS, Phoon MC, Engelward BP, Welsch RE, Rajapakse JC et al. Neutrophils infected with highly virulent influenza H3N2 virus exhibit augmented early cell death and rapid induction of type I interferon signaling pathways. Genomics 2012 pii: S0888-7543(12)00228-5.
Cho JL, Roche MI, Sandall B, Brass AL, Seed B, Xavier RJ et al. Enhanced Tim3 activity improves survival after influenza infection. J Immunol 2012; 189: 2879–2889.
Mukherjee S, Majumdar S, Vipat VC, Mishra AC, Chakrabarti AK . Non structural protein of avian influenza A (H11N1) virus is a weaker suppressor of immune responses but capable of inducing apoptosis in host cells. Virol J 2012; 9: 149.
Lam WY, Yeung AC, Chan PK . Apoptosis, cytokine and chemokine induction by non-structural 1 (NS1) proteins encoded by different influenza subtypes. Virol J 2011; 8: 554.
Han X, Li Z, Chen H, Wang H, Mei L, Wu S et al. Influenza virus A/Beijing/501/2009(H1N1) NS1 interacts with beta-tubulin and induces disruption of the microtubule network and apoptosis on A549 cells. PLoS One 2012; 7: e48340.
Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G . Viral control of mitochondrial apoptosis. PLoS Pathog 2008; 4: e1000018.
Altmann M, Hammerschmidt W . Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 2005; 3: e404.
Lum JJ, Badley AD . Resistance to apoptosis: mechanism for the development of HIV reservoirs. Curr HIV Res 2003; 1: 261–274.
Badley AD, Parato K, Cameron DW, Kravcik S, Phenix BN, Ashby D et al. Dynamic correlation of apoptosis and immune activation during treatment of HIV infection. Cell Death Differ 1999; 6: 420–432.
Lopez-Huertas MR, Mateos E, Sanchez Del Cojo M, Gomez-Esquer F, Diaz-Gil G, Rodriguez-Mora S et al. The presence of HIV-1 Tat second exon delays Fas-mediated apoptosis in CD4+ T lymphocytes: a potential mechanism for persistent viral production. J Biol Chem 2013; 288: 7626–7644.
Conti L, Rainaldi G, Matarrese P, Varano B, Rivabene R, Columba S et al. The HIV-1 vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J Exp Med 1998; 187: 403–413.
Berro R, de la Fuente C, Klase Z, Kehn K, Parvin L, Pumfery A et al. Identifying the membrane proteome of HIV-1 latently infected cells. J Biol Chem 2007; 282: 8207–8218.
Zheng L, Yang Y, Guocai L, Pauza CD, Salvato MS . HIV Tat protein increases Bcl-2 expression in monocytes which inhibits monocyte apoptosis induced by tumor necrosis factor-alpha-related apoptosis-induced ligand. Intervirology 2007; 50: 224–228.
Chehimi J, Papasavvas E, Tomescu C, Gekonge B, Abdulhaqq S, Raymond A et al. Inability of plasmacytoid dendritic cells to directly lyse HIV-infected autologous CD4+ T cells despite induction of tumor necrosis factor-related apoptosis-inducing ligand. J Virol 2010; 84: 2762–2773.
Schnepple DJ, Shepard B, Bren GD, Cummins NW, Natesampillai S, Trushin S et al. Isolation of a TRAIL antagonist from the serum of HIV-infected patients. J Biol Chem 2011; 286: 35742–35754.
Lopez-Huertas MR, Mateos E, Sanchez Del Cojo M, Gomez-Esquer F, Diaz-Gil G, Rodriguez-Mora S et al. The presence of HIV-1 Tat protein second exon delays Fas protein-mediated apoptosis in CD4+ T lymphocytes: a potential mechanism for persistent viral production. J Biol Chem 2013; 288: 7626–7644.
Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 1999; 5: 512–517.
Pierson T, McArthur J, Siliciano RF . Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol 2000; 18: 665–708.
Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis 2008; 197: 714–720.
Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K et al. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 1993; 362: 359–362.
Wightman F, Solomon A, Khoury G, Green JA, Gray L, Gorry PR et al. Both CD31(+) and CD31(−) naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J Infect Dis 2010; 202: 1738–1748.
Spiegel H, Herbst H, Niedobitek G, Foss HD, Stein H . Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am J Pathol 1992; 140: 15–22.
Tornatore C, Chandra R, Berger JR, Major EO . HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 1994; 44 (3 Pt 1): 481–487.
He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997; 385: 645–649.
Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol 2012; 12: 607–614.
Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC . NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 2006; 25: 139–149.
Treand C, du Chene I, Bres V, Kiernan R, Benarous R, Benkirane M et al. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J 2006; 25: 1690–1699.
Han Y, Lin YB, An W, Xu J, Yang HC, O’Connell K et al. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008; 4: 134–146.
Pace MJ, Agosto L, Graf EH, O’Doherty U . HIV reservoirs and latency models. Virology 2011; 411: 344–354.
Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB . Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci USA 1989; 86: 5974–5978.
Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS . Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987; 238: 800–802.
Jordan A, Bisgrove D, Verdin E . HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 2003; 22: 1868–1877.
Kutsch O, Benveniste EN, Shaw GM, Levy DN . Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 2002; 76: 8776–8786.
Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997; 387: 183–188.
Dokmanovic M, Marks PA . Prospects: histone deacetylase inhibitors. J Cell Biochem 2005; 96: 293–304.
Shan L, Yang HC, Rabi SA, Bravo HC, Shroff NS, Irizarry RA et al. Influence of host gene transcription level and orientation on HIV-1 latency in a primary-cell model. J Virol 2011; 85: 5384–5393.
Marini A, Harper JM, Romerio F . An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol 2008; 181: 7713–7720.
Burnett JC, Lim KI, Calafi A, Rossi JJ, Schaffer DV, Arkin AP . Combinatorial latency reactivation for HIV-1 subtypes and variants. J Virol 2010; 84: 5958–5974.
Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC . A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 2012; 7: e30176.
Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc Natl Acad Sci USA 2010; 107: 16934–16939.
Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR . CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 2007; 110: 4161–4164.
Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 2009; 119: 3473–3486.
Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM . Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 2009; 25: 207–212.
Ya-Chi Ho LS, Wang J, Hosmane N, Blankson J, Siliciano R Characterization of non-induced HIV-1 proviruses dampens the hope for HIV-1 eradication. 20th Conference on Retroviruses and Opportunistic Infections, Atlanta [abstract 43] 2013.
Chinnaiyan AM, Woffendin C, Dixit VM, Nabel GJ . The inhibition of pro-apoptotic ICE-like proteases enhances HIV replication. Nat Med 1997; 3: 333–337.
Aillet F, Masutani H, Elbim C, Raoul H, Chene L, Nugeyre MT et al. Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4(+) T- or monocytic cell lines. J Virol 1998; 72: 9698–9705.
Wang X, Ragupathy V, Zhao J, Hewlett I . Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem Biophys Res Commun 2011; 414: 20–24.
Bren GD, Whitman J, Cummins N, Shepard B, Rizza SA, Trushin SA et al. Infected cell killing by HIV-1 protease promotes NF-kappaB dependent HIV-1 replication. PLoS One 2008; 3: e2112.
Weng NP, Araki Y, Subedi K . The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol 2012; 12: 306–315.
Cummins NW, Badley A . Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis 2010; 1: e99.
Unsoeld H, Pircher H . Complex memory T-cell phenotypes revealed by coexpression of CD62L and CCR7. J Virol 2005; 79: 4510–4513.
Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C et al. CCR5 is characteristic of Th1 lymphocytes. Nature 1998; 391: 344–345.
Zaunders JJ, Ip S, Munier ML, Kaufmann DE, Suzuki K, Brereton C et al. Infection of CD127+ (interleukin-7 receptor+) CD4+ cells and overexpression of CTLA-4 are linked to loss of antigen-specific CD4 T cells during primary human immunodeficiency virus type 1 infection. J Virol 2006; 80: 10162–10172.
Trushin SA, Carena AA, Bren GD, Rizza SA, Dong X, Abraham RS et al. SDF-1alpha degrades whereas glycoprotein 120 upregulates Bcl-2 interacting mediator of death extralong isoform: implications for the development of T cell memory. J Immunol 2012; 189: 1835–1842.
Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog 2013; 9: e1003211.
Lodi S, Meyer L, Kelleher AD, Rosinska M, Ghosn J, Sannes M et al. Immunovirologic control 24 months after interruption of antiretroviral therapy initiated close to HIV seroconversion. Arch Intern Med 2012; 172: 1252–1255.
Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M, Chun T-W et al. Functional HIV cure after very early ART of an infected infant. 20th Conference on Retroviruses and Opportunistic Infections 3–6 March 2013 Atlanta, GA [abstract 48LB].
Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012; 487: 482–485.
Elliott J, Soloman A, Whightman F, Smith M, Palmer S, Prince M et al. The safety and effect of multiple doses of vorinostat on HIV transcription in HIV+ patients receiving cART. 20th Conference on Retroviruses and Opportunistic Infections Atlanta, GA [abstract 50LB] 2013.
Siliciano JD . Safety and feasibility of using disulfiram to enhance HIV transcription among long-term ARV-treated adults: preliminary results from a Pilot study 2012; 1.
Shehu-Xhilaga M, Rhodes D, Wightman F, Liu HB, Solomon A, Saleh S et al. The novel histone deacetylase inhibitors metacept-1 and metacept-3 potently increase HIV-1 transcription in latently infected cells. AIDS 2009; 23: 2047–2050.
Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012; 36: 491–501.
Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V . Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog 2011; 7: e1002288.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 2008; 26: 808–816.
Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL et al. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther 2012; 20: 849–859.
Lai Y . CCR5-targeted hematopoietic stem cell gene approaches for HIV disease: current progress and future prospects. Curr Stem Cell Res Ther 2012; 7: 310–317.
Stremlau M, Perron M, Welikala S, Sodroski J . Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 2005; 79: 3139–3145.
Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D . Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2012; 481: 81–84.
Veselinovic M, Neff CP, Mulder LR, Akkina R . Topical gel formulation of broadly neutralizing anti-HIV-1 monoclonal antibody VRC01 confers protection against HIV-1 vaginal challenge in a humanized mouse model. Virology 2012; 432: 505–510.
Sadelain M, Brentjens R, Riviere I . The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009; 21: 215–223.
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011; 118: 4817–4828.
Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 2012; 4: 132ra53.
Li MJ, Kim J, Li S, Zaia J, Yee JK, Anderson J et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther 2005; 12: 900–909.
DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med 2010; 2: 36ra43.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996; 273: 1856–1862.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996; 86: 367–377.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996; 382: 722–725.
Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 2010; 330: 1551–1557.
Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet 2007; 39: 733–740.
Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 2008; 29: 1009–1021.
Rajasuriar R, Khoury G, Kamarulzaman A, French MA, Cameron PU, Lewin SR . Persistent immune activation in chronic HIV infection: do any interventions work? AIDS 2013.
Hatano H, Jain V, Hunt PW, Lee TH, Sinclair E, Do TD et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J Infect Dis 2012; 208: 50–56.
Taiwo B, Hunt PW, Gandhi RT, Ellingson A, McKenna M, Jacobson JM et al. CD8+ T-cell activation in HIV-1-infected patients experiencing transient low-level viremia during antiretroviral therapy. J Acquir Immune Defic Syndr 2013; 63: 101–104.
Gandhi RT, Zheng L, Bosch RJ, Chan ES, Margolis DM, Read S et alon behalf of the ACTGAt. The effect of Raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med 2010; 7: e1000321.
Hiroyu Hatano MS, Scherzer R, Sinclair E, Palmer S, Busch M, Bacchetti P et alIncrease in 2-LTR circles after raltegravir intensification in HAART-suppressed patients with high CD4+ T cell counts: a randomized, controlled trial. 20th Conference on Retroviruses and Opportunistic Infections, 3-6 March 2013, Atlanta GA (Abstract 42).
Chun TW, Davey RT Jr., Engel D, Lane HC, Fauci AS . Re-emergence of HIV after stopping therapy. Nature 1999; 401: 874–875.
Levy Y, Lacabaratz C, Weiss L, Viard JP, Goujard C, Lelievre JD et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest 2009; 119: 997–1007.
Sereti I, Dunham RM, Spritzler J, Aga E, Proschan MA, Medvik K et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood 2009; 113: 6304–6314.
Levy Y, Sereti I, Tambussi G, Routy JP, Lelievre JD, Delfraissy JF et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis 2012; 55: 291–300.
Seder RA, Grabstein KH, Berzofsky JA, McDyer JF . Cytokine interactions in human immunodeficiency virus-infected individuals: roles of interleukin (IL)-2, IL-12, and IL-15. J Exp Med 1995; 182: 1067–1077.
Lum JJ, Schnepple DJ, Nie Z, Sanchez-Dardon J, Mbisa GL, Mihowich J et al. Differential effects of interleukin-7 and interleukin-15 on NK cell anti-human immunodeficiency virus activity. J Virol 2004; 78: 6033–6042.
Mueller YM, Do DH, Altork SR, Artlett CM, Gracely EJ, Katsetos CD et al. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J Immunol 2008; 180: 350–360.
Pallikkuth S, Parmigiani A, Pahwa S . Role of IL-21 and IL-21 receptor on B cells in HIV infection. Crit Rev Immunol 2012; 32: 173–195.
Pallikkuth S, Rogers K, Villinger F, Dosterii M, Vaccari M, Franchini G et al. Interleukin-21 administration to rhesus macaques chronically infected with simian immunodeficiency virus increases cytotoxic effector molecules in T cells and NK cells and enhances B cell function without increasing immune activation or viral replication. Vaccine 2011; 29: 9229–9238.
Conti L, Matarrese P, Varano B, Gauzzi MC, Sato A, Malorni W et al. Dual role of the HIV-1 vpr protein in the modulation of the apoptotic response of T cells. J Immunol 2000; 165: 3293–3300.
Fernandez Larrosa PN, Croci DO, Riva DA, Bibini M, Luzzi R, Saracco M et al. Apoptosis resistance in HIV-1 persistently-infected cells is independent of active viral replication and involves modulation of the apoptotic mitochondrial pathway. Retrovirology 2008; 5: 19.
Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH . Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin Emerg Drugs 2011; 16: 59–70.
Gyrd-Hansen M, Meier P . IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 2010; 10: 561–574.
Reddy N, Czuczman MS . Enhancing activity and overcoming chemoresistance in hematologic malignancies with bortezomib: preclinical mechanistic studies. Ann Oncol 2010; 21: 1756–1764.
Mita AC, Mita MM, Nawrocki ST, Giles FJ . Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res 2008; 14: 5000–5005.
Kim D, Cheng GZ, Lindsley CW, Yang H, Cheng JQ . Targeting the phosphatidylinositol-3 kinase/Akt pathway for the treatment of cancer. Curr Opin Investig Drugs 2005; 6: 1250–1258.
Sayers TJ . Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother 2011; 60: 1173–1180.
Nie Z, Bren GD, Vlahakis SR, Schimnich AA, Brenchley JM, Trushin SA et al. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J Virol 2007; 81: 6947–6956.
Strauss G, Knape I, Melzner I, Debatin KM . Constitutive caspase activation and impaired death-inducing signaling complex formation in CD95-resistant, long-term activated, antigen-specific T cells. J Immunol 2003; 171: 1172–1182.
Li X, Jiang S, Tapping RI . Toll-like receptor signaling in cell proliferation and survival. Cytokine 2010; 49: 1–9.
Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol 2005; 175: 1551–1557.
Thibault S, Imbeault M, Tardif MR, Tremblay MJ . TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology 2009; 389: 20–25.
Montefiori DC, Mitchell WM . Antiviral activity of mismatched double-stranded RNA against human immunodeficiency virus in vitro. Proc Natl Acad Sci USA 1987; 84: 2985–2989.
Guidi F, Puglia M, Gabbiani C, Landini I, Gamberi T, Fregona D et al. 2D-DIGE analysis of ovarian cancer cell responses to cytotoxic gold compounds. Mol Biosyst 2012; 8: 985–993.
Lewis MG, DaFonseca S, Chomont N, Palamara AT, Tardugno M, Mai A et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 2011; 25: 1347–1356.
Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N . Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J Biol Chem 2011; 286: 22211–22218.
Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V . Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS One 2009; 4: e6093.
Savarino A, Mai A, Norelli S, El Daker S, Valente S, Rotili D et al. "Shock and kill" effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 2009; 6: 52.
Choi BS, Lee HS, Oh YT, Hyun YL, Ro S, Kim SS et al. Novel histone deacetylase inhibitors CG05 and CG06 effectively reactivate latently infected HIV-1. AIDS 2010; 24: 609–611.
Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM . Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 2009; 23: 1799–1806.
Matalon S, Palmer BE, Nold MF, Furlan A, Kassu A, Fossati G et al. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J Acquir Immune Defic Syndr 2010; 54: 1–9.
Victoriano AF, Imai K, Togami H, Ueno T, Asamitsu K, Suzuki T et al. Novel histone deacetylase inhibitor NCH-51 activates latent HIV-1 gene expression. FEBS Lett 2011; 585: 1103–1111.
Rasmussen TA, Schmeltz Sogaard O, Brinkmann C, Wightman F, Lewin S, Melchjorsen J et al. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum Vaccin Immunother 2013; 9: 5.
Wightman F RS, Saleh S et alPotency and toxicity of HDACi and other immune activators in inducing HIV production using a primary resting T-cell model of HIV latency [abstract 198]. Poster presented at the 18th Conference on Retroviruses and Opportunistic Infections; 27 February–2 March 2011 Boston, MA.
Ying H, Zhang Y, Zhou X, Qu X, Wang P, Liu S et al. Selective histonedeacetylase inhibitor M344 intervenes in HIV-1 latency through increasing histone acetylation and activation of NF-kappaB. PLoS One 2012; 7: e48832.
Fernandez G, Zeichner SL . Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol J 2010; 7: 266.
Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1+ HAART-treated patients. AIDS 2012; 26: 1473–1482.
Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I . The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett 2011; 585: 3549–3554.
Wei G CV, Fyne E, Balakrishnan M, Stepan G, Tsai A et alHistone deacetylase inhibitor romidepsin induces HIV in CD4+ T cells from ART-suppressed subjects at concentrations achieved by clinical dosing. 201th Conference on Retroviruses and Opportunistic Infections, Atlanta 2013.
Saleh S, Wightman F, Ramanayake S, Alexander M, Kumar N, Khoury G et al. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 2011; 8: 80.
Perez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Munoz-Fernandez MA et al. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr HIV Res 2010; 8: 418–429.
Klichko V, Archin N, Kaur R, Lehrman G, Margolis D . Hexamethylbisacetamide remodels the human immunodeficiency virus type 1 (HIV-1) promoter and induces Tat-independent HIV-1 expression but blunts cell activation. J Virol 2006; 80: 4570–4579.
Shishido T, Wolschendorf F, Duverger A, Wagner F, Kappes J, Jones J et al. Selected drugs with reported secondary cell-differentiating capacity prime latent HIV-1 infection for reactivation. J Virol 2012; 86: 9055–9069.
Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Virol 2011; 85: 6060–6064.
Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 2012; 92: 1147–1154.
Wang FX, Xu Y, Sullivan J, Souder E, Argyris EG, Acheampong EA et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest 2005; 115: 128–137.
Vandergeeten C, Fonseca SD, Sereti I, Lederman M, Sekaly R-P, Chomont N Differential impact of IL-7 and IL-15 on HIV reservoir persistence. 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention; 17–20 July, Rome, 2011.
DaFonseca S, Chomont N, Far ME, Baulassel R, Routy J, Sekaly R Purging the HIV-1 reservoir through the disruption of the PD-1 pathway. J Int AIDS Soc 2010 XVII International AIDS Conference, Vienna, Austria.
Micheva-Viteva S, Kobayashi Y, Edelstein LC, Pacchia AL, Lee HL, Graci JD et al. High-throughput screening uncovers a compound that activates latent HIV-1 and acts cooperatively with a histone deacetylase (HDAC) inhibitor. J Biol Chem 2011; 286: 21083–21091.
Acknowledgements
ADB is supported by NIH R01 AIO40384, SRL is supported by the National Health and Medical Research Council (NHMRC) of Australia (APP1042654, APP1041795, APP1002761, and APP1009533) and the National Institutes of Health (1R56AI095073-01A1) and the Delaney AIDS Research Enterprise (DARE) to find a cure collaborator (U19 AI096109), the Danish Medical Council and the University of Malaya. SRL is an NHMRC Practitioner Fellow. We gratefully acknowledge the contribution to this work of the Victorian Operational Infrastructure Support Program received by the Burnet Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Badley, A., Sainski, A., Wightman, F. et al. Altering cell death pathways as an approach to cure HIV infection. Cell Death Dis 4, e718 (2013). https://doi.org/10.1038/cddis.2013.248
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.248
Keywords
This article is cited by
-
Targeted shock-and-kill HIV-1 gene therapy approach combining CRISPR activation, suicide gene tBid and retargeted adenovirus delivery
Gene Therapy (2024)
-
Prevention, treatment and cure of HIV infection
Nature Reviews Microbiology (2023)
-
Novel pathways of HIV latency reactivation revealed by integrated analysis of transcriptome and target profile of bryostatin
Scientific Reports (2020)
-
Heterogeneity in HIV and cellular transcription profiles in cell line models of latent and productive infection: implications for HIV latency
Retrovirology (2019)
-
Analysis of networks of host proteins in the early time points following HIV transduction
BMC Bioinformatics (2019)
