
Abstract
Wolbachia is the most widespread endosymbiotic bacterium of insects and other arthropods that can rapidly invade host populations. Deliberate releases of Wolbachia into natural populations of the dengue fever mosquito, Aedes aegypti, are used as a novel biocontrol strategy for dengue suppression. Invasion of Wolbachia through the host population relies on factors such as high fidelity of the endosymbiont transmission and limited immigration of uninfected individuals, but these factors can be difficult to measure. One way of acquiring relevant information is to consider mitochondrial DNA (mtDNA) variation alongside Wolbachia in field-caught mosquitoes. Here we used diagnostic mtDNA markers to differentiate infection-associated mtDNA haplotypes from those of the uninfected mosquitoes at release sites. Unique haplotypes associated with Wolbachia were found at locations outside Australia. We also performed mathematical and qualitative analyses including modelling the expected dynamics of the Wolbachia and mtDNA variants during and after a release. Our analyses identified key features in haplotype frequency patterns to infer the presence of imperfect maternal transmission of Wolbachia, presence of immigration and possibly incomplete cytoplasmic incompatibility. We demonstrate that ongoing screening of the mtDNA variants should provide information on maternal leakage and immigration, particularly in releases outside Australia. As we demonstrate in a case study, our models to track the Wolbachia dynamics can be successfully applied to temporal studies in natural populations or Wolbachia release programs, as long as there is co-occurring mtDNA variation that differentiates infected and uninfected populations.
Similar content being viewed by others

Introduction
Wolbachia is the most widespread endosymbiotic bacterium of insects and other arthropods (Zug and Hammerstein, 2012). It is vertically transmitted from mother to offspring. It also often exhibits effects on host reproductive biology allowing rapid invasion of host populations (cf, Turelli and Hoffmann, 1991). One such reproductive manipulation is cytoplasmic incompatibility (CI), manifested as death of embryos produced by a fusion of Wolbachia-infected sperm with uninfected eggs or eggs that carry an incompatible Wolbachia infection (Yen and Barr, 1971). This mechanism gives a reproductive advantage to Wolbachia-infected females whose eggs are protected from CI, driving the Wolbachia invasion through host populations (Hoffmann and Turelli, 1997).
Because Wolbachia infection also confers increased host resistance to many pathogens, it is considered as a novel biocontrol method for suppressing vector-borne human diseases such as dengue (Moreira et al., 2009). The main vector of dengue, the Aedes aegypti mosquito, does not carry Wolbachia infections in the wild (Kittayapong et al., 2000), but there are now several Wolbachia-infected A. aegypti strains created by laboratory transfection from other insect hosts (McMeniman et al., 2009; Walker et al., 2011). Wolbachia infections originating from Drosophila melanogaster (wMel and wMelPop) and Aedes albopictus (wAlbB) reduce dengue virus load significantly in A. aegypti (Bian et al., 2010; Moreira et al., 2009; Walker et al., 2011). In addition, a strain like wMelPop can reduce the adult lifespan and viability of quiescent eggs in A. aegypti (McMeniman et al., 2009; McMeniman and O'Neill, 2010b; Yeap et al., 2011). Life-shortening and virus interference effects in the mosquito host are expected to decrease dengue incidence, given that older mosquitoes are more likely to transmit diseases to humans (cf, Focks et al., 1995).
Wolbachia-based strategies for disease suppression rely on the release of mosquitoes with a particular Wolbachia infection into the field. The Wolbachia then invades and replaces the resident population with mosquitoes that have a reduced potential for disease transmission (Hoffmann et al., 2011). The successful introduction of Wolbachia into natural mosquito populations requires the release of high-quality material that can invade the population and persist over time. Such release material should contain a Wolbachia infection that induces very strong CI, (near) perfect maternal transmission (that is, very weak maternal transmission leakage) and a small fitness cost to the host (Hoffmann et al., 2015).
The quality of release material can be assessed in several ways: (1) via laboratory fitness tests (see, for example, McMeniman and O'Neill, 2010a; Hoffmann et al., 2011; Yeap et al., 2011; Ross et al., 2014; Turley et al., 2013, 2) through the monitoring of performance during trial releases (Yeap et al., 2013) as well as (3) by tracking the changes in Wolbachia infection frequency over time (Hoffmann et al., 2011; Hoffmann et al., 2014). The latter can indicate the stability of the infection under possible imperfect maternal transmission and/or immigration of uninfected individuals.
A different approach for tracking the dynamics of Wolbachia in invaded populations is to examine mitochondrial DNA (mtDNA) variants associated with the infection. Because both Wolbachia and mitochondria are maternally inherited, the mitochondrial genome will be in linkage disequilibrium with the endosymbiont (Figure 1, upper). The level of disequilibrium between the two genomes depends on the fidelity of maternal transmission and incidence of paternal transmission (Turelli et al., 1992; Johnstone and Hurst, 1996). Linkage disequilibrium between Wolbachia and mtDNA variation was first reported in Drosophila simulans (Hale and Hoffmann, 1990), following the spread of the Riverside infection (wRi) in California (Turelli and Hoffmann, 1991; Turelli and Hoffmann, 1995). Such co-occurrence of mtDNA and Wolbachia infection has been detected subsequently in other species (see, for example, Armbruster et al., 2003; Schuler et al., 2013).

Diagrams depicting individuals with associated mitochondrial haplotypes and Wolbachia infection status that would be sampled from the release site under: (a) complete CI and perfect maternal transmission of Wolbachia (null model); (b) imperfect maternal transmission of Wolbachia but complete CI. Incomplete CI would also lead to some uninfected offspring in the infected male × uninfected female cross; (c) immigration of uninfected individuals, assuming the immigrating mosquitoes are similar to the uninfected individuals, hence differentiated from the infected individuals; (d) paternal transmission of mitochondria (D1) or Wolbachia (D2). For (D1), if CI is incomplete, there will be presence of uninfected offspring that are heteroplasmic, and for (D2) if CI is complete, the profile will be identical to (a). Heteroplasmic individuals can either produce heteroplasmic gametes or randomly pass on only one haplotype.
As a consequence of CI and maternal co-transmission of Wolbachia and mtDNA, an isolated population invaded by Wolbachia would be likely to contain only the mitochondrial background associated with the infection, regardless of the Wolbachia maternal transmission rates (Turelli et al., 1992). In A. aegypti, Wolbachia induces very strong CI (>99.9%) (McMeniman et al., 2009; Walker et al., 2011; Yeap et al., 2011) and all transfected mosquitoes should share a common mitochondrion as they are descendants of a single female (McMeniman et al., 2009; Walker et al., 2011). This association provides an opportunity to study Wolbachia dynamics following the invasion of the A. aegypti field populations, as long as the local and release mosquitoes have divergent mitochondrial genomes. In particular, deviations from perfect Wolbachia transmission should generate uninfected mosquitoes with a mitochondrial background associated with the infection, whereas immigration of uninfected mosquitoes should lead to the occurrence/maintenance of uninfected mosquitoes with a background different from the infected mosquitoes.
Here, our first aim was to (1) provide a set of diagnostic mitochondrial molecular markers for tracking the Wolbachia dynamics in populations of the dengue fever mosquito A. aegypti, given the recent field releases of Wolbachia-infected mosquitoes in Australia, Vietnam, Indonesia and Brazil to supress dengue transmission. Furthermore, we aimed to (2) develop a model framework for the Wolbachia infection and haplotype counts. We define a null model with the assumptions of perfect maternal co-transmission of mitochondria and Wolbachia, complete CI and isolation of an invaded field population. Deviations from null expectations are then explored and used to generate hypothetical scenarios for transmission parameters based on known studies of A. aegypti invasion with approximated fitness and population parameters. We also apply our framework to elucidate parameters of a documented Wolbachia invasion in Drosophila. Finally, we discuss applications (such as sampling strategies) and challenges in using mitochondrial variation as a monitoring tool for understanding Wolbachia dynamics in host populations.
Materials and methods
Development of diagnostic mitochondrial molecular markers in A. aegypti
Samples
To obtain diagnostic markers that differentiate mitochondrial haplotypes associated with the Wolbachia infection from haplotypes of uninfected individuals, mtDNA variation was analysed in A. aegypti individuals from different sources. (1) From laboratory colonies with three different Wolbachia transfections: wMel and wMelPop (originating from D. melanogaster; McMeniman et al., 2008; Walker et al., 2011) and wAlbB (originating from A. albopictus; Xi et al., 2005). (2) From uninfected mosquitoes originating from field populations in Australia, Indonesia, Vietnam and Brazil. In total, we analysed amplicon sequence data in 82 individuals from the Wolbachia-infected colonies and 227 uninfected individuals from the field, and double-digest restriction associated DNA (ddRAD) sequences in 257 individuals from the field (including the wMel infected). Sample sizes and dates for each collection are summarised in Table 1.
Samples were screened for single-nucleotide polymorphisms (SNPs) in several regions of the A. aegypti mitochondrial genome using amplicon sequencing and ddRAD sequencing (ddRAD-seq). As the ddRAD sequences span a wider region, they could reveal additional variation to separate infected versus uninfected individuals.
Amplicon sequencing
We screened regions of three mitochondrial genes: (1) cyt b gene (307 bp; Mousson et al., 2005, 2) ND5 gene (~450 bp; Mousson et al., 2005) and (3) COI gene (Lunt et al., 1996). (Table 1). Details of primer sequences and reaction conditions are found in Supplementary Tables S1 and S2.
PCR amplicons were sequenced from both directions using Sanger sequencing (Macrogen Inc., Geumcheongu, Seoul, South Korea) and the chromatograms were analysed using Geneious version 6.1.5 (http://www.geneious.com, Kearse et al., 2012). The identity of amplicons was confirmed with the BLASTn sequence homology searches against the National Center for Biotechnology Information (NCBI) nonredundant nucleotide database.
Samples from Australia and overseas (n=223) were also screened at five additional mitochondrial regions: ND4 region (da Costa-da-Silva et al., 2005) and four mitochondrial regions (amplified by primer pairs 1, 4, 6 and 16) that amplified 900–1200 bp fragments across the mitochondrial genome (Behura et al., 2011) (Table 1 and Supplementary Tables S1 and S2).
The ddRAD-seq
Nucleotide variation in the mitochondrial genome was also screened in 257 individuals using the 100 bp paired-end Illumina sequencing (Randwick, NSW, Australia) of ddRAD-seq. Customised ddRAD libraries were constructed following the protocol described by Rašić et al. (2014b). In brief, 100 ng of DNA from each individual was digested with the restriction enzymes NlaIII and MluCI (NEB, Ipswich, MA, USA), and ligated to the Illumina P1 and P2 adapters with customised barcode sequences (Rašić et al., 2014b). After sample pooling and purification, size selection of fragments from 300 to 450 bp was completed with the Pippin Prep (Sage Sciences, Beverly, MA, USA) protocol. The final library was enriched using 12 PCR cycles with standard Illumina primers.
Raw Illumina sequences were filtered and trimmed to the length of 80 bp using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html; key: citeulike: 9103573). The majority of the ddRAD reads in these libraries represented nuclear sequences (Rašić et al., 2014b, and unpublished data), but these were not central to the current work where inferences were based only on mtDNA patterns. Therefore, we retained only reads that were uniquely aligned to the mitochondrial reference genome of A. aegypti (Genbank identifier: 164523399) (Behura et al., 2011) by the short read aligner Bowtie (Langmead et al., 2009). Variant and likelihood-based genotype calling was done on loci with a depth of at least five reads using the default parameters of the Stacks pipeline (Catchen et al., 2013).
Sequence analyses
All coding sequences were first checked for the presence of premature stop codons using Geneious version 6.1.5 (http://www.geneious.com, Kearse et al., 2012). Haplotype networks were generated in HapStar-0.7 written in Python (Teacher and Griffiths, 2011). HapStar generates the network graph as a minimum spanning tree between all haplotypes, that is, computing the lowest total Hamming distance (number of mismatches) between all haplotypes. All pairwise Hamming distances between haplotypes were generated using a custom Python 2.7 script. Missing nucleotides in the ddRAD data set were excluded from the distance calculation. Given that HapStar does not account for missing haplotype nodes, we also generated the network graph using TCS (Clement et al., 2000). We considered sequences to be potentially heteroplasmic if their chromatograms contained double peaks (50–100% height) that were present in both sequencing directions, and/or if they were heterozygous in the ddRAD analysis. The potential heteroplasmic sequences were excluded from the haplotype tree reconstruction.
Wolbachia dynamics modelling
We were interested in identifying haplotype frequency profiles in infected and uninfected populations under a combination of suboptimal Wolbachia transmission effects, such as maternal leakage and presence of immigration/emigration from neighbouring populations. We were particularly interested in two distinct Wolbachia invasion fates, namely (1) a Wolbachia infection persisting in a population and (2) a Wolbachia infection that fails to invade and is lost from a population.
We adapted the overlapping generation model of Turelli (2010) to incorporate maternal transmission leakage, emigration/immigration and paternal transmission of Wolbachia. We did not incorporate rare paternal transmission of mitochondria for which the mode of inheritance would probably be dependent on the assortment of a high density of maternally derived mtDNA and a very low density of paternally derived mtDNA.
Modelling the Wolbachia infection counts
Using the nomenclature of Turelli (2010), we denoted Infected and Uninfected mosquitoes as I and U, with the number of adult (A) infected and uninfected reproductive females at day t as IA,t and UA,t; and the number of embryonic (E) females produced at day t as IE,t and UE,t (see Table 2 for a list of symbols). Similarly, we used an average length of pre-reproductive period, τ, with τ+1 as the period before a given female starts to reproduce. We assumed that both infected and uninfected individuals spent similar amount of time as pre-reproductives, that is, τI=τU. We also defined bI and bU as the number of female embryos produced per day by an infected and uninfected female respectively. The parameters determining Wolbachia dynamics are μ (the probability of an infected female not transmitting the infection to the eggs), H (the hatch rate of uninfected eggs fertilised by sperm from infected males) and ξ (the probability of Wolbachia transmission from infected males). For any given time, t, the number of pre-reproductive infected and uninfected females (IE,t and UE,t) can be expressed as:

The part of the equation multiplied by IA,t denotes the number of offspring from an infected female, whereas the part multiplied by UA,t denotes the number of offspring from an uninfected female (see Table 2 for definitions and symbols and Table 3 for derivation).
To account for fitness differences in egg viability in infected and uninfected individuals, we set the average probabilities of an infected and uninfected egg surviving to the larval stage as δI and δU respectively, and average probabilities of infected and uninfected larvae surviving to sexually mature adults, irrespective of larvae competition, as sI and sU respectively. Then, the number of reproductive infected and uninfected females at time t+1, given τ days to develop into reproductive adults without larval competition and immigration, can be expressed as:

The number of resultant reproductive adults is also dependent on the survival of reproductive adults from the previous day (ν).
To incorporate larval competition with density-dependent larval mortality, we partitioned the life stages to egg, larval and post-larval pre-reproductives. Egg and post-larval pre-reproductives (average duration ω and κ days respectively) were considered as not being in competition for resources with larval stages. Eggs laid on day t–τ would have hatched on day t–τ+ω, and larvae on that day would have encountered larvae that were laid as eggs as far back as τ–κ+1 days previously, that is, on day t–2τ+ω+κ+1. Thus, the average number of larvae encountered per day by larvae that were laid as eggs on day t–τ (here denoted as L) was the sum of pre-reproductives from t–2τ+ω+κ+1 to t–κ divided by days spent as larvae (that is, τ–ω–κ):

Following previous studies (Hancock et al., 2011), we used an exponential decay function dependent on the average number of larvae encountered per day by eggs laid on t–τ day:

where a and c are larval density-dependent constants. The superscripts ‘**’ are referenced below.
Migration modelling
For emigration/immigration, we considered a simple scenario where only two populations interact, and assumed the same dispersal rate for both infected and uninfected individuals. We also assumed that only reproductive adult females are capable of emigrating/immigrating. We let IA,t(1), UA,t(1), IA,t(2) and UA,t(2) denote the number of infected and uninfected adults at time t in populations 1 and 2, respectively, and m represents the emigration/immigration rate per generation between the two populations. Then, the number of infected and uninfected adults at time t+1 in populations 1 and 2 can be expressed as:

The value of m was divided by τ (assumed to be the average generation time) to indicate a daily migration rate.
Modelling the mitochondrial haplotype counts
We designated two mitochondrial haplotypes: α, the haplotype prevalent in uninfected individuals, and β, the haplotype prevalent in infected individuals. We assume that the two haplotypes are strictly neutral variants. We let αI,t and βI,t be the proportion of Infected individuals at time t with haplotype α and β respectively. Similarly, we define αU,t and βU,t as equivalent proportions for the Uninfected individuals. We let Iα,t+1 and Iβ,t+1 be the number of Infected adult females with haplotype α or β at t+1, and IEα,t+1 and IEβ,t+1 be the equivalent embryonic individuals. Replacing the letter I with U gives the designations for their Uninfected equivalents.
If we assume no paternal transmission of mitochondria (that is, offspring will always have only the mother’s haplotype), the numbers of individuals with a given mitochondrial haplotype can be designated the same way as the number of individuals with or without Wolbachia infection (defined by Equations (1) and (4)). For example, we can replace IA,t+1 with Iα,t+1 or Iβ,t+1 to designate the number of Infected adult females with the mitochondrial haplotype α or β. Analogously, we can replace UE,t+1 with UEα,t+1 or UEβ,t+1 to indicate the number of Uninfected Embryonic individuals with the mitochondrial haplotype α or β (see Supplementary Table S3 for all probability combinations of offspring haplotypes).
Generating scenarios for varying transmission parameters
We first considered the models mathematically and qualitatively to identify effects of maternal transmission leakage (μ>0), incomplete CI (H>0) and immigration/emigration. We then considered known cases where Wolbachia successfully invaded Aedes populations (involving wMel releases) and where the infection did not establish (involving wMelPop releases). In these situations, most population parameters have been estimated, and we used this information to indicate expected haplotype frequencies when there is maternal transmission leakage, immigration/migration and/or incomplete CI. We did not include paternal transmission in our analyses because the phenomenon has not been clearly demonstrated in mosquitoes, but we discuss how it might influence results of the models.
We tested the power of mitochondrial variants to detect imperfect transmission events (μ>0) under different levels of CI by simulating a population with different infection and haplotype frequencies. We tested two methods for detecting maternal transmission: (1) the traditional approach (Turelli and Hoffmann, 1995) of field sampling of live females and their offspring before screening for Wolbachia infection (method 1) and (2) the alternative approach of sampling field uninfected mosquitoes for mitochondrial haplotype frequency (method 2). The models were run in R3.1 (R Core Team, 2014).
Results
Diagnostic markers for Wolbachia infection in A. aegypti
We identified one SNP in the mitochondrial COI region that differentiates the wMel- and wMelPop-infected haplotype from haplotypes found in uninfected field populations of A. aegypti (Table 4). The diagnostic SNP represents a G to A transition at the 531 bp position in the amplicon of the COI region. The Wolbachia-associated COI haplotype was absent from all investigated non-Australian mosquito populations (Supplementary Figure S1, see haplotype 8 in Supplementary Table S4), but was detected at low frequencies in uninfected mosquitoes from two Australian populations (8.3–13.2%). We did not find a diagnostic COI haplotype associated with the wAlbB infection
The region amplified by primer pair 4 contained a G to A transition at the 295 bp position that allowed differentiation of wMel- and wMelPop-infected mosquitoes from most non-Australian samples (Table 4; haplotype 1 versus others, Supplementary Table S5). Complete differentiation is achieved when one of position 235, 601 or 604 is considered in the case where position 295 shares the same base with the wMel- or wMelPop-infected mosquitoes (compare haplotypes 1 and 8 in Supplementary Table S5). The infection-associated SNP combination was detected in natural mosquito populations in Australia with frequencies ranging from 20 to 50% (Supplementary Figure S2).
In the ND4 region we found 15 haplotypes, one of which (G→A at 210 bp) (Table 4) was fully diagnostic for the wAlbB infection in all investigated populations (Supplementary Figure S3, see haplotype 5 of Supplementary Table S6). Amplicon sequencing of the ND5, primer pair 1 and primer pair 6 regions each revealed one haplotype associated with all three Wolbachia infections in A. aegypti, but this haplotype was fully diagnostic only in the Brazilian samples (Supplementary Figures S4–S6, see Supplementary Tables S7–S9 for haplotype combinations).
In summary, we found that wMel and wMelPop could be completely differentiated from the non-Australian populations with one COI SNP and a combination of at least two SNPs in primer pair 4 amplicons. The wAlbB infection can be completely differentiated from all investigated populations with only one ND4 SNP (Table 4).
With ddRAD-seq we found 92 SNPs in 28 RAD loci that spanned 11 mitochondrial genes and these were present in at least 80% of individuals (Supplementary Table S10). All RAD loci were concatenated to form superhaplotypes with a final length of 2246 bp. We found 59 unique superhaplotypes in 257 individuals. Haplotype networks for the ddRAD and amplicon data sets were concordant. Out of 28 ddRAD loci, 15 covered parts of mitochondrial genes that were not analysed via amplicon sequencing (ND2, COIII, ATP6, ND1 and ND6; Supplementary Table S10). However, we did not detect additional SNPs that would be fully diagnostic for wMel infection in Australia. Even though the infection-associated superhaplotype 1 did not appear in any of the overseas samples, it was found in 12.1% of the Australian samples (Figure 2).

Network of RAD-seq superhaplotypes. Each nonsolid black coloured circle represents observed haplotype with greater circle size indicating greater number of individuals with that haplotype. Solid black circles represent a single base change and the interrupted line with numbers represents a larger number of base differences. Haplotype with number 1 is the haplotype associated with the wMel infection.
Nuclear copies of the mitochondrial DNA sequences (NuMts) are present in the A. aegypti genome (Behura et al., 2011) and may complicate genotype assignment. However, diagnostic SNPs that were found via amplicon sequencing did not exhibit any form of heteroplasmy or ambiguous base calls. Furthermore, RAD-seq-generated SNPs that spanned 11 mitochondrial genes supported the haplotype networks generated with the single locus data. We also did not detect premature stop codons in any of the haplotypes or superhaplotypes. Therefore, it is unlikely that our haplotype analyses were confounded by NuMts.
Wolbachia dynamics modelling
We defined a set of initial conditions as follows:
-
1
Haplotype β was at a higher frequency in infected than uninfected mosquitoes (100 vs 0–13% based on molecular data). Hence, we start with βI,0>βU,0 (that is, αI,0<αU,0).
-
2
The site of Wolbachia release and the neighbouring population (population 2) had the same initial uninfected haplotype frequency, that is, αU,0(1)=αU,t(2).
-
3
The viability of eggs, δI and δU is seasonally dependent, particularly for the wMelPop infection. In the wet season, we assumed that all eggs, regardless of infection, have the same probability of being viable. Based on the average number of rainy days in Cairns, Queensland and viability data from Juliano et al. (2002), we estimated that viability would decrease to ∼0.75 in uninfected eggs. For infected eggs, we considered the same viability (no fitness difference) and viability decreasing to 0.1 (as in wMelPop).
-
4
We assumed no difference in daily fecundity between infected and uninfected mosquitoes, bI=bU=0.3 and no seasonal dependence. The value was chosen to correspond to survival of juveniles to reproductive age with a small cost for all infection types, sI=0.85 versus sU=0.9, such that bIsI and bUsU correspond to the values described by Turelli (2010).
-
5
The daily adult survival rate (vU) was set to 0.85, and assumed to be constant throughout both wet and dry seasons (Hugo et al., 2014). We considered two scenarios: (1) no effect of Wolbachia and (2) a much lower survival of 0.65 (corresponding to wMelPop) (Yeap et al., 2014).
-
6
The juvenile development time was dependent on season, but not infection. We set the overall development time, τ=19 and 36 days for wet and dry season, respectively. Egg development and post-larval pre-reproductive period were ω=3 and 14 days and κ=6 and 8 days, respectively. The increase in length of each period simulates time spent in quiescence (for eggs) during the dry season and/or reduction in average temperature.
-
7
Larval competition parameters were set to a=0.1 and c=0.2 to prevent the population from expanding beyond 10 000 individuals (Supplementary Figure S7).
-
8
We implemented augmentative releases of 10 000 infected individuals every 7 days from the start for 12 releases.
-
9
The two seasons, wet and dry season, were set to 183 days each.
We varied the maternal transmission leakage rate, μ, and the CI rate, 1–H, arbitrarily. For immigration/migration rates, we chose between an approximated value, m=0.03%, of the population per generation (Endersby et al., 2009), and an arbitrarily high rate, m=6%.
-
a)
Maternal transmission leakage (μ>0) by itself leads to decoupling of the Wolbachia infection status and associated mitochondrial variation, such that Wolbachia-associated mtDNA genetic background shows up in the uninfected fraction of the population (Figure 3a and Supplementary Text 1). If persisting indefinitely, the haplotype frequencies in uninfected mosquitoes will eventually become the same as those in infected mosquitoes in the population.
Figure 3 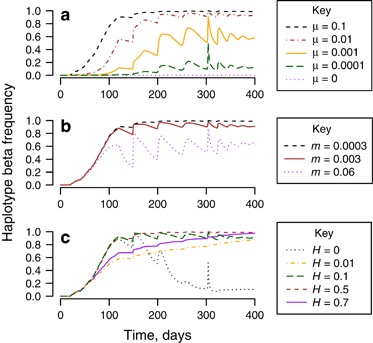
Frequency of haplotype β (infection-associated haplotype) in uninfecteds vs time in a modelled release. Unless stated, μ=0.1, m=0.0003, H=0.1. (a) Comparison of different magnitude of maternal transmission leakage, μ=0, 0.0001, 0.001, 0.01 and 0.1. (b) Comparison of different magnitude of immigration rates, m=0. (c) Comparison of variable levels of incomplete CI in the presence of transmission leakage, μ>0, and immigration, m>0 (H=0, 0.01, 0.1, 0.5, 0.7).
-
b)
Paternal transmission of Wolbachia (ξ>0) leads to the same result but working in the opposite direction: it causes haplotype frequencies in infected individuals to become the same as those in the uninfected mosquitoes of the population. The effects of both transmission parameters are limited by fitness of the infected/uninfected individuals (fecundity, bI and bU; egg survival, δI and δU; non-egg juvenile survival, sI and sU) and strength of CI (1–H) (Supplementary Text 1).
-
c)
Immigration/emigration allows for two (or more) interacting populations to equilibrate their haplotype frequencies. As a result, these processes potentially reduce or reverse the effect of maternal transmission leakage on haplotype frequency in uninfected individuals (Figure 3b and Supplementary Text 1).
-
d)
Cytoplasmic incompatibility does not affect haplotype frequency directly when there is no transmission leakage (μ=0 and ξ=0). However, in the presence of leakage, the magnitude of CI could limit the effects of leakage on haplotype frequencies when immigration is present (Figure 3c and Supplementary Text 1). For example, high levels of CI (H close to zero) reduce the effect of maternal transmission leakage at high infection frequencies (pt close to 1). As most mating pairs will be with infected males, any transmission leakage will be suppressed by CI, ultimately leaving only uninfected migrants if immigration occurs, or no uninfected individuals if immigration is absent. High levels of CI also completely suppress the effect of paternal transmission of Wolbachia on haplotype frequencies in uninfected and infected individuals. However, when CI levels become too low (for example, H=0.7), Wolbachia invasion will drastically slow down, reducing the effect of transmission leakage (Figure 3c, see H=0.7).

Inferences from successful Wolbachia invasions
Successful Wolbachia invasions result in a stable frequency of Wolbachia infection in a population. Invasions of wMel in A. aegypti populations from Australia have led to stable infections at a frequency close to 1 (Hoffmann et al., 2014). Here, we based fitness parameters on estimates obtained for the wMel infection in A. aegypti.
First, if an infection goes to fixation (pt=1), it can easily be inferred that immigration is absent, and that maternal transmission of infection is perfect (μ=0) and/or cytoplasmic incompatibility is complete (H=0) (Supplementary Text 2). Hence, any maternal transmission leakage can only be detected from haplotype frequencies during the invasion phase, that is, when uninfected individuals are still present.
Persistence of uninfected individuals in a stable infection implies that there is (1) immigration of uninfected mosquitoes into the invasion site (m>0); and/or (2) imperfect maternal transmission of Wolbachia (μ>0) and incomplete CI (H>0) as explained previously (Supplementary Text 2). Uninfected individuals have persisted in A. aegypti populations that were invaded by Wolbachia (Hoffmann et al., 2014).
After invasion, if (1) the frequency of the infection-associated haplotype β significantly increases in uninfected individuals and is moving towards the value present in infected individuals, we can infer that maternal transmission leakage and incomplete CI are likely to be present (Figure 4a). Under this scenario, however, the presence of immigration cannot be inferred. On the other hand, if (2) haplotype β in uninfected individuals is at a higher frequency during the invasion phase than after invasion, it is likely that immigration coupled with almost complete CI (H≈0) have countered the effects of transmission leakage (Figure 4b). (3) If no significant difference in haplotype frequencies is observed throughout the invasion and post-invasion periods, maternal transmission is probably perfect (μ=0) and immigration is present (m>0), allowing uninfected individuals to persist. Under such a scenario we cannot make any inferences about CI.

Plot of infection-associated haplotype frequency in uninfected: β, βU,t, infection frequency, pt and population size (shaded area). (a) Successful invasion with μ=0.05, m=0.0003, H=0.1 (incomplete CI). (b) Successful invasion with μ=0.05, m=0.0003 with incomplete CI, H=0. (c) Failed invasion with μ=0.05, m=0.0003. (d) Release that fails to establish with μ=0.05, m=0.06.
Inferences from failed Wolbachia invasions
Here, we approximated fitness parameters based on known releases of wMelPop infections that failed to invade an A. aegypti population (Yeap et al., 2014). When the infection fails to persist at high frequency, it is difficult to infer levels of CI and low/moderate levels of migration in the presence of maternal transmission leakage based on uninfected haplotype frequency (compare Figures 4c and d). Maternal transmission leakage will transform the haplotype frequencies of uninfected individuals as long as the infection is still present in the population. This change in haplotype frequency could still be detected even after the infection has been lost (Figure 4c). Thus, in this scenario, we would suggest first screening after releases are terminated as it would be redundant to screen samples during invasion to detect maternal transmission leakage. If the absence of maternal transmission leakage (μ=0) is suspected, individuals from invasion period could then be screened to test for a lack of change in uninfected haplotype frequency.
Workflow to detect maternal transmission leakage of Wolbachia
We provide a workflow that guides inferences about the processes underlying Wolbachia dynamics in host populations based on the analyses of infection and haplotype frequencies (Figure 5). The analyses are split into two sampling points: during the release phase and after releases (successful invasion or loss of infection). We also assess the power to detect maternal transmission leakage at these two sampling time points using the uninfected haplotype frequencies (method 2). We compare our method with the traditional approach (Turelli and Hoffmann, 1995) that assumes field sampling of infected gravid females to directly assess infection transmission to offspring (method 1).

List of possible observations (O), their respective inferences on model parameters (I) and sampling strategy to make further inferences on model parameters (S) based on three possible Wolbachia-invasion outcomes: (1) fixed at 100%; (2) stable coexistence of Wolbachia-infected and uninfected; and (3) infection extinction. We were interested in identifying whether certain parameters, particularly the maternal transmission leakage rate (μ), incompatible hatch rate (H) and immigration rate (m), differed from 0 based on observations of infection-associated haplotype frequencies at some time, t, in uninfected, βU,t and infected, βI,t individuals. βU,0 and βI,0 specify frequencies at time t=0, before introduction of Wolbachia-infected individuals. For scenarios where parameters are listed as ‘≥0’ or not mentioned, inference was not possible. We also included inferences on paternal transmission of Wolbachia rate (ξ), but this only applies when there is negligible paternal transmission of mitochondria. Stable infection frequency is denoted by pt=pt+k.
First, analyses of mitochondrial haplotype distributions enable detection of the presence or absence of maternal transmission leakage for both sampling regimes (during and after the Wolbachia invasion). Sampling during the invasion phase is most informative when the infection frequency lies between 0.5 and 0.8. If the infection frequency (pt) is too high, CI can hinder transmission leakage, and if pt is too low, there are too few transmission leakage events to be detected.
For post-release sampling, samples can be collected when the infection frequency has stabilised or when it drops to zero. For the traditional method 1, we simulated the situation where 100 and 200 offspring are obtained from the field-caught infected females. For method 2, we simulated the situation where mosquitoes are sampled over 5 weeks (assuming a trapping rate of 0.5% of the population per week) and leakage was detected through the presence of an uninfected individual with an infection-associated haplotype (β). The screening protocols were simulated 100 000 times, assuming a low rate of immigration/emigration at a rate of m=0.3% of the population per generation corresponding to a relatively isolated population (cf, Hoffmann et al., 2014).
With method 1, the ability to detect transmission leakage increases with leakage levels, but not with changing CI levels. This method is useful during and after an invasion, but cannot be used once an infection is lost. With method 2, relevant data on leakage can be obtained under all scenarios and the detection rate depends not only on the maternal transmission rate, but also on CI (Table 5). The two methods have comparable power in a situation where CI is not strong.
Case study
To highlight the utility of our approach, we considered the case of the Wolbachia infection wRi and its recent invasion and spread through the populations of D. simulans in eastern Australia (Kriesner et al., 2013). The infection was initially detected in California (Hoffmann et al., 1986) where it was tightly associated with a particular mtDNA variant (Hale and Hoffmann, 1990) that then spread in populations alongside the Wolbachia infection (Turelli et al., 1992). Based on the data presented by Kriesner et al. (2013), before the arrival of wRi infection to Australia, there was one haplotype (haplotype A) present in eastern Australian populations. This haplotype was present in both uninfected individuals and individuals carrying the wAu infection that is not invasive through cytoplasmic incompatibility. In 2004, in New South Wales, Australia, before wRi invading, New South Wales populations had a wAu frequency of 48.7%. By 2008, 7 sites had a new wRi infection with a frequency of 7.4±4.7%; wAu infection was present with a frequency of 56.1% and uninfected individuals comprised 36.4% of the total sample. Moreover, mitochondrial haplotype R was now detected for the first time and had a frequency of 14.8±14.8%, higher than the frequency of wRi because it was also detected in some uninfected individuals. By 2011, the frequency of wRi had increased to 91.4±3.5% and wAu was no longer detected. Moreover, both infected and uninfected individuals had haplotype R, consistent with patterns in other populations outside Australia (Turelli et al., 1992).
Applying the approach developed in the present study (Figure 5), the documented wRi invasion in Australia can be used to identify signatures of imperfect transmission and other processes. This is possible because the D. simulans population was sampled before invasion of wRi and the infection source population was known to be carrying a particular mtDNA variant. Based on Figure 5 (assuming we have no knowledge of laboratory estimates), it can be surmised that:
-
1)
The increase in frequency of haplotype R (the invading haplotype) in uninfected individuals immediately implicates maternal transmission leakage. This haplotype was not previously detected in any Australian population. If migration was involved, we would have expected uninfected individuals to carry haplotype A.
-
2)
Given that transmission leakage is present, we can infer incomplete CI because haplotype R has increased in frequency in uninfected individuals in New South Wales over time even when infection has reached high frequencies, instead of increasing initially and then dropping as the infection approaches high frequencies as expected under complete CI.
-
3)
There is no evidence for a high incidence of paternal transmission because no wRi-infected flies with haplotype A were detected. However, it would be difficult to detect a low rate of paternal transmission unless very large sample sizes were available from the invasion period.
Discussion
We discovered mtDNA variants associated with the wMel and wMelPop Wolbachia infections that allow complete differentiation of A. aegypti from the release stock and the wild samples collected in Brazil, Indonesia and Vietnam. These mtDNA markers, however, are not fully diagnostic in Australian populations. High similarity between the infected colonies and the Australian populations is not surprising given that all infected samples originate from a single isofemale line which has the Australian genetic background (McMeniman et al., 2008; McMeniman et al., 2009; Walker et al., 2011). We also detected a single diagnostic SNP that distinguishes the wAlbB infection from other Wolbachia strains and field samples of A. aegypti. The wAlbB was generated by the Wolbachia transfer from A. albopictus into the Waco A. aegypti strain that originated from Texas (Xi et al., 2005). The diagnostic markers we discovered can now be used to generate a high-throughput SNP genotyping assay that could be run in parallel with the detection of the Wolbachia infection (Lee et al., 2012). Such an assay would assist in ensuring successful Wolbachia invasions into the A. aegypti populations around the world in an effort to supress dengue transmission.
Our modelling results highlight the extent to which the presence of maternal leakage leads to increased numbers of uninfected mosquitoes with the mtDNA haplotype associated with the infection (that is, haplotype β). The usual method for studying maternal transmission of Wolbachia in the field involves directly testing the Wolbachia infection status in the offspring of field-caught females that are infected (Hoffmann et al., 1996, 1998). Although this approach can provide an estimate of transmission leakage, the major limitation is the substantial number of individuals that need to be tested. This is because useful information is obtained only from offspring of captured females that are infected. The mtDNA-based approach has several practical advantages in that live, field females are not required, and that the data can capture the average effects of leakage across time rather than only at the point when screening is undertaken. Although it might be less powerful when detecting low rates of maternal transmission leakage (μ<0.05) during the invasion phase, the mtDNA-based approach performs well after the release phase, particularly when there is no invasion or when some offspring hatch in incompatible crosses.
Based on the modelled power of detection, we propose that monitoring of mtDNA haplotype frequency in uninfected mosquitoes is a useful approach in understanding whether a failed or incomplete invasion is a result of maternal transmission leakage of Wolbachia. Nevertheless, estimating the maternal transmission leakage rate, μ, with this approach is not straightforward because of the number of variables that might affect such estimates. This is likely to require sampling of haplotypes from at least two time points.
Mitochondrial variation can also help in understanding the impact of other population processes such as migration patterns. Detection of immigrants is straightforward when high infection frequencies have been achieved. If we assume strong CI (H almost 0) or negligible paternal transmission of mitochondria, changes in the frequency of the haplotype associated with the infection in uninfected individuals after invasion provides a signature of immigration. Migration rates would still need to be directly estimated with approaches such as mark-release-recapture (see, for example, Marini et al., 2010) and population genetic studies (cf, Endersby et al., 2009; Endersby et al., 2011; Rašić et al., 2014a).
Monitoring of mitochondrial haplotype dynamics alone is not sufficient for inferring CI, but when coupled with maternal transmission of Wolbachia and other parameters, especially in the event of a successful invasion, it allows for the strength of CI to be inferred. We found that detecting maternal leakage of Wolbachia transmission via mtDNA haplotypes was affected by the strength of CI, and strong CI would have prevented any genetic material from the infected individuals leaking into the uninfected population via paternal transmission (cf, Hoffmann and Turelli, 2013). Strong CI allows re-establishment of the original haplotype frequency among uninfected individuals through immigration, as CI ensures the elimination of pre-existing uninfected individuals. However, the exact CI rate can only be inferred through observing hatch rate in gravid uninfected females captured during the release of infected mosquitoes.
In conclusion, we found sufficient differentiation between uninfected and infected haplotypes to allow qualitative detection of population parameters from mtDNA data during and after releases of Wolbachia-infected mosquitoes. Based on the scenarios presented, monitoring of haplotype frequencies in uninfected individuals should provide information on maternal transmission and the presence of immigrants in invaded populations. Our models could be applied not only to mosquito releases, but to any Wolbachia release program or temporal study of Wolbachia dynamics in natural populations, as long as there is mtDNA variation that differentiates infected and uninfected populations.
Data archiving
Molecular data used in this paper and R codes for the models can be accessed via DRYAD with accession numbers DOI: doi:10.5061/dryad.3f355. Raw Illumina sequences from the ddRAD libraries are deposited in NCBI SRA under the accession numbers PRJNA241150 and PRJNA273913.
Accession codes
References
Armbruster P, Damsky WE, Giordano R, Birungi J, Munstermann LE, Conn JE . (2003). Infection of new- and old-world Aedes albopictus (diptera: culicidae) by the intracellular parasite Wolbachia: implications for host mitochondrial DNA evolution. J Med Entomol 40: 356–360.
Behura SK, Lobo NF, Haas B, deBruyn B, Lovin DD, Shumway MF et al. (2011). Complete sequences of mitochondria genomes of Aedes aegypti and Culex quinquefasciatus and comparative analysis of mitochondrial DNA fragments inserted in the nuclear genomes. Insect Biochem Mol Biol 41: 770–777.
Bian GW, Xu Y, Lu P, Xie Y, Xi ZY . (2010). The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog 6: e1000833.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA . (2013). Stacks: an analysis tool set for population genomics. Mol Ecol 22: 3124–3140.
Clement M, Posada D, Crandall KA . (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1659.
da Costa-da-Silva AL, Capurro ML, Bracco JE . (2005). Genetic lineages in the yellow fever mosquito Aedes (Stegomyia) aegypti (Diptera: Culicidae) from Peru. Mem Inst Oswaldo Cruz 100: 539–544.
Endersby NM, Hoffmann AA, White VL, Lowenstein S, Ritchie S, Johnson PH et al. (2009). Genetic structure of Aedes aegypti in Australia and Vietnam revealed by microsatellite and exon primed intron crossing markers suggests feasibility of local control options. J Med Entomol 46: 1074–1083.
Endersby NM, Hoffmann AA, White VL, Ritchie SA, Johnson PH, Weeks AR . (2011). Changes in the genetic structure of Aedes aegypti (Diptera: Culicidae) populations in Queensland, Australia, across two seasons: implications for potential mosquito releases. J Med Entomol 48: 999–1007.
Focks DA, Daniels E, Haile DG, Keesling JE . (1995). A simulation model of the epidemiology of urban dengue fever: literature analysis, model development, preliminary validation, and samples of simulation results. Am J Trop Med Hyg 53: 489–506.
Hale LR, Hoffmann AA . (1990). Mitochondrial DNA polymorphism and cytoplasmic incompatibility in natural populations of Drosophila simulans. Evolution 44: 1383–1386.
Hancock PA, Sinkins SP, Godfray HCJ . (2011). Population dynamic models of the spread of Wolbachia. Am Nat 177: 323–333.
Hoffmann AA, Clancy D, Duncan J . (1996). Naturally-occurring Wolbachia infection in Drosophila simulans that does not cause cytoplasmic incompatibility. Heredity 76: 1–8.
Hoffmann AA, Hercus M, Dagher H . (1998). Population dynamics of the Wolbachia infection causing cytoplasmic incompatibility in Drosophila melanogaster. Genetics 148: 221–231.
Hoffmann AA, Iturbe-Ormaetxe I, Callahan AG, Phillips BL, Billington K, Axford JK et al. (2014). Stability of the wMel Wolbachia Infection following invasion into Aedes aegypti populations. PLoS Negl Trop Dis 8: e3115.
Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F et al. (2011). Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 476: 454–457.
Hoffmann AA, Ross PA, Rašić G . (2015). Wolbachia strains for disease control: ecological and evolutionary considerations. Evol Appl 8: 751–768.
Hoffmann AA, Turelli M . (1997). Cytoplasmic incompatibility in insects. In: ONeill S, Hoffmann A, Werren J (eds). Influential Passengers: Microbes and Invertebrate Reproduction. Oxford University Press. pp 42–80.
Hoffmann AA, Turelli M . (2013). Facilitating Wolbachia introductions into mosquito populations through insecticide-resistance selection. Proc R Soc B Biol Sci 280: 20130371.
Hoffmann AA, Turelli M, Simmons GM . (1986). Unidirectional incompatibility between populations of Drosophila simulans. Evolution 40: 692–701.
Hugo LE, Jeffery JAL, Trewin BJ, Wockner LF, Nguyen TY, Le NH et al. (2014). Adult survivorship of the dengue mosquito Aedes aegypti varies seasonally in central Vietnam. PLoS Negl Trop Dis 8: e2669.
Johnstone RA, Hurst GDD . (1996). Maternally inherited male-killing microorganisms may confound interpretation of mitochondrial DNA variability. Biol J Linnean Soc 58: 453–470.
Juliano SA, O'Meara GF, Morrill JR, Cutwa MM . (2002). Desiccation and thermal tolerance of eggs and the coexistence of competing mosquitoes. Oecologia 130: 458–469.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649.
Kittayapong P, Baisley KJ, Baimai V, O'Neill SL . (2000). Distribution and diversity of Wolbachia infections in southeast Asian mosquitoes (Diptera: Culicidea). J Med Entomol 37: 340–345.
Kriesner P, Hoffmann AA, Lee SF, Turelli M, Weeks AR . (2013). Rapid sequential spread of two Wolbachia variants in Drosophila simulans. PLoS Pathog 9: e1003607.
Langmead B, Trapnell C, Pop M, Salzberg SL . (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
Lee SF, White VL, Weeks AR, Hoffmann AA, Endersby NM . (2012). High-throughput PCR assays to monitor Wolbachia infection in the dengue mosquito (Aedes aegypti and Drosophila simulans. Appl Environ Microbiol 78: 4740–4743.
Lunt DH, Zhang DX, Szymura JM, Hewltt OM . (1996). The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol 5: 153–165.
Marini F, Caputo B, Pombi M, Tarsitani G, della Torre A . (2010). Study of Aedes albopictus dispersal in Rome, Italy, using sticky traps in mark-release-recapture experiments. Med Vet Entomol 24: 361–368.
McMeniman CJ, Lane AM, Fong AWC, Voronin DA, Iturbe-Ormaetxe I, Yamada R et al. (2008). Host adaptation of a Wolbachia strain after long-term serial passage in mosquito cell lines. Appl Environ Microbiol 74: 6963–6969.
McMeniman CJ, Lane RV, Cass BN, Fong AWC, Sidhu M, Wang YF et al. (2009). Stable introduction of a life-shortening Wolbachia infection into the mosquito Aedes aegypti. Science 323: 141–144.
McMeniman CJ, O'Neill SL . (2010a). A virulent Wolbachia infection decreases the viability of the dengue vector Aedes aegypti during periods of embryonic quiescence. PLoS Negl Trop Dis 4: e748.
McMeniman CJ, O'Neill SL . (2010b). A virulent Wolbachia infection decreases the viability of the dengue vector Aedes aegypti during periods of embryonic quiescence. PLoS Negl Trop Dis 4: e748.
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu GJ, Pyke AT, Hedges LM et al. (2009). A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell 139: 1268–1278.
Mousson L, Dauga C, Garrigues T, Schaffner F, Vazeille M, Failloux AB . (2005). Phylogeography of Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) (Diptera: Culicidae) based on mitochondrial DNA variations. Genet Res 86: 1–11.
R Core Team. (2014) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available from http://www.R-project.org/.
Rašić G, Endersby NM, Williams C, Hoffmann AA . (2014a). Using Wolbachia-based release for suppression of Aedes mosquitoes: insights from genetic data and population simulations. Ecol Appl 24: 1226–1234.
Rašić G, Filipović I, Weeks AR, Hoffmann AA . (2014b). Genome-wide SNPs lead to strong signals of geographic structure and relatedness patterns in the major arbovirus vector, Aedes aegypti. BMC Genomics 15: 275.
Ross PA, Endersby NM, Yeap HL, Hoffmann AA . (2014). Larval competition extends developmental time and decreases adult size of wMelPop Wolbachia-infected Aedes aegypti. Am J Trop Med Hyg 91: 198–205.
Schuler H, Bertheau C, Egan SP, Feder JL, Riegler M, Schlick-Steiner BC et al. (2013). Evidence for a recent horizontal transmission and spatial spread of Wolbachia from endemic Rhagoletis cerasi (Diptera: Tephritidae) to invasive Rhagoletis cingulata in Europe. Mol Ecol 22: 4101–4111.
Teacher AGF, Griffiths DJ . (2011). HapStar: automated haplotype network layout and visualization. Mol Ecol Resour 11: 151–153.
Turelli M . (2010). Cytoplasmic incompatibility in populations with overlapping generations. Evolution 64: 232–241.
Turelli M, Hoffmann AA . (1991). Rapid spread of an inherited incompatibility factor in California Drosophila. Nature 353: 440–442.
Turelli M, Hoffmann AA . (1995). Cytoplasmic incompatibility in Drosophila-simulans - dynamics and parameter estimates from natural-populations. Genetics 140: 1319–1338.
Turelli M, Hoffmann AA, McKechnie SW . (1992). Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics 132: 713–723.
Turley AP, Zalucki MP, O'Neill SL, McGraw EA . (2013). Transinfected Wolbachia have minimal effects on male reproductive success in Aedes aegypti. Parasit Vectors 6: 36.
Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ et al. (2011). The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476: 450–453.
Xi ZY, Khoo CCH, Dobson SL . (2005). Wolbachia establishment and invasion in an Aedes aegypti laboratory population. Science 310: 326–328.
Yeap HL, Axford JK, Popovici J, Endersby NM, Iturbe-Ormaetxe I, Ritchie SA et al. (2014). Assessing quality of life-shortening Wolbachia-infected Aedes aegypti mosquitoes in the field based on capture rates and morphometric assessments. Parasit Vectors 7: 58.
Yeap HL, Endersby NM, Johnson PH, Ritchie SA, Hoffmann AA . (2013). Body size and wing shape measurements as quality indicators of Aedes aegypti mosquitoes destined for field release. Am J Trop Med Hyg 89: 78–92.
Yeap HL, Mee P, Walker T, Weeks AR, O'Neill SL, Johnson P et al. (2011). Dynamics of the ‘popcorn’ Wolbachia infection in outbred Aedes aegypti informs prospects for mosquito vector control. Genetics 187: 583–595.
Yen JH, Barr AR . (1971). New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature 232: 657–658.
Zug R, Hammerstein P . (2012). Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7: e38544.
Acknowledgements
This work is funded by the program grants from the National Health and Medical Research Council, the Foundation for the National Institutes of Health through the Grand Challenges in Global Health Initiative of the Bill and Melinda Gates Foundation and the Tahija Foundation. The work is also supported by a fellowship from the Australian Research Council Australia to AAH and an Alfred-Nicholas Fellowship to HLY. Nectar Research Cloud and VIC Node (University of Melbourne) provided the bioinformatics resources. We are grateful to a number of researchers for providing field collections of A. aegypti, including Rafael Maciel-de-Freitas and Renato Máspero (Fiocruz, Brazil) and Brian Montgomery (Monash University). Additional thanks to Rosanna Powell, Anjali Goundar, Jason K. Axford, Katrina Billington and Iñaki Iturbe-Ormaetxe for laboratory samples and/or sample processing, Igor Filipovic for the bioinformatics support and Lucien Sankey for help with the figure design.
Author contributions
Planning of the experiment: HLY, NME, GR, SFL and AAH. Sample collection: EA and HLN. Performing the experiment: HLY. Running the analyses: HLY, GR and AAH. Writing the paper: HLY, GR, SFL, NME and AAH. All authors gave their final approval for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Yeap, H., Rašić, G., Endersby-Harshman, N. et al. Mitochondrial DNA variants help monitor the dynamics of Wolbachia invasion into host populations. Heredity 116, 265–276 (2016). https://doi.org/10.1038/hdy.2015.97
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.97
This article is cited by
-
Genomic databanks provide robust assessment of invasive mosquito movement pathways and cryptic establishment
Biological Invasions (2023)
-
Genetic stability of Aedes aegypti populations following invasion by wMel Wolbachia
BMC Genomics (2021)
-
Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok
Scientific Reports (2018)
-
Predicting Wolbachia invasion dynamics in Aedes aegypti populations using models of density-dependent demographic traits
BMC Biology (2016)
