
Key Points
-
All available genome data indicate that the various Shigella spp.have arisen from different ancestral Escherichia coli isolates on several independent occasions.
-
The acquisition of plasmids that encode virulence genes into numerous ancestral Shigella spp. were pivotal in their evolution as human pathogens. The possession and adaptation of these plasmids has shaped the current Shigella spp.
-
Convergent evolution, through the acquisition of mobile elements and loss of gene function, enabled Shigella spp. to become restricted to humans and exquisitely adapted to interact with the human intestinal mucosa.
-
The shift in dominance from Shigella flexneri to Shigella sonnei in economically transitioning nations warrants more in-depth studies of the evolution and epidemiology of these species.
-
There is a correlation between the global dissemination of Shigella spp. clones and acquisition of resistance to multiple antimicrobials. An antimicrobial-resistance phenotype is likely to be advantageous for the bacteria, as it would promote post-symptomatic shedding by the host and would sustain short-term transmission.
-
In comparison to S. sonnei and S. flexneri, comparatively little is currently understood about Shigella dysenteriae and Shigella boydii; this needs to be addressed to reveal the full evolutionary landscape of the genus.
-
Future laboratory research should be combined with genomics to address the survival, transmission and evolution of Shigella spp. in the environment, specifically focusing on how the environmental lifestyle can affect disease epidemiology and global public health.
Abstract
Shigella spp. are some of the key pathogens responsible for the global burden of diarrhoeal disease. These facultative intracellular bacteria belong to the family Enterobacteriaceae, together with other intestinal pathogens, such as Escherichia coli and Salmonella spp. The genus Shigella comprises four different species, each consisting of several serogroups, all of which show phenotypic similarity, including invasive pathogenicity. DNA sequencing suggests that this similarity results from the convergent evolution of different Shigella spp. founders. Here, we review the evolutionary relationships between Shigella spp. and E . coli, and we highlight how the genomic plasticity of these bacteria and their acquisition of a distinctive virulence plasmid have enabled the development of such highly specialized pathogens. Furthermore, we discuss the insights that genotyping and whole-genome sequencing have provided into the phylogenetics and intercontinental spread of Shigella spp.
Similar content being viewed by others

Main
Diarrhoea remains one of the main causes of mortality in young children in low-income countries1,2. Although the number of children aged <5 years who die owing to diarrhoea has declined steadily over the past decade, the incidence of diarrhoeal disease has remained comparatively stable over the same period, at ∼2.9 episodes per child per year in 2010 (Refs 1,3). In 2012, the WHO estimated that diarrhoeal disease contributes ∼3.6% of the global burden of disease in disability adjusted life years (DALY) and results in ∼1.5 million deaths annually.
The recent Global Enteric Multicenter Study (GEMS), an expansive case–control study of moderate-to-severe paediatric diarrhoeal disease, identified enterotoxigenic Escherichia coli (ETEC) and Shigella spp. as the most common bacterial pathogens in sub-Saharan Africa and South Asia4,5, and found Shigella spp. to be the most prevalent pathogens among children 24–59 months old5. Historical data suggest that there were ∼165 million cases of shigellosis annually mainly in low-income countries and in children <5 years old between 1966 and 1997, resulting in 1.1 million deaths worldwide6. More recently, it was estimated that Shigella spp. cause ∼125 million disease cases annually7, and that the incidence of shigellosis is 13.2 cases per 1,000 children per year in children aged <5 years in Asia8. Importantly, despite the continued high incidence of Shigella spp. infections, the mortality rate per case has dropped by 98%; this is probably due to the disappearance of epidemics associated with the highly pathogenic species Shigella dysenteriae and improved, more rapid treatment7.
The Gram-negative bacterial genus Shigella belongs to the family Enterobacteriaceae, which also encompasses other enteric pathogens, including ETEC, enteropathogenic E. coli (EPEC), enterohaemorrhagic E. coli (EHEC; also known as Shiga toxin-producing E. coli (STEC)), enteroaggregative E. coli (EAEC), diffusely adherent E. coli (DAEC) and enteroinvasive E. coli (EIEC)9,10. The pathogenesis and epidemiology of each of these E. coli pathovars are distinct and complex, and reflect the diverse catalogue of phenotypic traits that E. coli has acquired during its evolution from commensal to pathogen in humans and other mammals11. However, Shigella spp. stand out from other Enterobacteriaceae: their evolutionary history, mechanism of pathogenesis and human-restricted nature make them unique.
Shigella spp. are intracellular pathogens and are transmitted through the faecal–oral route. They can induce a symptomatic infection via an exceptionally low infectious dose (<10 bacteria), as opposed to Salmonella spp. and the various diarrhoeagenic E. coli pathovars, which have infectious doses of at least four orders of magnitude greater12. Shigella spp. cause bacillary dysentery, a severe form of diarrhoea in which blood and mucus can be observed in the stool as a consequence of epithelial cell damage in the lower gut (Box 1). The highly pathogenic and exotoxin-producing species S. dysenteriae was first described in 1897 by Kiyoshi Shiga, who isolated this species from the stool sample of a patient with epidemic dysentery in Japan13,14. The genus was expanded soon after: Shigella flexneri was identified in 1899, Shigella sonnei in 1906 and Shigella boydii in 1921 (Refs 15,16). Shigella bacteria are non-motile, non-sporulating and non- or late-lactose-fermenting, and classical taxonomy places all Shigella spp. into one major group, which is distantly related to E. coli17. However, even on their initial characterization, the biochemical and morphological proximity of members of the genus Shigella with E. coli was noted13. Biochemical differences exist between the two genera: S. dysenteriae is negative in an indole reaction and cannot ferment mannitol13, and all Shigella spp. are negative for lysine decarboxylation (LDC)18, whereas the opposites are true for E. coli. Current serological classification divides the genus Shigella into four species (also known as subgroups), which are further subdivided into serotypes according to type-specific antigens: S. dysenteriae (subgroup A) has 15 serotypes; S. flexneri (subgroup B) has 19 serotypes and subserotypes; S. boydii (subgroup C) has 20 serotypes; and S. sonnei (subgroup D) consists of a single serotype.
S. flexneri is currently the major cause of bacillary dysentery in low-income settings (in parts of Asia and sub-Saharan Africa, this species accounts for up to 62% of all Shigella spp. infections), whereas S. sonnei is the most common pathogen in transitional or high-income countries (up to 80% of all Shigella spp. infections in Europe and North America are caused by this species)19. S. boydii and S. dysenteriae cause <5% each of all cases of shigellosis globally. Notably, S. dysenteriae was the main cause of dysentery when it was first identified more than a century ago, but today it is infrequently isolated from patients with dysenteric diarrhoea7,19. It is thought that poor sanitation, malnutrition and unavailability of clean water, and an exceptionally low infectious dose, genomic plasticity and an ability to accept antimicrobial-resistance genes are all potential reasons why Shigella spp. are such successful pathogens and why particular human populations are specifically vulnerable to infection with these species.
The genomics revolution has revealed the dynamic genome plasticity of Shigella spp. and their close evolutionary history with E. coli20. The pathogenesis of Shigella spp. depends on a large virulence plasmid that, during its enigmatic evolutionary history, has acquired several factors that are essential for invasion and subversion of host defences21. Recent advances in high-throughput genomics and phylogenetics have detailed the emergence and spread of different Shigella spp. serogroups, and this information can in turn be used to inform control and public health polices for shigellosis22,23,24.
In this Review, we discuss the evolution of Shigella spp. to highly specialized, human-specific pathogens, taking into account both insights from traditional genotyping methods and current perspectives achieved from phylogenomics. We focus on the most commonly isolated Shigella spp., S. sonnei and S. flexneri, as these are the dominant species responsible for the global burden of shigellosis.

The evolutionary history of Shigella spp.
The acquisition of the Shigella virulence plasmid was the key event in the formation of the different Shigella spp.25, but the origins of this plasmid and the relationship between the species was contentious for a long time. The advent of DNA sequencing and accompanying phylogenetic analyses have led to a much clearer picture of the evolutionary relationships between the different Shigella spp. and their emergence from E. coli.
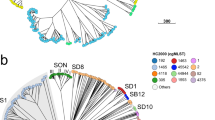
The phylogenetic relationships of Shigella spp. Pioneering research in the early genomics era, carried out by aligning and comparing the DNA sequences of eight chromosomal housekeeping genes, found the Shigella genus to contain three major clades or clusters (C1, C2 and C3) and a limited number of outliers, all of which are distinct from, but nested within lineages of E. coli26. A further examination of 23 chromosomal genes reached a similar phylogenetic conclusion, albeit with increased resolution, subdividing C1 into 3 subclusters (SC1, SC2 and SC3)27 (Fig. 1a). Most Shigella spp. serotypes are distributed over the three major clusters, demonstrating an incongruence between evolutionary history and the conventional serology-based nomenclature. Cluster C1 contains a combination of serotypes from S. dysenteriae and S. boydii, as well as S. flexneri serotype 6: SC1 includes only S. dysenteriae (serotypes 3, 4, 6, 9, 11, 12 and 13); SC2 contains mostly S. boydii (serotypes 1, 3, 6, 8, 10 and 18), as well as S. dysenteriae serotype 5; and SC3 is composed of three other S. boydii serotypes (2, 4 and 14) and S. flexneri serotype 6. Cluster C2 comprises S. boydii (serotypes 5, 7, 9, 11, 15, 16 and 17) and S. dysenteriae serotype 2. All S. flexneri serotypes except 6 (that is, 1, 2, 3, 4, 5, X and Y) fall into cluster C3, as well as S. boydii serotype 12. In this analysis, C2 and C3 were found to share a more recent common ancestor than their common ancestor with C1, thus emphasizing the close phylogenetic relationship between these two clusters.

a | A neighbour-joining phylogenetic tree generated by sequencing 23 chromosomal genes27. Strains are labelled by serotype and coloured by species: Shigella sonnei in red, Shigella flexneri in blue, Shigella boydii in green and Shigella dysenteriae in purple; Escherichia coli isolates are uncoloured. Bootstrap values of >50% are indicated at the major nodes, and the three major Shigella genus clusters (C) and subclusters (SC) are indicated. The carriage of the two specific isoforms of virulence plasmids is additionally indicated in the second column of coloured blocks: pINV A (grey), pINV B (black), either pINV A or pINV B (hatched black and grey), a unique form of pINV (pink), and either pINV B or a unique isoform (hatched black and pink). b | A comparative gene map of the Shigella virulence plasmid, using the S. sonnei virulence plasmid pSs_046 as a reference; the innermost ring represents pSs_046, with coordinates. The second ring (black) shows the GC content of the reference pSs_046 sequence. The following purple, pale green, teal, khaki, and blue rings show BLASTN comparisons between pSs_046 and the virulence plasmids of S. boydii str. BS512, S. boydii str. Sb227, S. dysenteriae str. Sd197, S. flexneri F2a str. 301 (pCP301) and S. flexneri F5a (pWR501), respectively. The outer ring represents annotations of genes or genetic clusters based on function: known virulence factor genes (red); plasmid replication, transfer and maintenance genes (black); transposon, phage-borne and insertion sequence elements (orange); genes encoding hypothetical proteins (teal); the S. sonnei-specific O antigen biosynthesis cluster (blue); and genes encoding proteins with other known functions (green). ipa, invasion plasmid antigen gene; icsP, also known as sopA; T3SS, type III secretion system. Part a is modified with permission from Ref. 27, Springer.
An analysis of short DNA sequences yielded an estimation of the age of the various clusters (50,000–270,000 years for each of C1 and C2; 35,000–170,000 years for C3)26; however, whole-genome sequencing and Bayesian phylogenetic tools are expected to provide a more accurate genome-wide dating of these clusters. Notable outliers not belonging to any of the three major clusters include S. sonnei, S. dysenteriae 1, 8 and 10, and S. boydii 13 (Fig. 1a). The position of S. boydii 13 on the tree topology indicates that it is also distant from the E. coli–Shigella clade. This genetic distance is consistent with the finding that S. boydii 13 and an Escherichia albertii group form a discrete lineage that separated from an E. coli ancestor ∼28 million years ago28.
As highlighted above, S. sonnei is an outlier from the other Shigella spp., and the precise phylogenetic relationship between S. sonnei and the other Shigella spp. remains ambiguous. It is assumed that S. sonnei emerged more recently than the other Shigella spp. and serotypes29. Unlike the other Shigella spp., S. sonnei expresses an O antigen, encoded by a genetic locus that is also found in the genetically distant Gram-negative organism, Plesiomonas shigelloides29. A sequence comparison of the O antigen loci from S. sonnei and P. shigelloides predicts that the O antigen genes diverged approximately 10,000 years ago, placing an upper limit on the age of the formation of S. sonnei29. However, a more recent study using whole-genome sequencing data from globally distributed isolates estimated that all extant strains of S. sonnei descend from a common ancestor that existed <400 years ago, implying that a historical evolutionary bottleneck might have resulted in the extinction of the pre-existing S. sonnei strains22.
The early sequence-based genotyping studies described above largely resolved the phylogenetic relationships of the different Shigella spp., but more recent studies have exploited Shigella spp. and E. coli whole-genome sequences to investigate the evolutionary relationship between these two taxa in more detail. Phylogenetic trees for the entire E. coli–Shigella group were constructed using an alignment-free feature frequency profile (FFP), which compares genomes based on the frequencies of oligonucleotide sequences with an optimal length for analysis (so-called features)30. These phylogenies, together with those deduced from other studies using core genetic features (present in all genomes and with low variability), have confirmed that the genus Shigella is composed of several clusters interspersed in the E. coli–Shigella phylogeny, strongly supporting the notion that Shigella spp. have emerged from several E. coli ancestors on multiple independent occasions31,32,33. The phylogenomic structure of the genus Shigella derived from a collection of 336 E. coli–Shigella isolates correlates with the grouping from the aforementioned studies based on a limited number of genetic markers34. In addition, whole-genome resolution phylogenomics also resolves the context for the origins of these major clades: it places C1 and S. sonnei in E. coli group B1; C2 and C3 in E. coli group A; and S. dysenteriae 1 in E. coli group E34. This supports the theory that the phenotypic similarity observed across the Shigella spp. is the result of convergent evolution, in which different Shigella founders independently gained genes that facilitate invasive pathogenicity. Only one E. coli pathovar, EIEC, has also acquired invasiveness; EIEC comprises several discrete lineages and exhibits pathogenic and biochemical features that are indistinguishable from those of Shigella spp. Notably, both EIEC and Shigella spp. harbour an analogous virulence plasmid, are non-motile and show a negative LDC test35. These similarities have led to the speculation that EIEC represents a distinct non-toxin-producing Shigella 'prototype', which could be a precursor for a 'complete' Shigella sp. if selective pressure favours further adaptation of this invasive E. coli pathovar36.
The Shigella virulence plasmid. The Shigella virulence plasmid, which can be as large as ∼220 kb, encodes essential virulence factors that facilitate the invasion and spread of Shigella spp. into human macrophages and enterocytes37 (Box 1). The virulence plasmid contains the conserved 30 kb mxi–spa locus, which encodes the Mxi–Spa type III secretion system (T3SS), and genes encoding invasion plasmid antigens (Ipas). The Mxi–Spa T3SS is a molecular syringe that injects effector proteins directly into host cells. This secretion apparatus enables a complex interaction between the bacterium and the host cell, ultimately resulting in a disruption of the intestinal mucosa and the distinctive symptoms of bacillary dysentery. Therefore, the virulence plasmid is the key molecular signature of Shigella spp. pathogenesis and is fundamental for initiating infection and manipulating the immune response of the host (Box 2).
Various DNA sequencing projects have been carried out across several different Shigella spp. lineages to elucidate the structure and functions of the virulence plasmid. These projects have uncovered a complex plasmid configuration with a mosaic nature, which is the result of numerous horizontal gene transfer and rearrangement events21,38,39 (Fig. 1b). The evaluation of three genes in the mxi–spa region (mxiA, mxiC and ipgD) revealed two isoforms of the Shigella virulence plasmid (pINV A and pINV B) with greater divergence in ipgD than in the two mxi genes25. pINV A and pINV B exhibited incompatibility grouping (plasmids of the same incompatibility group cannot be stably inherited in the same cell)40. When plasmid subtype is mapped onto the Shigella spp. phylogeny (Fig. 1a), all C1 isolates harbour pINV A, whereas all C3 isolates possess the pINV B isoform. Both forms of the plasmid can be found in C2 isolates. The outlier strains harbour either of the two plasmid forms, which is a sign of lateral gene transfer in their history. For example, S. dysenteriae 10 and most EIEC strains harbour pINV A, whereas S. sonnei retains pINV B36,41. By contrast, S. dysenteriae 1 harbours a unique mixed plasmid form (ipgD derived from pINV A, and mxiA and mxiC derived from pINV B). This suggests that several ancestral virulence plasmids, from an unknown source, have entered into a diverse background of E. coli founder strains. Such introductions include pINV A and pINV B into major Shigella clusters C1 and C3, respectively, thus giving rise to these two lineages. Independent acquisitions of either plasmid isoform by Shigella spp. isolates not belonging to the main clades, as well as lateral gene transfer in the C2 isolates, complicate the evolutionary history of Shigella spp.
Plasmid sequences have also been compared for five virulence genes (mkaD (also known as ospF), CP0014, parA, parB and repA) located outside the core entry region (defined as the ∼30 kb cluster encoding the T3SS and associated effector proteins that facilitate the mechanistic invasion of the bacteria into enterocytes). The constructed phylogeny was consistent with the one based on chromosomal genes27, except for a close relationship between C1 and C2 isolates in the plasmid phylogeny; by contrast, the C2 and C3 clusters showed close proximity in the chromosomal phylogeny. The authors of the plasmid phylogeny report argued that the virulence plasmid acquired by C1 and C2 isolates differed from the one obtained by C3 isolates. Interestingly, the virulence plasmids from the outliers S. dysenteriae 1 and S. sonnei share considerable homogeneity and can be grouped together outside of the three major clusters. These data suggest that Shigella spp. have arisen on several independent occasions owing to the transmission of multiple virulence plasmid forms to many E. coli ancestors. The authors suggested that the subsequent loss of the tra locus, which aids the exchange of plasmids between bacteria by conjugation, on the virulence plasmid restricted its transmissibility and enabled parallel evolution of the virulence plasmid and the bacterial chromosomes, thus creating the several discrete Shigella lineages observed today.
Gain and loss of gene function. The ability to invade host cells and escape the competitive environment of the gastrointestinal tract was pivotal in the emergence of Shigella spp. Although acquisition of the virulence plasmid is a 'foothold moment' in the evolution of this pathogen, it is not the only long-term evolutionary change. Numerous other plasmids with different functions have been crucial during the evolution of Shigella spp. (Box 3). In addition to the genes encoded on the virulence plasmid, several clusters of horizontally acquired genetic material, carrying genes that facilitate interactions with the host and contribute to pathogenesis, have been incorporated into the chromosome of Shigella spp.
These pathogenesis-associated genomic regions are pathogenicity islands (PAIs) and have various functions; the largest PAI encodes an enterotoxin (Shigella island 1 (SHI-1)) and it enables the sequestration of iron (SHI-2, SHI-3 and sitABCD), the ability to modify the O antigen (SHI-O) and resistance to antimicrobials (Shigella resistance locus (SRL))37,42,43,44,45,46. PAIs have enhanced the virulence and adaptability of Shigella spp. and are commonly associated with bacteriophage integrases, which highlights the fact that bacteriophages had a major role in the evolution of Shigella spp. One such bacteriophage-associated element is the Shiga toxin (Stx) prophage in S. dysenteriae 1; Stx expression can have severe complications, including haemolytic uraemic syndrome (HUS). Recently, an alternative prophage (φPOC-J13) encoding Stx1a was identified in several clinical isolates of S. flexneri and S. dysenteriae 4 from patients returning from or residing in Hispaniola47,48,49,50. Unlike the cryptic prophage in S. dysenteriae 1, φPOC-J13 seems to be capable of disseminating the stx1a gene into other Shigella spp. isolates by transduction50. Insertion sequence elements — small transposable DNA sequences that can 'jump' within bacterial genomes — are also highly abundant in Shigella spp. chromosomes and virulence plasmids. These elements have shaped the genome architecture of Shigella spp., causing gene inactivation and genome rearrangement20,21,51. An analysis of >400 genomes from a range of bacterial species found that, in relation to genome size, E. coli and Shigella spp. possess the highest number of insertion sequence elements52.
Linked to this insertion sequence expansion, Shigella spp. genomes have also undergone substantial functional gene loss53. Similar phenomena have been observed in other human-restricted pathogens, such as Yersinia pestis, Mycobacterium leprae and Salmonella enterica subsp. enterica serovar Typhi54,55,56. The modes of gene inactivation are variable in different Shigella spp. strains and range from the complete deletion of a locus, to missense point mutations, to insertions. However, gene inactivation has occurred preferentially in specific genetic regions and operons rather than being randomly distributed throughout the genome20,57. Independent inactivation of the same or functionally similar genes in different Shigella spp. represents a major pathway of convergent evolution, resulting in similar phenotypic changes that are associated with adaptation to new niches. For example, different mutations have resulted in the loss of flagellum biosynthesis and specific structures of the fimbriae20,58. Importantly, flagellum loss results in reduced immunogenicity and in evasion of the human immune system, as flagellin is a pathogen-associated molecular pattern (PAMP)59. In addition, computational reconstruction of metabolic functions (or their loss) based on genomic data groups the different Shigella spp. together and away from E. coli pathovars and commensals, purely by their catabolic function60. For example, the E. coli genes cadA and nadAB (responsible for the synthesis of cadaverine and the NAD precursor quinolinate, respectively) hinder intercellular spread, phagosomal escape and antigen secretion61,62,63. Likewise, ompT and argT inhibit intracellular motility and invasive capacity, respectively64,65. Therefore, the loss of these genes in Shigella spp. ensures patho-adaptation for an intracellular lifestyle. Alternatively, the loss of gene function can increase the survival rate in a new niche. Disruption of speG, which encodes spermidine acetyltransferase, leads to the accumulation of the polyamine spermidine, which acts as scavenger of free radicals and thereby provides resistance to oxidative stress in macrophages66.
In comparison to E. coli, Shigella spp. have lost more genes, which is attributed to a reduction in genome-wide purifying selection and the fixation of inactivating mutations without greatly compromising fitness. It has been suggested that this resulted from a decrease in effective population size when Shigella spp. became human-restricted pathogens, compared with their E. coli ancestor. In addition, the intracellular niche in which Shigella spp. began to thrive imposed a more relaxed selective pressure owing to abundant resources and a relative lack of competitors67,68.
Genomic insights into Shigella spp.
The advent of high-throughput whole-genome sequencing has permitted the detection of genomic variation in the form of single-nucleotide polymorphisms (SNPs) and accessory genome content. These techniques give us an unprecedented view of how Shigella spp. have emerged and been transmitted globally, and how antimicrobial resistance has swept through the population throughout the later part of the twentieth century.
Shigella sonnei. Although S. sonnei is the most common Shigella species in middle-income and high-income countries, the recent emergence of this species in transitional lower-income countries has highlighted the need for more effective surveillance systems and has opened new avenues of vaccine research69,70,71,72,73 (Box 4). In a key study, the genomes of 132 globally representative S. sonnei isolates were sequenced and analysed to investigate the recent evolution of the species22. Three main lineages of S. sonnei were identified (I, II and III), which share a most recent common ancestor <400 years ago. All lineages probably originated in Europe, as the oldest lineages, and the majority of all genetic diversity, were detected in European isolates (Fig. 2a). Although all three lineages were distributed in Europe, not all of them have spread globally. Isolates from Asia, Africa and South America were predominantly representatives of the more recently expanded lineage III. Lineage III (particularly clade Global III) emerged in the 1970s and spread internationally in the 1980s and 1990s, establishing more distant endemic populations in other regions of the world (Fig. 2a). Importantly, there was a correlation between global dissemination and the acquisition of resistance to multiple antimicrobials. Resistance was mediated through the gain of class II integrons and mutations in DNA gyrase subunit A (gyrA), which encodes the target protein for fluoroquinolones (a family of broad-spectrum antimicrobials). These modifications probably resulted from strong selective pressures induced by antimicrobial exposure; indeed, antimicrobial resistance may be advantageous in promoting post-symptomatic shedding of bacteria and sustained short-term transmission in the host population74.

a | A global map showing the spread of Shigella sonnei out of Europe, using data from Ref. 22. S. sonnei diverged into three main lineages (I, II and III) that have been circulating in Europe since the early nineteenth century (red). Years represent the estimated dates of introduction of these strains from Europe into new human populations. The most successful of these global lineages is lineage III, which harbours a combination of antimicrobial-resistance genes (bold dates indicate the introduction of the Global III clade). b | An unrooted phylogenetic tree showing the relationship between sequenced S. sonnei strains isolated in three different locations across Vietnam: Ho Chi Minh City in the south, Khanh Hoa province on the south-central coast and Hue in the central region; based on data from Ref. 75. The tree shows that strains from Ho Chi Minh City are frequently transferred to other Vietnamese cities and rarely form new populations. However, as highlighted by two clonal expansions in Khanh Hoa (Khanh Hoa 1 and Khanh Hoa 2) and one in Hue, pioneering S. sonnei strains can form new location-specific subpopulations. The ongoing selection of these organisms seems to be driven by antimicrobials, as there is evidence of homoplasies by the acquisition and maintenance of differing DNA gyrase subunit A (gyrA) mutations and of differing plasmids encoding extended-spectrum β-lactamases (ESBLs), which confer resistance to fluoroquinolones and third-generation cephalosporins, respectively. Strains harbouring ESBL-encoding plasmids are highlighted by background shading (blue for the incompatibility group I1 (IncI1) plasmid pKHSB1 encoding CTX-M-15, and red for the IncA/C plasmid encoding CTX-M-14). Part a is reproduced from Ref. 169, Nature Publishing Group.
A study that investigated more than 250 S. sonnei samples in Vietnam expanded the observations of the global study75. Genomic and phylogeographical analyses showed that the Global III lineage became established in Ho Chi Minh City following the reunification of the country in 1975. The founder clone later spread north to other provinces, where it established, albeit after multiple introductions, further discrete endemic populations (Fig. 2b). Clonal expansion in these regions contributed to the increase in S. sonnei-associated dysentery in Vietnam. In larger human populations, such as in Ho Chi Minh City, a series of bottlenecks in the bacterial population and the stepwise accumulation of antimicrobial resistance were observed, probably as a consequence of the strong selective pressure exerted by the high use of antimicrobials in the country. Furthermore, plasmid pDPT1, encoding an E5 type colicin (a bactericidal toxin with RNA degradation potency) and an associated immunity protein (protecting the producer from the activity of the corresponding colicin), became fixed in the Ho Chi Minh City population following the first selective sweep in 1994, providing a crucial selective advantage over other non-immune Shigella spp. and E. coli strains. In the 2006 selective sweep, the population acquired plasmid pKHSB1, which harbours an extended-spectrum β-lactamase (ESBL) gene. This explains the sudden increase in the isolation rate of cephalosporin-resistant S. sonnei in the following years in the region76. The acquisition of a plasmid conferring resistance to third-generation cephalosporins (Fig. 2b) reoccurred in satellite populations in the central region of Vietnam, namely in the Khanh Hoa province. Similarly, other signs of convergent evolution included the independent emergences of gyrA mutations in Ho Chi Minh City and other provinces, reducing the susceptibility to fluoroquinolones. With such a detailed understanding of the S. sonnei population in Vietnam, the authors suggested that S. sonnei could act as a sentinel organism for the surveillance of human enteric bacterial pathogens by providing a tractable window onto the circulating antimicrobial-resistance elements in other Gram-negative enteric bacteria in a specific region. Indeed, the transfer of third-generation cephalosporin resistance plasmids between S. sonnei and commensal E. coli in the human gut might occur, as the expansion of the S. sonnei population during an episode of infection greatly increases the chance of contact between these two organisms77.
Shigella flexneri. Alongside S. sonnei, S. flexneri remains a major aetiological agent of bacillary dysentery, particularly in low-income and middle-income countries. Much of our epidemiological knowledge about S. flexneri comes from serotyping data. S. flexneri serotypes differ in their O antigens, and there is experimental evidence that the O antigen conformation is important for invasion and the evasion of innate immunity78. However, serotype conversion (that is, the modification of the serotype in a clonal population) is well documented in S. flexneri and mediated by bacteriophages and plasmids carrying genes that contribute to variation of the O antigen structure. The bacteriophages often integrate as prophages into the chromosomal thrW tRNA site, for prophages carrying the glycosylation (gtr) operon, or into the argW tRNA site, for those carrying the O-acetylation (oac) gene, and lead to changes in the O antigen structure79,80. Many O antigen-modifying bacteriophages have been identified to date, including SfI, SfII, Sf6, SfIV, SfV and SfX, which convert S. flexneri Y into serotypes 1a, 2a, 3b, 4a, 5a and X, respectively81,82,83,84,85,86. Furthermore, several novel S. flexneri serotypes have been discovered in the past decade, which complicates the epidemiology and potential protective efficacy of any potential O antigen-based vaccines (Box 4).
The emergence of novel S. flexneri serotypes has been widely observed. For example, S. flexneri 1c was first identified in Bangladesh in the 1980s, and an unrelated clone of this serotype was then also found to be prevalent in rural northern Vietnam and several other Asian countries8,87,88. Furthermore, the emergence of S. flexneri 1d, X variant (Xv) and 4s has been reported in China89,90,91. Many of these novel serotypes harbour more than one O antigen-modifying operon, resulting in additional modifications in the already highly modified tetrasaccharide. For example, the introduction of gtr1C into S. flexneri 1a leads to the addition of a glucosyl group on the glucosyl-linked N-acetylglucosamine, effectively converting this serotype into the novel serotype 1c92. Unpredictably, the gtr1C cluster shares similarities with genes from Citrobacter koseri rather than with previously characterized orthologues in other S. flexneri serotypes. This suggests that S. flexneri can sample from a large pool of O antigen-modifying genes. Plasmid-mediated serotype conversion has also been reported in S. flexneri Xv, 4s and Yv. The plasmid-borne O antigen phosphoethanolamine transferase (opt) gene was found to be essential for the transfer of phosphoethanolamine (PEtN) to the second rhamnose (RhaII) and RhaIII of the O antigen in S. flexneri Xv and S. flexneri Yv, respectively93,94.
Molecular typing of S. flexneri has, to date, largely relied on pulsed-field gel electrophoresis (PFGE) and/or multilocus sequence typing (MLST), using the sequences of the seven housekeeping genes: adk, fumC, gyrB, icd, mdh, purA and recA95,96. MLST of more than 100 Asian S. flexneri isolates revealed that serotypes 1–5, X and Y belong to a discrete clonal complex (ST245 of the ST245 complex), whereas serotype 6 forms a distinct clonal complex (ST145 of the ST243 complex)97,98. Although the resolution of MLST for S. flexneri is limited because of an inadequate number of differentiating mutations in the selected housekeeping genes, especially for investigating local clonal expansion or fine-scaled phylogenetic relationships, this method has provided insights into the genetic relationship between major S. flexneri serotypes. For example, studies examining the spread of the epidemic S. flexneri clone ST91 in China have low resolution, but have aided the tracking of this pathogen across the region90. S. flexneri clone ST91, which was typed using another E. coli genotyping scheme90, is actually typed ST245 using the Shigella spp. MLST approach described above95. The alternative E. coli typing scheme relies on 15 housekeeping genes — aspC, clpX, fadD, icdA, lysP, mdh, uidA, arcA, aroE, cyaA, dnaG, grpE, mtlD, mutS and rpoS — and provides better resolution for MLST, especially for clonal populations, such as the S. flexneri ST245 complex. To obtain even higher resolution, this expanded MLST scheme was combined with PFGE to investigate the expansion of S. flexneri clone ST91. Somewhat atypically for members of the genus Shigella, S. flexneri clone ST91 underwent at least 57 independent serotype switching events during its clonal expansion in China90, illustrating the potential problem with using serotyping as a proxy for genetic relatedness. A major serotype conversion in the S. flexneri ST245 complex led to the rise of a novel variant, S. flexneri Xv, which then rapidly spread and became one of the most prevalent serotypes in China since 2000 (Ref. 90). The spread of S. flexneri Xv is concerning, as this serotype is resistant to several antimicrobials (see below).
Extensive serotype switching and the success of specific clones highlight the need for higher-resolution tracking and monitoring of S. flexneri. Whole-genome sequencing provides such higher-resolution data; for example, this method showed that S. flexneri ST91 serotype Xv had acquired a plasmid carrying opt, leading to O antigen modification, on three independent occasions94. Before the opt-harbouring plasmid was introduced, clone ST91 had already carried antimicrobial-resistance genes, including the SRL locus (a multidrug-resistance (MDR) genomic island harbouring resistance genes against tetracycline (tetACDR), streptomycin (aadA2), ampicillin (oxa1) and chloramphenicol (cat)), Tn7 (an MDR transposon carrying resistance genes against trimethoprim (dfrA1), streptothricin (sat1) and streptomycin (aadA1)) and two mutations in gyrA facilitating resistance against nalidixic acid. The rapid expansion of the ST91 clone in different geographical locations can be explained by O antigen switching and the evasion of pre-existing immunity in host populations, and by the ineffectiveness of antimicrobials owing to the MDR background, which promotes prolonged faecal shedding and sustained circulation74.
In addition to the substantial species shift observed in developing countries, S. flexneri epidemiology has also changed in certain populations in developed countries. The isolation rate of S. flexneri 3a has increased steadily in men who have sex with men (MSM) communities in Canada, England and Wales99,100. This increased isolation rate is not attributable to an introduction (or introductions) from the low-income countries, suggesting that the ecology of this particular variant may now be better adapted to transmission within MSM populations99. A recent genomic analysis of a global collection of this serotype indicated the emergence of an S. flexneri 3a lineage attributed to infections in MSM populations in higher-income countries101. This lineage has spread globally since its emergence in 1998, and as is common for current populations of Shigella spp., has acquired resistance to multiple antimicrobials, most notably azithromycin, a frequent antimicrobial treatment for sexually transmitted diseases, including gonorrhoea, syphilis and chlamydia. This change in antimicrobial susceptibility, seen as the response to selective pressure exerted by azithromycin treatment for comorbid infections, has contributed to the dominance of this organism in MSM populations101.
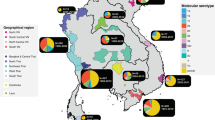
Studying the evolution and epidemiology of S. flexneri has proved complicated owing to serotype diversity, until a recent study of 351 whole-genome sequences from different serotypes of this species24. This study concluded that S. flexneri, with the exclusion of the diverging serotype 6, consists of seven phylogenetic groups (Fig. 3). Notably, these phylogenetic groups are inconsistent with serotype groupings and have arisen on several occasions between the 1300s and the 1800s24. The presence of numerous serotypes in all phylogenetic groups suggests that serotype switching is common, consistent with previous research90.

The figure shows a maximum likelihood phylogeny of Shigella flexneri serotypes, created from genome sequences of a representative global collection of 351 isolates of S. flexneri, spanning serotypes F1–F5, FX, FXv, FY and FYv. The isolates were collected from the main foci of endemic disease today (Africa, Asia, and South and Central America), and historical isolates from reference collections dating back to 1914 were also used. Phylogenetic groups (PGs) determined by Bayesian analysis of population structure clustering are boxed, and the geographical and serotypic composition of isolates in each PG are inlaid as pie charts. This figure is reproduced from Ref. 24.
This study also revealed substantial variability in the composition of S. flexneri virulence factors (for example, the genomic island SHI-1, and genes encoding iron uptake systems, such as the enterobactin genes and the ferric dicitrate transport (fec) locus) and antimicrobial-resistance genes (for example, the SRL island). SHI-1, SRL and enterobactin genes exclusively co-occur in phylogenetic group 3 (PG3), which is composed predominantly of S. flexneri serotype 2a, and this may account for the enhanced virulence and international dominance of this serotype24. The accumulation of antimicrobial-resistance genes in S. flexneri over the past three decades is considered to be essential for maintaining successful lineages. However, unlike for S. sonnei, this has neither led to the displacement of pre-existing antimicrobial-susceptible lineages nor resulted in substantial international transmission, with the exception of the global spread of the MSM-associated serotype S. flexneri 3a22,101. This finding supports the concept of longer-term colonization, in which diverse populations of both antimicrobial-resistant and antimicrobial-susceptible lineages co-circulate in endemic locations. These data also imply that S. flexneri is persisting in the environment, where selection for antimicrobial resistance may be less favourable.
Shigella dysenteriae and Shigella boydii. As S. dysenteriae and S. boydii account for <10% of the cases of shigellosis, research into these organisms is less of a priority for global health research19. Furthermore, research is complicated by the sheer diversity of serotypes in these species (15 for S. dysenteriae, and 20 for S. boydii) and by a lack of large, well-characterized, geographically diverse collections of isolates.
One of the best studied S. dysenteriae serotypes is S. dysenteriae serotype 1, which induces a more severe disease phenotype than other Shigella spp. and serotypes. The hypervirulence of S. dysenteriae 1 can be explained by the release of Stx, which inhibits protein synthesis102, and could also be partly attributed to the presence of the shu cluster, which is upregulated in response to the host body temperature and uses haem as an iron source, leading to better adaptation in the human host103,104. Notably, EHEC O157:H7 also carries the stx-encoding prophage and the shu cluster and can also cause severe complications, demonstrating that S. dysenteriae 1 and EHEC have inherited and maintained these virulence factors from a common ancestor103. A detailed study of the proteomic profile of S. dysenteriae 1 revealed several proteins that are expressed preferentially in the host environment, including the Mxi–Spa T3SS, and proteins that are involved in anaerobic energy metabolism, acid resistance, modulation of the outer membrane and modification of peptidoglycan structure105. The last reported dysentery outbreak caused by S. dysenteriae 1 occurred in Sierra Leone in 1999 (Ref. 106); since then, the prevalence of disease caused by this serotype has become negligible7. The most recent pandemic clone of S. dysenteriae 1 emerged from a common ancestor at the beginning of the twentieth century, which is much more recent than the common ancestor of the major current clones of both S. sonnei and S. flexneri23. Two lineages of S. dysenteriae 1 rapidly disseminated intercontinentally, facilitated by poor sanitation and excessive human migration during the two world wars23. Furthermore, these lineages independently acquired antimicrobial-resistance genes, seemingly through selection during outbreaks. However, directional selection in the chromosome is unlikely to occur, as inactivating mutations equally affected all metabolic functions23. Such unbiased mutations and a generally high mutation rate suggest that S. dysenteriae 1 could be maintained and transmitted through long-term human carriage, similarly to S. Typhi107. This theory may explain the infrequent isolation rate of S. dysenteriae 1 but its ability to cause devastating outbreaks in vulnerable populations.
S. boydii was first isolated in the Indian subcontinent and seems to be restricted to this region, as it is rarely isolated elsewhere108. However, a new serotype, S. boydii serotype 20, was discovered in travellers to Central America, which demonstrates that the epidemiology of S. boydii is more complicated than previously described or predicted109,110. In developing countries, S. boydii serotype 2 is the most prevalent and clinically relevant serotype, with an isolation rate of ∼50% of all S. boydii isolates111,112,113. Other S. boydii serotypes are rare, but several O antigen clusters from S. boydii have been transferred to different members of the genus Escherichia; for example, S. boydii O antigens 10 and 15 can be found in EHEC and Escherichia fergusonii, respectively114,115.
Linking genomics and pathogenesis. As members of the genus Shigella do not form a single monophyletic group, distinct Shigella spp. can differ in both physiology and pathogenesis. Shigella pathogenesis mainly relies on the Mxi–Spa T3SS and its effector proteins, so the subtle phenotypic variation seen in host–pathogen interactions could be caused by the gain and/or loss of other genetic material. Alternatively, convergent evolution has enabled several Shigella spp. to adopt an intracellular lifestyle, exemplified by the independent loss of flagella, fimbriae, and metabolic pathways, such as LDC, carbon utilization and transporters (of carbohydrates, amino acids and amines)20,57,58.
Information related to pathogenic differences between and in the various Shigella spp. is scarce, because most experimental studies have used S. flexneri. The other species are used less frequently for experiments owing to the instability of their virulence plasmid (S. sonnei), their unavailability or simply the fact that they are less of a global health priority (S. dysenteriae and S. boydii). Nevertheless, recent findings have shed more light on variation between the different species. For example, it has been shown that bacteriophage-borne glycosylation of the O antigen in S. flexneri 5 optimizes its length, enhancing the exposure of the T3SS apparatus without making it more of a target for host antibodies78. There is a fine balance between virulence and immune protection: in S. flexneri serotype 2a, the plasmid pHS-2 carries a gene that results in very long O antigen chains which mask the cell from serum killing, whereas the chromosomally determined chains are short and unmask the T3SS structure to enhance functionality116,117. S. sonnei uses a different mechanism: this species expresses a group 4 capsule composed of pINV-borne O antigen sugars118. Removal of the capsule increases invasiveness and inflammation, but decreases the capacity to spread from cell to cell and increases susceptibility to immune killing, thus showing that the capsule is crucial for the balance between virulence and immune evasion. The g4c operon, which encodes this capsule, is intact in S. sonnei but is lacking in S. flexneri serotype 2a owing to a frame-shift deletion118. This variance may explain, in part, the differential virulence and immunogenicity of Shigella spp. Differences also exist in the use of adhesins for attachment to host cells. In S. flexneri, the T3SS-dependent protein IcsA (also known as VirG) mediates both adhesion and actin-based motility, facilitating invasion into the host cell119. In S. sonnei, an additional multivalent adhesion molecule (MAM), SSO1327, has been shown to function as a non-redundant adhesin to IcsA120. Deletion of either of these proteins in S. sonnei reduces attachment and invasion in vivo. The gene encoding SSO1327 is intact in isolates of S. sonnei, S. dysenteriae and S. boydii, but is a pseudogene in S. flexneri120. This difference in adhesin composition may explain the differential interaction of S. flexneri and S. sonnei with the host; for example, bile salts stimulate the attachment of S. flexneri but impede the attachment of S. sonnei119,120.
Conclusions and outlook
The evolutionary history of the bacterial genus Shigella is shaped by three key processes. First, Shigella spp. have arisen from different ancestral E. coli strains on several independent occasions. Second, the acquisition of plasmids that encode virulence genes into numerous ancestral Shigella strains were 'foothold moments' in their evolution; similar observations have been made for other enteric human pathogens, such as Y. pestis and, more recently, Yersinia enterocolitica121. The acquisition and adaptation of these plasmids has shaped all existing Shigella spp. Third, convergent evolution, by the independent acquisition of mobile elements and loss of gene function, has further transformed these organisms to become restricted to humans and exquisitely customized to interact with the human intestinal mucosa.
The shift in dominance from S. flexneri to S. sonnei in economically transitioning nations should prompt more in-depth studies of the evolution and epidemiology of these two species. Although whole-genome analyses of S. sonnei and S. flexneri provided insights into their evolution and spread, comparatively little is currently understood about S. dysenteriae and S. boydii. As genome sequencing becomes more accessible and affordable, it will be essential to apply this tool to investigate the evolution of other Shigella spp. locally and globally. Greater insights into the epidemiology of these species should aid their control in disease-burdened regions as well as facilitate vaccine development and distribution. Conserved proteins across all Shigella spp., such as the T3SS proteins IpaB and IpaD, have been identified as promising candidates for a serotype-independent pan-Shigella vaccine. Preclinical testing in mice indicates that IpaB and IpaD are safe and provide substantial protection against challenges with S. flexneri and S. sonnei122,123,124. However, the utility of these antigens needs to be further validated in human studies. Owing to the multiple serotypes of S. flexneri, their complex evolutionary history and the extent of horizontal gene transfer, studying this species is more challenging. Further, S. boydii and S. dysenteriae research has been neglected owing to their lower disease burdens. S. dysenteriae serotype 1, in particular, warrants more attention because it can cause severe disease and has the potential to cause major epidemics. Future laboratory research should be integrated with genomics to address the survival, transmission and evolution of Shigella spp., focusing on how their lifestyle in the environment can affect disease epidemiology and global public health.
References
World Health Organization. World Health Statistics 2014. (WHO, 2014).
Liu, L. et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 379, 2151–2161 (2012).
Fischer Walker, C. L., Perin, J., Aryee, M. J., Boschi-Pinto, C. & Black, R. E. Diarrhea incidence in low- and middle-income countries in 1990 and 2010: a systematic review. BMC Publ. Health 12, 220 (2012).
Kotloff, K. L. et al. The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: epidemiologic and clinical methods of the case/control study. Clin. Infect. Dis. 55 (Suppl. 4), S232–S245 (2012).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case–control study. Lancet 382, 209–222 (2013).
Kotloff, K. L. et al. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77, 651–666 (1999).
Bardhan, P., Faruque, A. S., Naheed, A. & Sack, D. A. Decrease in shigellosis-related deaths without Shigella spp.-specific interventions, Asia. Emerg. Infect. Dis. 16, 1718–1723 (2010).
von Seidlein, L. et al. A multicentre study of Shigella diarrhoea in six Asian countries: disease burden, clinical manifestations, and microbiology. PLoS Med. 3, e353 (2006).
Nataro, J. P. & Kaper, J. B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201 (1998).
Croxen, M. A. & Finlay, B. B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38 (2010).
Croxen, M. A. et al. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26, 822–880 (2013).
Kothary, M. & Babu, U. Infective dose of foodborne pathogens in volunteers: a review. J. Food Saf. 21, 49–68 (2001).
Shiga, K. Ueber den erreger der dysenterie in Japan. Zentralbl. Bakteriol. Mikrobiol. 23, 599–600 (1898).
Trofa, A. F., Ueno-olsen, H., Oiwa, R. & Yoshikawa, M. Dr. Kiyoshi Shiga: discoverer of the dysentery Bacillus. Clin. Infect. Dis. 29, 1303–1306 (1999).
Flexner, S. On the etiology of tropical dysentery. Philadelphia Med. J. 6, 414–421 (1900).
Barceloux, D. G. Shigella species (Shiga enterotoxins) in Medical Toxicology of Natural Substances: Foods, Fungi, Medicinal Herbs, Plants, and Venomous Animals. Ch. 20, 150–155. (Wiley & Sons, 2008).
Dodd, C. E. & Jones, D. A numerical taxonomic study of the genus Shigella. J. Gen. Microbiol. 128, 1933–1957 (1982).
Falkow, S. Activity of lysine decarboxylase as an aid in the identification of Salmonellae and Shigellae. Am. J. Clin. Pathol. 29, 598–600 (1958).
Gu, B. et al. Comparison of the prevalence and changing resistance to nalidixic acid and ciprofloxacin of Shigella between Europe–America and Asia–Africa from 1998 to 2009. Int. J. Antimicrob. Agents 40, 9–17 (2012).
Yang, F. et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 33, 6445–6458 (2005). A seminal paper aimed at genomic comparison of four Shigella spp.: S. flexneri, S. sonnei, S. dysenteriae and S. boydii . The analysis provides evidence for convergent evolution among Shigella spp., by gene gain and gene loss.
Buchrieser, C. et al. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38, 760–771 (2000).
Holt, K. E. et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 44, 1056–1059 (2012). This key investigation reconstructed the phylogenetic structure and geographical spread of S. sonnei on a global scale, emphasizing that the successful introduction of this species from Europe into other distant regions is facilitated by antimicrobial resistance.
Rohmer, L. et al. Genomic analysis of the emergence of 20th century epidemic dysentery. BMC Genomics 15, 355 (2014). The first work to use phylogenomics to understand the evolutionary history of the pandemic S. dysenteriae 1, highlighting the fact that its recent emergence is coupled with independent acquisitions of antimicrobial-resistance genes.
Connor, T. R. et al. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. eLife 4, e07335 (2015). The original whole-genome study on the diversity of S. flexneri globally, showing that serotype switching is common among phylogenetic groups. The study suggests that the long-term success of this species is linked to its local persistence.
Lan, R., Lumb, B., Ryan, D. & Reeves, P. R. Molecular evolution of large virulence plasmid in Shigella clones and enteroinvasive Escherichia coli. Infect. Immun. 69, 6303–6309 (2001).
Pupo, G. M., Lan, R. & Reeves, P. R. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl Acad. Sci. USA 97, 10567–10572 (2000). The first article to demonstrate the multiple origins of Shigella spp. from E. coli , on the basis of a phylogenetic analysis of eight housekeeping genes.
Yang, J. et al. Revisiting the molecular evolutionary history of Shigella spp. J. Mol. Evol. 64, 71–79 (2007).
Hyma, K. E. et al. Evolutionary genetics of a new pathogenic Escherichia species: Escherichia albertii and related Shigella boydii strains. J. Bacteriol. 187, 619–628 (2005).
Shepherd, J. G., Wang, L. & Reeves, P. R. Comparison of O-antigen gene clusters of Escherichia coli (Shigella) sonnei and Plesiomonas shigelloides O17: sonnei gained its current plasmid-borne O-antigen genes from P. shigelloides in a recent event. Infect. Immun. 68, 6056–6061 (2000).
Sims, G. E. & Kim, S. H. Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs). Proc. Natl Acad. Sci. USA 108, 8329–8334 (2011).
Touchon, M. et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5, e1000344 (2009).
Kaas, R. S., Friis, C., Ussery, D. W. & Aarestrup, F. M. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13, 577 (2012).
Zhang, Y. & Lin, K. A phylogenomic analysis of Escherichia coli / Shigella group: implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol. Biol. 12, 174 (2012).
Sahl, J. W. et al. Defining the phylogenomics of Shigella species: a pathway to diagnostics. J. Clin. Microbiol. 53, 951–960 (2015). This paper utilizes the most complete collection of Shigella and E. coli isolates for phylogenomic analysis, the results of which both consolidate the evidence for there being multiple origins of Shigella spp. from E. coli , and point to reliable genetic markers for differentiating major Shigella clades.
van den Beld, M. J. & Reubsaet, F. A. Differentiation between Shigella, enteroinvasive Escherichia coli (EIEC) and noninvasive Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 31, 899–904 (2012).
Lan, R. et al. Molecular evolutionary relationships of enteroinvasive Escherichia coli and Shigella spp. Infect. Immun. 72, 5080–5088 (2004).
Schroeder, G. N. & Hilbi, H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156 (2008). This extensive review details the molecular pathogenesis of Shigella spp.
Venkatesan, M. M. et al. Complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri. Infect. Immun. 69, 3271–3285 (2001). Together with reference 21, this report describes the first sequence analysis of the Shigella virulence plasmid, highlighting the rich repertoire of virulence factors encoded by this plasmid.
Jiang, Y. et al. The complete sequence and analysis of the large virulence plasmid pSS of Shigella sonnei. Plasmid 54, 149–159 (2005).
Makino, S., Sasakawa, C. & Yoshikawa, M. Genetic relatedness of the basic replicon of the virulence plasmid in shigellae and enteroinvasive Escherichia coli. Microb. Pathog. 5, 267–274 (1988).
Lan, R. & Reeves, P. R. Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect. 4, 1125–1132 (2002).
Al-Hasani, K. et al. The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect. Immun. 68, 2457–2463 (2000).
Ingersoll, M., Groisman, E. A. & Zychlinsky, A. Pathogenicity islands of Shigella. Curr. Top. Microbiol. Immunol. 264, 49–65 (2002).
Luck, S. N., Turner, S. A., Rajakumar, K., Sakellaris, H. & Adler, B. Ferric dicitrate transport system (Fec) of Shigella flexneri 2a YSH6000 is encoded on a novel pathogenicity island carrying multiple antibiotic resistance genes. Infect. Immun. 69, 6012–6021 (2001).
Vokes, S. A., Reeves, S. A., Torres, A. G. & Payne, S. M. The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol. Microbiol. 33, 63–73 (1999).
Fisher, C. R. et al. Genetics and virulence association of the Shigella flexneri Sit iron transport system. Infect. Immun. 77, 1992–1999 (2009).
Gupta, S. K. et al. Short report: emergence of Shiga toxin 1 genes within Shigella dysenteriae type 4 isolates from travelers returning from the Island of Hispañola. Am. J. Trop. Med. Hyg. 76, 1163–1165 (2007).
Gray, M. D. et al. Clinical isolates of Shiga toxin 1a-producing Shigella flexneri with an epidemiological link to recent travel to Hispañiola. Emerg. Infect. Dis. 20, 1669–1677 (2014).
Gray, M. D. et al. Prevalence of Shiga toxin-producing Shigella species isolated from French travellers returning from the Caribbean: an emerging pathogen with international implications. Clin. Microbiol. Infect. 21, 765.e9–765.e14 (2015).
Gray, M. et al. Stx-producing Shigella species from patients in Haiti: an emerging pathogen with the potential for global spread. Open Forum Infect. Dis. 2, ofv134 (2015).
Siguier, P., Gourbeyre, E. & Chandler, M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891 (2014).
Wagner, A. & de la Chaux, N. Distant horizontal gene transfer is rare for multiple families of prokaryotic insertion sequences. Mol. Genet. Genom. 280, 397–408 (2008).
Ochman, H. & Davalos, L. M. The nature and dynamics of bacterial genomes. Science 311, 1730–1733 (2006).
Eiglmeier, K. et al. The decaying genome of Mycobacterium leprae. Lepr. Rev. 72, 387–398 (2001).
Parkhill, J. et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523–527 (2001).
Holt, K. E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40, 987–993 (2008).
Feng, Y., Chen, Z. & Liu, S.-L. Gene decay in Shigella as an incipient stage of host-adaptation. PLoS ONE 6, e27754 (2011).
Bravo, V., Puhar, A., Sansonetti, P., Parsot, C. & Toro, C. S. Distinct mutations led to inactivation of type 1 fimbriae expression in Shigella spp. PLoS ONE 10, e0121785 (2015).
Ramos, H. C., Rumbo, M. & Sirard, J. C. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 12, 509–517 (2004).
Monk, J. M. et al. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc. Natl Acad. Sci. USA 110, 20338–20343 (2013).
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K. & Fasano, A. 'Black holes' and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl Acad. Sci. USA 95, 3943–3948 (1998).
Prunier, A. L. et al. nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153, 2363–2372 (2007).
Prosseda, G. et al. Shedding of genes that interfere with the pathogenic lifestyle: the Shigella model. Res. Microbiol. 163, 399–406 (2012).
Nakata, N. et al. The absence of a surface protease, OmpT, determines the intercellular spreading ability of Shigella: the relationship between the ompT and kcpA loci. Mol. Microbiol. 9, 459–468 (1993).
Zhao, G. et al. A novel anti-virulence gene revealed by proteomic analysis in Shigella flexneri 2a. Proteome Sci. 8, 30 (2010).
Barbagallo, M. et al. A new piece of the Shigella pathogenicity puzzle: spermidine accumulation by silencing of the speG gene. PLoS ONE 6, e27226 (2011).
Hershberg, R., Tang, H. & Petrov, D. A. Reduced selection leads to accelerated gene loss in Shigella. Genome Biol. 8, R164 (2007).
Balbi, K. J., Rocha, E. P. C. & Feil, E. J. The temporal dynamics of slightly deleterious mutations in Escherichia coli and Shigella spp. Mol. Biol. Evol. 26, 345–355 (2009).
Vinh, H. et al. A changing picture of shigellosis in southern Vietnam: shifting species dominance, antimicrobial susceptibility and clinical presentation. BMC Infect. Dis. 9, 204 (2009).
Ud-Din, A. I. M. S. et al. Changing trends in the prevalence of Shigella species: emergence of multi-drug resistant Shigella sonnei biotype g in Bangladesh. PLoS ONE 8, e82601 (2013).
Bangtrakulnonth, A. et al. Shigella from humans in Thailand during 1993 to 2006: spatial-time trends in species and serotype distribution. Foodborne Pathog. Dis. 5, 773–784 (2008).
Wei, H. L., Wang, Y. W., Li, C. C., Tung, S. K. & Chiou, C. S. Epidemiology and evolution of genotype and antimicrobial resistance of an imported Shigella sonnei clone circulating in central Taiwan. Diagn. Microbiol. Infect. Dis. 58, 469–475 (2007).
Nygren, B. L. et al. Foodborne outbreaks of shigellosis in the USA, 1998–2008. Epidemiol. Infect. 141, 233–241 (2013).
Vinh, H. et al. Treatment of bacillary dysentery in Vietnamese children: two doses of ofloxacin versus 5-days nalidixic acid. Trans. R. Soc. Trop. Med. Hyg. 94, 323–326 (2000).
Holt, K. E. et al. Tracking the establishment of local endemic populations of an emergent enteric pathogen. Proc. Natl Acad. Sci. USA 110, 17522–17527 (2013). The first study to detail the microevolution of S. sonnei in a developing region in high resolution, pointing out that antimicrobial resistance is a crucial factor for understanding the clonal expansion of this species.
Nguyen, N. T. K. et al. The sudden dominance of blaCTX–M harbouring plasmids in Shigella spp. circulating southern Vietnam. PLoS Negl. Trop. Dis. 4, e702 (2010).
Rashid, H. & Rahman, M. Possible transfer of plasmid mediated third generation cephalosporin resistance between Escherichia coli and Shigella sonnei in the human gut. Infect. Genet. Evol. 30, 15–18 (2015).
West, N. P. et al. Optimization of virulence functions through glucosylation of Shigella LPS. Science 307, 1313–1317 (2005).
Allison, G. E. & Verma, N. K. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol. 8, 17–23 (2000).
Bastin, D. A., Lord, A. & Verma, N. K. Cloning and analysis of the glucosyl transferase gene encoding type I antigen in Shigella flexneri. FEMS Microbiol. Lett. 156, 133–139 (1997).
Sun, Q. et al. Isolation and genomic characterization of SfI, a serotype-converting bacteriophage of Shigella flexneri. BMC Microbiol. 13, 39 (2013).
Mavris, M., Manning, P. A. & Morona, R. Mechanism of bacteriophage SfII-mediated serotype conversion in Shigella flexneri. Mol. Microbiol. 26, 939–950 (1997).
Clark, C., Beltrame, J. & Manning, P. The oac gene encoding a lipopolysaccharide O-antigen acetylase maps adjacent to the integrase-encoding gene on the genome of Shigella flexneri bacteriophage Sf6. Gene 107, 43–52 (1991).
Jakhetia, R., Talukder, K. A. & Verma, N. K. Isolation, characterization and comparative genomics of bacteriophage SfIV: a novel serotype converting phage from Shigella flexneri. BMC Genomics 14, 677 (2013).
Huan, P. T., Bastin, D. A., Whittle, B. L., Lindberg, A. A. & Verma, N. K. Molecular characterization of the genes involved in O-antigen modification, attachment, integration and excision in Shigella flexneri bacteriophage SfV. Gene 195, 217–227 (1997).
Guan, S., Bastin, D. & Verma, N. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology 145, 1263–1273 (1999).
Wehler, T. & Carlin, N. I. Structural and immunochemical studies of the lipopolysaccharide from a new provisional serotype of Shigella flexneri. Eur. J. Biochem. 176, 471–476 (1988).
Stagg, R. M., Cam, P. D. & Verma, N. K. Identification of newly recognized serotype 1c as the most prevalent Shigella flexneri serotype in northern rural Vietnam. Epidemiol. Infect. 136, 1134–1140 (2008).
Luo, X. et al. Emergence of a novel Shigella flexneri serotype 1d in China. Diagn. Microbiol. Infect. Dis. 74, 316–319 (2012).
Ye, C. et al. Emergence of a new multidrug-resistant serotype X variant in an epidemic clone of Shigella flexneri. J. Clin. Microbiol. 48, 419–426 (2010).
Qiu, S. et al. Emergence of a novel Shigella flexneri serotype 4s strain that evolved from a serotype X variant in China. J. Clin. Microbiol. 49, 1148–1150 (2011).
Stagg, R. M. et al. A novel glucosyltransferase involved in O-antigen modification of Shigella flexneri serotype 1c. J. Bacteriol. 191, 6612–6617 (2009).
Sun, Q. et al. Identification and characterization of a novel Shigella flexneri serotype Yv in China. PLoS ONE 8, e70238 (2013).
Zhang, N. et al. Genomic portrait of the evolution and epidemic spread of a recently emerged multidrug-resistant Shigella flexneri clone in China. J. Clin. Microbiol. 52, 1119–1126 (2014).
Lacher, D. W., Steinsland, H., Blank, T. E., Donnenberg, M. S. & Whittam, T. S. Molecular evolution of typical enteropathogenic Escherichia coli: clonal analysis by multilocus sequence typing and virulence gene allelic profiling. J. Bacteriol. 189, 342–350 (2007).
Ribot, E. M. et al. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog. Dis. 3, 59–67 (2006).
Wirth, T. et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151 (2006).
Choi, S. Y. et al. Multilocus sequence typing analysis of Shigella flexneri isolates collected in Asian countries. J. Med. Microbiol. 56, 1460–1466 (2007).
Borg, M. L. et al. Ongoing outbreak of Shigella flexneri serotype 3a in men who have sex with men in England and Wales, data from 2009–2011. Euro Surveill. 17, 20137 (2012).
Ratnayake, R. Allard, R. & Pilon, P. A. Shifting dominance of Shigella species in men who have sex with men. Epidemiol. Infect. 140, 2082–2086 (2012).
Baker, K. S. et al. Intercontinental dissemination of azithromycin-resistant shigellosis through sexual transmission: a cross-sectional study. Lancet Infect. Dis. 15, 913–921 (2015).
Khan, W. A., Griffiths, J. K. & Bennish, M. L. Gastrointestinal and extra-intestinal manifestations of childhood shigellosis in a region where all four species of Shigella are endemic. PLoS ONE 8, e64097 (2013).
Wyckoff, E. E. et al. Structure of the Shigella dysenteriae haem transport locus and its phylogenetic distribution in enteric bacteria. Mol. Microbiol. 28, 1139–1152 (1998).
Kouse, A. B., Righetti, F., Kortmann, J., Narberhaus, F. & Murphy, E. R. RNA-mediated thermoregulation of iron-acquisition genes in Shigella dysenteriae and pathogenic Escherichia coli. PLoS ONE 8, e63781 (2013).
Kuntumalla, S. et al. In vivo versus in vitro protein abundance analysis of Shigella dysenteriae type 1 reveals changes in the expression of proteins involved in virulence, stress and energy metabolism. BMC Microbiol. 11, 147 (2011).
Guerin, P. J. et al. Shigella dysenteriae serotype 1 in west Africa: intervention strategy for an outbreak in Sierra Leone. Lancet 362, 705–706 (2003).
Roumagnac, P. et al. Evolutionary history of Salmonella Typhi. Science 314, 1301–1304 (2006).
Khan, M. U., Roy, N. C., Islam, R., Huq, I. & Stoll, B. Fourteen years of shigellosis in Dhaka: an epidemiological analysis. Int. J. Epidemiol. 14, 607–613 (1985).
Kalluri, P. et al. Epidemiological features of a newly described serotype of Shigella boydii. Epidemiol. Infect. 132, 579–583 (2004).
Woodward, D. L. et al. Identification and characterization of Shigella boydii 20 serovar nov., a new and emerging Shigella serotype. J. Med. Microbiol. 54, 741–748 (2005).
Smith, A. M. et al. Analysis of a temporal cluster of Shigella boydii isolates in Mpumalanga, South Africa, November to December 2007. J. Infect. Dev. Ctries 3, 65–70 (2009).
El-Gendy, A. M. et al. Genetic diversity and antibiotic resistance in Shigella dysenteriae and Shigella boydii strains isolated from children aged <5 years in Egypt. Epidemiol. Infect. 140, 299–310 (2012).
Livio, S. et al. Shigella isolates from the global enteric multicenter study inform vaccine development. Clin. Infect. Dis. 59, 933–941 (2014). Using data from the largest prospective paediatric diarrhoea study, this paper provides an up-to-date description for the prevalence of different Shigella spp. in developing countries.
Iguchi, A., Iyoda, S., Seto, K. & Ohnishi, M. Emergence of a novel Shiga toxin-producing Escherichia coli O serogroup cross-reacting with Shigella boydii type 10. J. Clin. Microbiol. 49, 3678–3680 (2011).
Azmuda, N. et al. Evidence of interspecies O antigen gene cluster transfer between Shigella boydii 15 and Escherichia fergusonii. Apmis 120, 959–966 (2012).
Hong, M. & Payne, S. M. Effect of mutations in Shigella flexneri chromosomal and plasmid-encoded lipopolysaccharide genes on invasion and serum resistance. Mol. Microbiol. 24, 779–791 (1997).
Morona, R., Daniels, C. & Van Den Bosch, L. Genetic modulation of Shigella flexneri 2a lipopolysaccharide O antigen modal chain length reveals that it has been optimized for virulence. Microbiology 149, 925–939 (2003).
Caboni, M. et al. An O antigen capsule modulates bacterial pathogenesis in Shigella sonnei. PLoS Pathog. 11, e1004749 (2015).
Brotcke Zumsteg, A., Goosmann, C., Brinkmann, V., Morona, R. & Zychlinsky, A. IcsA is a Shigella flexneri adhesin regulated by the type III secretion system and required for pathogenesis. Cell Host Microbe 15, 435–445 (2014).
Mahmoud, R. Y. et al. The multivalent adhesion molecule SSO1327 plays a key role in Shigella sonnei pathogenesis. Mol. Microbiol. 99, 658–673 (2016).
Reuter, S. et al. Parallel independent evolution of pathogenicity within the genus Yersinia. Proc. Natl Acad. Sci. USA 111, 6768–6773 (2014).
Martinez-Becerra, F. J. et al. Broadly protective Shigella vaccine based on type III secretion apparatus proteins. Infect. Immun. 80, 1222–1231 (2012).
Heine, S. J. et al. Intradermal delivery of Shigella IpaB and IpaD type III secretion proteins: kinetics of cell recruitment and antigen uptake, mucosal and systemic immunity, and protection across serotypes. J. Immunol. 192, 1630–1640 (2014).
Heine, S. J. et al. Shigella IpaB and IpaD displayed on L. lactis bacterium-like particles induce protective immunity in adult and infant mice. Immunol. Cell Biol. 93, 641–652 (2015).
Carayol, N. & Tran Van Nhieu, G. Tips and tricks about Shigella invasion of epithelial cells. Curr. Opin. Microbiol. 16, 32–37 (2013).
Sansonetti, P. J. et al. Infection of rabbit Peyer's patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect. Immun. 64, 2752–2764 (1996).
Zychlinsky, A., Prevost, M. & Sansonetti, P. Shigella flexneri induces apoptosis in infected macrophages. Nature 358, 167–169 (1992).
Hilbi, H. et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 273, 32895–32900 (1998).
Lafont, F., Tran Van Nhieu, G., Hanada, K., Sansonetti, P. & van der Goot, F. G. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44–IpaB interaction. EMBO J. 21, 4449–4457 (2002).
Skoudy, A. et al. CD44 binds to the Shigella IpaB protein and participates in bacterial invasion of epithelial cells. Cell. Microbiol. 2, 19–33 (2000).
Watarai, M., Funato, S. & Sasakawa, C. Interaction of Ipa proteins of Shigella flexneri with α5β1 integrin promotes entry of the bacteria into mammalian cells. J. Exp. Med. 183, 991–999 (1996).
Mounier, J. et al. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 5, e1000271 (2009).
Handa, Y. et al. Shigella IpgB1 promotes bacterial entry through the ELMO–Dock180 machinery. Nat. Cell Biol. 9, 121–128 (2007).
Niebuhr, K. et al. Conversion of PtdIns(4,5)P2 into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J. 21, 5069–5078 (2002).
Park, H., Valencia-Gallardo, C., Sharff, A., Tran Van Nhieu, G. & Izard, T. Novel vinculin binding site of the IpaA invasin of Shigella. J. Biol. Chem. 286, 23214–23221 (2011).
Yoshida, S. et al. Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 21, 2923–2935 (2002).
Romero, S. et al. ATP-mediated Erk1/2 activation stimulates bacterial capture by filopodia, which precedes Shigella invasion of epithelial cells. Cell Host Microbe 9, 508–519 (2011).
Fernandez-Prada, C. M. et al. Shigella flexneri IpaH7.8 facilitates escape of virulent bacteria from the endocytic vacuoles of mouse and human macrophages. Infect. Immun. 68, 3608–3619 (2000).
Harrington, A., Darboe, N. & Kenjale, R. Characterization of the interaction of single tryptophan containing mutants of IpaC from Shigella flexneri with phospholipid membranes. Biochemistry 45, 626–636 (2006).
Egile, C., Loisel, T. & Laurent, V. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146, 1319–1332 (1999).
Iwai, H. et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130, 611–623 (2007).
Pendaries, C. et al. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034 (2006).
Kim, M. et al. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459, 578–582 (2009).
Mounier, J. et al. Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 12, 381–389 (2012).
Dong, N. et al. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150, 1029–1041 (2012).
Fasano, A., Noriega, F. R., Liao, F. M., Wang, W. & Levine, M. M. Effect of Shigella enterotoxin 1 (ShET1) on rabbit intestine in vitro and in vivo. Gut 40, 505–511 (1997).
Nataro, J. P. et al. Identification and cloning of a novel plasmid-encoded enterotoxin of enteroinvasive Escherichia coli and Shigella strains. Infect. Immun. 63, 4721–4728 (1995).
Faherty, C. S. et al. Chromosomal and plasmid-encoded factors of Shigella flexneri induce secretogenic activity ex vivo. PLoS ONE 7, e49980 (2012).
Ashida, H. et al. Shigella deploy multiple countermeasures against host innate immune responses. Curr. Opin. Microbiol. 14, 16–23 (2011).
Sansonetti, P. J. et al. Caspase-1 activation of IL-1β and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12, 581–590 (2000).
Sansonetti, P. & Arondel, J. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect. Immun. 67, 1471–1480 (1999).
Kim, D. W. et al. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl Acad. Sci. USA 102, 14046–14051 (2005).
Arbibe, L. et al. An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8, 47–56 (2007).
Newton, H. J. et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6, e1000898 (2010).
Salgado-Pabón, W., Konradt, C., Sansonetti, P. J. & Phalipon, A. New insights into the crosstalk between Shigella and T lymphocytes. Trends Microbiol. 22, 192–198 (2014).
Konradt, C. et al. The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe 9, 263–272 (2011).
Salgado-Pabón, W. et al. Shigella impairs T lymphocyte dynamics in vivo. Proc. Natl Acad. Sci. USA 110, 4458–4463 (2013).
Nothelfer, K. et al. B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection. J. Exp. Med. 211, 1215–1229 (2014).
Sansonetti, P. J., Kopecko, D. J. & Formal, S. B. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 35, 852–860 (1982).
Sansonetti, P. J., Kopecko, D. J. & Formal, S. B. Shigella sonnei plasmids: evidence that a large plasmid is necessary for virulence. Infect. Immun. 34, 75–83 (1981). This is the first study to demonstrate that the large plasmid is necessary for virulence in Shigella spp.
Rehel, N. & Szatmari, G. Characterization of the stable maintenance of the Shigella flexneri plasmid pHS-2. Plasmid 36, 183–190 (1996).
Stieglitz, H. & Lipsky, P. Association between reactive arthritis and antecedent infection with Shigella flexneri carrying a 2-MD plasmid and encoding an HLA-B27 mimetic epitope. Arthritis Rheum. 36, 1387–1391 (1993).
Gaston, J. Shigella induced reactive arthritis. Ann. Rheum. Dis. 64, 517–518 (2005).
Hannu, T., Mattila, L., Siitonen, A. & Leirisalo-Repo, M. Reactive arthritis attributable to Shigella infection: a clinical and epidemiological nationwide study. Ann. Rheum. Dis. 64, 594–598 (2005).
Calcuttawala, F., Hariharan, C., Pazhani, G. P., Ghosh, S. & Ramamurthy, T. Activity spectrum of colicins produced by Shigella sonnei and genetic mechanism of colicin resistance in conspecific S. sonnei strains and Escherichia coli. Antimicrob. Agents Chemother. 59, 152–158 (2015).
The, H. C. et al. The introduction and establishment of fluoroquinolone resistant Shigella sonnei into Bhutan. Microb. Genom. http://dx.doi.org/10.1099/mgen.0.000042 (2015).
Ferreccio, C. et al. Epidemiologic patterns of acute diarrhea and endemic Shigella infections in children in a poor periurban setting in Santiago, Chile. Am. J. Epidemiol. 134, 614–627 (1991).
Noriega, F. R. et al. Strategy for cross-protection among Shigella flexneri serotypes. Infect. Immun. 67, 782–788 (1999).
Feil, E. J. The emergence and spread of dysentery. Nat. Genet. 44, 964–965 (2012).
Levine, M., Kotloff, K. L, Barry, E. M., Pasetti, M. F. & Sztein, M. B. Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat. Rev. Microbiol. 5, 540–553 (2007).
Barry, E., Pasetti, M. & Sztein, M. Progress and pitfalls in Shigella vaccine research. Nat. Rev. Gastroenterol. Hepatol. 10, 245–255 (2013).
Walker, R. I. An assessment of enterotoxigenic Escherichia coli and Shigella vaccine candidates for infants and children. Vaccine 33, 954–965 (2015). A comprehensive and up-to-date review evaluating the current approaches for a Shigella spp. vaccine.
Martinez-Becerra, F. J. et al. Characterization of a novel fusion protein from IpaB and IpaD of Shigella spp. and its potential as a pan-Shigella vaccine. Infect. Immun. 81, 4470–4477 (2013).
Riddle, M. S. et al. Safety and immunogenicity of an intranasal Shigella flexneri 2a Invaplex 50 vaccine. Vaccine 29, 7009–7019 (2011).
Acknowledgements
H.C.T., D.P.T. and N.R.T. are funded by the Wellcome Trust (grant 098051); K.E.H is funded by the Australian National Health and Medical Research Council (NHMRC); and S.B. is a Sir Henry Dale Fellow, funded by the Wellcome Trust and the Royal Society (grant 100087/Z/12/Z).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Disability adjusted life years
-
(DALY). A measure of overall disease burden, expressed as the cumulative number of years lost owing to ill health, disability or early death.
- Pathovars
-
Groups of bacterial strains that have similar characteristics and are differentiated at the subspecies level on the basis of their distinctive pathogenicity in one or more hosts.
- Lysine decarboxylation
-
(LDC). A reaction that is used in a biochemical test to determine the ability of a microorganism to use lysine as a source of carbon for growth. In a positive LDC test, lysine is metabolized into the amine cadaverine through the activity of the enzyme lysine decarboxylase.
- Microfold cell
-
(M cell). A specialized epithelial cell type found in the follicle-associated epithelium of the gastrointestinal tract. Their function is to transport macromolecules and microorganisms across the epithelial barriers to the immune cells, thus inducing mucosal immunity.
- Transcytosis
-
The selective vesicular transport of macromolecules from one side of the cell to the other while maintaining the unique compositions of these vesicular environments.
- O antigen
-
A repeating glycan polymer attached to the outer core in lipopolysaccharide. This structure is on the very outer surface of the bacterial cell and is therefore a target for recognition by the host immune system.
- Conjugation
-
The transfer of genetic material between bacterial cells through cell-to-cell contact or by a bridge-like connection between two cells.
- Transduction
-
A mode of horizontal gene transfer whereby genetic material is transferred from one bacterium to another by a virus.
- Flagellum
-
A multiprotein thread-like structure protruding from prokaryotic or eukaryotic cells that is used for motility and for the sensory perception of extracellular chemicals and temperature.
- Fimbriae
-
Appendages composed of the protein curlin and found on many Gram-negative and some Gram-positive bacteria. Fimbriae are used mainly for adherence to bacterial cells, host cells and abiotic surfaces.
- Pathogen-associated molecular pattern
-
(PAMP). A set of specific molecules that are present on groups of pathogens and are recognized by the innate immune system. These conserved molecular motifs in bacteria, such as lipopolysaccharide and flagellin, are usually recognized by Toll-like receptors and other pattern-recognition receptors.
- Cadaverine
-
A diamine with a putrid odour. It is the product of lysine decarboxylation.
- Quinolinate
-
A dicarboxylic acid generated as the downstream product of tryptophan catabolism. It acts as a substrate for the biosynthesis of nicotinic acid mononucleotide and, ultimately, the formation of the coenzyme NAD.
- Class II integrons
-
Mobile genetic elements that are capable of carrying genes, including antimicrobial-resistance genes, and integrating into bacterial chromosomes by site-specific recombination. An integron contains at least an integrase, an attachment site and a promoter. Classification is based on the type of integrase.
- Pulsed-field gel electrophoresis
-
(PFGE). A molecular typing technique based on the migration pattern of DNA fragments of variable lengths, generated by restriction enzyme treatment, in an electrical field.
- Multilocus sequence typing
-
(MLST). A DNA sequence-based molecular typing scheme in which each isolate is distinguished by a combination of unique alleles of housekeeping genes (by comparing their genetic variations).
- Homoplasies
-
Phenotypic or genotypic characteristics that are shared by a set of organisms but not inherited from a common ancestor.
Rights and permissions
About this article

Cite this article
The, H., Thanh, D., Holt, K. et al. The genomic signatures of Shigella evolution, adaptation and geographical spread. Nat Rev Microbiol 14, 235–250 (2016). https://doi.org/10.1038/nrmicro.2016.10
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro.2016.10
This article is cited by
-
Epistatic interactions between the high pathogenicity island and other iron uptake systems shape Escherichia coli extra-intestinal virulence
Nature Communications (2023)
-
Identification and characterization of a novel lytic peptidoglycan transglycosylase (MltC) in Shigella dysenteriae
Brazilian Journal of Microbiology (2023)
-
The population genetics of pathogenic Escherichia coli
Nature Reviews Microbiology (2021)
-
Shigella sonnei: virulence and antibiotic resistance
Archives of Microbiology (2021)
-
Historical, current, and emerging tools for identification and serotyping of Shigella
Brazilian Journal of Microbiology (2021)
