
Abstract
Genetic heterogeneity is a difficulty frequently encountered in the search for genes conferring susceptibility to prostate cancer. To circumvent this issue, we selected a large prostate cancer pedigree for genome-wide linkage analysis from a population that is genetically homogeneous. Selected cases and first-degree relatives were genotyped with Affymetrix 10K SNP arrays, identifying a 14 Mb haplotype on chromosome 5 (5p13–q12) inherited identical-by-descent (IBD) by multiple cases. Microsatellite genotyping of additional deceased case samples confirmed that a total of eight cases inherited the common haplotype (P=0.0017). Re-sequencing of eight prioritised candidate genes in the region in six selected individuals identified 15 SNPs segregating with the IBD haplotype, located within the ITGA2 gene. Three of these polymorphisms were selected for genotyping in an independent Tasmanian data set comprising 127 cases with familial prostate cancer, 412 sporadic cases and 319 unaffected controls. Two were associated with prostate cancer risk: rs3212649 (OR=1.67 (1.07–2.6), P=0.0009) and rs1126643 (OR=1.52 (1.01–2.28), P=0.0088). Significant association was observed in both familial and sporadic prostate cancer. Although the functional SNP remains to be identified, considerable circumstantial evidence, provided by in vivo and in vitro studies, supports a role for ITGA2 in tumour development.
Similar content being viewed by others

Introduction
In many Western countries prostate cancer (MIM176807) is the most commonly diagnosed cancer after skin cancer.1 Familial aggregation of the disease indicates that it has a genetic component.2, 3 Evidence presented by the International Consortium for Prostate Cancer Genetics (ICPCG)4 highlighted five suggestive linkage regions supporting putative susceptibility loci reported earlier, including one on chromosome 5q.5 More recently, several genome-wide association studies (GWAS) in large independent case–control data sets have provided compelling evidence for multiple new susceptibility loci. Common sequence variants on chromosomes 8q24 and 17q have been significantly and independently associated with prostate cancer risk and these findings have been replicated in several populations.6, 7, 8, 9, 10 Subsequently, evidence for additional loci on chromosomes 2, 3, 6, 7, 8, 10, 11, 17 and X10, 11, 12 has been provided. Although many of these SNPs are located in or close to known genes, a functional role for these SNPs in prostate cancer is yet to be demonstrated.
Although complex diseases are probably influenced by a mixture of common and rare genetic variants, there is evidence that rarer variants play a larger role in cancer than in other diseases.13, 14 Large multiplex pedigrees are a powerful approach for identifying rare variants. They can also be used to help address the problem of genetic heterogeneity and are therefore potentially advantageous when studying a complex disease such as prostate cancer. Here, we present the linkage analysis of an extended Tasmanian pedigree (PcTas9) in which 25 prostate cancer cases can be traced back seven generations to a common founding pair. Evidence for a prostate cancer susceptibility locus on chromosome 5 (5p13–q12) is presented, adding to the mounting evidence that this region harbours a putative prostate cancer susceptibility gene.4, 5, 15, 16, 17, 18 In addition, although the recently published GWAS have not highlighted specific candidate susceptibility SNPs on chromosome 5, Thomas et al12 noted the likelihood that chromosome 5 may also harbour prostate cancer susceptibility loci.
Resequencing within the region of interest on chromosome 5 identified sequence variants that co-segregated with disease in this large pedigree. Genotyping of these variants in an independent data set, comprising both familial and sporadic prostate cancer cases and age-matched controls, revealed two polymorphisms that are significantly associated with prostate cancer risk in the wider Tasmanian population.
Subjects and methods
Study subjects and preparation
Ethics approval for this study was obtained from the Southern Tasmanian Human Research Ethics Committee and written informed consent was obtained from all participating individuals. Using a genealogical database at the Menzies Research Institute (MRI) and the records of the Tasmanian Cancer Registry (TCR), families with multiple cases of prostate cancer were identified. All families had at least two affected close relatives; with the largest family comprising 37 affected men, and 10 families with more than 11 cases. Blood samples were available for 131 familial cases in total.
A seven-generation pedigree was selected to conduct genome-wide linkage analysis, comprising 25 patients diagnosed with histologically confirmed prostate cancer (average age of diagnosis 72, range 50–89) (Figure 1). A total of 16 DNA samples were obtained from PcTas9 prostate cancer patients; 9 were extracted from blood samples and 7 from paraffin-embedded prostate tumour tissue. In addition, 57 DNA samples were available from relatives. DNA was extracted from whole blood or buccal mucosa swabs using the Nucleon Bacc3 (Amersham Biosciences AB) and PureGene DNA Isolation Kits (Gentra Systems), respectively. Seven paraffin-embedded tumour blocks for the individuals shown in Figure 1 were obtained from two pathology laboratories located in Southern Tasmania. Using Method E as described by Sato et al,19 DNA was isolated from selected tissue sections comprising a significant proportion of normal tissue.

PcTas9 prostate cancer pedigree showing the segregating haplotype across 5p13–q12. Only cases and individuals providing genotyping information pertinent to the cases are presented due to pedigree size. Two cases are not represented here (9.6-brother of 9.132; 9.13-uncle of 9.1, 9.2 and 9.474). The affected status of ‘older’ generations is unknown as the Tasmanian Cancer Registry only lists patients’ records since 1978. Shaded boxed areas represent the shared haplotype. Individuals genotyped with the Affymetrix 10K array are indicated by an asterisk (*). Individuals genotyped from paraffin-embedded tissue are indicated by a cross (+). # Denotes age at diagnosis, followed by Gleason score presented in parentheses.
A prostate cancer case–control study is also being conducted concurrently by the MRI and is recruiting sporadic prostate cancer cases and unaffected controls. Blood samples, serum samples, physical measures and environmental exposure data are being collected from participating individuals. A total of 412 sporadic prostate cancer cases were identified from the TCR and recruited. Eligible cases were men under the age of 70 years diagnosed with histologically confirmed cancer of the prostate during the period 1996–2005. Controls were randomly selected from the electoral roll. Eligible controls (319 in total) were age-matched within 5-year age groups to the sporadic cases and self-reported as unaffected with prostate cancer.
Genotyping
Seven prostate cancer patients in family PcTas9 were genotyped with Affymetrix Human Mapping 10K arrays (indicated with an asterisk in Figure 1), along with six first-degree relatives to provide phase information. Genotyping was performed as described by Kennedy et al20 using 250 ng of genomic DNA. The mean genotype call rate was 95%.
Twenty microsatellites across 5p13–q12 (chosen from the Genome Database – http://www.gdb.org/ – see Figure 1) were genotyped in all 73 available DNA samples from PcTas9 using standard fluorescent PCR techniques and an ABI PRISM 310 Genetic Analyzer (Applied Biosystems). Microsatellite allele frequencies were estimated from 32 Tasmanian samples comprising spouses of affected cases and unaffected family members.
Linkage analysis
PEDCHECK21 was used to identify Mendelian errors in the genotype data. The 10K SNP data were analysed using the genetic map and Caucasian allele frequencies provided by Affymetrix.20 A total of 954 markers in strong linkage disequilibrium (LD) with other markers were removed from the data set. The family was too large for exact multipoint linkage analysis with the Lander–Green algorithm,22 and the Markov chain Monte Carlo linkage algorithm in Simwalk223 was slow to converge with this number of markers. Linkage analysis was performed using a recently published method that breaks pedigrees into smaller subunits for inference of patterns of identical-by-descent (IBD) sharing between case pairs.24 We have performed simulations, some using the PcTas9 pedigree (Figure 1), to confirm the accuracy of the pedigree-splitting approach for dense marker sets.24
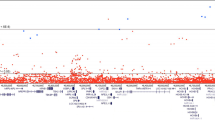
Microsatellite haplotypes were inferred using Simwalk2.23 Genotyping so many microsatellites allowed the determination of patterns of IBD sharing with virtually complete certainty – hence we assumed that patterns of IBD sharing were known with complete certainty for assessing significance. The significances of NPLpairs scores (SNP data) and patterns of IBD sharing (microsatellite data) were computed by simulating fully informative markers on the complete pedigree under the hypothesis of no linkage (gene dropping).23 Nominal (single-point) P-values were calculated using 108 simulations of a single marker, and genome-wide and ICPCG-region-wide corrected P-values were calculated using 105 simulations of the entire genome or the ICPCG linkage regions respectively, with recombinations simulated using the Haldane map function. The suggestive linkage threshold plotted in Figure 2 corresponds to the value of NPLpairs expected to occur on average once in a genome-wide scan with fully informative markers.25

Results of the Affymetrix 10K array genome-wide scan. An NPL score of 5.58 (suggestive linkage threshold expected to occur once by chance in a genome-wide scan with fully informative markers41) is indicated by a horizontal dashed line.
Prioritising candidate genes
Over 90 genes in the region spanned by the chromosome 5 haplotype were prioritised for sequencing using the program ‘GeneSniffer’ (www.genesniffer.org; Autogen Limited, Australia). The program builds a list of ranked candidate genes within the specified region using publicly available human and mouse genetic databases, and a supplied list of key words (available on request).
Sequencing candidate genes
The coding regions of eight candidate genes including all exon/intron boundaries and 500 bp upstream of the transcription start site were resequenced in PcTas9.4, 9.5, 9.8 and 9.12, all of whom carried the chromosome 5 haplotype, and in two unrelated controls. Resequencing was performed using PCR primers designed using the software packages ‘Exon Primer’ (http://ihg.gsf.de/cgi-bin/snps/seq1.pl) and ‘Primer 3’ (http://frodo.wi.mit.edu). Primer sequences and conditions are available on request. Following PCR amplification, samples were sequenced on an ABI310 Genetic Analyzer (Applied Biosystems) in accordance with the manufacturer's instructions.
Genotyping of selected sequence variants
All familial prostate cancer cases (excluding PcTas9.4, 9.5, 9.8 and 9.12), sporadic cases and controls were genotyped for three sequence variants – rs28095, rs1126643 and rs3212649. Allele-specific PCR was used to detect the rs28095 polymorphism (herein referred to as C-52T), using forward primers 5′-aatcaggaggggcgggct-3′, 5′-aatcaggaggggcgggcc-3′ and reverse primer 5′-gcgctgggtttgcagaggtt-3′ (Promega). Rs1126643 (herein referred to as C807T) was amplified using primers 5′-gatgccttaaagctaccggc-3′ and 5′-taactttcccagctgccttc-3′ (Promega). Genotype was resolved by digestion with Hyp188 1. Rs3212649 (herein referred to as 3′UTR in/del) was amplified using primers, 5′FAM-gcaactacagaagtggaagtgc-3′ and 5′-tctgtggcaactttggatga-3′ (Promega). Genotype was resolved by sizing on an ABI310 Genetic Analyser. The insertion allele was 137 bp and the deletion allele was 134 bp. Detailed conditions are available on request. For all three SNPs, 15% of all samples were regenotyped to validate genotyping quality.
Statistical analysis to detect association for selected sequence variants
The association analysis excluded all PcTas9 samples with the known chromosome 5 haplotype (cases PcTas9.4, 9.5, 9.8 and 9.12). The MQLS test26 was used to test for differences in allele frequencies at polymorphisms C-52T, C807T and 3′UTR in/del between familial and sporadic prostate cancer cases and controls. This test allows for the non-independence of the genotypes of related individuals, regardless of the complexities of the relationships, and also exploits the fact that cases with affected relatives are generally more likely to carry disease susceptibility alleles than cases without affected relatives.
We then performed further exploratory analyses, making no allowance for the non-independence of the familial cases. Logistic regression was used to calculate genotypic odds ratios for the two polymorphisms showing significant association, both adjusted and unadjusted for genotypes at the other polymorphism. Haploview 3.2 (www.broad.mit.edu/mpg/haploview/) was used to examine LD between the polymorphisms, and to perform haplotypic tests of association.
Results
The results of the non-parametric linkage analysis of available, high-quality DNA using the 10K SNP genome-wide scan are displayed in Figure 2. There was one suggestive linkage peak at 5p13–q12 (NPLpairs=6.72; P=0.005 using gene dropping),23 where there was evidence that four out of seven cases had inherited a common haplotype. Along the rest of the genome, there was no evidence anywhere of more than three cases inheriting a common haplotype.
Microsatellite genotyping was possible in 15 of the remaining 17 cases, where SNP genotyping was not possible. This provided data on the 5p13–q12 region in PcTas9 prostate cancer patients and their children in a total of 23 of the 25 cases. Of these 23 cases, it was confirmed that 8 shared a common haplotype (including 4 cases genotyped with SNPs). Individuals 9.4, 9.5, 9.8 and 9.12 were confirmed as sharing the common IBD haplotype by both SNP and microsatellite genotyping; individuals 9.7 and 9.20 were confirmed by microsatellite genotyping alone; and for individuals 9.6 and 9.13 haplotype sharing was inferred by genotyping offspring with microsatellites. Twelve individuals did not carry this same haplotype, and the sharing status of three cases (individuals 9.10, 9.22, 9.517) could not be determined (Figure 1). Given this configuration of genotyped and un-genotyped cases and relatives, the probability of observing a common haplotype shared by eight or more cases by chance at a single locus is P=0.0017. The chromosome 5 haplotype extends across 14 Mb between markers D5S2506 and D5S664 (Figure 1).
The eight patients carrying the chromosome 5 haplotype are clustered into a single branch of the pedigree descendant from individuals 9.101 and 9.102. The probability of identifying the same haplotype in eight or more affected descendants of these individuals is P=0.0003. Although this linkage is not of genome-wide significance, this region overlaps substantially with one of five suggestive linkage regions identified in a large study conducted by the ICPCG.4 Gene-dropping simulations of the 1-LOD drop intervals in these five regions showed that the probability of observing segregation in eight or more cases in one of these regions by chance is P=0.043.
The mean age at diagnosis for PcTas9 cases sharing the chromosome 5 haplotype was 69.0 years compared to 71.6 years for those cases not sharing the haplotype (P=0.53, t-test). Similarly, there was no significant difference in Gleason scores between cases sharing the risk haplotype and those that did not (mean of 5.83 and 5.75, respectively; P=0.95, t-test).
Candidate genes within the chromosome 5 interval were prioritised using the bioinformatics tool, GeneSniffer (www.genesniffer.org; Autogen Limited). Eight genes were resequenced from the GeneSniffer prioritised list. A diagrammatic representation of the 14 Mb region of chromosome 5 including the selected genes is shown in Figure 3a.

Diagrammatic representation of the 14 Mb region of interest on chromosome 5. (a) Shows the location and relative size of prioritised genes for resequencing. (b) Shows an expanded view of the ITGA2 gene. Exons are represented as tall black rectangles. Short grey lines represent SNPs identified from the HapMap and SeattleSNPs databases that are in strong LD (r2>0.72) with 3′UTR in/del. The relative positions of the five exonic SNPs discussed in the text are shown.
The coding regions, intron/exon boundaries and promoter sequences of FGF10, GHR, DAB2, PPAP2A, PTGER4, EMB and GZMA were resequenced in four PcTas9 cases carrying the common chromosome 5 haplotype (PcTas9.4, 9.8, 9.5, 9.12 in Figure 1), and two unrelated controls. In these genes no variants were detected that were unique to the identified haplotype (data not shown). In contrast, within the ITGA2 gene 15 polymorphisms were detected that segregated with the chromosome 5 haplotype (Table 1). The ITGA2 gene (hg18 location chr5: 52320913–52426366) comprises 30 exons and spans 105288 bp (Figure 3b). Although 10 of the identified SNPs lie in intronic regions of ITGA2, the remaining 5 are located in the promoter (C-52T; rs28095), exon 7 (C807T; rs1126643), exon 8 (G873A; rs1062535), exon 27 (C3300T; rs2303122) and the 3′UTR of exon 30 (3′UTR in/del; rs3212649; Table 1).
Three of the SNPs in ITGA2 (C-52T, C807T and the 3′UTR in/del) were selected for further examination using association testing in an independent data set. The PcTas9 individuals used to identify these SNPs were excluded from this analysis.
The C-52T SNP was selected for its potential functional role in regulation of the ITGA2 gene. Located 52 bases upstream of the transcription start site, it is positioned between two tandem Sp1/Sp3-binding elements and the presence of the T allele has been shown, by in vitro studies, to reduce ITGA2 transcription.27 The C807T polymorphism lies within exon 7, and whilst it does not alter the amino-acid sequence of the protein, there is circumstantial evidence that it is associated with altered expression of ITGA2.28 Functional modelling software identifies a predicted enhancer splice element (ESE) at C807T that is disrupted in the presence of the T allele (http://pupasuite.bioinfo.cipf.es/).
The synonymous G873A polymorphism in exon 8 is in strong LD with C807T (r2=0.97 in the HapMap CEU population of North-West European ancestry; www.hapmap.org) and was therefore not genotyped.
The remaining two polymorphisms, C3300T and 3′UTR in/del, have no known or putative function. The C3300T is perfectly correlated with the 3′UTR in/del as reported in the PGA European Panel from the SeattleSNP database (r2=1, http://pga.gs.washington.edu/) and consequently only the 3′UTR in/del was genotyped.
Thus the three SNPs were genotyped in 127 cases ascertained as part of other large pedigrees (ie, cases with strong evidence of family history), 412 sporadic cases and 319 controls. After adjusting for multiple testing using the Bonferroni method, the whole-sample unadjusted P-values quoted below remain significant.
No association with disease was seen with the C-52T SNP for sporadic cases (P=0.36), familial cases (P=1.0) or when all cases were combined (P=0.49). In contrast, the C807T polymorphism was significantly associated with prostate cancer (P=0.0088; Table 2) for combined prostate cancer cases and unaffected controls. There was a more significant difference in allele frequencies between familial cases and controls (P=0.020) than between sporadic cases and controls (P=0.070; Table 2). The 3′UTR deletion allele showed a significant association with prostate cancer (P=0.0009; Table 2) in combined prostate cancer cases and unaffected controls. This significant association was observed in both familial cases and controls (P=0.0018) and between sporadic cases and controls (P=0.015; Table 2).
Our data suggest that the 3′UTR deletion allele and the 807T allele both confer risk in a dominant manner, with similar estimated odds ratios for heterozygous and homozygous carriers of these alleles (Table 3). Odds ratios are higher in familial cases than in sporadic cases for both polymorphisms. There is strong LD between the two polymorphisms (r2=0.72; D′=0.96) with the 3′UTR deletion allele almost always occurring with the 807T allele. In the absence of very large sample sizes, this strong LD makes it difficult to separate the individual effects of these alleles, and using a haplotypic test for association, the results were inconclusive (data not shown). However, after adjusting for the 3′UTR in/del and assuming a dominant disease model, the association between the C807T polymorphism and disease disappears (Table 3). This suggests that the risk from the 807T allele may only occur because of LD between the two SNPs.
Associations between clinical characteristics of disease (age at diagnosis, survival and Gleason score) and the risk genotypes (3′UTR deletion and 807T) were examined in both sporadic and familial data sets. Overall, there was little evidence to suggest there was a difference in age at diagnosis between sporadic cases carrying at least one risk 3′UTR deletion allele (age range 44–69 years, mean=61.8 years) compared to those with no risk allele (age range 47–69 years, mean=62.5 years; P=0.31, t-test). Similarly, no significant association between the risk 3′UTR deletion allele and age at diagnosis was observed in familial cases (P=0.27, t-test). Survival analysis of both familial and sporadic cases indicated that there was no decrease in overall survival associated with carrying the 3′UTR deletion allele (P=0.11, log-rank test).
For sporadic cases where Gleason score was available (n=92), there was no significant difference between cases carrying at least one risk 3′UTR deletion allele (mean Gleason score 5.41) and those not carrying a risk allele (mean Gleason score 5.26; P=0.66, t-test). Similar results were obtained for familial cases (n=50), with a mean Gleason score of 5.57 for those carrying at least one risk allele compared to a mean of 5.25 with no risk allele (P=0.67).
Owing to the LD between the C807T and the 3′UTR in/del SNP, almost identical results were obtained when associations between the 807T risk allele and age at diagnosis, survival and Gleason score were examined in both prostate cancer data sets (data not shown).
It remains to be determined which of the SNPs identified in ITGA2 are functionally significant, or whether another, as yet unidentified SNP, is involved. Samples of European ancestry from the HapMap and SeattleSNPs databases were examined to search for other candidate SNPs in LD with the 3′UTR in/del and C807T SNPs. An examination of these databases revealed 30 and 14 SNPs, respectively, in LD (r2>0.72) with C3300T (ie, LD greater than that observed between 3′UTR in/del and C807T). A review of these SNPs identified six SNPs within the 3′UTR in significant LD, with the 3′UTR in/del polymorphism. Use of modelling software that predicts miRNA-binding sequences revealed that two of these SNPs (rs6880055 and rs57674800) are contained within predicted miRNA-target motifs (http://regrna.mbc.nctu.edu.tw/). The presence of the alternative genoytpe at these sites is predicted to disrupt miRNA binding to the corresponding mRNA sequence. No SNP outside the ITGA2 gene was correlated with an r2>0.35.
Discussion
Suggestive evidence of linkage at 5p13–q12 was obtained following analysis of a genome-wide scan using Affymetrix 10K SNP arrays in a large Tasmanian prostate cancer pedigree. Subsequent microsatellite genotyping of further cases confirmed and extended this finding. Eight related cases shared a common haplotype (P=0.0017). Nominal evidence for linkage to chromosome 5 has been provided by several previous studies,5, 15, 16, 17 including the ICPCG study of 1233 prostate cancer families.4 Through the use of a gene prioritisation tool, resequencing and a follow-up association study, two polymorphisms within the ITGA2 gene, 3′UTR in/del and C807T, were found to be associated with prostate cancer risk in independent datasets comprising familial cases, sporadic cases and controls.
The ITGA2 gene encodes for the α2 subunit of the α2β1 integrin receptor, a cell adhesion molecule, responsible for interaction and mediation of signalling events with the extracellular matrix (ECM).29 The α2β1 integrin receptor is expressed on many epithelial cell types, and in normal prostate tissue expression is restricted to basal epithelial cells.30, 31 High levels of α2β1 integrin expression characterise prostate cancer stem cells,32 and upregulation of α2β1 integrin may potentially explain the observation that over 80% of prostate cancer metastases are to bone,29, 31 as collagen is the main component of bone ECM and the preferred ligand for α2β1 integrin.33
Candidate gene association studies have also linked the ITGA2 gene with cancer risk. Two separate studies of breast cancer and oral cancer have reported an association of the C807T ITGA2 polymorphism with disease risk.34, 35 In a study comprising 500 sporadic breast cancer cases and 500 controls, the 807C–1648G haplotype was found to decrease risk compared to non-carriers, and higher grade breast tumours were significantly associated with the 807T/T genotype.34 In addition, the 807T allele was significantly associated with increased risk of oral cancer (P<0.001).35 Studies conducted in platelets have demonstrated that the 807T allele is associated with a two-fold higher density of the ITGA2 receptor on the platelet cell surface and may function to alter mRNA splicing as it is contained within a predicted ESE. However, it remains to be determined whether this polymorphism is directly influencing gene expression.36
Given the evidence presented here, the 3′UTR in/del polymorphism remains of particular interest, as it may be important in regulating ITGA2 mRNA. Although we have not identified a functional role for the 3′UTR in/del polymorphism, we have identified two SNPs (rs6880055 and rs57674800) in high LD with this SNP. These SNPs lie within predicted miRNA-binding sites and the presence of the alternative genoytpe at these sites is predicted to disrupt miRNA binding to this sequence. Thus, presence of one or both of the alternate alleles at these SNPs may result in loss of miRNA-mediated transcriptional regulation. It is also possible that other SNPs in LD with the 3′UTR in/del identified here are causally related to prostate cancer.
Although variants within ITGA2 have been significantly associated with other cancers, to our knowledge this is the first reported association between ITGA2 and prostate cancer risk. The cancer genetic markers of susceptibility (CGEMS) prostate cancer genome-wide association scan (www.caintegrator.nci.gov/cgems/browse.do) reported no significant associations between prostate cancer and the 1569 SNPs genotyped within the 14 Mb interval identified here. However, attempts to replicate significant associations of risk variants to previously identified prostate cancer susceptibility loci frequently fail to support the original findings (ELAC2:37, 38MSR1:39RNASEL:40). Reasons for this have been widely discussed and include: the heterogeneity of genes contributing to prostate cancer risk in different populations; different selection criteria for each of the study populations examined; and the variable contribution of environmental factors interacting with multiple genetic factors.
In summary, we have presented evidence for a prostate cancer susceptibility gene on chromosome 5p13–q12, with two ITGA2 polymorphisms showing a significant association with prostate cancer risk in the Tasmanian population. Although the functional SNP is still to be identified, considerable circumstantial evidence, provided by in vivo and in vitro studies, supports a role for this gene in tumour development. Replication of this result in independent data sets and studies elucidating the role of this gene will be of fundamental interest to the field of prostate cancer biology.
References
Parkin DM : Global cancer statistics in the year 2000. Lancet Oncol 2001; 2: 533–543.
Gronberg H, Damber L, Damber JE, Iselius L : Segregation analysis of prostate cancer in Sweden: support for dominant inheritance. Am J Epidemiol 1997; 146: 552–557.
Schaid DJ, McDonnell SK, Blute ML, Thibodeau SN : Evidence for autosomal dominant inheritance of prostate cancer. Am J Hum Genet 1998; 62: 1425–1438.
Xu J, Dimitrov L, Chang BL et al: A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet 2005; 77: 219–229.
Wiklund F, Gillanders EM, Albertus JA et al: Genome-wide scan of Swedish families with hereditary prostate cancer: suggestive evidence of linkage at 5q11.2 and 19p13.3. Prostate 2003; 57: 290–297.
Amundadottir LT, Sulem P, Gudmundsson J et al: A common variant associated with prostate cancer in European and African populations. Nat Genet 2006; 38: 652–658.
Gudmundsson J, Sulem P, Manolescu A et al: Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007; 39: 631–637.
Haiman CA, Patterson N, Freedman ML et al: Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 2007; 39: 638–644.
Yeager M, Orr N, Hayes RB et al: Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007; 39: 645–649.
Eeles RA, Kote-Jarai Z, Giles GG et al: Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008; 40: 316–321.
Gudmundsson J, Sulem P, Rafnar T et al: Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet 2008; 40: 281–283.
Thomas G, Jacobs KB, Yeager M et al: Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 2008; 40: 310–315.
Risch N : The genetic epidemiology of cancer: interpreting family and twin studies and their implications for molecular genetic approaches. Cancer Epidemiol Biomarkers Prev 2001; 10: 733–741.
Wang WY, Barratt BJ, Clayton DG, Todd JA : Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 2005; 6: 109–118.
Smith JR, Freije D, Carpten JD et al: Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science 1996; 274: 1371–1374.
Goddard KA, Witte JS, Suarez BK, Catalona WJ, Olson JM : Model-free linkage analysis with covariates confirms linkage of prostate cancer to chromosomes 1 and 4. Am J Hum Genet 2001; 68: 1197–1206.
Hsieh CL, Oakley-Girvan I, Balise RR et al: A genome screen of families with multiple cases of prostate cancer: evidence of genetic heterogeneity. Am J Hum Genet 2001; 69: 148–158.
Slager SL, Zarfas KE, Brown WM et al: Genome-wide linkage scan for prostate cancer aggressiveness loci using families from the University of Michigan Prostate Cancer Genetics Project. Prostate 2006; 66: 173–179.
Sato Y, Sugie R, Tsuchiya B, Kameya T, Natori M, Mukai K : Comparison of the DNA extraction methods for polymerase chain reaction amplification from formalin-fixed and paraffin-embedded tissues. Diagn Mol Pathol 2001; 10: 265–271.
Kennedy GC, Matsuzaki H, Dong S et al: Large-scale genotyping of complex DNA. Nat Biotechnol 2003; 21: 1233–1237.
O’Connell JR, Weeks DE : PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63: 259–266.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Thomson R, Quinn S, McKay J et al: The advantages of dense marker sets for linkage analysis with very large families. Hum Genet 2007; 121: 459–468.
Lander E, Kruglyak L : Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 1995; 11: 241–247.
Thornton T, McPeek MS : Case–control association testing with related individuals: a more powerful quasi-likelihood score test. Am J Hum Genet 2007; 81: 321–337.
Jacquelin B, Tarantino MD, Kritzik M et al: Allele-dependent transcriptional regulation of the human integrin alpha2 gene. Blood 2001; 97: 1721–1726.
Kunicki TJ : The influence of platelet collagen receptor polymorphisms in hemostasis and thrombotic disease. Arterioscler Thromb Vasc Biol 2002; 22: 14–20.
Koistinen P, Heino J : Integrins in Cancer Cell Invasion; in Heino J (ed): Cancer Invasion. Austin, USA, 2006, Vol. 19. Available online at: http://www.landesbioscience.com
Knox JD, Cress AE, Clark V et al: Differential expression of extracellular matrix molecules and the alpha 6-integrins in the normal and neoplastic prostate. Am J Pathol 1994; 145: 167–174.
Knudsen BS, Miranti CK : The impact of cell adhesion changes on proliferation and survival during prostate cancer development and progression. J Cell Biochem 2006; 99: 345–361.
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ : Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005; 65: 10946–10951.
Bonkhoff H, Stein U, Remberger K : Differential expression of alpha 6 and alpha 2 very late antigen integrins in the normal, hyperplastic, and neoplastic prostate: simultaneous demonstration of cell surface receptors and their extracellular ligands. Hum Pathol 1993; 24: 243–248.
Langsenlehner U, Renner W, Yazdani-Biuki B et al: Integrin alpha-2 and beta-3 gene polymorphisms and breast cancer risk. Breast Cancer Res Treat 2006; 97: 67–72.
Vairaktaris E, Yapijakis C, Derka S et al: Association of platelet glycoprotein Ia polymorphism with minor increase of risk for oral cancer. Eur J Surg Oncol 2006; 32: 455–457.
Kunicki TJ, Kritzik M, Annis DS, Nugent DJ : Hereditary variation in platelet integrin alpha 2 beta 1 density is associated with two silent polymorphisms in the alpha 2 gene coding sequence. Blood 1997; 89: 1939–1943.
Camp NJ, Tavtigian SV : Meta-analysis of associations of the Ser217Leu and Ala541Thr variants in ELAC2 (HPC2) and prostate cancer. Am J Hum Genet 2002; 71: 1475–1478.
Severi G, Giles GG, Southey MC et al: ELAC2/HPC2 polymorphisms, prostate-specific antigen levels, and prostate cancer. J Natl Cancer Inst 2003; 95: 818–824.
Sun J, Hsu FC, Turner AR et al: Meta-analysis of association of rare mutations and common sequence variants in the MSR1 gene and prostate cancer risk. Prostate 2006; 66: 728–737.
Nupponen NN, Wallen MJ, Ponciano D et al: Mutational analysis of susceptibility genes RNASEL/HPC1, ELAC2/HPC2, and MSR1 in sporadic prostate cancer. Genes Chromosomes Cancer 2004; 39: 119–125.
Lander ES, Schork NJ : Genetic dissection of complex traits. Science 1994; 265: 2037–2048.
Acknowledgements
We thank Annette Banks, Kris Hazelwood, Sally Inglis and Annette Edwards for their assistance in creating genealogical records and/or sample collection; and Melanie Bahlo, Terence Speed and David Goldgar for useful suggestions and discussions about methods of linkage analysis. The Royal Hobart Hospital Research Foundation, The Cancer Council of Tasmania, CaPCure, The Mazda Foundation and Perpetual Charitable Trusts and the Australian Cancer Research Fund supported this study. Dr Dickinson is a Cancer Council of Tasmania Research Fellow. We are also greatly indebted to the participants of the TasPac study, the TCR staff, the Tasmanian Urologists and Pathologists, and the wider Tasmanian clinical and research community.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
FitzGerald, L., Patterson, B., Thomson, R. et al. Identification of a prostate cancer susceptibility gene on chromosome 5p13q12 associated with risk of both familial and sporadic disease. Eur J Hum Genet 17, 368–377 (2009). https://doi.org/10.1038/ejhg.2008.171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.171
Keywords
This article is cited by
-
Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets
Nature Genetics (2018)
-
The genetic epidemiology of prostate cancer and its clinical implications
Nature Reviews Urology (2014)
-
Predictive value in the analysis of RNASEL genotypes in relation to prostate cancer
Prostate Cancer and Prostatic Diseases (2012)
