
Abstract
CHD7 variants are a well-established cause of CHARGE syndrome, a disabling multi-system malformation disorder that is often associated with deafness, visual impairment and intellectual disability. Less severe forms of CHD7-related disease are known to exist, but the full spectrum of phenotypes remains uncertain. We identified a de novo missense variant in CHD7 in a family presenting with musculoskeletal abnormalities as the main manifestation of CHD7-related disease, representing a new phenotype. The proband presented with prominent scapulae, mild shoulder girdle weakness and only subtle dysmorphic features. Investigation revealed hypoplasia of the trapezius and sternocleidomastoid muscles and semicircular canal defects, but he did not fulfill diagnostic criteria for CHARGE syndrome. Although the shoulders are often sloping and anteverted in CHARGE syndrome, the underlying neuromuscular cause has never been investigated. This report expands the phenotypes associated with CHD7 mutations to include a musculoskeletal presentation, with hypoplasia of the shoulder and neck muscles. CHD7 should be considered in patients presenting in childhood with stable scapular winging, particularly if accompanied by dysmorphic features and balance difficulties.
Similar content being viewed by others

Introduction
CHARGE syndrome is a disabling complex of congenital malformations associated with variants in CHD7 in 60–90% of patients.1, 2, 3 Various clinical diagnostic criteria have been proposed,4, 5, 6 but musculoskeletal abnormalities outside the face are uncommonly reported. Nonsense and frameshift variants are most common, but missense variants are also reported.7, 8, 9 A small number of patients with mild phenotypes, who do not fulfill the diagnostic criteria, have been reported in association with missense variants.9 Commonly these patients have had ear abnormalities, with or without one or two other minor criteria.
We studied a proband with scapular winging who had been extensively investigated for genetic myopathies. He was found to have a novel de novo missense variant in CHD7 associated with hypoplasia of shoulder and neck muscles, and hypoplasia of the semicircular canals. Review of a large cohort of patients with CHD7-related disease revealed a further patient with atrophic shoulder muscles and cavovarus foot deformities. This report enlarges the spectrum of CHD7-related disease to include patients with musculoskeletal presentations.
Methods
Patient 1 was referred with proximal muscle weakness and scapular winging, suggestive of an inherited myopathy and was included in a whole-exome sequencing (WES) study. Patient 2 is the mildly affected son of Patient 1. The database of the University Medical Center Groningen (UMCG) CHARGE clinic was screened for patients with neuromuscular problems. Patient 3 was identified as presenting with a suspected myopathy.
WES was performed in Patient 1 and his unaffected parents using a well-established pipeline at the Broad Institute.10 The CHD7 variant was confirmed by Sanger sequencing in the proband and close family members. The coding regions of CHD7 were sequenced in Patient 3. All variants were submitted to the Leiden Open Variant CHD7 database (www.LOVD.nl/CHD7; patient IDs 51495 and 51531). The variants and phenotypes will also be available in updated CHD7 database (www.chd7.org).
Results
WES in Patient 1 showed a heterozygous missense variant in CHD7 c.3398C>T, p.(Thr1133Met); (NM_017780.3). Sanger sequencing confirmed that this variant arose de novo in Patient 1, and was also present in his mildly affected son (Patient 2). The p.(Thr1133Met) variant has not been previously reported in the CHD7 database (www.chd7.org, accessed July 2015) or the Exome Aggregation Consortium (ExAC) database (www.exac.broadinstitute.org, accessed July 2015). The variant is located in the helicase ATP-binding domain.11 Four in silico prediction software packages predicted the variant to be pathogenic (PolyPhen-2 1.0;12 SIFT deleterious (0.000); Provean deleterious (−5.610);13 MutationTaster disease causing (0.9990)14). On the basis of the prediction programs (score +2) and de novo occurrence (score +3), this missense variant probably affects function according to the scoring classification published by Bergman.15
In Patient 3, CHD7 sequencing revealed an intronic variant predicted to disrupt the exon 6 donor splice site (c.2442+5G>C; NM_017780.3; g.127819G>C; NG_007009.1). The same variant was present in his affected older brother9 and their mildly affected father. The variant was not present in the ExAC database.
Clinical features
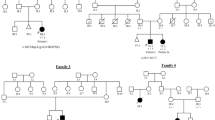
Patient 1 had prominent scapulae, shoulder girdle weakness and pain from 7 years of age. He was assessed in his early 20 s with a suspected inherited myopathy. He had no history of gross motor delay or poor balance, but had mild learning difficulties at school. At 27 years he had subtle dysmorphic features, including small ears and hockey-stick palmar creases. His neck was short and broad, and his shoulders had an abnormal anterior position. His scapulae were laterally rotated, and with shoulder abduction they showed cranial elevation and dorsal winging, more pronounced on the right (Figures 1a and c). He had mild weakness (MRC grade 4/5) of shoulder abduction and neck flexion. He had no facial asymmetry or weakness. Sense of smell, genital development, ophthalmology review, audiology assessment, echocardiogram and respiratory examinations were normal. Scoliosis was not present. Creatine kinase (CK) level was normal. A deltoid muscle biopsy taken at age 23 years was non-specific. Southern blot analysis for facio–scapular–humeral dystrophy (FSHD) type 1 was negative. Nerve conduction studies (NCS) were normal. At 27 years of age, stimulation of the spinal accessory nerves elicited normal amplitude compound muscle action potentials (CMAP). Electromyography of both trapezii showed polyphasic motor units with reduced recruitment.

(a) Pedigrees illustrating the relationships of Patient 1 and 2, and Patient 3. (b) Posterior view of Patient 1 showing sloping shoulders, dorsal winging and lateral displacement of the scapulae during arm abduction. (c) Lateral view of Patient 1 showing anterior position of the shoulders, small ears and mild retrognathia. (d) Posterior view of Patient 2 showing laterally displaced scapulae and bilateral acromial dimples. (e) Patient 3 showing mild prominence of the right scapula. (f) The lower legs of Patient 3 showing calf atrophy and pes cavovarus. A full colour version of this figure is available at the European Journal of Human Genetics journal online.
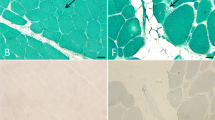
An MRI brain scan, with fine cuts through the semicircular canals, showed aplasia of the superior semicircular canal bilaterally and dysplastic horizontal canals. The brain and cervical spinal cord were normal, although the XIth cranial nerve was not seen. Muscle MRI showed reduced trapezius muscle bulk with changes more marked on the right (Figure 2a). The sternocleidomastoid and paravertebral muscle bulk (C3–C4 level) were reduced bilaterally (Figure 2b). Serratus anterior and the lower limb musculature were normal.

(a) Coronal T1 muscle MRI image from Patient 1. The arrows point to the trapezius muscle, which is hypoplastic bilaterally, with the right more severely affected. (b) Axial T1 section through the neck of Patient 1. The arrow indicates hypoplasia of the paraspinal muscles. (c) Axial T2 section from Patient 3. The arrows point to the trapezius muscles showing asymmetry with the left smaller than the right. A full colour version of this figure is available at the European Journal of Human Genetics journal online.
Patient 2, the 18 month-old son of Patient 1, had normal developmental milestones, no dysmorphic features and no muscle weakness. He had an abnormal shoulder shape with hypermobile, laterally positioned scapulae with bilateral dimples over the acromial processes. Genital development, echocardiogram, audiology and ophthalmology assessments were normal. He was not investigated with MRI imaging.
Patient 3, and his older brother, have been previously described (as family 1) in a paper on familial CHARGE.9 Both fulfilled diagnostic criteria for a typical CHARGE syndrome. Their father was recently available for testing and review. He was mildly affected with a square face, small earlobes and a hockey-stick palmar crease. Patient 3 was referred for a neuromuscular evaluation at 8 years of age because of pes cavovarus and fatigue. He was re-evaluated at age 13 because of mildly progressive pes cavovarus and calf atrophy. He had an abnormal gait with inversion of the ankle and impaired dorsiflexion of the right foot. He had bilateral scapular winging and an atrophic appearance to the shoulder muscles, with neck webbing (Figures 1b and d). Scoliosis was not present. CK was normal. NCS showed reduction of the right peroneal nerve CMAP amplitude. EMG was normal. MRI showed mild asymmetry of the trapezius muscles (Figure 2c) and preserved sternocleidomastoids. There was a slight reduction in the size of the right calf compared with the left, and mild atrophic fatty change in the right flexor halluces longus and tibialis anterior.
To investigate the frequency of musculoskeletal features affecting the shoulder region and limbs in CHD7-related disease, we searched the database of the UMCG CHARGE expert clinic (n=104). Clinical photographs of the shoulder region were available for 41 patients with CHARGE syndrome. Abnormal shoulder posture was present in 31 out of 41 (76%) patients, involving anteverted (n=18), sloping (n=29) and/or asymmetric (n=18) shoulder posture. In addition to patient 4, underlying neuromuscular pathology was suspected in 6 out of 104 patients. In these six patients, neuromuscular symptoms involved reduced exercise tolerance (n=2), proximal muscle weakness (n=2), pes adductus varus (n=1), shortened pectoral muscles (n=1), limited arm abduction (n=2) and atrophic shoulder muscles (n=1).
Discussion
Patient 1 was investigated for a suspected inherited myopathy over several years due to abnormal scapular position and movements. Limb girdle muscular dystrophy and FSHD were considered; however, a specific diagnosis could not be established. We were surprised to identify a de novo heterozygous missense variant in CHD7 with WES.
Patient 1 did not fulfill diagnostic criteria for CHARGE syndrome but the presence of dysplastic semicircular canals, an uncommon finding that is strongly associated with CHD7-related disease, makes it highly likely that the p.(Thr1133Met) de novo variant affects function. Patient 2, his son, also had a mild phenotype with an abnormal shoulder shape. Missense variants in CHD7 are relatively uncommon in the literature, and it is possible that mild or atypical CHARGE phenotypes, such as we report, have been under-recognized. Variable expressivity is common in autosomal dominant transmission of CHD7 variants,9 and it is uncertain whether the p.(Thr1133Met) variant predisposes to a mild phenotype or could lead to typical CHARGE syndrome in some individuals.
Patient 1 had hypoplastic trapezius and sternocleidomastoid muscles on muscle MRI explaining his prominent scapulae, abnormal shoulder shape and shoulder girdle weakness. Although scapular winging was noted in Patient 3, this was less marked than in Patient 1 and the trapezius muscles were only mildly asymmetric in bulk, suggesting there is a spectrum of severity of neuromuscular involvement. Patient 3 also demonstrates that abnormal neuromuscular features can accompany typical CHARGE syndrome. In addition to the shoulder abnormalities, Patient 3 had pes cavovarus and mild calf muscle atrophy. These observations of muscle hypotrophy have not been previously described in association with CHD7 mutations.
Sloping, asymmetric and anteverted shoulders are a common feature of CHARGE syndrome.5 A retrospective evaluation of the UMCG database showed this to be present in 31 out of 41 patients. A systematic analysis of these findings has never been performed and the cause for this phenomenon has not been previously identified. More interestingly, we have demonstrated in Patient 1 that muscle hypoplasia can be the most prominent clinical abnormality associated with CHD7 mutations.
The CHD7 protein is a transcriptional regulator, which regulates nuclear gene expression and rRNA biogenesis.16 It has an essential role in the formation of multipotent neural crest cells and their migration, and development into a range of head and neck structures.17 Chick embryos have high CHD7 protein expression in the optic, otic and nasal placodes, and branchial arches, which corresponds with the structures classically affected in CHARGE syndrome.18
The sternocleidomastoid and trapezius muscles are both innervated by the XIth cranial nerve, which has its embryological origins in the neural crest. The EMG findings in Patient 1 were suggestive of chronic denervation and the spinal accessory nerves were not visualized on MRI imaging, raising the possibility that the primary defect is in the development of and innervation from the XIth cranial nerve. The abnormalities of the VIIth and other cranial nerves are common in CHARGE syndrome. In zebrafish models, CHD7 is important in neuronal development and axonal projections. Zebrafish with reduced CHD7 expression showed disrupted organization of cranial motor neurons.19 However, the involvement of the paraspinal muscles, and the peroneal weakness seen in Patient 3 is not easily explained by this hypothesis.
An alternative explanation is that the abnormalities may result from abnormal patterning, migration and differentiation of muscle progenitor cells, a process dependent on normal cranial neural crest cell function. In chick embryo models, myogenesis was initiated in the absence of neural crest cells, but the migratory pathways and anterior–posterior registration of the paraxial mesoderm was severely impaired and overall myofiber organization was disrupted.20,21
Making a diagnosis of CHD7-related disease has implications for health surveillance and genetic counseling, given the autosomal dominant inheritance and well-described risk of germline mosaicism.8 CHD7-related disease should be considered in the differential diagnosis of children with early-onset prominent scapulae and a stable clinical course, particularly if associated with dysmorphic facial features and balance difficulties, and in our clinic has lead to the identification of a further unpublished patient. If suspected, investigation with a CT or MRI of the brain for semicircular canal malformations is recommended, which if present give strong support to this diagnosis. A next-generation sequencing approach to the genetic diagnosis of scapular winging is advantageous given the heterogeneity of this presentation and CHD7 should be considered for inclusion in neuromuscular gene panels.
References
Vissers LELM, van Ravenswaaij CMA, Admiraal R et al: Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004; 36: 955–957.
Jongmans MCJ, Admiraal RJ, van der Donk KP et al: CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 2006; 43: 306–314.
Colin E, Bonneau D, Boussion F et al: Prenatal diagnosis of CHARGE syndrome by identification of a novel CHD7 mutation in a previously unaffected family. Prenat Diagn 2012; 32: 692–694.
Lalani SR, Hefner MA, Belmont JW et al. CHARGE Syndrome. 2006 Oct 2 [Updated 2012 Feb 2], in: Pagon RA, Adam MP, Bird TD et al (eds): GeneReviews™ [Internet]. University of Washington: Seattle, WA, USA, 1993–2014.
Blake KD, Davenport SL, Hall BD et al: CHARGE association: an update and review for the primary pediatrician. Clin Pediatr 1998; 37: 159–173.
Verloes A : Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet 2005; 133A: 306–308.
Janssen N, Bergman JEH, Swertz MA et al: Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 2012; 33: 1149–1160.
Bergman JEH, Janssen N, Hoefsloot LH et al: CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 2011; 48: 334–342.
Jongmans MCJ, Hoefsloot LH, van der Donk KP et al: Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet 2007; 146A: 43–50.
Menezes MP, Waddell LB, Lenk GM et al: Whole exome sequencing identifies three recessive FIG4 mutations in an apparently dominant pedigree with Charcot-Marie-Tooth disease. Neuromus Dis 2014; 24: 666–670.
Uniprot Consortium: Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 2014; 42: D191–D198.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Schwarz JM, Rödelsperger C, Schuelke M et al: MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Bergman JE, Janssen N, Van der Sloot AM et al: A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat 2012; 33: 1251–1260.
Zentner GE, Hurd EA, Schnetz MP et al: CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet 2010; 19: 3491–3501.
Bajpai R, Chen DA, Rada-Iglesias A et al: CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2011; 463: 958–962.
Kosaki K : Role of rare cases in deciphering the mechanisms of congenital anomalies: CHARGE syndrome research. Congenit Anom 2011; 51: 12–15.
Patten SA, Jacobs-McDaniels NL, Zaouter C et al: Role of Chd7 in Zebrafish: a model for CHARGE syndrome. PLoS One 2012; 7: e31650.
Rinon A, Lazar S, Marshall H et al: Cranial neural crest cells regulate head muscle patterning and differentiation during vertebrate embryogenesis. Development 2007; 134: 3065–3075.
Grenier J, Teillet M-A, Grifone R et al: Relationship between neural crest cells and cranial mesoderm during head muscle development. PLoS One 2009; 4: e4381.
Acknowledgements
We thank the families for their invaluable contributions. This work was funded by the National Health and Medical Research Council of Australia grants 1022707 and 1031893 (NFC and KNN), and 1056285 (GO). Exome sequencing was supported by grants from the National Human Genome Research Institute of the US National Institutes of Health (Medical Sequencing Program grant U54 HG003067 to the Broad Institute principal investigator, Lander). Grant 1202-023 was provided by Fund Nuts Ohra to MTYW.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
O'Grady, G., Ma, A., Sival, D. et al. Prominent scapulae mimicking an inherited myopathy expands the phenotype of CHD7-related disease. Eur J Hum Genet 24, 1216–1219 (2016). https://doi.org/10.1038/ejhg.2015.276
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.276
This article is cited by
-
Multiple muscular abnormalities in a fetal cadaver with CHARGE syndrome
Surgical and Radiologic Anatomy (2019)
