
Abstract
Ciliopathies comprise a large number of hereditary human diseases and syndromes caused by mutations resulting in dysfunction of either primary or motile cilia. Both types of cilia share a similar architecture. While primary cilia are present on most cell types, expression of motile cilia is limited to specialized tissues utilizing ciliary motility. We characterized protein complexes of ciliopathy proteins and identified the conserved AAA-ATPase Ruvbl1 as a common novel component. Here, we demonstrate that Ruvbl1 is crucial for the development and maintenance of renal tubular epithelium in mice: both constitutive and inducible deletion in tubular epithelial cells result in renal failure with tubular dilatations and fewer ciliated cells. Moreover, inducible deletion of Ruvbl1 in cells carrying motile cilia results in hydrocephalus, suggesting functional relevance in both primary and motile cilia. Cilia of Ruvbl1-negative cells lack crucial proteins, consistent with the concept of Ruvbl1-dependent cytoplasmic pre-assembly of ciliary protein complexes.
Similar content being viewed by others

Introduction
Cilia are microtubule-based, highly conserved organelles that project from the cellular surface and play a role in extracellular signal-sensing, cell motility, and the movement of extracellular fluids. According to their structure, localization, number, and main function, two distinct types of cilia can be distinguished. Primary monocilia can be found on the apical membrane of many cell types, including renal tubular cells, are usually immotile and act primarily as sensory organelles that receive signals from the cellular micro-milieu and modulate multiple intracellular signaling cascades. Motile cilia are typically found as bundles on the cell surface and actively contribute to the movement of extracellular fluids and cells by performing a coordinated beat pattern. This phenomenon is of critical importance for clearance of the airways, movement of oocytes through the oviduct, the movement of cerebrospinal fluid through the ventricular system in the brain, and for other tissues1,2,3,4. Molecular and genetic studies from the past 15 years have established a strong link between ciliary dysfunction and numerous diseases, although the exact contributions of primary cilia to the corresponding phenotypes remain unclear5,6. Prominent examples of ciliopathies are hereditary forms of polycystic kidney disease (PKD) as a consequence of the dysfunction of renal primary cilia and primary ciliary dyskinesia (PCD) as a disorder of motile cilia. Recently, there has been remarkable progress in the identification of genetic causes of these so-called ciliopathies. The majority of pathophysiological relevant genes encode proteins that are involved in the formation, elongation, or resorption of cilia, or in the regulation of cilia-associated signaling pathways1,2,3,5,7. These proteins are frequently organized in multi-protein complexes at the ciliary base, either localized at the basal body or the transition zone, as recently demonstrated for the Bardet–Biedl syndrome protein complex (BBSome), or nephronophthisis protein (NPHP) complexes in renal tubules8,9. To identify novel components of the NPHP complex, we have previously performed several protein-interaction studies10,11,12. Interestingly, we identified the chaperone-associated protein Ruvbl1 as a novel candidate interactor in several screens using NPH proteins as bait.
Ruvbl1 (Pontin) and its paralogue Ruvbl2 (Reptin) are evolutionarily highly conserved and belong to the AAA+ superfamily of ATPases13,14,15,16. They are components of various protein supercomplexes that link them to diverse cellular functions including chromatin remodeling, DNA damage repair, or mitotic spindle assembly13,14. Mechanistically, Ruvbl1, Ruvbl2, and the two Hsp90-interactors RNA polymerase II-associated protein 3 (Rpap3) and protein interacting with Hsp90 1 domain containing 1 (Pih1d1) have been proposed to comprise the R2TP complex. This complex functions as co-chaperone for Hsp9014 and is important for the assembly and function of large signaling protein complexes15,16. Interestingly, Pih1 family proteins and the Pih1-related protein Kintoun (Ktu) have been linked to the function of motile cilia, and a role in the cytoplasmic pre-assembly of dynein arm complexes has been suggested17,18,19. Recently, mutations in PIH1D3 were identified as a cause of PCD20,21. Moreover, recent proteomics studies using murine photoreceptors and isolated primary cilia from murine renal cell lines have suggested that Ruvbl1 is a bona fide ciliary protein22,23. Therefore, we generated conditional mouse models to investigate the in vivo function of Ruvbl1 in different epithelia carrying either primary or motile cilia. In this study, we demonstrate that Ruvbl1 is essential in the monociliated renal tubular epithelium as well as in the multiciliated ependymal epithelium within the ventricular system. Mechanistically, our in vivo data implicate a novel function of the Ruvbl1 protein complex in the assembly and targeting of ciliary protein complexes.
Materials and methods
Mice
Transgenic mouse stem cells from a C57BL/6N background were received from EUCOMM (European Conditional Mouse Mutagenesis) and injected into SV129 blastocysts as previously described to obtain transgenic Ruvbl1frt-fl mice24. Mice expressing the FLPe recombinase in the germline (ß-actin-FLPe, MGI: 2448985) and mT/mG (MGI: 3716464) reporter mice were obtained from the Jackson Laboratory and have been previously described25,26. Ksp:Cre mice (MGI: 2665300) expressing the Cre recombinase under the Cadherin-16 promotor in the distal tubules and collecting ducts of the kidney were kindly provided by Peter Igarashi and have previously been described27. Tamoxifen-inducible Ksp:Cre mice (Ksp:Cre:ERT2, MGI: 3641108) have previously been described28. Tamoxifen-inducible FoxJ1:Cre mice (FoxJ1:Cre:ERT2, MGI: 3808149) were kindly provided by Brigid Hogan29. Genotyping for the Ruvbl1 locus was carried out using the following primers: 5′-gaagtgatgccttccctgag-3′ (fp), 5′-ccacaacgggttcttctgtt-3′ (rp1), 5′-ttggactgcagtggtgtctc-3′ (rp2), and 5′-tgtgaaaccataccccacct-3′ (rp3). Genotyping of the other mouse lines was performed as described previously25,26,27. The mice were housed according to the standardized specific pathogen-free conditions in the animal facility of the University of Cologne. Newborn mice were killed by decapitation, and adult mice were killed by cardiac perfusion. Tissue was processed by freezing or fixation, and sera were analyzed in the Department of Clinical Chemistry at the University Hospital of Cologne or the clinical laboratory of the Department of Pediatrics of the University Hospital in Erlangen, Germany. The biochemical measurements were carried out on a Cobas Integra system (Roche) and included a kinetic assay for urea and an enzymatic assay for creatinine. The electrolytes were measured using ion-selective electrodes. Tamoxifen was administered to tamoxifen-inducible knockout and control mice via intraperitoneal injection with a dose of 1 mg per 10 g body weight at the age of 10–12 weeks or 8–20 weeks to Ksp:Cre:ERT2 or FoxJ1:Cre:ERT2 mice, respectively. Induced mice were weighed twice a week and killed after reaching a maximal weight loss of 20%. For calculation of the cystic index, the cystic area and total kidney area were measured using Fiji30 and then divided by each other.
Histology
For histological analysis of the kidney, periodic acid-Schiff (PAS) staining was performed using standard methods. Briefly, formalin-fixed, paraffin-embedded tissue was cut in 3-µm-thick sections. Paraffin was removed by xylene treatment following rehydration in graded ethanol. After 10 min of incubation in 0.9% periodic acid (Roth), the slides were stained with Schiff reagent (Merck). For nuclear counter staining, the slides were treated with Mayer’s Haemalaun (Sigma-Aldrich).
Plasmids
The full-length coding sequences of human and murine Ruvbl1, human NPHP5, human NPHP10, and murine Dpcd were cloned by standard PCR amplification techniques from human HEK293T or murine NIH-3T3 cDNA. The GFP, EPS, and NPHP1-4 plasmids have previously been described10,31,32,33. Murine Dnaaf1 was cloned by standard PCR from an EST clone (ID 6774345), and the sequence was confirmed.
For the generation of FlpIn NIH-3T3 stably expressing GFP.Ruvbl1, murine Ruvbl1 CDS was amplified from cDNA obtained from NIH-3T3 cells, first cloned into a modified pENTR Gateway vector (Invitrogen) and subsequently recombined into pgLAP3, which was a gift from Peter Jackson34 (Addgene #19704).
Antibodies
The NPHP1 antibody has been described previously12. Companies and dilutions of antibodies are listed in the following table.
Antigen | Company | Catalog number | Dilutions |
|---|---|---|---|
Acetylated tubulin | Sigma-Aldrich | T6793 | IF 1:2000 |
Aqp2 | BD Biosciences | 550649 | IF 1:500 |
Arl13b | Proteintech | 17711-1-AP | IF 1:500 |
DBA-biotinylated | Vector Labs | B-1035 | IP 1:10 |
DBA-FITC | Vector Labs | FL-1031 | IF 1:500 |
Dnai2 | Abnova | H00064446-M01 | IF 1:100 |
Dnali1 | Santa Cruz | sc-160296 | IF 1:500 |
FLAG | Sigma-Aldrich | F3165 | WB 1:10,000 |
GFP | Santa Cruz | sc-8334 | WB 1:1000 |
HA | Covance | PRB-101P | IP 1:1000 |
Ift88 | Ptglab | 13967-1-AP | IF 1:500 |
Ki67 | Abcam | ab16667 | IF 1:500 |
NPHP1 | Homemade | Clone #62; 0.6 µg/µl | IF 1:20; IP 1:50 |
RUVBL1 | ProteinTech | 10210 | IF 1:500; WB 1:1000 |
V5 | Serotec | MCA1360 | IP 1:1000; WB 1:10,000 |
Cell culture
HEK293T and FlpIn NIH-3T3 cells were cultured in DMEM (Sigma-Aldrich) containing 10% FBS under standard conditions (37 °C, 5% CO2).
FlpIn cells
For stable expression of a murine GFP.Ruvbl1 fusion protein, FlpIn NIH-3T3 cells (Thermo Fisher Scientific) were transfected with the pgLAP3 vector containing murine Ruvbl1 CDS and Flp-recombinase expression vector (pOG44, Invitrogen) using Lipofectamine LTX (Invitrogen)34. Stably integrated cells were selected by Hygromycin B (InvivoGen) treatment. Genomic DNA was isolated from original FlpIn NIH-3T3 or GFP.Ruvbl1 cells using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s specifications. Stable integration of the GFP.Ruvbl1 fusion construct was tested via standard PCR methods using construct-specific primers and internal control primers.
Co-immunoprecipitation
HEK293T cells were transfected with plasmid DNA (as indicated) using a modified calcium phosphate method. After 24 h, the proteins were isolated by lysing the cells in lysis buffer (1% Triton X-100, 20 mM Tris pH 7.5, 25 mM NaCl, 50 mM NaF, 15 mM Na4P2O7, 1 mM EDTA, 0.25 mM PMSF, and 5 mM Na3VO4) on ice for 15 min and centrifugation (20,000 × g, 4 °C, 15 min). Flag-tagged proteins were pulled out of the supernatant using anti-Flag antibody-coupled sepharose beads (M2-beads, Sigma-Aldrich); V5-tagged proteins were precipitated using an anti-V5 antibody and Protein G beads. After a binding time of 2 h, the beads were washed three times with lysis buffer and boiled in 2× Laemmli containing 0.1 M DTT for protein denaturation. For endogenous co-immunoprecipitation, the binding time was increased to 16–20 h.
Western blot
Protein samples were separated by 10% SDS-PAGE (Invitrogen system) and blotted onto a PVDF membrane (Millipore). The membrane was blocked for 30 min in 5% BSA (PAA) and incubated with the indicated primary antibodies overnight at 4 °C. After incubation with the appropriate horseradish peroxidase-coupled secondary antibodies (Dako), the proteins were visualized using homemade-enhanced chemiluminescence technology (1:1 mixture of solution A (100 mM Tris, 2.5 mM luminol, 0.4 mM coumaric acid) and solution B (100 mM Tris, 0.01% H2O2))35.
Immunofluorescence staining
Cryo-conserved kidneys were sliced into 7-µm-thick sections and fixed in 4% PFA. The tissue sections were blocked in 5% normal donkey serum (NDS) and 1% bovine serum albumin (BSA) for 1 h at RT and stained with the indicated antibodies overnight at 4 °C. After incubation with fluorescent secondary antibodies, the slides were washed and mounted in Prolong Gold antifade containing DAPI (Invitrogen). Paraffin-embedded tissue was processed in the same manner after deparaffinizing and rehydrating the slides using xylene and incubation with a descending series of ethanol concentrations.
For immunofluorescence staining of FlpIn NIH-3T3 cells, the cells were seeded on coverslips and labeled using standard staining techniques with the indicated antibodies. Briefly, the cells were fixed in 4% PFA for 10 min at RT and blocked in 5% NDS for 30 min at RT. Subsequent incubation with the indicated primary and secondary antibodies was followed by mounting in Prolong Gold antifade containing DAPI (Invitrogen). Immunofluorescence stainings were analyzed with an Axiovert 200M microscope (Zeiss) or a Meta 710 confocal microscope (Zeiss).
Immunoprecipitation of DBA-positive renal tubules
We used a slightly modified protocol of this method that has previously been described36. A whole kidney was minced using a scalpel and then digested with Collagenase II (Worthington) and RNaseI (Applichem). The obtained tubular fragments were pelleted, resuspended in 1× HBSS and incubated with biotinylated DBA for 1 h at 4 °C on a shaker. Streptavidin magnetic beads (Pierce) were then added, and the samples were incubated for another hour on the shaker at room temperature. The magnetic beads were collected using a magnetic particle concentrator (Life Technologies) and washed several times with 1× HBSS. The concentrated DBA-positive renal tubules were then analyzed via western blot or quantitative real-time PCR analysis.
Quantitative real-time PCR
RNA isolation (Direct-zolTM RNA MiniPrep Kit, Zymo Research) and reverse transcription (High-Capacity cDNA Reverse Transcription Kit, ABI) of the indicated tissues were performed according to the manufacturer’s protocols. Quantitative real-time PCR was performed with Power SYBR® Green Master Mix (ABI) on the QuantStudioTM 12K Flex (ABI) system using the following primers (5′ to 3′): Ruvbl1: GGGGCCTCCTGGAACTGGCA (fp) and ACCTCGCTACCCACCATCGGG (rp); Pih1d1: CAGCGTCGTGAAAATCTCCC (fp) and GTGCCAAAAACGTCGAGTCC (rp); Rpap3: GAAGTTCCCCAGAGATGCTGT (fp) and AGCTGGCTTTTCCCTCTCAA (rp); Ift88: GGACGACCTTTACTCTGGTTTCA (fp) and AAAACCCGTGTCATTCTCCAA (rp); Kif3a: CCACCATGCCGATCAATAAGT (fp) and AGAGGCCGGCACCTAACC (rp); Wdr34: TGAACTGGACTGAGCAGCAG (fp) and CATCTAGCCGACCATAGGCA (rp); Actb: AAGAGCTATGAGCTGCCTGA (fp) and TACGGATGTCAACGTCACAC (rp).
Sample preparation for interactome analysis
FlpIn NIH-3T3 cells expressing GFP.Ruvbl1 (murine), GFP. RUVBL2 (human) and GFP.GFP fusion proteins were lysed in lysis buffer (1% Triton X-100, 20 mM Tris pH 7.5, 25 mM NaCl, 50 mM NaF, 15 mM Na4P2O7, 1 mM EDTA, 1xPIM, and 5 mM Na3VO4) and by sonication (30% amplitude, 9 s, 0.1 s sonic, 0.9 s pause). Lysates were cleared by centrifugation (17,000 × g, 15 min, 4 °C) and ultracentrifugation (165,000 × g, 20 min, 4 °C). Supernatants were incubated with GFP µMACS magnetic beads (Miltenyi) for 1 h. Then, the lysates were loaded on a µMACS column, the proteins were reduced and alkylated, and on-column digestion was performed overnight as previously described37. Eluates were acidified the next day using 1–2% formic acid, stage-tip clean-up was performed as previously described38 and samples were dried using a vacuum centrifuge.
nLC-MS/MS
Peptides were resuspended in 0.1% FA and separated using a 1-h gradient on an nLC coupled to a Q Exactive Plus tandem mass spectrometer (Thermo scientific) or an LTQ orbitrap XL mass spectrometer. Separation was carried out on an in-house packed 50-cm column with 1.7-μm C18 beads (Dr Maisch GmbH). The gradient consisted of two buffers: Buffer A: 0.1% formic acid and B: 80% acetonitrile, 0.1% formic acid. Linear gradients from 7 to 38% B in 60 min were used for separation with a subsequent increase to 80% B for 5 min and a re-equilibration to 5% B. Peptides were then sprayed into a tandem mass spectrometer, the Q Exactive plus (60). The resolution for MS1 spectra was 70,000 (mass range, 200–1200 mz−1). MS1 spectra were acquired using 1E6 as an AGC target. MS/MS spectra of the top ten most intense peaks were obtained by higher-energy collisional dissociation fragmentation. The resolution for MS/MS spectra was 35,000 at 200 mz−1, the AGC target was 5E5, and the max. injection time was 120 ms.
Bioinformatics analysis
RAW files generated by the Q Exactive tandem mass spectrometer were processed using MaxQuant, v.1.4.1.2 with default settings. Fixed modifications were carbamidomethylation on cysteines. The variable modification was methionine oxidation. The peptide spectrum matches (PSM) and site and protein false discovery rate (FDR) were 0.01. The minimal peptide length was 7. The match between run option and label-free quantification option was enabled. Data were searched against a mouse uniprot database in fasta format from the Mus musculus, and the reference proteome was downloaded from uniprot.org on 23 February 2014. MaxQuant output (proteingroups.txt file) was uploaded in Perseus, v. 1.4.0.11. Contaminants and reverse hits were removed. Determination of significant interactors was performed using a two-tailed t-test followed by an approach similar to SAM as proposed by Tusher et al.39. As previously described40, determination of significant interactors was performed using the FDR, and s0 cutoff values are indicated in the figure legends. Stoichiometry analysis was performed using intensity-based absolute quantification (iBAQ) values as previously described41.
Magnetic resonance imaging and quantification of ventricle volumes
Magnetic resonance imaging (MRI) was performed on a clinical 3.0 Tesla MR system (Achieva, Philips, the Netherlands) using a dedicated small animal solenoid coil with a diameter of 40 mm and an implemented heating system to keep the body temperature constant during MR examination (Philips Research Europe, Hamburg, Germany). Animals were anesthetized by isoflurane inhalation (1.5–2.5%). High-resolution transversal and coronal T2-weighted MR images of the brain were acquired with the following parameters: Turbo Spin Echo (TSE) factor of 15, repetition time (TR) of 2000 ms, echo time (TE) of 110 ms, number of acquisition (NSA) of 1, slice thickness of 1 mm (without gap), matrix of 256 × 256, field of view (FOV) of 50 × 50 mm with a reconstructed voxel size 0.39 × 0.39 × 0.52, and an acquisition time of 2:36 min for 20 slices. After MRI examination, the mice were killed.
Ventricle volumes were measured using Fiji30. Ventricles were segmented manually, and volumes were subsequently measured using the 3D objects counter plugin42.
Electron microscopy
Mice were killed, and the ovary ducts were perfused with electron microscopy fixation buffer (4% paraformaldehyde, 2% glutaraldehyde, and 0.2% picric acid in PBS, pH 7.25–7.35). The samples were osmicated with 2% OsO4 in 0.1 M cacodylate buffer (pH 7.2–7.3) and dehydrated in a graduated ethanol series. Epon infiltration and flat embedding were performed following standard procedures. Semithin sections (500 nm) were stained with toluidine blue. Ultrathin sections (50 nm) were cut using an Ultramicrotome Ultracut EM UC6 (Leica) and then stained with 1.5% aqueous uranylic acetate and lead citrate. Samples were analyzed with a Zeiss EM 902 electron microscope (Zeiss).
Statistics
Data are expressed as the mean ± SEM of n experiments. Statistical evaluation was performed using a two-tailed Student’s t test, one-way ANOVA with Tukey’s post hoc analyses or two-way ANOVA with Bonferroni correction. P values less than 0.05 were considered significant.
Study approval
All animal procedures were conducted in accordance with European (EU directive 86/609/EEC), national (TierSchG), and institutional guidelines and were approved by local governmental authorities (LANUV NRW).
Results
Deletion of Ruvbl1 in renal tubular epithelial cells leads to acute kidney injury and high perinatal mortality
To study the in vivo function of Ruvbl1, we generated Ruvbl1 knockout mice by blastocyst injection of stem cells containing a “knockout-first allele”43,44. We observed neither any homozygous whole-body knockout mice nor embryos at day E8.5, E11.5, or E12.5 in 35 tested embryos from seven different pregnant mothers, consistent with a previous study demonstrating that Ruvbl1 is essential during early embryogenesis45. Next, we generated a Ruvbl1fl/fl line with a conditional Cre-dependent allele by crossing with a ß-actin-FLPe line followed by back-breeding to remove the Flipase transgene (Fig. 1a, b). Since cysts in NPH derive from distal parts of the nephron, we crossed Ruvbl1fl/fl mice with a Ksp:Cre mouse line, which drives Cre expression in the distal tubules and collecting ducts starting at day E14.527. The specificity and efficacy of Ksp:Cre mice were confirmed using a mT/mG reporter line, in which Cre activity results in a switch from tdTomato to GFP expression (Fig. 1c)26. Newborn Ruvbl1fl/flKsp:Cre+ (tubular knockout) mice were born according to the Mendelian ratio (Fig. 1d), did not show any macroscopic phenotype (Fig. 1e), and did not express Ruvbl1 in the distal tubules (Fig. 1f). Surprisingly, neonatal mortality within the first 24 h after birth was high in Ruvbl1fl/flKsp:Cre+ (Fig. 1g) compared with Ruvbl1fl/flKsp:Cre− or Ruvbl1fl/wtKsp:Cre+ animals, with only four survivors out of 21 tubular knockout animals in the entire cohort thus far. The phenotypes of two of the four survivors are presented in Suppl. Figure 1. The stomach of newborn tubular Ruvbl1 knockouts was visibly filled with milk, indicating normal feeding. Interestingly, the weight of the Ruvbl1 knockouts 12 h after birth was significantly higher than that of littermates (Fig. 1h). The Ruvbl1 knockouts did not show any signs of urine production, as the urine bladder was always found completely empty. Further analyses of the knockout animals at P1 revealed significantly increased serum creatinine (Fig. 2a) and serum urea concentrations (Fig. 2b), as well as significant hyponatremia (Fig. 2c) indicative of acute kidney injury with subsequent fluid overload. There were no histologic changes in the major non-renal visceral or thoracic organs (Suppl. Figure 2). As oligohydramnios might have affected pulmonary development, we paid special attention to the lungs. There was no evidence of pulmonary hypoplasia (Suppl. Figure 2). Renal histology was performed at day P0; it is well known that, in contrast to humans, murine kidneys at P0 are still immature (Fig. 2d, upper panel). Ruvbl1 knockout kidneys displayed considerable medullary tubular dilatations and small cystic lesions (Fig. 2d, lower panel). Consistently, the cystic index in knockout animals was significantly increased (Fig. 2e). To confirm that the observed cysts in Ruvbl1fl/flKsp:Cre+ mice were derived from the distal part of the nephron, we performed immunofluorescence staining using Aquaporin-2 and Dolichos biflorus agglutinin (DBA) as markers for collecting ducts. Cyst-lining epithelia in knockout mice at day P1 stained positively for Aquaporin-2 (Fig. 2f) and DBA (Suppl. Figure 3) and negatively for the proximal tubule marker Lotus tetragonolobus lectin (LTL; Suppl. Figure 3). To ultimately confirm that cystogenesis was not the result of cell death due to Ruvbl1 depletion, we crossed Ruvbl1fl/flKsp:Cre+ animals with the mT/mG reporter mouse line. Strikingly, cyst-lining epithelia were positive for GFP, thus proving Cre activity in these cells (Fig. 2g), and showed only few ciliated cells (~30%, data not shown). We did not detect cyst-lining epithelia positive for Tomato. The first two animals to escape neonatal death developed signs of renal disease later in life, and both had to be killed at the age of 4 or 26 weeks, respectively. In each of these animals, one kidney was grossly cystic-dysplastic, whereas the other kidney was less severely affected (Suppl. Figure 1). Taken together, deficiency of Ruvbl1 in the distal nephron leads to neonatal kidney injury with cystic dysplasia.

a The EUCOMM strategy of the Ruvbl1FRT-FL mouse allows use as a conventional (frt-fl) or a conditional (fl) knockout. A premature stop codon can be eliminated by crossing with an FLPe deleter mouse line, resulting in a conditional floxed knockout model (fl) with exon 4 flanked by loxP sites. Tissue or cell-specific knockout of Ruvbl1 (deletion allele, del) was achieved by crossing with the respective Cre line. b Genotyping of mice, including detection of the Ruvbl1 deletion allele from murine kidneys. c To validate the efficacy of the Ksp:Cre mouse line, it was crossed to the mT/mG reporter mouse line (R26mTmG). This line expresses a membrane-tagged tomato (magenta), which is replaced by expression of a membrane-tagged GFP (green) after Cre recombination. Kidneys of newborn mice were fixed in 4% formalin, treated with increasing amounts of sucrose and cryo-conserved. Next, 7-µm-thick slides were analyzed for GFP expression. Distal tubules and collecting ducts were GFP-positive. Scale bar: 1 mm. d Ruvbl1 tubular knockout mice (fl/fl, Cre+; n = 33) were born according to the expected Mendelian ratios (total, n = 103). e Ruvbl1 tubular knockout mice (tubular KO) did not show a macroscopic phenotype in comparison to control siblings. f Immunofluorescence staining of control or Ruvbl1 tubular knockout kidneys showing no expression of Ruvbl1 (magenta) in cystic tubules, marked by aquaporin-2 (Aqp2, green) in Ruvbl1 knockout animals (tubular KO). Scale bars: 20 µm. g Eighty percent of the newborn Ruvbl1 tubular knockout animals (fl/fl, Cre+; n = 21) died within the first days of life, whereas control animals did not (total, n = 90). h Newborn Ruvbl1 tubular knockout animals (tKO; n = 7) weighed significantly more than control (co; n = 15) or heterozygous (het; n = 3) littermates on day P0. Mean weight ± SEM. (F(2,22) = 6.243, p = 0.0071; Tukey’s post hoc *p < 0.05)

a–c On day P1, newborn Ruvbl1 tubular knockout animals (tKO; n = 8) showed overall significantly increased serum creatinine levels (F(2, 16) = 4.158, p = 0.04; Tukey’s post hoc *p < 0.05) (a), significantly increased serum urea levels (F(2, 16) = 9.953, p = 0.002; Tukey’s post hoc *p < 0.05, **p < 0.01) (b), and significantly reduced serum sodium levels (F(2, 16) = 5.472, p = 0.02; Tukey’s post hoc *p < 0.05) (c) compared with control (co; n = 6) or heterozygous (het; n = 5) littermates. Mean ± SEM. d Histological PAS staining of control and Ruvbl1 tubular knockout kidneys of newborn mice showing dilated renal tubules in Ruvbl1 tubular knockout animals. Scale bars: 500 µm (left) and 100 µm (right). e The cystic Index was significantly increased in Ruvbl1 tubular knockout (tKO) kidneys (t(8) = 4.280, **p < 0.01). Mean cystic index ± SEM. f Immunofluorescence staining showing that the cystic tubules in tubular Ruvbl1 knockout animals were restricted to Aqp2-positive (collecting duct) sections of the nephron (*). Scale bars: 1 mm (left) and 500 µm (right). g After crossing in the mTmG reporter mouse line, GFP-expressing dilated tubules showed few ciliated (arrow heads) epithelial cells compared with neighboring GFP-negative tubules (arrows) in tubular knockout mice (Ruvbl1fl/flKsp:Cre+R26mTmGtg). Scale bar: 20 µm
Ruvbl1 is required for maintenance of kidney function and tubular architecture
To understand whether Ruvbl1 is primarily required for renal development or whether Ruvbl1 is also important for the maintenance of renal function and tubular structure, we crossed Ruvbl1fl/fl mice with a previously described tamoxifen-inducible Ksp:Cre line46. By crossing the Ksp:Cre:ERT2 line to the mT/mG reporter mouse line, we validated its specificity and efficacy (Suppl. Figure 4). Upon tamoxifen injection at 10–12 weeks of age, Ruvbl1fl/flKsp:Cre:ERT2+ animals, but not control mice, showed progressive weight loss over time and had to be killed approximately 7 weeks after induction (Fig. 3a). Serum creatinine and serum urea levels of Ruvbl1-deficient animals were significantly increased at the time of killing (Fig. 3b, c). To confirm the knockout of Ruvbl1 in the induced knockout animals, we precipitated DBA-positive renal tubules out of the kidney and tested for the expression of Ruvbl1 by western blot and quantitative real-time PCR analyses, which revealed a significant reduction in both Ruvbl1 protein expression (Suppl. Figure 5a) and Ruvbl1 mRNA abundance (Suppl. Figure 5b) in induced knockout compared with control animals. In addition, we examined the expression of different Ruvbl1-associated and ciliary proteins at the mRNA level and demonstrated a significant downregulation of the known R2TP component Pih1d1 and the crucial ciliary proteins Ift88, and Kif3a in Ruvbl1 knockout mice (Suppl. Figure 5b). mRNA levels of Rpap3, another subunit of the RT2P complex, were unaffected. Furthermore, we observed mild alterations in renal histology with tubular dilatations, a significantly increased kidney-to-body weight ratio and a significantly increased cystic index at the time of killing (Fig. 3d, f). Strikingly, there were significantly fewer ciliated cells in the distal nephron in Ruvbl1-deficient animals than in control mice (48% vs 84%; Fig. 3g and Suppl. Figure 6). The remaining cilia in cyst-lining epithelial cells in induced knockout animals were significantly longer compared with control animals (4.08 ± 0.28 µm vs. 1.40 ± 0.06 µm; Fig. 3g). Interestingly, the staining pattern of DBA was shifted within epithelial cells with enhanced signal intensity at the apical membrane, suggesting alterations in tubular apico-basal polarity (Fig. 3g). These data demonstrate the impact of Ruvbl1 on the maintenance of tubular architecture, ciliary structure, and renal function in adult mice, even after completion of renal development. Loss of renal function in Ruvbl1 knockout mice may be related to the observed tubular dilatations and to reduced numbers of ciliated cells; however, neither the constitutive nor the inducible Ruvbl1 knockout animals displayed a clear and exclusive ciliopathy phenotype. Thus, we cannot exclude the possibility that Ruvbl1 affects additional cellular events in renal epithelium, resulting in loss of renal function.

a Loss of Ruvbl1 in adult mice was achieved by using a tamoxifen-inducible Ksp:Cre mouse line. Induced loss of tubular Ruvbl1 (n = 4) resulted in significant body weight loss at 32 days (**p < 0.01) and at 35 days (****p < 0.0001) after induction, while control mice consistently gained weight (n = 5). Repeated measures two-way ANOVA with Bonferroni-corrected post hoc analyses (F(10,70) = 5.807, p < 0.0001). Mean body weight ± SEM. b, c Induced loss of tubular Ruvbl1 (iKO) resulted in increased serum creatinine (t(7) = 4.255, **p < 0.01) (b) and serum urea (t(7) = 4.232, **p < 0.01) (c) levels compared with controls (co). Mean values at sacrifice ± SEM. d–f Induced loss of tubular Ruvbl1 (iKO) resulted in a significantly increased kidney-to-body-weight ratio (t(7) = 2.905, *p < 0.05) (d), dilated tubules as shown in the histological PAS staining (e), and a significantly increased cystic index (t(7) = 4.255, **p < 0.01) (f) compared with controls. Scale bars, e: 1 mm (upper panel) and 500 µm (lower panel). g Induced loss of tubular Ruvbl1 (induced KO) resulted in significantly fewer and longer ciliated cells (magenta, arrowheads pointing to cilia) and an altered apico-basal localization of the collecting duct marker DBA (green) compared with controls. The graph on the right depicts the number of ciliated epithelial cells in black (t(7) = 8.435, ****p < 0.0001) and cilia length in red (t(143) = 11.87, ****p < 0.0001). Z-stacks: 1 µm. Scale bars: 10 µm
Interactomic analyses of Ruvbl1
To evaluate the function of Ruvbl1 in ciliated cells, we generated FlpIn NIH-3T3 cells expressing a GFP.Ruvbl1 fusion protein (Fig. 4a). Here, stable single-copy genomic integration led to nearly physiological levels of protein expression, as confirmed by specific immunoblotting (Fig. 4b) and quantitative real-time PCR (Suppl. Fig. 7a). Immunofluorescence staining showed that GFP.Ruvbl1 was mainly localized to the nucleus, but also showed a cytosolic signal, thus co-localizing with endogenous Ruvbl1 (Suppl. Fig. 7b). Furthermore, FlpIn NIH-3T3 cells expressing GFP.Ruvbl1 did not show substantial changes in cilia compared with GFP.GFP-expressing cells (Suppl. Fig. 7b). Although there was no clear ciliary signal in the cellular immunofluorescence staining for Ruvbl1 (not shown), GFP.Ruvbl1 was specifically co-precipitated from these cells with endogenous Nphp1 (Suppl. Fig. 8a). Additional co-immunoprecipitation experiments in HEK293T cells suggested that Ruvbl1 was also associated with NPHP2, NPHP3, NPHP4, NPHP5, and NPHP10 (Suppl. Fig. 8b–c), emphasizing the association between Ruvbl1 and the different NPHP complexes8,9. To gain an unbiased insight into Ruvbl1 functions, we performed an interactome analysis by label-free quantitative nLC MS/MS analyses after immunoprecipitation of GFP.Ruvbl1 from stable cell lines to identify components of the Ruvbl1 protein complex in mammalian cells40. Hierarchical clustering of protein intensities from the data set revealed a clear separation between Ruvbl1 and control immunoprecipitations (Suppl. Fig. 7c, d). Using stringent cutoff criteria to identify significant interactors, we found 76 proteins that co-precipitated with Ruvbl1 based on five independent experiments ((FDR = 0.01 and s0 = 1), Fig. 4c and Suppl. Table 1). As previous reports have suggested common and distinct functions of Ruvbl1 compared with its partner protein Ruvbl2, we performed the same experiments with FlpIn cells expressing GFP.Ruvbl2 (Fig. 4d and Suppl. Table 2). The proteins that were co-purified with Ruvbl2 almost exclusively contained proteins from the Ruvbl1 interactome, consistently with similar functions (Fig. 4e) and confirmed previous interactome analyses in different species13,14,15,16,47. Estimation of complex stoichiometry using iBAQ values demonstrated a predominance of Ruvbl1 and Ruvbl2 protein copy numbers in the protein complex. Ruvbl1 and Ruvbl2 occurred at a similar molar ratio, consistent with the recently described dodecameric conformation of the Ruvbl1/2 complex composed of two heterohexameric rings13,14,15,16,48. Among the most abundant identified proteins were many previously known components of the Ruvbl1/2 interactomes, including the R2TP components Rap3a3 and Pih1d1 as well as components of the prefoldin chaperone complex (Fig. 4f, g). Consistent with a possible mechanistic link to cilia function and cilia morphogenesis, one-third (25 of the 76 proteins pulled down with Ruvbl1) were mapped onto the centrosome–cilium interface Database (CCDB) of proteins obtained by proximity labeling (Fig. 4g)49. Furthermore, the analysis identified known ciliopathy-associated proteins, including WD-repeat domain 34 (Wdr34). Wdr34 is associated with Jeune syndrome, a chondrodysplasia associated with dysfunction of primary cilia50,51,52. Immunofluorescence staining showed normal expression and localization of Wdr34 in GFP.Ruvbl1-expressing FlpIn NIH-3T3 cells (Suppl. Fig. 7e). Interestingly, however, Wdr34 mRNA expression was significantly reduced in renal epithelium after induced loss of Ruvbl1 (Suppl. Fig. 7f). Additional candidates in our analysis were the motile ciliopathy protein deleted in primary ciliary dyskinesia homolog (Dpcd)53, as well as proteins involved in cytosolic and prefoldin-associated pre-assembly of ciliary protein complexes in multiciliated cells54. Remarkably, we did not identify any Nph proteins in our MS/MS data sets, suggesting that expression of Nph proteins was below detection levels. Within the increased protein population, the GO terms “chromatin modification” as well as “Ino80 complex” and “microtubular transport” were statistically overrepresented (Fig. 4h). In summary, the interactome analyses support the conclusion that the observed renal phenotype after Ruvbl1 deletion may be the consequence of an imbalance of various cellular mechanisms influenced by Ruvbl1 and suggest that the cellular function of Ruvbl1 includes cilia-associated processes, in addition to its known extraciliary functions.

a Cloning strategy to generate an FlpIn NIH-3T3 cell line stably expressing a GFP.Ruvbl1 transgene. b The GFP.Ruvbl1 transgene showed nearly endogenous Ruvbl1 protein expression levels in stably integrated FlpIn NIH-3T3 cells compared with cells expressing a GFP.GFP transgene. c, d Analysis of the Ruvbl interactome (n = 5). GFP.Ruvbl1 or GFP.Ruvbl2 were immunoprecipitated and analyzed by label-free quantitative proteomics. Seventy-six proteins had a significantly increased abundance in the GFP.Ruvbl1 immunoprecipitation (Ruvbl1) compared with the control (GFP) pulldown (c). Thirty-three proteins had a significantly increased abundance in the GFP.Ruvbl2 immunoprecipitation (Ruvbl2) compared with the control (GFP) pulldown (d). In the volcano plots, the significance between groups (−log10 p value) is plotted against the log2 fold change between groups. Dots outside the curved line represent proteins with significantly increased abundance (FDR 0.01, s0 = 1). The corresponding data for the Ruvbl1/2 interactome can be found in Suppl. Tables S1 and S2. e Comparison of the Ruvbl1 and Ruvbl2 interactome. The Ruvbl2 interactome overlapped with proteins identified as Ruvbl1 interactors. f Stoichiometry analysis of Ruvbl1 and Ruvbl2 interactors. iBAQ values were normalized to Ruvbl2 to obtain an estimation of the absolute abundances of proteins within the purified complex. The ten most abundant proteins in the Ruvbl1 interactome are depicted. # marks proteins previously associated with cilia. g STRING network visualization of the Ruvbl1 interactome demonstrated a prominent representation of ciliary proteins (violet = identified in Centrosome and Cilium Database (CCDB), blue = not identified in CCDB). h Gene ontology (GO) term overrepresentation analysis of Ruvbl1 interactors compared with non-enriched proteins (“background binding proteins”). Fold enrichment of the significantly enriched GO terms is plotted for GO-MF (molecular function, black bars), GO-CC (cellular compartment, gray bars), and GO-BP (biological process, white bars) terms. Statistical enrichment was performed using a Fisher’s exact test (FDR < 0.02)
Ruvbl1 deletion in cells with motile cilia results in hydrocephalus and failed localization of ciliary dynein components
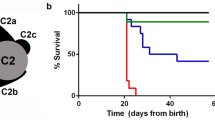
The detection of Dpcd in our interactomic analysis suggested a link between Ruvbl1 and the function of motile cilia. To uncover the role of Ruvbl1 in cells expressing motile cilia, we crossed Ruvbl1fl/fl mice to a previously described tamoxifen-inducible FoxJ1:Cre line29. The conserved FoxJ1 transcription factor induces expression of genes that control the generation of motile cilia in multiple tissues, including the epithelia of airways, oviducts, or brain ventricles55,56. The specificity and efficacy of the FoxJ1:Cre:ERT2 line were confirmed through the mT/mG reporter line (Suppl. Fig. 9) and by immunofluorescence staining in brain (Suppl. Fig. 10) and lung (Suppl. Fig. 11). Importantly, efficacy appeared to be stronger in ventricular ependymal cells than in bronchial epithelial cells in the lung (Suppl. Fig. 9). After Cre-induction, the Ruvbl1fl/flFoxJ1:Cre:ERT2+ animals, but not control mice, showed progressive weight loss over time and had to be killed approximately 7 weeks after induction (Fig. 5a). While thoracic computer tomography and histologic examination of the lungs did not show a dramatic pulmonary phenotype (Suppl. Fig. 11), these animals displayed significant widening of brain ventricles as a sign of hydrocephalus and hence ciliary dysfunction (Fig. 5b). Consistently, cerebral magnetic resonance imaging with subsequent quantification revealed a significant enlargement of ventricular volumes in Ruvbl1fl/flFoxJ1:Cre:ERT2+ animals compared with various controls (Fig. 5c and Suppl. Fig. 10b). This phenotype implicates an important function of Ruvbl1 in cells with motile cilia.

a Induction of Ruvbl1 knockout (iKO; n = 34) in multiciliated cells by tamoxifen injection (“day 0”) led to significant weight loss compared with control (co; n = 31) or heterozygous (het; n = 8) littermates. Repeated measures two-way ANOVA (F(26,906) = 8.821, p < 0.0001) with Bonferroni-corrected post hoc analyses (****p < 0.0001). b Histological PAS staining of brain tissue showing that the induction of Ruvbl1 knockout in ependymal cells led to widening of brain ventricles (asterisks). Scale bars: 1 mm. c MRI scan of brain showing significantly increased ventricle volumes in the induced knockout mice (iKO; n = 4) compared with control (co; n = 3) or heterozygous (het; n = 3) littermates (F(2, 7) = 34.47, p = 0.0002; Tukey’s post hoc ***p < 0.001). Mean ± SEM. d Dnaaf1 specifically co-precipitated with Ruvbl1, but not with the control protein. FLAG-tagged Ruvbl1 or control protein (EPS) was precipitated, and precipitates were analyzed for co-precipitating V5-tagged Dnaaf1. e Dnaaf1 specifically co-precipitated with Dpcd, but not with a control protein. FLAG-tagged Dpcd or control protein (EPS) was precipitated, and precipitates were analyzed for co-precipitating V5-tagged Dnaaf1. f Ruvbl2 specifically co-precipitated with Dpcd, but not with a control protein. FLAG-tagged Dpcd or control protein (EPS) was precipitated, and precipitates were analyzed for co-precipitating V5-tagged Ruvbl2
Interestingly, Ruvbl1 and the motile ciliopathy-associated protein Dpcd, which were detected in our interactome analysis, were also found independently in pulldown experiments using dynein axonemal assembly factor 1 (Dnaaf1) as bait (data not shown); our data confirmed the association of Ruvbl1 with both Dnaaf1 and Dpcd (Fig. 5d–f). Mutations in DNAAF132 and Dpcd53 in humans and mice are known causes of primary ciliary dyskinesia syndromes. Thus, we assumed that a targeted deletion of Ruvbl1 in multiciliated cells would mimic a phenotype of dysfunction of motile cilia. Together with the established association of Ruvbl1 with Pih family proteins, some of which have been associated with pre-assembly of dynein arms, this finding suggests that Ruvbl1 could be a scaffold protein for machinery involved in the assembly of ciliary protein complexes. An important indicator of deficient pre-assembly of protein complexes in motile cilia is failed localization of dynein-associated proteins17,32,57. Therefore, we investigated ciliary and further subcellular localization of several ciliary components, including Dnali1 and Dnai2, as main dynein components. The expression and localization of Arl13b and Ift88 were not changed in cells depleted for Ruvbl1 compared with control cells (Suppl. Fig. 12). Strikingly however, when staining the epithelium of the ependyma and the oviduct, the inner dynein arm component dynein axonemal light intermediate chain-1 (Dnali1, Fig. 6a) and the outer dynein arm component dynein axonemal intermediate chain 2 (Dnai2, Fig. 6b) were not detectable in cilia of Ruvbl1 knockout cells, suggesting an important role of Ruvbl1 in the normal protein composition and function of motile cilia. These data were supported by transmission electron microscopy analyses of multiciliated cells from ovary duct (Fig. 6c). In cilia from Ruvbl1-proficient tissues, electron-dense material representing radial spokes and outer dynein arms could frequently be observed. However, these signals could not be detected in cilia of induced Ruvbl1fl/flFoxJ1:Cre:ERT2+ mice. Instead, undefined protein clusters that might originate from mislocalized ciliary proteins were visible only in knockout cilia. Moreover, whereas in control cilia the microtubule doublets were arranged regularly in an almost perfect circle, microtubule doublets were less organized in knockout cilia (Fig. 6c).

a Immunofluorescence staining of ependymal cells revealed that expression of the inner dynein arm complex member Dnali1 and its ciliary localization was substantially reduced in induced Ruvbl1 knockout mice. Scale bars: 10 µm. b Immunofluorescence staining of the ovary duct clearly demonstrated that Dnai2, a compartment of the outer dynein arm complex, lost ciliary localization in multiciliated cells, showing a complete Ruvbl1 knockout (*). Cells still expressing Ruvbl1 showed Dnai2 localization to cilia (+). Scale bars: 10 µm. c Transmission electron microscopy of motile cilia in the ovary duct revealed structural ciliary changes after induction of Ruvbl1 knockout. Electron-dense material corresponding to, e.g., radial spokes (marked with arrows) and outer dynein arms (marked with arrowheads) in control mice only. However, in induced Ruvbl1 knockout mice, these components could not be found. Instead, undefined protein clusters were identified in motile cilia of knockout mice (asterisks). In addition, in control mice, the nine microtubule doublets were arranged around the central pair in a nearly perfect ring shape, whereas in knockout animals the microtubule doublets were less organized (yellow circle). Scale bars: 100 nm (left) and 250 nm (right)
Discussion
Cilia research has revolutionized our mechanistic understanding of a large number of important genetic syndromes and diseases1,2. Genetic studies and a variety of model organisms combined with multi-dimensional cell culture systems have provided fundamental new cell biological and pathophysiological insights. However, the precise molecular mechanisms that control intracellular trafficking to cilia and generate its very specific protein composition are still not entirely understood. Substantial insight has been obtained from studies in the green algae Chlamydomonas reinhardtii and in zebrafish, as well as from ciliopathy-oriented genetics1,32,58,59,60,61,62. Cilia, as highly specialized microtubule-based organelles, require the proper composition and arrangement of multi-protein supercomplexes and structural components to fulfill their diverse functions. Therefore, ciliary protein components must be delivered and transported within cilia since protein biosynthesis is absent within the cilium. Ruvbl1 is involved in multiple cellular processes, and the observed phenotype of Ruvbl1 deficiency in the renal tubule is most likely the result of a complex interplay of various affected signaling pathways. However, our study adds an important novel mechanism that contributes to this field: We describe Ruvbl1 as a ciliopathy-associated protein involved in the pre-assembly of ciliary protein complexes. The evolutionarily highly conserved AAA-ATPase Ruvbl1 has previously been linked to ciliary biology and ciliopathies in different ways63,64,65,66,67,68. Most importantly, it has been shown that the Ruvbl1 homolog in Chlamydomonas reinhardtii is upregulated during flagellar regeneration and that knockdown of ruvbl1 in the zebrafish Danio rerio is associated with a phenotype consistent with ciliary dysfunction that includes cystic kidneys66,67,68. The closely related family member ruvbl2 is associated with comparable phenotypes that have been attributed to both cilia-specific and cilia-independent processes and a role in cytosolic dynein arm formation in zebrafish66,67,68,69. Of note, zebrafish pronephric cilia are motile cilia, in contrast to primary cilia in mammalian metanephric kidneys70. Our in vivo studies demonstrate a link between Ruvbl proteins and ciliary biology in mammals. We observe a renal phenotype with tubular dilatations and fewer cilia in Ruvbl1 renal knockout mice, supporting the importance of Ruvbl1 and most likely also Ruvbl1-mediated pre-assembly in immotile, primary cilia. Furthermore, we describe a murine phenotype typical for motile cilia dysfunction and a common Ruvbl1–Dnaaf1 protein complex suggestive of a role for Ruvbl1 in the cytosolic pre-assembly of ciliary protein complexes. Pre-assembly of dynein complexes in the cytoplasm has previously been suggested for motile cilia and has been linked to proteins of the Pih family18,19,20,21,71.
Our study adds two novel important aspects to our understanding of ciliogenesis and ciliary maintenance.
First, we find Ruvbl1 to be crucial in motile cilia. Ruvbl1 is the scaffold protein of the R2TP complex consisting of Ruvbl1 and Ruvbl2, as well as the two Hsp90-interactors Rpap3 and the PIH protein Pih1d115, and many of the functions of Ruvbl1 have been attributed to its role as a co-chaperone. The current work has identified multiple known PCD-related proteins that are involved in the cytosolic pre-assembly machinery of dyneins as part of the Ruvbl1 interactome, including the human PCD-protein DNAAF132,72. Another very recent study in zebrafish could demonstrate that Ruvbl1 is essential for the motility of cilia and stabilization of dynein arm assembly components, which is consistent with our findings73. Knockdown of ruvbl1 leads to a reduction of dynein arms in motile cilia and subsequent defects in the motility of cilia in several ciliated tissues in zebrafish, while Ruvbl1 is essential for the stabilization of two dynein arm assembly factors, Dnai1 and Dnai2, in mouse testis73. Interestingly, the function of Pih1 has previously been described for two proteins involved in dynein assembly in flagella or cilia, Ktu/Pf13 and Mot4817,74. Mutations in the human KTU, also called DNAAF2, cause primary ciliary dyskinesia due to abnormal axonemal dynein arms, resulting in the same human phenotype as DNAAF1 mutations. Mutants of the homolog of ktu in medaka fish, Oryzias latipes, results in a ciliopathy phenotype including PKD17 and thus the same phenotype as deficiency of dnaaf1 and ruvbl1 in zebrafish67,75. Functional studies of pf13, the homolog of the highly conserved ktu in Chlamydomonas reinhardtii, have revealed that this protein is part of a cytosolic pre-assembly complex of dynein arms, and it has been suggested that Ktu can function as a co-chaperone for Hsp7017. Based on our findings, Ruvbl1 and the R2TP-prefoldin complex may act as comparable Dnaaf1-associated co-chaperones for Hsp90 in ciliary trafficking in mice. The observed phenotypes suggest that Ruvbl1 may be a major organizer of murine dynein arm complex assembly prior to delivery into the restricted ciliary compartment. Additional experimental analyses are required to examine the role of Ruvbl1 in the assembly of other ciliary protein complexes. Further components of this complex likely include other proteins such as Dpcd, prefoldin proteins, and the prefoldin-associated Wdr92, which has also been linked to the cytoplasmic assembly of dyneins54. Second, our in vivo data demonstrate that in addition to its role in motile cilia, Ruvbl1 is also important for murine primary cilia in vivo. It is tempting to speculate that the cilia-related function of the R2TP–Hsp90 complex goes beyond modifying dyneins in motile cilia and that similar pre-assembly mechanisms exist in primary cilia to ensure the proper orchestration of this sensory organelle. Primary cilia coordinate multiple signaling pathways, sense the environment, may act as flow sensors and have a crucial function in cell cycle regulation1,2,3,4,5. The entire organelle is reabsorbed and reconstructed in a cyclic fashion during the cell cycle and can contribute to intercellular signaling via exocytic vesicles76. As these different ciliary functions may occur simultaneously, a perfect functional organization within this organelle is crucial. Thus, “outsourcing” of quality control and of assembly steps for multi-protein supercomplexes into the cytoplasm may be required to ensure proper ciliary multitasking. Interestingly, the HSP component Hsp90α localizes to the ciliary base77, and inhibition of Hsp90 slows renal and hepatic cyst growth in a model of autosomal-dominant polycystic kidney disease, a bona fide ciliopathy78,79. A functional link between Ruvbl1 and primary cilia is also supported by our finding that Wdr34 forms part of the Ruvbl1 protein complex and is downregulated in Ruvbl1-deficient renal tubular epithelium. Mutations in WDR34 in humans have been described as a cause of Jeune syndrome, a disease of primary cilia51. Furthermore, Ruvbl1 has recently been identified as a regulator of ciliogenesis in an siRNA-based functional genomics screen80. Most recent independent work has suggested a role for Ruvbl1 and its homolog in ciliogenesis in human epithelial cells and in Chlamydomonas reinhardtii81 and in the co-localization of Dnaaf1 and Ift8882. Our data also demonstrate a downregulation of the crucial ciliogenesis components Ift88 and Kif3a in Ruvbl1-deficient renal tubular epithelium. The phenotype of renal Ruvbl1-deficient mice with tubular dilatations, but without pronounced polycystic kidney disease, may not be exclusively due to ciliary dysfunction, yet our findings point to an important function of Ruvbl1 in ciliary structure and function, including an important role for the R2TP–Hsp90 complex in the pre-assembly of a subset of protein complexes targeted to cilia. In summary, we describe the highly conserved AAA-ATPase Ruvbl1 as a ciliopathy-linked protein in mice. Functionally, our data suggest a role for Ruvbl1 in the cytosolic pre-assembly of ciliary protein complexes and link the R2TP component Ruvbl1 to the structural integrity of both primary and motile cilia.
References
Hildebrandt, F., Benzing, T. & Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533–1543 (2011).
Brown, J. M. & Witman, G. B. Cilia and diseases. Bioscience 64, 1126–1137 (2014).
Reiter, J. F. & Leroux, M. R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547 (2017).
Satir, P. CILIA: before and after. Cilia 6, 1 (2017).
Vivante, A. & Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 12, 133–146 (2016).
Ma, M., Tian, X., Igarashi, P., Pazour, G. J. & Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 45, 1004–1012 (2013).
Renkema, K. Y., Stokman, M. F., Giles, R. H. & Knoers, N. V. A. M. Next-generation sequencing for research and diagnostics in kidney disease. Nat. Rev. Nephrol. 10, 433–444 (2014).
Nachury, M. V. et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213 (2007).
Sang, L. et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528 (2011).
Otto, E. A. et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 34, 413–420 (2003).
Olbrich, H. et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 34, 455–459 (2003).
Liebau, M. C. et al. Nephrocystin-4 regulates Pyk2-induced tyrosine phosphorylation of nephrocystin-1 to control targeting to monocilia. J. Biol. Chem. 286, 14237–14245 (2011).
Grigoletto, A., Lestienne, P. & Rosenbaum, J. The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim. Biophys. Acta 1815, 147–157 (2011).
Nano, N. & Houry, W. A. Chaperone-like activity of the AAA+proteins Rvb1 and Rvb2 in the assembly of various complexes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20110399 (2013).
Rosenbaum, J. et al. The emergence of the conserved AAA+ATPases Pontin and Reptin on the signaling landscape. Sci. Signal. 6, mr1 (2013).
Matias, P. M. et al. The AAA+proteins Pontin and Reptin enter adult age: from understanding their basic biology to the identification of selective inhibitors. Front. Mol. Biosci. 2, 17 (2015).
Omran, H. et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 456, 611–616 (2008).
Dong, F. et al. Pih1d3 is required for cytoplasmic pre-assembly of axonemal dynein in mouse sperm. J. Cell Biol. 204, 203–213 (2014).
Yamamoto, R., Hirono, M. & Kamiya, R. Discrete PIH proteins function in the cytoplasmic preassembly of different subsets of axonemal dyneins. J. Cell Biol. 190, 65–71 (2010).
Paff, T. et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am. J. Hum. Genet. 100, 160–168 (2017).
Olcese, C. et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 8, 14279 (2017).
Liu, Q. et al. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell Proteomics 6, 1299–1317 (2007).
Ishikawa, H., Thompson, J., Yates, J. R. & Marshall, W. F. Proteomic analysis of mammalian primary cilia. Curr. Biol. 22, 414–419 (2012).
Hövelmeyer, N. et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J. Exp. Med. 204, 2615–2627 (2007).
Rodríguez, C. I. et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25, 139–140 (2000).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genes 45, 593–605 (2007).
Shao, X., Somlo, S. & Igarashi, P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 (2002).
Lantinga-van Leeuwen, I. S. et al. Transgenic mice expressing tamoxifen-inducible Cre for somatic gene modification in renal epithelial cells. Genes 44, 225–232 (2006).
Rawlins, E. L., Ostrowski, L. E., Randell, S. H. & Hogan, B. L. M. Lung development and repair: contribution of the ciliated lineage. Proc. Natl Acad. Sci. USA 104, 410–417 (2007).
Schindelin, J. et al. Fiji - an open source platform for biological image analysis. Nat. Methods 9, 676–682 (2012).
Benzing, T. et al. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc. Natl Acad. Sci. USA 98, 9784–9789 (2001).
Loges, N. T. et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 85, 883–889 (2009).
Borgal, L. et al. The ciliary protein nephrocystin-4 translocates the canonical Wnt regulator Jade-1 to the nucleus to negatively regulate β-catenin signaling. J. Biol. Chem. 287, 25370–25380 (2012).
Torres, J. Z., Miller, J. J. & Jackson, P. K. High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics 9, 2888–2891 (2009).
Mruk, D. D. & Cheng, C. Y. Enhanced chemiluminescence (ECL) for routine immunoblotting: An inexpensive alternative to commercially available kits. Spermatogenesis 1, 121–122 (2011).
Outeda, P. et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 92, 1130–1144 (2017).
Rinschen, M. M. et al. The ubiquitin ligase Ubr4 controls stability of podocin/MEC-2 supercomplexes. Hum. Mol. Genet. 25, 1328–1344 (2016).
Rappsilber, J., Ishihama, Y. & Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 (2003).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Kohli, P. et al. Label-free quantitative proteomic analysis of the YAP and TAZ interactome. Am. J. Physiol. Cell Physiol. 306, C805–C818 (2014).
Smits, A. H., Jansen, P. W. T. C., Poser, I., Hyman, A. A. & Vermeulen, M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 41, e28 (2013).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011).
Pettitt, S. J. et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495 (2009).
Bereshchenko, O. et al. Pontin is essential for murine hematopoietic stem cell survival. Haematologica 97, 1291–1294 (2012).
Lantinga-van Leeuwen, I. S. et al. Transgenic mice expressing tamoxifen-inducible Cre for somatic gene modification in renal epithelial cells. Genes 44, 225–232 (2006).
Lakshminarasimhan, M. et al. Proteomic and genomic analyses of the Rvb1 and Rvb2 interaction network upon deletion of R2TP complex components. Mol. Cell Proteomics 15, 960–974 (2016).
Gorynia, S. et al. Structural and functional insights into a dodecameric molecular machine - the RuvBL1/RuvBL2 complex. J. Struct. Biol. 176, 279–291 (2011).
Gupta, G. D. et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163, 1484–1499 (2015).
Schmidts, M. et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 93, 932–944 (2013).
Huber, C. et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am. J. Hum. Genet. 93, 926–931 (2013).
Schmidts, M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J. Pediatr. Genet. 3, 46–94 (2014).
Zariwala, M. et al. Investigation of the possible role of a novel gene, DPCD, in primary ciliary dyskinesia. Am. J. Respir. Cell Mol. Biol. 30, 428–434 (2004).
Patel-King, R. S. & King, S. M. A prefoldin-associated WD-repeat protein (WDR92) is required for the correct architectural assembly of motile cilia. Mol. Biol. Cell 27, 1204–1209 (2016).
Chen, J., Knowles, H. J., Hebert, J. L. & Hackett, B. P. Mutation of the mouse hepatocyte nuclear factor/forkhead homologue 4 gene results in an absence of cilia and random left-right asymmetry. J. Clin. Invest. 102, 1077–1082 (1998).
Brody, S. L., Yan, X. H., Wuerffel, M. K., Song, S. K. & Shapiro, S. D. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. Am. J. Respir. Cell Mol. Biol. 23, 45–51 (2000).
Mitchison, H. M. et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 44, 381–389 (2012). S1-2.
Wessely, O. & Obara, T. Fish and frogs: models for vertebrate cilia signaling. Front. Biosci. 13, 1866–1880 (2008).
Diniz, M. C., Pacheco, A. C. L., Farias, K. M. & de Oliveira, D. M. The eukaryotic flagellum makes the day: novel and unforeseen roles uncovered after post-genomics and proteomics data. Curr. Protein Pept. Sci. 13, 524–546 (2012).
Huang, B., Piperno, G. & Luck, D. J. Paralyzed flagella mutants of Chlamydomonas reinhardtii. Defective for axonemal doublet microtubule arms. J. Biol. Chem. 254, 3091–3099 (1979).
Sullivan-Brown, J. et al. Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Dev. Biol. 314, 261–275 (2008).
Sung, C.-H. & Leroux, M. R. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat. Cell Biol. 15, 1387–1397 (2013).
Jha, S. & Dutta, A. RVB1/RVB2: running rings around molecular biology. Mol. Cell 34, 521–533 (2009).
Giorgio, G. et al. Functional characterization of the OFD1 protein reveals a nuclear localization and physical interaction with subunits of a chromatin remodeling complex. Mol. Biol. Cell 18, 4397–4404 (2007).
Ducat, D., Kawaguchi, S.-I., Liu, H., Yates, J. R. 3rd & Zheng, Y. Regulation of microtubule assembly and organization in mitosis by the AAA+ATPase Pontin. Mol. Biol. Cell 19, 3097–3110 (2008).
Stolc, V., Samanta, M. P., Tongprasit, W. & Marshall, W. F. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc. Natl Acad. Sci. USA 102, 3703–3707 (2005).
Sun, Z. et al. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Dev. Camb. Engl. 131, 4085–4093 (2004).
Amsterdam, A. et al. Identification of 315 genes essential for early zebrafish development. Proc. Natl Acad. Sci. USA 101, 12792–12797 (2004).
Zhao, L. et al. Reptin/Ruvbl2 is a Lrrc6/Seahorse interactor essential for cilia motility. Proc. Natl Acad. Sci. USA 110, 12697–12702 (2013).
Kramer-Zucker, A. G. et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Dev. Camb. Engl. 132, 1907–1921 (2005).
Kakihara, Y. & Houry, W. A. The R2TP complex: discovery and functions. Biochim. Biophys. Acta 1823, 101–107 (2012).
Duquesnoy, P. et al. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 85, 890–896 (2009).
Li, Y., Zhao, L., Yuan, S., Zhang, J. & Sun, Z. Axonemal dynein assembly requires the R2TP complex component Pontin. Dev. Camb. Engl. 144, 4684–4693 (2017).
Yamamoto, R., Hirono, M. & Kamiya, R. Discrete PIH proteins function in the cytoplasmic preassembly of different subsets of axonemal dyneins. J. Cell Biol. 190, 65–71 (2010).
van Rooijen, E. et al. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J. Am. Soc. Nephrol. 19, 1128–1138 (2008).
Baldari, C. T. & Rosenbaum, J. Intraflagellar transport: it’s not just for cilia anymore. Curr. Opin. Cell Biol. 22, 75–80 (2010).
Wang, H. et al. Hsp90α forms a stable complex at the cilium neck for the interaction of signalling molecules in IGF-1 receptor signalling. J. Cell Sci. 128, 100–108 (2015).
Seeger-Nukpezah, T. et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc. Natl Acad. Sci. USA 110, 12786–12791 (2013).
Smithline, Z. B. et al. Inhibiting heat shock protein 90 (HSP90) limits the formation of liver cysts induced by conditional deletion of Pkd1 in mice. PLoS ONE 9, e114403 (2014).
Wheway, G. et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087 (2015).
Tammana, D. & Tammana, T. V. S. Human DNA helicase, RuvBL1 and its Chlamydomonas homologue, CrRuvBL1 plays an important role in ciliogenesis. Cytoskelet. Hoboken 74, 251–259 (2017).
Hartill, V. L. et al. DNAAF1 links heart laterality with the AAA+ATPase RUVBL1 and ciliary intraflagellar transport. Hum. Mol. Genet. 27, 529–545 (2018).
Acknowledgements
The authors thank Stefanie Keller, Ruth Herzog, and Martyna Brütting for excellent technical assistance. We thank the CECAD Imaging facility for outstanding technical support. We thank Oliver Rinner (Department of Biology, Institute of Molecular Systems Biology, ETH, 8092 Zurich, Switzerland; Biognosys AG, 8952 Schlieren, Switzerland) and Ruedi Aebersold (Department of Biology, Institute of Molecular Systems Biology, ETH, 8092 Zurich, Switzerland) for a very fruitful collaboration that led to the initial identification of RUVBL1 and RUVBL2 as components of the NPHP complex. This study was supported by the Deutsche Forschungsgemeinschaft (SCHE 1562/6 to B.S.). M.C.L. was supported by a Koeln Fortune Grant and the GEROK program of the Medical Faculty of the University of Cologne, and by the Marga and Walter Boll-Stiftung. M.C.L. and B.S. are supported by the German Federal Ministry for Education and Research (NEOCYST consortium FKZ 01GM1515E). C.D. was supported by a Koeln Fortune Grant. R.H.G. and S.G.B. acknowledge funding from the EU FP7 “SYSCILIA” consortium (no. 241955) and the Dutch Kidney Foundation “KOUNCIL” (CP11.18).
Author information
Authors and Affiliations
Contributions
C.D., M.M.R., L.B., C.E., S.G.B., M.F., M.R., W.B., F.T.W., D.T., T.M., S.H., and M.C.L. designed and conducted experiments. M.R., H.G., W.B., F.T.W., D.J.M.P., J.D., R.U.M., J.C.B., T.P., R.H.G., T.B., B.S., and M.C.L. provided reagents. C.D., M.M.R., L.B., M.H., M.R., H.G., W.B., F.T.W., R.U.M., J.C.B., T.P. R.H.G., T.B., B.S., and M.C.L. analyzed data. M.C.L., B.S., and C.D. wrote the manuscript, which was critically reviewed by L.B., M.M.R., M.H., D.J.M.P., R.U.M., R.G.H., and T.B. M.C.L. and B.S. directed the research, analyzed data, and had overall oversight over the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dafinger, C., Rinschen, M.M., Borgal, L. et al. Targeted deletion of the AAA-ATPase Ruvbl1 in mice disrupts ciliary integrity and causes renal disease and hydrocephalus. Exp Mol Med 50, 1–17 (2018). https://doi.org/10.1038/s12276-018-0108-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0108-z
This article is cited by
-
Primary cilia suppress Ripk3-mediated necroptosis
Cell Death Discovery (2022)
-
β-Catenin Deletion in Regional Neural Progenitors Leads to Congenital Hydrocephalus in Mice
Neuroscience Bulletin (2022)
-
Targeted deletion of Ruvbl1 results in severe defects of epidermal development and perinatal mortality
Molecular and Cellular Pediatrics (2021)
-
Motile cilia genetics and cell biology: big results from little mice
Cellular and Molecular Life Sciences (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}