
Abstract
Borrone Dermato-Cardio-Skeletal (BDCS) syndrome is a severe progressive autosomal recessive disorder characterized by coarse facies, thick skin, acne conglobata, dysmorphic facies, vertebral abnormalities and mitral valve prolapse. We identified a consanguineous kindred with a child clinically diagnosed with BDCS. Linkage analysis of this family (BDCS1) identified five regions homozygous by descent with a maximum LOD score of 1.75. Linkage analysis of the family that originally defined BDCS (BDCS3) identified an overlapping linkage peak at chromosome 5q35.1. Sequence analysis identified two different homozygous mutations in BDCS1 and BDCS3, affecting the gene encoding the protein SH3 and PX domains 2B (SH3PXD2B), which localizes to 5q35.1. Western blot analysis of patient fibroblasts derived from affected individuals in both families demonstrated complete loss of SH3PXD2B. Homozygosity mapping and sequence analysis in a second published BDCS family (BDCS2) excluded SH3PXD2B. SH3PXD2B is required for the formation of functional podosomes, and loss-of-function mutations in SH3PXD2B have recently been shown to underlie 7 of 13 families with Frank-Ter Haar syndrome (FTHS). FTHS and BDCS share some overlapping clinical features; therefore, our results demonstrate that a proportion of BDCS and FTHS cases are allelic. Mutations in other gene(s) functioning in podosome formation and regulation are likely to underlie the SH3PXD2B-mutation-negative BDSC/FTHS patients.
Similar content being viewed by others


INTRODUCTION
In 1993, Borrone et al1 described a family in which two brothers presented with a multisystemic disorder affecting the skin, bone, joints and heart. The severe progressive disorder was characterized by thick skin, acne conglobata, osteolysis, gingival hypertrophy, brachydactyly, camptodactyly and mitral valve prolapse. There was no family history of the disorder nor reported consanguinity; inheritance was consistent with an X-linked or autosomal recessive model. Subsequently, a second family was described with parental consanguinity, suggesting autosomal recessive inheritance.2 The two affected brothers had severe acne, mitral valve prolapse and left ventricular dilation requiring valvuloplasty. Clinical features shared with the original family included brachydactyly and vertebral abnormalities. Gingival hypertrophy was not observed and initial presentation was later (17 and 19 years) than observed in the original family; however, the facial phenotype was noted to be very similar between the four affected individuals.1, 2 The disorder was classified as a distinct clinical entity called Borrone Dermato-Cardio-Skeletal syndrome (BDCS, MIM 211170), although to date the underlying genetic etiology has remained unknown.
In this study, we identified a new family with a clinical diagnosis of BDCS. We performed exome sequencing and identified homozygous loss-of-function mutations in the gene encoding the protein SH3 and PX domains 2B (SH3PXD2B), also known as Tyrosine Kinase Substrate with 4 Src homology3 (SH3) domains (TKS4).
MATERIALS AND METHODS
Patients
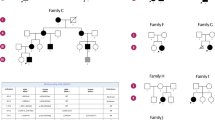
The parents and proband of the family diagnosed with BDCS (BDCS1, Figure 1a) were referred to the Victorian Clinical Genetics Services by their specialist. After receiving institutional ethics approval (Royal Children’s Hospital, Melbourne, Australia) and informed consent of participants, blood and tissue samples were obtained. The clinical descriptions of families BDCS2 (Figure 1b)2 and BDCS3 (Figure 1c)1 were reported previously. Genomic DNA was extracted from whole blood using the BACC DNA extraction kit (GE Healthcare Life Sciences, Uppsala, Sweden) according to the manufacturer’s protocols. Primary fibroblast cultures were established using standard procedures and maintained in RPMI supplemented with 10% fetal bovine serum (FBS).

Pedigree structure of BDCS families. Previously unreported Australian kindred with one male affected by BDCS (a) and previously published1, 2 Dutch (b) and Italian kindreds (c) that defined BCDS as a distinct clinical syndrome. Affected individuals are represented by black symbols. DNA from individuals that was used for linkage analysis on the Affymetrix (*) or Illumina platform (**) is indicated.
Linkage analysis
DNA was genotyped using high-density SNP array (Affymetrix Genome Wide 6 SNP chip (Santa Clara, CA, USA) for BDCS1, Illumina 660 quad chip (San Diego, CA, USA) for BDCS2 and BDCS3). Genotypes were called using commercial software, and markers incompatible with Mendelian inheritance were removed using LINKDATAGEN.3 A subset of ∼12000 informative SNPs were selected and parametric linkage analysis was performed using MERLIN.4 The analysis assumed a fully penetrant autosomal recessive disease model with a disease allele frequency of 0.0001.
Exome sequencing
To identify the underlying genetic cause of BDCS, gDNA (5 μg) was analyzed by whole exome capture and massively parallel sequencing (MPS). Approximately 62 Mb of coding sequence and exon-flanking regions were enriched using the Illumina TruSeq capture kit (San Diego, CA, USA) and 101-bp pair-end reads were sequenced on a HiSeq2000 (Axeq Technologies, Macrogen, Seoul, Korea). Reads were aligned to the reference genome (UCSC hg19) by Novoalign (version 2.07.09); those aligning to multiple locations or marked as duplicates by PicardTools 1.43 were discarded. Variants were detected with the SAMtools package, version 0.1.16,5 and annotated using ANNOVAR.6 Variants were filtered against the May 2011 release of the 1000 Genomes database and mutations were validated by standard PCR amplification and Sanger sequence analysis. Primer sequences are described in Supplementary Table S2. The reference sequences for SH3PXD2B used were NM_001017995.2 and NG_027746.1. Variants identified in this study were submitted to dbSNP at NCBI (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Western blot analysis
Primary fibroblast cells were harvested in extraction buffer containing 10 mM Tris-HCl, pH 7.5, 2% SDS and protease inhibitors (P8340, Sigma-Aldrich, St Louis, MO, USA). Lysates were sonicated before protein estimation by the BCA method (Thermo Fisher Scientific, Rockford, IL, USA). Western blot analysis was performed with 60 μg of total cell protein separated on 4–20% SDS-PAGE gradient gels that were subsequently transferred to Hybond-Low Fluorescent PVDF membranes (GE Healthcare Life Sciences, Uppsala, Sweden). Membranes were incubated in blocking buffer (5% BSA in PBS) with anti-SH3PXD2B antibody and monoclonal anti-β-actin antibody (1:1000 HPA036471/1:5000 A5441, Sigma-Aldrich). Antibody binding was revealed using fluorescent-conjugated secondary antibodies, and images were captured using a Typhoon imager (GE Healthcare Life Sciences, Uppsala, Sweden).
RESULTS
Clinical description
We identified a male child with a progressive multisystemic disorder whose parents are first cousins once removed (Figure 1a). The affected individual (BDCS1-17) displayed the craniofacial characteristics including a coarse face, broad forehead and broad nasal bridge, hypertelorism with prominent subocular folds, flat supraorbital ridges and a long double row of eyelashes (Figure 2a–g). An ophthalmological examination excluded eye abnormalities such as megalocornea and glaucoma. The observed clinical features and typical craniofacial characteristics were most consistent with the diagnosis of BDCS. BDCS1-17 also displayed a prominent maxilla, broad mouth, long flat philtrum, thin lips and broad alveolar ridges. Tooth eruption did not occur spontaneously and required surgical intervention owing to gingival hyperplasia. Histological analysis of the gingival lesions revealed that the fibroblasts had accumulated cytoplasmic membrane–bound structures containing collagen fibers. However, in vitro analysis revealed normal Type I and Type III collagen synthesis and secretion. Ultrastructural analysis of the skin and biochemical and enzymatic analysis excluded the diagnosis of a known metabolic disorder in BDCS1-17 (data not shown).

Clinical phenotype of family BDCS1. The facial features of BDCS1-17 at 3 months (a, b) and 5 years (c, d) display typical BDCS characteristics including a coarse face, broad forehead, broad nasal bridge, hypertelorism with prominent subocular folds and flat supraorbital ridges. At 5 years, brachydactyly, lateral deviation of the digits, thickened interphalangeal joints and a flexion deformity of the metacarpal joints are clearly evident (e, f). The skeletal dysplasia of BDCS1-17 also included dislocation of the radial head and genu valgum (g).
The skeletal dysplasia observed in BDCS1-17 was evident from birth and was more severe than described for previously reported individuals with BDCS (BDCS22 and BDCS31). There was brachydactyly with lateral deviation of the digits, thickened interphalangeal joints and a flexion deformity of the metacarpal joints. There was bilateral dislocation of the radial heads, genu valgum and club foot deformity. Premature fusion of the sagittal and lambdoid sutures resulted in abnormal cranial morphology. By the age of 5 years, the patient displayed normal cognitive development, but the progressive severity of multiple fixed contractures of large and small joints resulted in confinement to a wheelchair. X-rays at 7 years of age revealed generalized osteopenia of the radius and ulna, with marked widening of the medullary cavity of the bones and thinning of the overlaying cortex. Multiple anomalies were evident in the vertebral column, including kyphoscoliosis, gibbus deformity, anterior beaking of L2 vertebra and scalloping of the lower thoracic and upper lumbar vertebrae. As a consequence of a thoracic wall deformity, the patient also suffered from a restrictive lung deficit.
Linkage analysis and gene identification
We estimated the inbreeding coefficient of genotyped individuals from all three families with FEstim7 (Supplementary Table S1). This analysis validated the documented consanguinity in BDCS1 and BDCS2 and identified undocumented consanguinity in BDCS3, where the two patients were predicted to be the offspring of second cousins. We used FEstim to adjust LOD scores generated by MERLIN for the estimated degree of inbreeding.
The analysis of BDCS1 identified six linkage regions (Table 1) that all achieved the maximum possible adjusted LOD (FLOD) score of 1.75. Five linkage peaks attaining a maximum FLOD score of 1.74 were identified in BDCS2 (Table 1). None of these regions corresponded to any of the linkage peaks in BDCS1, suggesting locus heterogeneity in BDCS. For BDCS3, we identified a single homozygous peak at hg19 chr5:169352662-172043315. This region was located entirely within the Chromosome 5 peak observed in BDCS1 (Supplementary Figure S1A) and achieved a maximum FLOD score of 4.1. These results suggested disease locus homogeneity in BDCS1 and BDCS3. However, we were unable to detect any relatedness between individuals from BDCS1 and BDCS3 using PLINK,8 suggesting allelic heterogeneity between these two families.
To identify the underlying genetic cause of BDCS, gDNA from BDCS1-17 was analyzed by whole exome capture and massively parallel sequencing (MPS). Within the shared linkage region, 114 variants were identified, of which 15 candidates remained after filtering to identify rare variants (absent from the May 2011 release of the 1000 Genomes database or present with a frequency of less than 0.01). None of these variants were in the coding sequence of a gene. Only one variant, a novel homozygous alteration affecting the canonical GT splice donor site of intron 5 in SH3PXD2B, was predicted to have a functional consequence, namely frameshift and premature termination of the protein (NM_001017995.2:c.401+1G>A, p.(Glu134Glufs*1)). Sanger sequencing confirmed the splice-site alteration and segregation within the BDCS1 pedigree (data not shown). This alteration was absent in 534 unrelated control samples tested, and it has not been reported in public databases including dbSNP137 and 6503 exomes from the NHLBI GO Exome Sequencing Project (ESP6500).
Sequence analysis of SH3PXD2B in BDCS3 did not identify any variants in the 5'UTR and first 12 coding exons. However, exon 13, which encodes 1545 bp of the open reading frame and the entire 4873 bp 3′ UTR, consistently failed to amplify in the affected individuals (Figure 3a). This result suggested a homozygous deletion, and close re-examination of the BDCS3 SNP genotyping data identified a reduced LogR ratio affecting three consecutive markers in exon 13 spanning hg19 chr5:171753489-171772333 (Supplementary Figure S1B). PCR and sequence analysis defined the proximal break point within intron 12 and the distal break point 1718 bp 3′ of the SH3PXD2B polyadenylation site. The 12583-bp deletion [c.1188+1773_2733+6592del] included the hg19 reference sequence chr5:171758785-171771367. The breakpoints are located within two separate Alu repeat sequences, and the deletion results in the generation of a single contiguous Alu repeat sequence, suggesting that the deletion has occurred by homologous recombination between these elements.

Molecular characterization of SH3PXD2B. PCR analysis of genomic DNA spanning exon 12, the coding region of exon 13, the 3′UTR and a downstream intronic region of SH3PXD2B in BDCS3-affected patients (BDCS3-3, BDCS3-4) confirmed the deletion of exon 13. Reaction products for control and no template (NTC) are shown (a). Immunoblot analysis with a SH3PXD2B-specific antibody (1:200 Sigma-Aldrich #HPA036471) identified an ∼120-kDa protein in control fibroblasts (C1, C2, C3) that was absent in extracts from patient-derived fibroblasts (BDCS1-17, BDCS3-3/4). Analysis with an antibody directed against β-actin (1:5000 Sigma-Aldrich #A5441) confirmed equivalent protein loading (b).
The 911-amino-acid protein encoded by SH3PXD2B is SH3 and PX domains 2B, also known as Tyrosine Kinase Substrate with 4 Src homology3 (SH3) domains (TKS4). The protein structure consists of a conserved Phox homology (PX) domain at the amino-terminal, connected via linker regions to four conserved SH3 domains.9 The splice donor mutation in BDCS1 was predicted to result in a truncated protein encoding 134 amino acids or ∼15% of the full-length protein (Supplementary Figure S1C). Similarly, the deletion mutation in BDSC3 was predicted to result in a truncated protein encoding 396 amino acids or ∼40% of the full-length protein. Both mutations were expected to result in the complete loss of full-length functional SH3PXD2B, and this was confirmed by western blot analysis of patient-derived fibroblast cells. In control fibroblast cells, a single band of the expected size (approximately 120 kDa) was detected using an anti-SH3PXD2B antibody. However, this protein was not detected in patient fibroblasts derived from BDCS1 or BDCS3, confirming a complete loss of SH3PXD2B in the BDCS patients (Figure 3b).
DISCUSSION
In this study, we identified and characterized a previously unreported individual with the clinical diagnosis of Borrone Dermato-Cardio-Skeletal syndrome (BDCS, [MIM 211170]). The clinical features were generally consistent with those previously reported for BDCS with a more pronounced skeletal phenotype. However, the proband did not develop the characteristic acne conglobata observed in the other reported BDCS patients, and ultrastructural analysis of a skin biopsy revealed no underlying dermal pathology. The absence of these features might result from the prepubertal death of the patient. This occurred at 8 years of age because of the combined consequence of mitral valve prolapse leading to acute congestive cardiac failure and respiratory failure associated with the restrictive lung deficit.
Linkage and sequence analysis demonstrated that mutations in SH3PXD2B underlie the BDCS phenotype in the family that originally defined BDCS (BDCS3) and the family newly described herein (BDCS1). Recent studies have demonstrated that SH3PXD2B is an adaptor protein required for podosome formation and function.9 Podosomes are actin-rich membrane protrusions that facilitate cell adhesion, migration and extracellular matrix (ECM) remodeling.10 They are required for the normal function of a variety of cell types including macrophages, lymphocytes, dendritic cells, endothelial cells, myoblasts and osteoclasts.11, 12, 13, 14, 15, 16 Dysfunction of podosomes in these cell types is likely to mediate the BDCS phenotype. For example, osteoclast podosomes are essential drivers in the migration of osteoclasts during bone development and homeostasis. SH3PXD2B facilitates podosome ECM remodeling via the recruitment of matrix metalloproteases and members of the ADAM (a disintegrin and metalloprotease) metalloprotease family. The murine homolog Sh3pxd2b is highly expressed in the eye, heart and bone during embryonic development. Sh3pxd2b null mice replicate many of the clinical features of BDCS, including severe craniofacial and skeletal deformities such as hypertelorism and kyphosis and ventricular and mitral valve defects.17, 18
Interestingly, SH3PXD2B missense and frameshift mutations have been recently shown to underlie approximately 50% (7/13 families analyzed) of patients with Frank-Ter Haar Syndrome (FTHS, (MIM249420)).17 FTHS is a progressive autosomal recessive condition displaying several clinical features directly comparable to those observed in BDCS including the craniofacial anomalies, skeletal dysplasia and cardiac defects.17, 19, 20, 21, 22, 23, 24 However, some phenotypic traits differ between the described syndromes, perhaps explaining why only a single report has speculated that the two syndromes might represent a single clinical entity.21 For example, eye abnormalities such as megalocornea are commonly observed in both SH3PXD2B-mediated FTHS and Sh3pxd2b null mice where deletion or frameshift mutation results in the complete loss of SH3PXD2B.17, 25 In contrast, eye anomalies have either not been observed or not been reported in patients with BDCS. Glaucoma was absent in individuals with BDCS and was also absent in three of seven individuals with FTHS owing to complete loss of SH3PXD2B.17
Our results demonstrate that the phenotype in two families, including the family that originally defined BDCS, can be explained by loss of SH3PXD2B and therefore are equivalent to FTHS. The differential description of the two syndromes was probably due to the phenotypic variation observed between BDCS and FTHS patients. This phenotypic variability occurs despite common truncating loss of protein mutations (Table 2) and is likely to be the result of differences in genetic background, environmental influences and also the progressive nature of the disorder. For instance, the cardiac manifestations contribute to the early childhood death of many FTHS patients. Symptoms with a later onset, such as the pubertal appearance of severe acne, may not have developed because of the premature death of the patients.
Despite the similarity in symptoms with the other BDCS (and FTHS) families, linkage analysis excluded linkage to SH3PXD2B for BDCS2 (LOD<−5), and we did not identify any potential mutations in affected individuals by direct sequencing of SH3PXD2B (data not shown). Linkage analysis also excluded the SH3PXD2B paralog SH3PXD2A as a candidate in BDCS2 (LOD<−2), consistent with the exclusion of this gene in FTHS families.17 This confirms that locus heterogeneity underlies BDCS2 and the six SH3PXD2B-mutation-negative FTHS families17 and genes that function in the same pathway are likely candidates. Therefore, future studies investigating the unresolved BDCS/FTHS families should initially focus on pathways involved in podosome function and SH3PXD2B interacting proteins.
References
Borrone C, Di Rocco M, Crovato F, Camera G, Gambini C : New multisystemic disorder involving heart valves, skin, bones, and joints in two brothers. Am J Med Genet 1993; 46: 228–234.
van Steensel MA, Ceulen RP, Delhaas T, de Die-Smulders C : Two Dutch brothers with Borrone dermato-cardio-skeletal syndrome. Am J Med Genet A 2007; 143A: 1223–1226.
Bahlo M, Bromhead CJ : Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics 2009; 25: 1961–1962.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Li H : Improving SNP discovery by base alignment quality. Bioinformatics 2011; 27: 1157–1158.
Leutenegger AL, Prum B, Genin E et al: Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet 2003; 73: 516–523.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, Courtneidge SA : The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol Biol Cell 2009; 20: 1302–1311.
Gimona M, Buccione R, Courtneidge SA, Linder S : Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol 2008; 20: 235–241.
Binks M, Jones GE, Brickell PM, Kinnon C, Katz DR, Thrasher AJ : Intrinsic dendritic cell abnormalities in Wiskott-Aldrich syndrome. Eur J Immunol 1998; 28: 3259–3267.
Carman CV, Sage PT, Sciuto TE et al: Transcellular diapedesis is initiated by invasive podosomes. Immunity 2007; 26: 784–797.
Lehto VP, Hovi T, Vartio T, Badley RA, Virtanen I : Reorganization of cytoskeletal and contractile elements during transition of human monocytes into adherent macrophages. Lab Invest 1982; 47: 391–399.
Marchisio PC, Cirillo D, Naldini L, Primavera MV, Teti A, Zambonin-Zallone A : Cell-substratum interaction of cultured avian osteoclasts is mediated by specific adhesion structures. J Cell Biol 1984; 99: 1696–1705.
Moreau V, Tatin F, Varon C, Genot E : Actin can reorganize into podosomes in aortic endothelial cells, a process controlled by Cdc42 and RhoA. Mol Cell Biol 2003; 23: 6809–6822.
Thompson O, Kleino I, Crimaldi L, Gimona M, Saksela K, Winder SJ : Dystroglycan, Tks5 and Src mediated assembly of podosomes in myoblasts. PLoS One 2008; 3: e3638.
Iqbal Z, Cejudo-Martin P, de Brouwer A et al: Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of Frank-Ter Haar Syndrome. Am J Hum Genet 2010; 86: 254–261.
Mao M, Thedens DR, Chang B et al: The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development. Mamm Genome 2009; 20: 462–475.
Frank Y, Ziprkowski M, Romano A et al: Megalocornea associated with multiple skeletal anomalies: a new genetic syndrome? J Genet Hum 1973; 21: 67–72.
Hamel BC, Draaisma JM, Pinckers AJ et al: Autosomal recessive Melnick-Needles syndrome or ter Haar syndrome? Report of a patient and reappraisal of an earlier report. Am J Med Genet 1995; 56: 312–316.
Maas SM, Kayserili H, Lam J, Apak MY, Hennekam RC : Further delineation of Frank-ter Haar syndrome. Am J Med Genet A 2004; 131: 127–133.
Megarbane A, Tomey K, Wakim G : Congenital glaucoma, limb deformities, skeletal dysplasia, and facial anomalies: report of another family. Am J Med Genet 1997; 73: 67–71.
ter Haar B, Hamel B, Hendriks J, de Jager J : Melnick-Needles syndrome: indication for an autosomal recessive form. Am J Med Genet 1982; 13: 469–477.
Wallerstein R, Scott CI Jr., Nicholson L : Extended survival in a new case of ter Haar syndrome: further delineation of the syndrome. Am J Med Genet 1997; 70: 267–272.
Mao M, Hedberg-Buenz A, Koehn D, John SW, Anderson MG : Anterior segment dysgenesis and early-onset glaucoma in nee mice with mutation of Sh3pxd2b. Invest Ophthalmol Vis Sci 2011; 52: 2679–2688.
Acknowledgements
We thank the families involved in this research and are grateful to Hayley Mountford and Greta Gillies for assistance with patient samples. We thank John Bateman for collagen analysis in BDCS1. Skin fibroblasts from BDCS3 were obtained from the Cell Line and DNA Biobank from patients affected by genetic diseases (G.Gaslini Institute), Telethon Network of Genetic Biobank (Project GTB07001A). This work was funded in part by National Health and Medical Research Council Australia Program Grant 490037 to DJA and MB. KRS is supported by a PhD scholarship funded by the Pratt Foundation. MB is supported by an ARC Future Fellowship (FT100100764). MBD is supported by an NHMRC Practitioner Fellowship (546452) and PJL is supported by an NHMRC Career Development Fellowship (APP1032364). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Wilson, G., Sunley, J., Smith, K. et al. Mutations in SH3PXD2B cause Borrone dermato-cardio-skeletal syndrome. Eur J Hum Genet 22, 741–747 (2014). https://doi.org/10.1038/ejhg.2013.229
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.229
Keywords
This article is cited by
-
Hyaline fibromatosis syndrome: a case presenting with gingival enlargement as the only clinical manifestation and a report of two new mutations in the ANTXR2 gene
BMC Oral Health (2021)
-
Enhanced endothelial motility and multicellular sprouting is mediated by the scaffold protein TKS4
Scientific Reports (2019)
-
The scaffold protein Tks4 is required for the differentiation of mesenchymal stromal cells (MSCs) into adipogenic and osteogenic lineages
Scientific Reports (2016)
-
Accumulation of the PX domain mutant Frank-ter Haar syndrome protein Tks4 in aggresomes
Cell Communication and Signaling (2015)
-
Closing the Gap: Genetic and Genomic Continuum from Syndromic to Nonsyndromic Craniosynostoses
Current Genetic Medicine Reports (2014)
