
Abstract
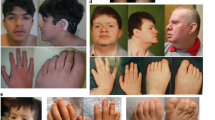
Zimmermann-Laband syndrome (ZLS) is a developmental disorder characterized by facial dysmorphism with gingival enlargement, intellectual disability, hypoplasia or aplasia of nails and terminal phalanges, and hypertrichosis1,2,3,4. We report that heterozygous missense mutations in KCNH1 account for a considerable proportion of ZLS. KCNH1 encodes the voltage-gated K+ channel Eag1 (Kv10.1). Patch-clamp recordings showed strong negative shifts in voltage-dependent activation for all but one KCNH1 channel mutant (Gly469Arg). Coexpression of Gly469Arg with wild-type KCNH1 resulted in heterotetrameric channels with reduced conductance at positive potentials but pronounced conductance at negative potentials. These data support a gain-of-function effect for all ZLS-associated KCNH1 mutants. We also identified a recurrent de novo missense change in ATP6V1B2, encoding the B2 subunit of the multimeric vacuolar H+ ATPase, in two individuals with ZLS. Structural analysis predicts a perturbing effect of the mutation on complex assembly. Our findings demonstrate that KCNH1 mutations cause ZLS and document genetic heterogeneity for this disorder.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Accession codes
Primary accessions
NCBI Reference Sequence
Referenced accessions
NCBI Reference Sequence
Protein Data Bank
References
Laband, P.F., Habib, G. & Humphreys, G.S. Hereditary gingival fibromatosis. Report of an affected family with associated splenomegaly and skeletal and soft-tissue abnormalities. Oral Surg. Oral Med. Oral Pathol. 17, 339–351 (1964).
Alavandar, G. Elephantiasis gingivae. Report of an affected family with associated hepatomegaly, soft tissue and skeletal abnormalities. J. All India Dent. Assoc. 37, 349–353 (1965).
Chacon-Camacho, O.F., Vazquez, J. & Zenteno, J.C. Expanding the phenotype of gingival fibromatosis–mental retardation–hypertrichosis (Zimmermann-Laband) syndrome. Am. J. Med. Genet. 155A, 1716–1720 (2011).
Dávalos, I.P. et al. Zimmermann-Laband syndrome: further clinical delineation. Genet. Couns. 16, 283–290 (2005).
Castori, M. et al. Clinical and genetic study of two patients with Zimmermann-Laband syndrome and literature review. Eur. J. Med. Genet. 56, 570–576 (2013).
Abo-Dalo, B. et al. Extensive molecular genetic analysis of the 3p14.3 region in patients with Zimmermann-Laband syndrome. Am. J. Med. Genet. 143A, 2668–2674 (2007).
Kim, H.G. et al. Candidate loci for Zimmermann-Laband syndrome at 3p14.3. Am. J. Med. Genet. 143A, 107–111 (2007).
Stefanova, M. et al. Zimmermann-Laband syndrome associated with a balanced reciprocal translocation t(3;8)(p21.2;q24.3) in mother and daughter: molecular cytogenetic characterization of the breakpoint regions. Am. J. Med. Genet. A. 117A, 289–294 (2003).
Abo-Dalo, B. et al. No mutation in genes of the WNT signaling pathway in patients with Zimmermann-Laband syndrome. Clin. Dysmorphol. 17, 181–185 (2008).
Petrovski, S. et al. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013).
Samocha, K.E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
Bauer, C.K. & Schwarz, J.R. Physiology of EAG K+ channels. J. Membr. Biol. 182, 1–15 (2001).
Garg, V., Sachse, F.B. & Sanguinetti, M.C. Tuning of EAG K+ channel inactivation: molecular determinants of amplification by mutations and a small molecule. J. Gen. Physiol. 140, 307–324 (2012).
Thouta, S. et al. Proline scan of the HERG channel S6 helix reveals the location of the intracellular pore gate. Biophys. J. 106, 1057–1069 (2014).
Wynia-Smith, S.L., Gillian-Daniel, A.L., Satyshur, K.A. & Robertson, G.A. hERG gating microdomains defined by S6 mutagenesis and molecular modeling. J. Gen. Physiol. 132, 507–520 (2008).
Veeramah, K.R. et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54, 1270–1281 (2013).
Yang, Y. et al. Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2-R327H demonstrate the role of this residue in stabilizing the channel closed state. J. Neurosci. 33, 16586–16593 (2013).
Forgac, M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 (2007).
Marshansky, V., Rubinstein, J.L. & Gruber, G. Eukaryotic V-ATPase: novel structural findings and functional insights. Biochim. Biophys. Acta 1837, 857–879 (2014).
Arai, S. et al. Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature 493, 703–707 (2013).
Fuster, D.G., Zhang, J., Xie, X.S. & Moe, O.W. The vacuolar-ATPase B1 subunit in distal tubular acidosis: novel mutations and mechanisms for dysfunction. Kidney Int. 73, 1151–1158 (2008).
Martin, S. et al. Eag1 potassium channel immunohistochemistry in the CNS of adult rat and selected regions of human brain. Neuroscience 155, 833–844 (2008).
Hemmerlein, B. et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 5, 41 (2006).
Pardo, L.A. & Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 14, 39–48 (2014).
Ouadid-Ahidouch, H. & Ahidouch, A. K+ channel expression in human breast cancer cells: involvement in cell cycle regulation and carcinogenesis. J. Membr. Biol. 221, 1–6 (2008).
Weber, C. et al. Silencing the activity and proliferative properties of the human EagI potassium channel by RNA interference. J. Biol. Chem. 281, 13030–13037 (2006).
Zhang, Y.Y. et al. BKCa and hEag1 channels regulate cell proliferation and differentiation in human bone marrow–derived mesenchymal stem cells. J. Cell. Physiol. 229, 202–212 (2014).
Ufartes, R. et al. Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum. Mol. Genet. 22, 2247–2262 (2013).
Harakalova, M. et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat. Genet. 44, 793–796 (2012).
van Bon, B.W. et al. Cantú syndrome is caused by mutations in ABCC9. Am. J. Hum. Genet. 90, 1094–1101 (2012).
Brownstein, C.A. et al. Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities—support for the role of KATP channels in this condition. Eur. J. Med. Genet. 56, 678–682 (2013).
Cooper, P.E. et al. Cantú syndrome resulting from activating mutation in the KCNJ8 gene. Hum. Mutat. 35, 809–813 (2014).
Lipscombe, D., Helton, T.D. & Xu, W. L-type calcium channels: the low down. J. Neurophysiol. 92, 2633–2641 (2004).
Corrêa, J.D. et al. Phenytoin-induced gingival overgrowth: a review of the molecular, immune, and inflammatory features. ISRN Dent. 2011, 497850 (2011).
Mantegazza, M. et al. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424 (2010).
Wynn, R.L. Calcium channel blockers and gingival hyperplasia—an update. Gen. Dent. 57, 105–107 (2009).
Jacquinet, A. et al. Temple-Baraitser syndrome: a rare and possibly unrecognized condition. Am. J. Med. Genet. 152A, 2322–2326 (2010).
White, S.M. & Fahey, M. Report of a further family with dominant deafness-onychodystrophy (DDOD) syndrome. Am. J. Med. Genet. 155A, 2512–2515 (2011).
Campeau, P.M. & Hennekam, R.C. DOORS syndrome: phenotype, genotype and comparison with Coffin-Siris syndrome. Am. J. Med. Genet. 166C, 327–332 (2014).
Simons, C. et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 47, 73–77 (2015).
Nelson, R.D. et al. Selectively amplified expression of an isoform of the vacuolar H+-ATPase 56-kilodalton subunit in renal intercalated cells. Proc. Natl. Acad. Sci. USA 89, 3541–3545 (1992).
Puopolo, K. et al. Differential expression of the “B” subunit of the vacuolar H+-ATPase in bovine tissues. J. Biol. Chem. 267, 3696–3706 (1992).
van Hille, B. et al. Heterogeneity of vacuolar H+-ATPase: differential expression of two human subunit B isoforms. Biochem. J. 303, 191–198 (1994).
Yuan, Y. et al. De novo mutation in ATP6V1B2 impairs lysosome acidification and causes dominant deafness-onychodystrophy syndrome. Cell Res. 24, 1370–1373 (2014).
Mortensen, L.S. et al. KV10.1 opposes activity-dependent increase in Ca2+ influx into the presynaptic terminal of the parallel fibre–Purkinje cell synapse. J. Physiol. (Lond.) 593, 181–196 (2015).
Poëa-Guyon, S. et al. The V-ATPase membrane domain is a sensor of granular pH that controls the exocytotic machinery. J. Cell Biol. 203, 283–298 (2013).
Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 (2012).
Steinberg, B.E. et al. A cation counterflux supports lysosomal acidification. J. Cell Biol. 189, 1171–1186 (2010).
Van Dyke, R.W. Acidification of rat liver lysosomes: quantitation and comparison with endosomes. Am. J. Physiol. 265, C901–C917 (1993).
Kohl, T., Lörinczi, E., Pardo, L.A. & Stühmer, W. Rapid internalization of the oncogenic K+ channel KV10.1. PLoS ONE 6, e26329 (2011).
Ninkovic, M. et al. Physical and functional interaction of KV10.1 with Rabaptin-5 impacts ion channel trafficking. FEBS Lett. 586, 3077–3084 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, E2393–E2402 (2013).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010).
Pettersen, E.F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Schuster, A.M., Glassmeier, G. & Bauer, C.K. Strong activation of ether-à-go-go–related gene 1 K+ channel isoforms by NS1643 in human embryonic kidney 293 and Chinese hamster ovary cells. Mol. Pharmacol. 80, 930–942 (2011).
Clay, J.R. Determining K+ channel activation curves from K+ channel currents often requires the Goldman-Hodgkin-Katz equation. Front. Cell. Neurosci. 3, 20 (2009).
Schönherr, R. et al. Individual subunits contribute independently to slow gating of bovine EAG potassium channels. J. Biol. Chem. 274, 5362–5369 (1999).
Acknowledgements
We are grateful to the patients and their families who contributed to this study. We thank I. Jantke, S. Cecchetti and S. Venanzi for skillful technical assistance, T. Kock for site-directed mutagenesis, A. Hasse for CHO cell transfection and injection, R. Bähring, J.M. Schröder and E. Neumann for help with the oocyte experiments, P. Meinecke for discussing clinical phenotypes and A. Podolska for help with ATP6V1B2 sequencing. G.B., L.S. and M.T. acknowledge CINECA for computational resources (whole-exome sequencing data and structural analyses). The KCNH1/heag1 clone was kindly provided by S.H. Heinemann (Friedrich Schiller University Jena). This work was supported by grants from the Deutsche Forschungsgemeinschaft (KO 4576/1-1 to F.K. and KU 1240/5-1 to K.K.), Istituto Superiore di Sanità (Ricerca Corrente 2013 to M.T.), Ministero della Salute (Ricerca Finalizzata RF-2010-2312766 to B.D.) and Ospedale Pediatrico Bambino Gesù (Gene-Rare to B.D.).
Author information
Authors and Affiliations
Contributions
F.K. performed whole-exome sequencing data analysis and validation, molecular screening and genotyping and wrote the manuscript. V.C. performed whole-exome sequencing data analysis and validation and wrote the manuscript. C.K.B. contributed the electrophysiological studies and wrote the manuscript. L.S. and G.B. performed the homology modeling and structural analysis. A.C. and M.A. contributed to the whole-exome sequencing data processing and analysis. E.F., S.P., M.L.D. and T.T.M.N. carried out the molecular screening and/or genotyping. P.G., G.C.K., V.L., D.M., L.D.V.N., P.T., S.M.W., B.D. and A.P. recruited and clinically characterized the study subjects and collected the biological samples. P.M.C. performed whole-exome sequencing data analysis, validation and genotyping. M.T. and K.K. conceived the project, analyzed and interpreted the data, and wrote the manuscript. All authors contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Sequence electropherograms of individuals with KCNH1 or ATP6V1B2 mutation.
Sequence electropherograms showing the de novo origin of the identified KCNH1 and ATP6V1B2 missense mutations in subjects 1–8 (upper and lower panels, indicated by red arrows). The heterozygous state of three mutations was documented in peripheral leukocytes, hair bulb and/or buccal cells of subjects 4, 5 and 7, indicating germline origin. An additional previously annotated (ExAC database) heterozygous KCNH1 variant, c.125T>C (p.Ile42Thr), was present in subject 5 and his healthy mother (indicated by blue arrows). By cloning the KCNH1 exon 7–containing amplicon of subject 2 followed by sequencing, we determined the haplotypes and found that the two identified de novo changes c.974C>A and c.1066G>C are in cis (wild-type allele and mutated KCNH1 allele in the middle panel; mutated nucleotides are framed).
Supplementary Figure 2 Multiple protein sequence alignments around the KCNH1 and ATP6V1B2 amino acid substitutions from different species.
Alignment of the regions flanking the detected missense variants in orthologous proteins, showing the evolutionary conservation of amino acids S325, G348, L352, V356, I467 and G469 in human KCNH1 (NP_002229.1) and of R485 in human ATP6V1B2 (NP_001684.2). Multiple alignments were gathered from http://www.ncbi.nlm.nih.gov/homologene/. Conserved residues have a red background, and non-conserved residues have a gray background. Amino acid sequence alignments demonstrate high (S325 and V356 in human KCNH1) or complete (G348, L352, I467 and G469 in human KCNH1 and R485 in human ATP6V1B2) evolutionary conservation of the altered residues.
Supplementary Figure 3 Voltage dependence of wild-type and mutant KCNH1 channel activation.
(a) KCNH1 channels were expressed in CHO cells, and families of wild-type (WT) and L352V current traces recorded with the depicted pulse protocols are shown. Zero current is indicated by dashed lines and arrowheads. (b) Mean (±s.e.m.) normalized instantaneous tail current amplitudes as a function of the preceding test pulse potential. Lines represent first-order Boltzmann functions fitted to the data points. The voltage dependence of channel activation was analyzed from instantaneous tail current measurements at +40 mV (for KCNH1 WT and I467V for all experiments and for G348R and S325Y/V356L for four experiments each) or at –20 mV (for L352V, G348R and S325Y/V356L). No significant differences were found between the potentials for half-maximal G348R or S325Y/V356L channel activation determined with the two different constant pulse potentials. (c) Table with means ± s.e.m. of the potential of half-maximal channel activation and the slope factor k derived from fits of first-order Boltzmann functions to the normalized instantaneous tail current amplitudes of the individual experiments. n, number of experiments; *, significantly different from WT with P < 0.05; ***, significantly different from WT with P < 0.001. Values were tested for significant differences compared to WT data with one-way ANOVA and post-hoc Bonferroni t test.
Supplementary Figure 4 Analysis of the activation and deactivation kinetics of wild-type and mutant KCNH1 channels expressed in CHO cells.
(a) The time course of channel activation was analyzed at +40 mV from experiments as shown in Figure 4 by fitting a double-exponential function to the current traces, yielding the fast and the slow time constant of current activation at +40 mV as well as the amplitudes of the two current components (Af and As). Note that the preceding holding potential was –80 mV, except for the mutant L352V, where a holding potential of –100 mV was used. Compared to wild-type KCNH1 channels, the time course of current activation was accelerated for all mutant channels. For G348R, I467V and L352V, both activation time constants were significantly decreased, and for the double mutant S325Y/V356L, the slow time constant decreased significantly in combination with a higher relative contribution of the faster activating current component. (b) Families of wild-type and G348R current traces recorded with the depicted deactivation protocol. Zero current is indicated by a dashed line. (c) The time course of KCNH1 channel deactivation was analyzed at –120 mV from experiments as shown in b. Compared to wild-type channel, the deactivation time course of all mutant channels was significantly slowed. Current decay upon hyperpolarization to –120 mV was fitted with a double-exponential function, yielding the fast and the slow time constant of current deactivation as well as the amplitudes of the two current components (Af and As). Af and As were extrapolated to segment start, because the first 0.5 to 1 ms of the pulse segment at –120 mV was not used for fitting to minimize contributions of capacitive currents. (a,c) The number of experiments is given in the lower bar plots; **, significantly different from wild-type channel with P < 0.01; ***, significantly different from wild-type channel with P < 0.001. Values were tested for significant differences compared to wild-type data with a two-tailed heteroscedastic t test and Bonferroni correction for multiple testing.
Supplementary Figure 5 Voltage dependence of S325Y and V356L KCNH1 channel activation.
(a,b) Mean values (±s.e.m.) of normalized current amplitudes (a) and whole-cell conductance (b) for S325Y (n = 14) and V356L (n = 11). Data points in a are connected by lines; the dark gray lines in b represent fits to the data points using equation (2). Corresponding data for the double mutant S325Y/V356L and for wild-type (WT) channels are shown for comparison as light gray and black lines, respectively (data from Fig. 4). Fit parameters are given in Supplementary Table 4.
Supplementary Figure 6 Expression and coexpression of wild-type (WT) and mutant (G469R) KCNH1 channels in Xenopus laevis oocytes.
(a) Families of current traces recorded with the depicted pulse protocol. For the G469R mutant, depolarizing pulses failed to induce voltage-dependent outward currents and coexpression of the mutant with wild-type channels suppressed the amplitude of KCNH1 outward currents at more positive potentials. Zero current is indicated by dashed lines and arrowheads. (b) Means (+s.e.m.) of the current amplitude recorded at the end of the test pulse to +40 mV. Absolute current values were normalized to the average current amplitude obtained for wild-type KCNH1 channels. ***, significantly different from WT with P < 0.001 (one-way ANOVA and post-hoc Bonferroni t test); n.s., not significantly different. The amount of injected cRNA is indicated, and the number of experiments is given in parentheses. Similar results were obtained in experiments using other batches of oocytes.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6 and Supplementary Tables 1–5. (PDF 3899 kb)
Rights and permissions
About this article

Cite this article
Kortüm, F., Caputo, V., Bauer, C. et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet 47, 661–667 (2015). https://doi.org/10.1038/ng.3282
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3282
This article is cited by
-
Establishment and characterization of ZJUCHi003: an induced pluripotent stem cell line from a patient with Temple–Baraitser/Zimmermann–Laband syndrome carrying KCNH1 c.1070G > A (p.R357Q) variant
Human Cell (2024)
-
Zimmermann-Laband syndrome and infantile systemic hyalinosis: an enigma with two separate terms with overlapping features: a case report
BMC Pediatrics (2023)
-
14-3-3 proteins regulate cullin 7-mediated Eag1 degradation
Cell & Bioscience (2023)
-
Low expression of SLC34A1 is associated with poor prognosis in clear cell renal cell carcinoma
BMC Urology (2023)
-
Channelopathy of small- and intermediate-conductance Ca2+-activated K+ channels
Acta Pharmacologica Sinica (2023)
