
Abstract
Based on the discovery by the Resilience Project (Chen R. et al. Nat Biotechnol 34:531–538, 2016) of rare variants that confer resistance to Mendelian disease, and protective alleles for some complex diseases, we posited the existence of genetic variants that promote resilience to highly heritable polygenic disorders1,0 such as schizophrenia. Resilience has been traditionally viewed as a psychological construct, although our use of the term resilience refers to a different construct that directly relates to the Resilience Project, namely: heritable variation that promotes resistance to disease by reducing the penetrance of risk loci, wherein resilience and risk loci operate orthogonal to one another. In this study, we established a procedure to identify unaffected individuals with relatively high polygenic risk for schizophrenia, and contrasted them with risk-matched schizophrenia cases to generate the first known “polygenic resilience score” that represents the additive contributions to SZ resistance by variants that are distinct from risk loci. The resilience score was derived from data compiled by the Psychiatric Genomics Consortium, and replicated in three independent samples. This work establishes a generalizable framework for finding resilience variants for any complex, heritable disorder.
Similar content being viewed by others

Introduction
Our progress in understanding the genetic basis for mental disorders has accelerated over the last decade due to improved methods and the increased sample sizes collated by the Psychiatric Genomics Consortium (PGC) [1]. Dissecting the genetic risk for these disorders is, in itself, extraordinarily valuable for guiding mechanistic studies, developing better diagnostics, and formulating therapeutics. But an understanding of risk states also has the benefit of allowing research on resilience. Knowing how some people avoid illness despite being at elevated risk should shed light on novel avenues for early intervention or treatment that could not be illuminated by studying affected individuals alone.
The psychological, sociological, and biological constructs of resilience, commonly defined as positive adaptation to extreme adversity [2], have been studied extensively. This paradigm of resilience focuses on “active coping” mechanisms; e.g., high emotionality, flexibility of thinking, having social support, and a sense of purpose, among others [3]. The way we are conceptualizing resilience differs from the traditional view in that we attribute resilience in part to heritable variation that increases resistance to disease, which closely relates to the paradigm that was invoked by the Resilience Project [4]. This new paradigm of genetic resilience focuses on the discovery of genetic variants that help unaffected individuals cope with a relatively large genetic burden of disease-associated variants. Our model has two postulates: (1) genetic resilience is in part mediated by common genetic variants that act by lowering the penetrance of risk variants, and (2) resilience variants are orthogonal to risk loci. The meaning of the second postulate is that resilience as defined here is not simply the inverse of risk. As opposed to “protective” variants, which are simply the alternate alleles at each risk-associated locus that have higher frequency in controls than cases, resilience alleles are hypothesized to ameliorate the effects of the risk loci and reduce the likelihood of the disorder. Research on the genetic basis of resilience (i.e., resistance to onset of illness) is contingent upon and necessarily lags behind the discovery of bona fide risk states. For schizophrenia (SZ), the Psychiatric Genomic Consortium (PGC) has identified a “polygenic risk score” that accounts for ~20% of the heritability on the observed scale (or ~6–7% on the liability scale) in risk through the additive effects of thousands of common variants [5, 6]. The reliability of this risk score continues to increase as additional samples contribute to its derivation; however, the genetic variance accounted for by additive effects of individual alleles appears to have recently reached an asymptote [7]. With the allelic-additive common-variant landscape of SZ coming into view, we have arrived at a point where a systematic and risk-informed study of the genetic basis for resilience to SZ is both possible and warranted, capitalizing on this estimate of polygenic risk.
The pursuit of genetic resilience factors for complex neuropsychiatric disorders is nascent, but not unprecedented. For example, the APOE ε2 allele is a well-known protective factor for Alzheimer’s disease that has been studied for its effects on biological and psychological features that may insulate carriers from the risk for the disorder. Results from prospective studies of psychiatric disorders related to trauma exposure, such as post-traumatic stress disorder (PTSD) in U.S. Marines who experienced extreme combat stress, suggest that genetic resilience factors may mitigate environmental vulnerability to mental illness [8]. Furthermore, studies of genetically modified mice shed light on promising candidate genes involved in glutamatergic synaptic transmission that increase resilience to phenotypes related to SZ [9]. The first bona fide study of genetic resilience was published by Chen et al. [4], which focused on rare diseases that manifest in childhood, and emerged from the Resilience Project undertaken by Mount Sinai and Sage Bionetworks [4]. Although our study is completely separate from the work by the Resilience Project, we consider both branches of work to parallel one another. In the Chen et al. [4], study, nearly 600,000 healthy adults were surveyed for highly penetrant mutations associated with Mendelian diseases that typically manifest during childhood (i.e., c.1558 G > T;p.V520F [cystic fibrosis], c.964-1 G > C [Smith-Lemil-Optiz syndrome], c.2204 + 6 T > T [Familial dysautonomia], etc.) [4]. The results from the Chen et al. [4] study suggest that a small number of individuals (~0.0022%) are genetically resilient to rare and devastating forms of childhood disease . Their study demonstrates that genetic resilience is an important avenue for understanding disease etiology. Investigating genetic and environmental factors that counteract inherited or exposed sources of adversity may help illuminate mechanisms that can be modulated to divert or reverse pathophysiological processes.
Here we present a general framework for identifying common variants that promote genetic resilience and for computing a “polygenic resilience score” that moderates the penetrance of known genetic risk factors. This is a proof-of-concept study that has the potential to increase understanding about the resilience to complex polygenic disorders. In essence, the strategy is to identify unaffected individuals at the highest levels of genetic risk, match them to affected individuals at equivalent levels of risk, and contrast these two subgroups to find residual variation associated with resilience. We present the results of applying this method in the largest available sample of SZ from the PGC and in three independent replication samples. We also describe general principles, specific parameters, limitations, and future applications of the approach, which may be useful for studying resilience to any heritable, complex, polygenic disorder.
Methods
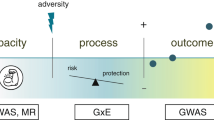
Our approach to derive polygenic resilience scores for SZ is presented schematically in Fig. 1. Supplementary Table 1 outlines decision points that occurred in our analysis and parameters used at each point, including steps for truncating samples and variants for the GWAS and deriving an informative SNP set for resilience scoring. Wherever possible, we adhered to the precedent set by the PGC-SZ Working Group [5].

An Illustration of our method for deriving polygenic resilience scores for a complex disorder
Description of case–control GWAS samples
A description of sample ascertainment procedures used in each study is available in the supplementary text published by the PGC-SZ Working Group [5]. All subjects were confirmed to be independent based on relatedness tests using directly genotyped SNPs. Cases had a clinical diagnosis of SZ-spectrum disorders or SZ based on Diagnostic and Statistical Manual for Mental Disorder (DSM)-Version IV or International Classification of Diseases (ICD), 10th revision. Control ascertainment varied across studies. Our study inherited the same issues that the PGC-SZ faced in that some of the studies comprised control sets that were not screened for SZ, which to the best of our knowledge included the Gottingen Research Association for Schizophrenia (GRAS, controls n = 1232) and the Icelandic deCODE Genetics, Inc. sample which served as an out-of-sample replication set (controls n = 138,761). If there happen to be controls affected with SZ in our discovery sample (unlikely to be a large number as population prevalence of SZ is ~1%), then the loss of power potentially incurred in our analysis should be proportional to published GWAS by the PGC, which in that study was deemed reasonable [5].
Meta-analysis of 51 GWASs in PGC2-SZ
We obtained GWAS summary statistics per study for the same 45 European-ancestry case–control studies, three East Asian-ancestry case–control studies, and three European trio-based studies assembled by the PGC Schizophrenia Working Group for their published wave 2 data set (PGC2-SZ) [5]. We then meta-analyzed single-variant association results genome-wide using inverse-variance fixed-effect summary statistics for these 51 studies using the inverse-variance fixed-effect method in the software METAL [10].
LD-clumped SNPs and weights for risk scoring
We obtained from the PGC-SZ a list of 103,129 linkage-disequilibrium (LD)-independent SNPs and effect-size weights derived from the 51-study GWAS meta-analysis of SZ [5], along with imputed GWAS data for 45 of the 51 studies analyzed by Ripke et al. [5] (excluding six studies privately owned by pharmaceutical companies and unavailable to us for secondary analysis: four Johnson & Johnson and Roche samples, the Pfizer sample, and the Eli Lilly sample).
Identifying subjects with high levels of polygenic risk
The variance in SZ explained by polygenic risk score (PRS) was maximized at a p-value bin of p < 0.05 [5], thus we used that threshold for selecting risk alleles for our PRS analysis. PRS were standardized using z-score scaling within each study. Subjects were then ranked by PRS and a percentile-based threshold was used to identify the highest-risk controls, i.e., “resilient” controls, along with cases with similarly high PRS. We set an arbitrary threshold at the 90th percentile of PRS in controls, and called controls above that threshold ‘resilient’ (different thresholds could be chosen). SZ cases whose PRS was between the 90th percentile for controls and the maximum PRS for controls were retained as the comparison group (Fig. 1). This method produced 3786 high-risk resilient controls (includes 83 pseudo-controls from trio studies) and 18,619 equal-risk cases (includes 494 cases from trio studies) for analysis. The total number of cases and controls retained per study after risk score matching is provided in Supplementary Table 2.
Ripke et al., reported that subjects in the highest PRS decile exhibit an increased risk for SZ (OR = 8–20) compared with the baseline rate of SZ in the lowest PRS decile [5], which is on-par with the estimated increase in risk for SZ among persons with affected first-degree relatives [11]. Building on these findings, Supplementary Table 3 shows that the population in the upper 10th percentile of PRS (who were included in our analysis) have an absolute risk of 40% and an increase in relative risk of 1.91 compared with all subjects above the lowest 10th percentile of PRS, which is similar to the relative increase in risk for those with an affected aunt or uncle [11].
Derivation of polygenic resilience scores
Marginal SNP effects on resilience were computed using logistic regression models with Plink v1.9 [12] including adjustment for four principal components derived from autosome-wide SNP data that were significantly different between high-risk controls and equal-risk cases at a significance threshold of p < 0.1 to correct for population stratification (Supplementary Table 4). Any variant that showed an association with SZ risk from our meta-analysis of the 51 PGC2-SZ studies at a p < 0.05, or variants that were in LD with risk variants (at a R2 > 0.2 in a 1 megabase window), were discarded from this GWAS of resilience as a conservative measure, to avoid re-discovering risk variants and to ensure that any polygenic resilience score derived from our analysis was independent of the polygenic risk score used to stratify the samples. Our choice of threshold for pruning away SNPs in LD was used by the PGC and others [13,14,15]. We considered setting a stricter threshold of R2 ≤ 0.0, but this would have removed almost the entire genome and left only six variants for evaluation. Marginal SNP effects were obtained per study, and then pooled using inverse-variance fixed effect meta-analysis [10] to arrive at a final GWAS summary statistic per SNP.
Generating a polygenic score formula for resilience was done in a series of steps. Clumping the GWAS summary statistics output (Plink command: --clump-kb 500 --clump-r2 0.2 --clump-p1 1.0 --clump-p2 1.0) was performed using a reference panel that represents the predominant ancestry found in the GWAS sample (i.e., 1000 Genomes European phase 1 version 3, n = 379). We removed variants according to the following exclusion criteria referenced by the PGC-SZ working group in ref. [5] to retain an informative SNP set for polygenic resilience scoring: (i) variants in the MHC region (chr6:25 Mb-34 Mb), (ii) variants in the chromosome 8 inversion region (chr8:7 Mb–14 Mb), (iii) variants with an imputation quality score <0.9, and (iv) variants that are strand-ambiguous (AT/CG genotypes) or were small insertions/deletions. In addition, we removed variants with a minor allele frequency <0.05 and variants present in 10 or fewer studies. A total of 80,822 LD-independent SNPs associated with resilience to SZ were available for inclusion in calculating “resilience scores” in our training and test sets.
Resilience scores were determined by counting the number of protective alleles for sets of variants defined by p-value cutoffs (p < 0.0001, <0.001, <0.01, <0.05, <0.1, <0.2, <0.3, <0.5, <0.7, <1.0) and multiplying allele counts by the natural logarithm of the resilience odds ratio for each variant. We adopted a method used by the PGC [5] to estimate the amount of variance in resilience status that can be attributed to resilience scores. The approach is a two-model regression. In the first regression, resilience scores were specified as a predictor variable to estimate the odds of being a high-risk resilient control versus a matched-risk case (treated as the reference) per standard unit increase in resilience score. Four principal components derived from genome-wide SNP data that were significantly different between high-risk controls and high-risk cases were included in this regression as covariates to control for population stratification. A second logistic regression model was fit to estimate the amount of additive variance that the four principal component covariates that were specified in the first model contributed to resilience status. The variance in resilience status explained by each model was (based on Nagelkerke’s pseudo-R2) was calculated using the R package fmsb (version 0.6.1). We calculated the difference in R2 values between in the two models, yielding a value of R2 that was attribute to the amount of variance in resilience status explained by resilience scores.
Correlation and interaction analyses of risk and resilience scores
We calculated Pearson’s correlation coefficients for risk and resilience scores in four separate groups: (1) SZ cases, (2) controls, (3) subjects with low-risk scores for SZ grouped by case–control status, and (4) and subjects with high-risk scores grouped by case–control status. Ultra-high-risk cases were excluded from the analysis (i.e., SZ cases with a risk score exceeding the maximum risk score of controls) as they lacked a matching set of controls. In addition, we performed a logistic regression analysis using all PGC samples (excluding ultra-high-risk cases) to test for a non-linear effect of risk and resilience scores on case–control status. Subjects were then split into deciles based on risk score. With logistic regressions, we computed the odds of a being a case between the bottom decile compared with each of the other deciles, as well as comparing the estimate the change in odds of being a control versus a case within each decile based on the unit increase in resilience score. The top 10 principal components for ancestry were included as covariates in the regression models.
Replication
We had direct access to imputed GWAS data for two independent studies that were withheld entirely from all discovery analyses and used exclusively in our replication analysis (i.e., the Molecular Genetics of Schizophrenia (MGS), and the Danish iPSYCH study) along with the summary results from a third fully Icelandic sample from deCODE Genetics [16,17,18]. Subjects in the MGS (n controls = 2,482, n cases = 2,638) sample were ascertained from the US and Australia, which included cases with a DSM-IV diagnosis of SZ or schizoaffective disorder, and controls with no known history of mood, anxiety, substance use, psychotic, or bipolar disorders. Cases from the iPSYCH Consortium sample (n controls = 10,175, n cases = 3634) were ascertained from the Danish Civil Registration System and linked to the Danish Psychiatric Central Research Register to obtain diagnoses of SZ, whereas controls were ascertained by random sampling from the Danish Civil Registration System and removing individuals with a diagnosis of SZ or bipolar disorder. The third case–control replication set was the Icelandic population-based sample generated by deCODE Genetics, Inc [18]. comprised of 138,761 controls and 873 cases. Risk scores were calculated in the replication samples using SNP rsIDs and weights derived from the 51-study GWAS meta-analysis of risk. We applied our 90th percentile threshold method to identify high-risk controls and equal-risk cases. Resilience scores were calculated in the replication samples using the formulae derived in the discovery sample, and logistic regression models were used to estimate the effect of resilience scores on affection status and the proportion of variance in resilience explained by resilience scores after adjusting for select principal components to control for population stratification. For the MGS and iPSYCH samples, we selected principal components that significantly differed between high-risk controls and equal-risk cases at a significance threshold of p < 0.05 (three for MGS and two for iPSYCH). The top 10 principal components were used for the deCODE sample. We performed an inverse-variance fixed-effect meta-analysis with the R package metafor (version 2.0.0) using the natural logarithm of odds ratios and standard errors for resilience scores obtained from the MGS, Danish iPSYCH, and Icelandic deCODE samples, in order to assess the aggregate predictive capacity of resilience scores. Using code adapted from Ricopili (https://github.com/Nealelab/ricopili), the proportion of variance in resilience status explained by resilience scores was transformed to the liability scale assuming a population prevalence of 10% based on the 90% cutoff used to define resilience [19].
Gene annotations
We downloaded a GTF file containing the positions of 57,820 protein- and non-coding genes, RNAs, and pseudogenes from the human reference genome version GRCh37.p13 from GENCODE [20]. The mapping of variants to genes was performed using the R package GenomicRanges. We retained annotations for protein-coding genes, non-coding genes (microRNAs, snoRNAs, snRNA, lincRNA), and antisense genes.
Enrichment of resilience SNPs in risk genes
After annotating SNPs to genes, we computed association scores per gene for both risk and resilience to SZ by averaging the z-scores of intragenic SNPs within a given gene obtained from our meta-analyses. Gene scores were determined using 480,469 intragenic risk SNPs and 1,681,145 intragenic resilience SNPs. We did not extend the coordinates of genes beyond the intragenic region when mapping SNPs to genes. Only risk SNPs at a p < 0.05 significance level were included to ensure that risk SNPs were mostly LD-independent (R2 < 0.2) from resilience SNPs found within the same gene. A linear regression model was used to predict the per-gene risk association score using the per-gene resilience association score while simultaneously adjusting for gene length (in kilobases), the number of SNPs per kilobase of gene length, the average minor allele frequency of variants in each gene, and the chromosome the gene was located on. Cluster-robust standard errors were computed to correct for heteroscedasticity potentially caused by LD between genes.
Results
Polygenic resilience scores
As expected, resilience scores were significantly associated with resilience status in our discovery set (top panel of Fig. 2) with a maximum of 46.9% of the variance in resilience explained by these scores (two-tailed p < 1.0 × 10−300). To replicate the association of resilience scores with resilience status, we performed a meta-analysis using results from three fully independent data sets: European ancestry subsample of the Molecular Genetics of Schizophrenia study (MGS; high-risk controls n = 244, high-risk cases n = 811), the Danish iPSYCH study (high-risk controls n = 931, high-risk cases n = 465), and Icelandic deCODE Genetics samples (high-risk controls n = 6944, high-risk cases n = 161). These samples yielded a total of 8,119 high-risk controls and 1,437 high-risk cases. Our meta-analysis replicated the significant association of resilience scores with resilience status across five p-value bins (p < 0.2, p < 0.3, p < 0.5, p < 0.7, and p < 1.0), with the most significant effect found at the p < 0.3 bin (OR = 1.12 per standardized unit increase in resilience score, SE = 0.041, two-tailed p = 0.0044; bottom panel of Fig. 2). The replicated effects of resilience scores survived multiple-testing correction for each p-value bin at the Benjamini-Hochberg FDR < 0.05 level. In the replication sets, resilience scores explained an average 0.042% (SD = 0.0001) of the variance in resilience status or 0.07% (SD = 0.0002) under the liability-threshold model (i.e., the SNP-heritability of resilience, or h2SNP).

(Top panel) Polygenic resilience scores were computed in our discovery sample (high-risk controls = 3,786, high-risk cases = 18,619) based on results obtained from GWAS meta-analysis of resilience to SZ. The barplot shows the amount of variance in resilience explained by resilience scores (i.e., high-risk controls versus high-risk cases) explained by resilience scores across ten p-value bins. The dot plot shows corresponding Odds Ratios (OR) for resilience scores, wherein OR > 1.0 represents that high-risk controls have higher resilience scores compared with high-risk cases. (Bottom panel) The predictive performance of polygenic resilience scores is shown based on a meta-analysis of results obtained from three independent replication samples (Molecular Genetics of Schizophrenia, iPSYCH, and deCODE Genetics; high-risk controls = 7,653, high-risk cases = 1,903). Average Nagelkerke’s pseudo-R2 values calculated using arithmetic means and 95% confidence intervals are shown in the bottom left panel. Meta-analysis was used to pool natural log of OR and standard errors with an inverse-variance fixed effect model using the R package metafor
Risk and resilience scores showed a small but significant positive correlation in the full discovery sample (Pearson’s r = 0.08, 95% CI = 0.068–0.083, degrees of freedom [df] = 66,615, two-tailed p < 8.2 × 10−95), but this differed by affection status. Risk and resilience scores were not significantly correlated among SZ cases (Pearson’s r = −0.003, 95% CI = −0.015–0.008, df = 28867, two-tailed p = 0.54), but were strongly and significantly positively correlated in controls (Pearson’s r = 0.47, 95% CI = 0.461–0.477, df = 37746, two-tailed p < 2.2 × 10−16), which validates the notion that, as risk score increases, so too must the resilience score in order for an at-risk individual to remain unaffected (Supplementary Fig. 3).
In the discovery sample, resilience scores were significantly higher in controls than cases (two-tailed p < 2.2 × 10−308), in high-risk controls than high-risk cases (two-tailed p < 2.2 × 10−308), and in high-risk controls than low-risk controls (two-tailed p < 2.2 × 10−308, Supplementary Table 5). Upon meta-analysis of the three replication samples, these difference in resilience scores replicated between controls and cases (two-tailed p = 0.013), high-risk controls and matched-risk cases (two-tailed p = 0.002), and high-risk controls and low-risk controls (two-tailed p = 8.3 × 10−8) (Supplementary Table 5). Resilience scores yielded significant associations only when higher p-value thresholds were used, thus we examined the level of LD between risk SNPs and resilience SNPs. We found a significant negative correlation between a SNP’s association with resilience and its level of LD with a risk SNP within 1 Mb (r = −0.032, p = 3.7 × 10−17), indicating that stronger resilience SNPs tend to have less LD with risk SNPs than weaker resilience SNPs. In addition, we found that the average LD between risk SNPs and resilience SNPs was low and relatively uniform across the ten p-value bins used to derive polygenic scores for resilience (range R2: 0.061–0.070). A significant negative interaction effect of risk and resilience scores on case–control status was found in the full PGC sample (β = −0.71, SE = 0.021, p = 1.6 × 10−246) in the presence of a significant main effect of risk score (β = 3.013, SE = 0.024, p < 1.0 × 10−300) and resilience score (β = −0.66, SE = 0.16, p < 1.0 × 10−300). As shown in Supplementary Table 6, an increased odds of being a case was found in higher deciles compared with the bottom decile of risk. Furthermore, controls showed significantly higher resilience scores compared with cases in the upper half of deciles (Supplementary Table 6).
Top loci associated with resilience to SZ
Across the ~1.9 million variants (MAF ≥ 5%) examined in our meta-analysis, we compared the genome-wide resilience p-values with an expected (i.e., null) distribution of p-values, revealing that the observed values fit closely with expected values as shown in the quantile–quantile plot (Supplementary Fig. 1). The median χ2 value from our GWAS deviated slightly from the expected χ2 value as given by the genomic inflation factor (i.e., λGC = 1.03), which was negligible compared with the level of inflation seen in the GWAS meta-analysis of SZ risk by (Ripke et al. [5]) (i.e., λGC = 1.468). None of the individual SNPs displayed a genome-wide significant association with resilience to SZ (Supplementary Fig. 2), which was not unexpected due to the small size of the subsamples of resilient controls and risk-matched cases relative to the full sample from which they were drawn. Results for the top seven resilience loci (p < 1.0 × 10−5) are provided in Supplementary Table 7. As shown in Supplementary Table 7, the top seven resilience SNPs exhibited low LD with risk SNPs (range r2 = 0.0063–0.089), confirming that top resilience loci are largely independent from risk loci. The magnitude of effect sizes for the top seven variants associated with resilience in the present analysis markedly exceeded effect sizes of those same variants on SZ risk in the full GWAS meta-analysis of SZ cases and controls (Fig. 3). One was nominally associated with risk in the full MGS replication sample (rs66718632, p = 0.035), and none in the other replication data sets.

A pair of odds ratios and confidence intervals are plotted for the top seven common variants associated with resilience to SZ (p < 1× 10−05). This demonstrates that variants associated with resilience to SZ were not significantly associated with SZ risk (i.e., p > 0.05). Associations in red are for resilience obtained from the sample of individuals at the upper tail of the distribution of risk scores (i.e., high-risk controls, n = 3,775, and equal-risk cases, n = 18,581), whereas associations in blue were obtained from the full sample of SZ cases and controls in the published PGC-SZ2 data set (51 studies, n cases = 32,838, n controls = 44,357). A dotted line denoted no effect (i.e., OR = 1.0). An OR > 1.0 for red dots indicates that the allele was observed more frequently in high-risk controls than high-risk cases (i.e., increases resilience), whereas a OR > 1.0 for blue dots indicates that the allele was observed more frequently in cases than controls (i.e., increases risk)
We found qualitative evidence for this at the gene level as four of the top seven resilience SNPs and liberally defined risk SNPs (p < 0.05) map to the same genes but different LD blocks (Supplementary Table 8). A regression analysis of per-gene association scores for risk and resilience revealed a small but statistically significant positive linear relationship between a gene’s association with resilience and risk for SZ after adjustment for confounding variables described in the Methods section (regression β = 0.049, robust SE = 0.011, two-tailed p = 4.35 × 10−6).
Discussion
Genetic analyses of resilience may help us understand factors that moderate the risk of mental illness among those at elevated risk. We present here a generalizable strategy to investigate genetic factors involved in resilience to complex polygenic disorders, as well as the first application of the method to demonstrate feasibility. SZ and several other psychiatric disorders have high heritability, and GWAS studies show that common variation is reliably associated with risk [1, 5, 21,22,23,24,25,26,27]. Our approach illuminates the role of common variation in buffering the penetrance of risk alleles associated with complex disease, and may aid in unraveling the sources of their etiologic heterogeneity. We assessed the validity of our method using the large publicly available genome-wide data assembled by the PGC for SZ and replicated both the direction of effect and the significance of a polygenic resilience score in three independent samples.
Our study produced the first estimate of the SNP-based heritability for resilience to SZ, which was relatively low (i.e., h2SNP < 1%) compared with previous estimates of SNP-based heritability reported for SZ [5, 6]. Note that our approach did not look for resilience variants close to the risk variants, and so likely missed a portion. Deriving an estimate of the total heritability of resilience (i.e., H2) from family or twin-based studies will be important for interpretation of our SNP-heritability results, and for future studies that will explore the sharing of genetic predictors between resilience and other phenotypes. We showed that resilience scores produce conditionally independent effects on caseness relative to risk scores, confirming that our hypothesis that resilience alleles are not simply the inverse of the risk-associated alleles. Our resilience-scoring algorithm can be applied to GWAS data sets for other complex medical disorders, especially those for which polygenic risk scores have previously been derived in samples independent of those in which resilience is to be examined (such as our replication samples in the present study). These resilience scores can in turn be used to investigate heritable mediators of resilience, and potentially to identify factors that exert a risk-buffering effect across diagnostic boundaries. Cross-disorder meta-analyses of psychiatric GWAS data have already identified shared polygenic mediators of risk common to several disorders; this approach could be revisited in the context of resilience [22]. Another important question future studies may seek to address is: are there common genetic variants that influence resilience to SZ and also contribute to personality traits (i.e., extraversion, conscientiousness, openness, agreeableness) or constructs of cognitive well-being (i.e., educational attainment, genetic cognitive function) associated with the psychosocial construct of resilience? New methods could be devised to expand the capabilities of our approach or address shortcomings. For instance, because we could not derive a meaningful estimate of heritability from our results using LD score regression (LDSC) due to weak signals from regions of low LD, deriving new models for LDSC to allow for greater flexibility could be valuable. An alternative way to conceptualize resilience is as a form of gene–gene interaction wherein the penetrance of a risk variant can change based on the effect of a resilience variant. We modeled a relatively simple type of interaction by computing the multiplicative effect between risk and resilience score on case–control status and found that a significant interaction emerged between these scores (Supplementary Table 6). In future work, extensive simulation work could be performed to evaluate various types of interaction models to determine which contributes the strongest effects to resilience. Using knowledge about gene pathways and/or protein–protein interaction networks may help to discern epistatic effects between risk and resilience loci, and yield novel insight into the biology of disease. In short, there are multiple ways our method could be used and adapted to revisit previous single- and cross-disorder analyses to uncover genetic factors that mitigate vulnerability with shared or disorder-specific effects, and to account for the decreased penetrance and missing heritability in polygenic risk scores.
Because we focus on the tail of the risk score distribution, the sample size for our resilience analyses was an order of magnitude lower than the corresponding analyses of risk. Nevertheless, seven loci reached a ‘suggestive’ level of association (p < 1.0 × 10−5) with resilience to SZ. Assuming effect sizes hold in larger samples (despite the possibility of winner’s curse), adding about 2,030 high-risk controls to our analysis would be expected to yield sufficient power (>80%) to drive the lowest-ranking SNP in the 5.0 × 10−8 < p < 1.0 × 10−5 bin (e.g., rs11595156(T), OR = 1.32, allele frequency = 0.94) to genome-wide significance. However, it is important for independent replication in a much larger sample, which may be accomplished with the forthcoming phase-3 release of GWAS data from the PGC-SZ Working Group. Following replication, it will be appropriate to build on our genome-wide findings using bioinformatic tools, databases of molecular pathways, and functional annotations to identify and characterize the genetically driven biological pathways that mediate heritable effects on resilience.
To be conservative, we imposed some limitations on our approach, We used a conservative variant-filtering strategy to identify resilience-associated loci that were independent of risk variants. However, it is plausible biological that some resilience variants might be in the same regions or genes as risk variants. Risk-mediating common variants are more likely to occur in regions with broad LD [28], but the scope of the present study, after filtering out risk loci, restricted us to areas with relatively low LD. One caveat with our GWAS results was that resilience variants did not have strong LD support, because resilience variants were largely restricted to regions of low LD. Our approach was designed to identify resilience SNPs that are LD-independent of risk SNPs based on liberal definitions of risk (p < 0.05) and of LD (R2 < 0.2 with a risk-conferring variant) so that we avoided simply detecting additional risk SNPs; yet, biologically, it is expected that resilience SNPs can reduce the penetrance of nearby risk SNPs, even those within the same gene or LD block, such as by counteracting effects on gene expression levels (i.e., risk allele increases expression of gene while resilience allele decreases expression). Future work using Mendelian randomization or conditional association testing in much larger samples could be better suited to test the hypothesis that resilience signals are more likely to co-localize with loci and genes harboring risk variants for SZ.
There was a dramatic reduction in predictive performance of resilience scores when our model was applied to the much smaller, independent replication samples, as expected. Nevertheless, the resilience model was robust enough to yield significant prediction in the meta-analysis of replication samples. The sharp drop-off in variance explained indicates overfitting, which happens when training a model with an inadequate sample size or when too many noisy parameters are included in the model [29]. A challenge to our study design was statistical power, but we expect that the strength of our results in terms of variance explained and p-values of individuals SNPs will increase with the addition of more samples. The sample size for our study actually exceeded the discovery samples for recent PGC studies for anorexia nervosa [30], autism [21], and obsessive-compulsive disorder [31], as well the initial SZ study by the PGC in 2011 [32]. Looking back at the past decade of GWAS for SZ [5, 6, 26, 32, 33], our sample size is at the midpoint studies published between 2008 and 2009, and thus we might anticipate that, as in the study of SZ, the significance of individual resilience loci and their collective phenotypic variance explained will grow exponentially after the addition of more samples.
In conclusion, we have presented evidence for the validity of a method to identify individual loci and polygenic scores associated with resilience to a highly heritable complex disorder. This supports the idea that common variants that are not in LD with known SZ risk alleles exert a protective effect. Further replication will be key for validation of our method and findings. If our results are substantiated, for example in the forthcoming third wave of data from PGC-SZ, we anticipate a new wave of research on the genetics of resilience, the biology associated with resilience variants, and interventions that can foster resilience in at-risk populations.
Code availability
Custom written R scripts used for statistical analyses can be provided upon request.
Data availability
Genome-wide association summary statistics for all 1.9 million SNPs can be downloaded from: https://www.dropbox.com/sh/qhqbkcwvgzuwho3/AAClnqCvIdxIRsaklOLMGDLra?dl=0. The data used in this study were provided under restricted access by the Psychiatric Genomics Consortium (PGC), Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH), and deCODE Genetics, Inc. Only summary statistics were made available to us from deCODE Genetics, Inc. We were granted access to imputed genome-wide SNP genotypes by PGC and iPSYCH. The data sets from PGC and iPSYCH remained on the Genetic Cluster Computer and GenomeDK cluster, respectively, where statistical analyses were performed.
References
Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, et al. Psychiatric genomics: an update and an Agenda. Am J Psychiatry. 2017;175:15–27. appi.ajp.2017.1
Luthar SS, Cicchetti D, Becker B. The construct of resilience: a critical evaluation and guidelines for future work. Child Dev. 2000;71:543–62.
Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009;10:446–57.
Chen R, Shi L, Hakenberg J, Naughton B, Sklar P, Zhang J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531–8.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018. https://doi.org/10.1038/s41588-018-0059-2.
Ripke S and the SWG of the PGC. PGC SCZ Workgroup: GWAS with Over 70,000 Cases and 100,000 Controls. In: World Congress of Psychiatric Genetics. Orlando, Florida; 2017.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW, et al. Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: a genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology. 2015;51:459–71.
Mihali A, Subramani S, Kaunitz G, Rayport S, Gaisler-Salomon I. Modeling resilience to schizophrenia in genetically modified mice: a novel approach to drug discovery. Expert Rev Neurother. 2012;12:785–99.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Mortensen PB, Pedersen CB, Westergaard T, Wohlfahrt J, Ewald H, Mors O, et al. Effects of family history and place and season of birth on the risk of schizophrenia. N Engl J Med. 1999. https://doi.org/10.1056/NEJM199902253400803.
Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015. https://doi.org/10.1186/s13742-015-0047-8.
Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–9.
Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31:1466–8.
Palla L, Dudbridge F. A fast method that uses polygenic scores to estimate the variance explained by genome-wide marker panels and the proportion of variants affecting a trait. Am J Hum Genet. 2015. https://doi.org/10.1016/j.ajhg.2015.06.005.
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–7.
Pedersen CB, Bybjerg-Grauholm J, Pedersen MG, Grove J, Agerbo E, Bækvad-Hansen M, et al. The iPSYCH2012 case-cohort sample: New directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry. 2018;23:6–14.
Hakonarson H, Gulcher JJR, Stefansson K. deCODE genetics, Inc. Pharmacogenomics. 2003;4:209–15.
Lee SH, Goddard ME, Wray NR, Visscher PM. A better coefficient of determination for genetic profile analysis. Genet Epidemiol. 2012;36:214–24.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: The reference human genome annotation for the ENCODE project. Genome Res. 2012;22:1760–74.
The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21.
Smoller JW, Kendler K, Craddock N, Writing JW, Smoller N, Craddock K, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis Cross-Disorder Group of the Psychiatric Genomics Consortium. Lancet. 2013;3811016:1371–9.
Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–6.
Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52.
Richards AL, Jones L, Moskvina V, Kirov G, Gejman PV, Levinson DF, et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Mol Psychiatry. 2012;17:193–201.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7.
Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, et al. A Role for Noncoding Variation in Schizophrenia. Cell Rep. 2014;9:1417–29.
Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Consortium SWG of the PG. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Babyak MA. What you see may not be what you get: a brief, nontechnical introduction to overfitting in regression-type models. Psychosom Med. 2004;66:411–21.
Duncan L, Yilmaz Z, Gaspar H, Walters R, Goldstein J, Anttila V, et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am J Psychiatry. 2017. https://doi.org/10.1176/appi.ajp.2017.16121402.
Arnold PD, Askland KD, Barlassina C, Bellodi L, Bienvenu OJ, Black D, et al. Revealing the complex genetic architecture of obsessive–compulsive disorder using meta-analysis. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.154.
Ripke S, Sanders AR, Kendler KS, Levinson D, Sklar P, Holmans PA. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76.
O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–5.
Acknowledgements
SJG is supported by grants from the U.S. National Institutes of Health (5R01MH101519, 5R01AG054002), the Sidney R. Baer, Jr. Foundation, and NARSAD: The Brain & Behavior Research Foundation. SVF is supported by the K.G. Jebsen Centre for Research on Neuropsychiatric Disorders, University of Bergen, Bergen, Norway, the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement number 602805, the European Union’s Horizon 2020 research and innovation programme under grant agreement number 667302 and NIMH grants 5R01MH101519 and U01 MH109536-01. HJE is supported by grants from the U.S. National Institutes of Health (U10 AA008401; U01 MH109532). Statistical analyses were conducted on the Genetic Cluster Computer, which is financially supported by the Netherlands Scientific Organization (NOW; 480-05-003) along with a supplement from the Dutch Brain Foundation and VU University. The Danish iPSYCH (The Lundbeck Foundation Initiative for Integrative Psychiatric Research) and GEMS2 teams acknowledge funding from The Lundbeck Foundation (grant no R102-A9118 and R155-2014-1724), the Stanley Medical Research Institute, an Advanced Grant from the European Research Council (project no: 294838), the Danish Strategic Research Council and grants from Aarhus University to the iSEQ and CIRRAU centers. The Danish National Biobank resource at Statens Serum Institut was supported by the Novo Nordisk Foundation. Computational resources for handling and statistical analysis of iPSYCH data on the GenomeDK HPC facility were provided by the iSEQ center, Aarhus University, Denmark (grant to ADB).
List of authors in the Schizophrenia Working Group of the Psychiatric Genomics Consortium
Rolf Adolfsson, Department of Clinical Sciences, Psychiatry, Umeå University, Umeå, Sweden; Ingrid Agartz, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Esben Agerbo, National Centre for Register-based Research, Aarhus University, Aarhus, Denmark ; Margot Albus, State Mental Hospital, Haar, Germany; Madeline Alexander, Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, USA; Farooq Amin, Department of Psychiatry and Behavioral Sciences, Emory University, Atlanta, GA, USA; Ole A Andreassen, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Maria J Arranz, Fundació de Docència i Recerca Mútua de Terrassa, Universitat de Barcelona, Spain; Silviu A Bacanu, Virginia Institute for Psychiatric and Behavioral Genetics, Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA; Steven Bakker, Department of Psychiatry, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, Utrecht, The Netherlands; Gavin Band, Wellcome Trust Centre for Human Genetics, Oxford, UK; Ines Barroso, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Martin Begemann, Clinical Neuroscience, Max Planck Institute of Experimental Medicine, Göttingen, Germany; Céline Bellenguez, Wellcome Trust Centre for Human Genetics, Oxford, UK; Richard A Belliveau Jr, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Stephan Bender, Child and Adolescent Psychiatry, University of Technology Dresden, Dresden, Germany; Judit Bene, Department of Medical Genetics, University of Pécs, Pécs, Hungary; Sarah E Bergen, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; Elizabeth Bevilacqua, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Tim B Bigdeli, Virginia Institute for Psychiatric and Behavioral Genetics, Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA; Donald W Black, Department of Psychiatry, University of Iowa Carver College of Medicine, Iowa City, IA, USA; Hannah Blackburn, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Jenefer M Blackwell, Cambridge Institute for Medical Research, University of Cambridge School of Clinical Medicine, Cambridge, UK; Douglas HR Blackwood, Division of Psychiatry, University of Edinburgh, Edinburgh, UK; Elvira Bramon, University College London, UK; Matthew A Brown, Diamantina Institute of Cancer, Immunology and Metabolic Medicine, Princess Alexandra Hospital, University of Queensland, Brisbane, Queensland, Australia; Richard Bruggeman, University Medical Center Groningen, Department of Psychiatry, University of Groningen, The Netherlands; Nancy G Buccola, School of Nursing, Louisiana State University Health Sciences Center, New Orleans, LA, USA; Randy L Buckner, Center for Brain Science, Harvard University, Cambridge MA, USA; Brendan Bulik-Sullivan, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Suzannah J Bumpstead, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Joseph D Buxbaum, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; William Byerley, Department of Psychiatry, University of California at San Francisco, San Francisco, CA, USA; Wiepke Cahn, University Medical Center Utrecht, Department of Psychiatry, Rudolf Magnus Institute of Neuroscience, The Netherlands; Guiqing Cai, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Dominique Campion, Centre Hospitalier du Rouvray and INSERM U1079 Faculty of Medicine, Rouen, France; Rita M Cantor, Department of Human Genetics, David Geffen School of Medicine, University of California, Los Angeles, CA, USA; Vaughan J Carr, Schizophrenia Research Institute, Sydney, Australia; Noa Carrera, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Juan P Casas, Department of Epidemiology and Population Health, London School of Hygiene and Tropical Medicine, London, UK; Stanley V Catts, Royal Brisbane and Women's Hospital, University of Queensland, Brisbane, Australia; Kimberley D Chambert, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Ronald YL Chan, Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Raymond CK Chan, Institute of Psychology, Chinese Academy of Science, Beijing, PR China; Eric YH Chen, Department of Psychiatry and State Ket Laboratory for Brain and Cognitive Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Wei Cheng, Department of Computer Science, University of North Carolina, Chapel Hill, NC, USA; Eric FC Cheung, Castle Peak Hospital, Hong Kong SAR, PR China; Siow Ann Chong, Institute of Mental Health, Singapore; Sven Cichon, Division of Medical Genetics, Department of Biomedicine, University of Basel, Basel, Switzerland; C Robert Cloninger, Department of Psychiatry, Washington University, St. Louis, MO, USA; David Cohen, Department of Child and Adolescent Psychiatry, Pierre and Marie Curie Faculty of Medicine and Brain and Spinal Cord Institute (ICM), Paris, France; Nadine Cohen, Formerly of Neuroscience Therapeutic Area, Janssen Research and Development, LLC, Raritan, NJ, USA; David A Collier, Eli Lilly and Company Limited, Erl Wood Manor, Sunninghill Road, Windlesham, Surrey, UK; Paul Cormican, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity College Dublin, Ireland; Aiden Corvin, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity College Dublin, Ireland; Nick Craddock, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Benedicto Crespo-Facorro, University Hospital Marqués de Valdecilla, Instituto de Formación e Investigación Marqués de Valdecilla, University of Cantabria, Santander, Spain; James J Crowley, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Mark J Daly, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Ariel Darvasi, Department of Genetics, The Hebrew University of Jerusalem, Jerusalem, Israel; Michael Davidson, Sheba Medical Center, Tel Hashomer, Israel; Kenneth L Davis, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Franziska Degenhardt, Institute of Human Genetics, University of Bonn, Bonn, Germany; Jurgen Del Favero, Applied Molecular Genomics Unit, VIB Department of Molecular Genetics, University of Antwerp, Antwerp, Belgium; Panos Deloukas, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Ditte Demontis, Department of Biomedicine, Aarhus University, Aarhus, Denmark; Dimitris Dikeos, First Department of Psychiatry, University of Athens Medical School, Athens, Greece; Timothy Dinan, Department of Psychiatry, University College Cork, Ireland; Srdjan Djurovic, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Enrico Domenici, Neuroscience Discovery and Translational Area, Pharma Research and Early Development, F.Hoffman-La Roche, Basel, Switzerland; Peter Donnelly, Wellcome Trust Centre for Human Genetics, Oxford, UK; Gary Donohoe, Cognitive Genetics and Therapy Group, School of Psychology and Discipline of Biochemistry, National University of Ireland Galway, Ireland; Elodie Drapeau, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Serge Dronov, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Jubao Duan, Department of Psychiatry and Behavioral Sciences, NorthShore University HealthSystem, Evanston, IL, USA; Frank Dudbridge, Department of Non-Communicable Disease Epidemiology, London School of Hygiene and Tropical Medicine, London, UK; Audrey Duncanson, Molecular and Physiological Sciences, The Wellcome Trust, London, UK; Naser Durmishi, Department of Child and Adolescent Psychiatry, University Clinic of Psychiatry, Skopje, Republic of Macedonia; Sarah Edkins, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Hannelore Ehrenreich, Clinical Neuroscience, Max Planck Institute of Experimental Medicine, Göttingen, Germany; Peter Eichhammer, Department of Psychiatry, University of Regensburg, Regensburg, Germany; Johan Eriksson, Folkhälsan Research Center, Helsinki, Finland; Valentina Escott-Price, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Tõnu Esko, Estonian Genome Center, University of Tartu, Tartu, Estonia; Laurent Essioux, Translational Technologies and Bioinformatics, Pharma Research and Early Development, F.Hoffman-La Roche, Basel, Switzerland; Ayman H Fanous, Mental Health Service Line, Washington VA Medical Center, Washington DC, USA; Kai-How Farh, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Martilias S Farrell, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Josef Frank, Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Heidelberg, Germany; Lude Franke, Department of Genetics, University of Groningen, University Medical Centre Groningen, The Netherlands; Robert Freedman, Department of Psychiatry, University of Colorado Denver, Aurora, CO, USA; Colin Freeman, Wellcome Trust Centre for Human Genetics, Oxford, UK; Nelson B Freimer, Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, CA, USA; Marion Friedl, Department of Psychiatry, University of Halle, Halle, Germany; Joseph I Friedman, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Menachem Fromer, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Pablo V Gejman, Department of Psychiatry and Behavioral Sciences, NorthShore University HealthSystem, Evanston, IL, USA; Giulio Genovese, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Lyudmila Georgieva, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Eleni Giannoulatou, Wellcome Trust Centre for Human Genetics, Oxford, UK; Ina Giegling, Department of Psychiatry, University of Halle, Halle, Germany; Michael Gill, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity College Dublin, Ireland; Matthew Gillman, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Paola Giusti-Rodríguez, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Stephanie Godard, Departments of Psychiatry and Human and Molecular Genetics, INSERM, Institut de Myologie, Hôpital de la Pitiè-Salpêtrière, Paris, France; Jacqueline I Goldstein, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Vera Golimbet, Mental Health Research Centre, Russian Academy of Medical Sciences, Moscow, Russia; Srihari Gopal, Neuroscience Therapeutic Area, Janssen Research and Development, LLC, Raritan, NJ, USA; Jacob Gratten, Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; Emma Gray, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Hugh Gurling, Molecular Psychiatry Laboratory, Division of Psychiatry, University College London, UK; Rhian Gwilliam, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Lieuwe de Haan, Academic Medical Centre University of Amsterdam, Department of Psychiatry, Amsterdam, The Netherlands; Jeremy Hall, Division of Psychiatry, University of Edinburgh, Edinburgh, UK; Christian Hammer, Clinical Neuroscience, Max Planck Institute of Experimental Medicine, Göttingen, Germany; Naomi Hammond, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Marian L Hamshere, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Mark Hansen, Illumina, Inc., La Jolla, CA, USA; Thomas Hansen, Institute of Biological Psychiatry, MHC Sct. Hans, Mental Health Services Copenhagen, Denmark; Vahram Haroutunian, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Annette M Hartmann, Department of Psychiatry, University of Halle, Halle, Germany; Garrett Hellenthal, Wellcome Trust Centre for Human Genetics, Oxford, UK; Frans A Henskens, School of Electrical Engineering and Computer Science, University of Newcastle, Newcastle, Australia; Stefan Herms, Division of Medical Genetics, Department of Biomedicine, University of Basel, Basel, Switzerland; Joel N Hirschhorn, Department of Genetics, Harvard Medical School, Boston, MA, USA; Per Hoffmann, Division of Medical Genetics, Department of Biomedicine, University of Basel, Basel, Switzerland; Andrea Hofman, Institute of Human Genetics, University of Bonn, Bonn, Germany; Mads V Hollegaard, Section of Neonatal Screening and Hormones, Department of Clinical Biochemistry, Immunology and Genetics, Statens Serum Institut, Copenhagen, Denmark; Hailiang Huang, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Christina M Hultman, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; Sarah E Hunt, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Masashi Ikeda, Department of Psychiatry, Fujita Health University School of Medicine, Toyoake, Aichi, Japan; Nakao Iwata, Department of Psychiatry, Fujita Health University School of Medicine, Toyoake, Aichi, Japan; Conrad Iyegbe, Institute of Psychiatry at King's College London, London, UK; Assen V Jablensky, School of Psychiatry and Clinical Neurosciences, The University of Western Australia, Perth, Australia; Janusz Jankowski, Peninsula School of Medicine and Dentistry, Plymouth University, Plymouth, UK; Alagurevathi Jayakumar, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Inge Joa, Regional Centre for Clinical Research in Psychosis, Department of Psychiatry, Stavanger University Hospital, Stavanger, Norway; Erik G Jönsson, Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden; Antonio Julià, Rheumatology Research Group, Vall d'Hebron Research Institute, Barcelona, Spain; Anna K Kähler, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; René S Kahn, Department of Psychiatry, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, Utrecht, The Netherlands; Luba Kalaydjieva, Centre for Medical Research, The University of Western Australia, Perth, Western Australia, Australia; Sena Karachanak-Yankova, Department of Medical Genetics, Medical University, Sofia, Bulgaria; Juha Karjalainen, Department of Genetics, University of Groningen, University Medical Centre Groningen, The Netherlands; David Kavanagh, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Matthew C Keller, Department of Psychology, University of Colorado Boulder, Boulder, CO, USA; Kenneth S Kendler, Virginia Institute for Psychiatric and Behavioral Genetics, Departments of Psychiatry and Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, USA; James L Kennedy, Campbell Family Mental Health Research Institute, Centre for Addiction and Mental Health, Toronto, Ontario, Canada; Andrey Khrunin, Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, Russia; Yunjung Kim, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; George Kirov, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Janis Klovins, Latvian Biomedical Research and Study Centre, Riga, Latvia; Jo Knight, Lancaster University Medical School and Data Science Institute, Bailrigg, Lancaster, LA1 4YG, UK; James A Knowles, Department of Psychiatry and Zilkha Neurogenetics Institute, Keck School of Medicine at University of Southern California, Los Angeles, CA, USA; Bettina Konte, Department of Psychiatry, University of Halle, Halle, Germany; Vaidutis Kucinskas, Faculty of Medicine, Vilnius University, Vilnius, Lithuania; Zita Ausrele Kucinskiene, Faculty of Medicine, Vilnius University, Vilnius, Lithuania; Hana Kuzelova-Ptackova, Department of Biology and Medical Genetics, Charles University Prague, Prague, Czech Republic; Cordelia Langford, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Claudine Laurent, Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, USA; Stephen Lawrie, Division of Psychiatry, University of Edinburgh, Edinburgh, UK; S Hong Lee, Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; Phil Lee, Psychiatric and Neurodevelopmental Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Jimmy Lee Chee Keong, Institute of Mental Health, Singapore; Sophie E Legge, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Todd Lencz, The Zucker Hillside Hospital, Glen Oaks, NY, USA; Bernard Lerer, Department of Psychiatry, Hadassah-Hebrew University Medical Center, Jerusalem, Israel; Douglas F Levinson, Department of Psychiatry and Behavioral Sciences, Stanford University, Stanford, CA, USA; Cathryn M Lewis, Institute of Psychiatry at King's College London, London, UK; Tao Li, Mental Health Centre and Psychiatric Laboratory, West China Hospital, Sichuan University, Chendu, Sichuan, PR China; Qingqin S Li, Neuroscience Therapeutic Area, Janssen Research and Development, LLC, Raritan, NJ, USA; Miaoxin Li, Centre for Genomic Sciences and Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Kung-Yee Liang, Department of Biostatistics, Johns Hopkins University Bloomberg School of Public Health, Baltimore, Maryland, USA; Jennifer Liddle, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Jeffrey Lieberman, Department of Psychiatry, Columbia University, New York, NY, USA; Svetlana Limborska, Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, Russia; Kuang Lin, Institute of Psychiatry at King's College London, London, UK; Don H Linszen, Department of Psychiatry, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands; Jianjun Liu, Human Genetics, Genome Institute of Singapore, A*STAR, Singapore; Jouko Lönnqvist, Department of Mental Health and Substance Abuse Services, National Institute for Health and Welfare, Helsinki, Finland; Carmel M Loughland, Schizophrenia Research Institute, Sydney, Australia; Jan Lubinski, Department of Genetics and Pathology, International Hereditary Cancer Center, Pomeranian Medical University in Szczecin, Szczecin, Poland; Milan Macek, Department of Biology and Medical Genetics, Charles University Prague, Prague, Czech Republic; Patrik KE Magnusson, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden; Brion S Maher, Department of Mental Health, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA; Wolfgang Maier, Department of Psychiatry, University of Bonn, Bonn, Germany; Anil K Malhotra, The Zucker Hillside Hospital, Glen Oaks, NY, USA; Jacques Mallet, Centre National de la Recherche Scientifique, Laboratoire de Génétique Moléculaire de la Neurotransmission et des Processus Neurodégénératifs, Hôpital de la Pitié Salpêtrière, Paris, France; Hugh S Markus, Clinical Neurosciences, St George's University of London, London, UK; Sara Marsal, Rheumatology Research Group, Vall d'Hebron Research Institute, Barcelona, Spain; Ignacio Mata, University Hospital Marqués de Valdecilla, Instituto de Formación e Investigación Marqués de Valdecilla, University of Cantabria, Santander, Spain; Christopher G Mathew, Department of Medical and Molecular Genetics, School of Medicine, King's College London, Guy's Hospital, London, UK; Morten Mattingsdal, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Owen T McCann, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Robert W McCarley, VA Boston Health Care System, Brockton, MA, USA; Steven A McCarroll, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Mark I McCarthy, Oxford Centre for Diabetes, Endocrinology and Metabolism, Churchill Hospital, Oxford, UK; Colm McDonald, Department of Psychiatry, National University of Ireland Galway, Ireland ; Andrew M McIntosh, Division of Psychiatry, University of Edinburgh, Edinburgh, UK; Andrew McQuillin, Molecular Psychiatry Laboratory, Division of Psychiatry, University College London, UK; Sandra Meier, Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Heidelberg, Germany; Carin J Meijer, Academic Medical Centre University of Amsterdam, Department of Psychiatry, Amsterdam, The Netherlands; Bela Melegh, Department of Medical Genetics, University of Pécs, Pécs, Hungary; Ingrid Melle, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Raquelle I Mesholam-Gately, Massachusetts Mental Health Center Public Psychiatry Division of the Beth Israel Deaconess Medical Center, Boston, MA, USA; Andres Metspalu, Estonian Genome Center, University of Tartu, Tartu, Estonia; Patricia T Michie, School of Psychology, University of Newcastle, Newcastle, Australia; Lili Milani, Estonian Genome Center, University of Tartu, Tartu, Estonia; Vihra Milanova, First Psychiatric Clinic, Medical University, Sofia, Bulgaria; Younes Mokrab, Eli Lilly and Company Limited, Erl Wood Manor, Sunninghill Road, Windlesham, Surrey, UK; Jennifer L Moran, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Derek W Morris, Cognitive Genetics and Therapy Group, School of Psychology and Discipline of Biochemistry, National University of Ireland Galway, Co. Galway, Ireland; Bryan J Mowry, Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; Bertram Müller-Myhsok, Max Planck Institute of Psychiatry, Munich, Germany; Kieran C Murphy, Department of Psychiatry, Royal College of Surgeons in Ireland, Ireland; Robin M Murray, King's College London, UK; Inez Myin-Germeys, Maastricht University Medical Centre, South Limburg Mental Health Research and Teaching Network, EURON, Maastricht, The Netherlands; Benjamin M Neale, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Mari Nelis, Estonian Genome Center, University of Tartu, Tartu, Estonia; Igor Nenadic, Department of Psychiatry and Psychotherapy, Jena University Hospital, Jena, Germany; Deborah A Nertney, Department of Psychiatry, Queensland Brain Institute and Queensland Centre for Mental Health Research, University of Queensland, Brisbane, Queensland, Australia; Gerald Nestadt, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA; Kristin K Nicodemus, Department of Psychiatry, Trinity College Dublin, Ireland; Liene Nikitina-Zake, Latvian Biomedical Research and Study Centre, Riga, Latvia; Laura Nisenbaum, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN, USA; Annelie Nordin, Department of Clinical Sciences, Psychiatry, Umeå University, Umeå, Sweden; Markus M Nöthen, Institute of Human Genetics, University of Bonn, Bonn, Germany; Eadbhard O'Callaghan, DETECT Early Intervention Service for Psychosis, Blackrock, Dublin, Ireland; Michael C O'Donovan, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Colm O'Dushlaine, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; F Anthony O'Neill, Centre for Public Health, Institute of Clinical Sciences, Queens University Belfast, Belfast, UK; Sang-Yun Oh, Lawrence Berkeley National Laboratory, University of California at Berkeley, Berkeley, CA, USA; Ann Olincy, Department of Psychiatry, University of Colorado Denver, Aurora, CO, USA; Line Olsen, Institute of Biological Psychiatry, MHC Sct. Hans, Mental Health Services Copenhagen, Denmark; Roel A Ophoff, Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, CA, USA; Jim Van Os, Institute of Psychiatry at King's College London, London, UK; Michael J Owen, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Colin N A Palmer, Biomedical Research Centre, Ninewells Hospital and Medical School, Dundee, UK; Aarno Palotie, Institute for Molecular Medicine Finland, FIMM, Helsinki, Finland; Christos Pantelis, Melbourne Neuropsychiatry Centre, University of Melbourne & Melbourne Health, Melbourne, Australia; George N Papadimitriou, First Department of Psychiatry, University of Athens Medical School, Athens, Greece; Sergi Papiol, Clinical Neuroscience, Max Planck Institute of Experimental Medicine, Göttingen, Germany; Elena Parkhomenko, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Michele T Pato, Department of Psychiatry and Zilkha Neurogenetics Institute, Keck School of Medicine at University of Southern California, Los Angeles, CA, USA; Carlos N Pato, Department of Psychiatry and Zilkha Neurogenetics Institute, Keck School of Medicine at University of Southern California, Los Angeles, CA, USA; Tiina Paunio, Public Health Genomics Unit, National Institute for Health and Welfare, Helsinki, Finland; Richard Pearson, Wellcome Trust Centre for Human Genetics, Oxford, UK; Murray J Cairns, Schizophrenia Research Institute, Sydney NSW 2010, Australia; Lynn E DeLisi, VA Boston Health Care System, Brockton, Massachusetts 02301, USA; Elliot S Gershon, Departments of Psychiatry and Human Genetics, University of Chicago, Chicago, Illinois 60637 USA; Brian J Kelly, Priority Centre for Translational Neuroscience and Mental Health, University of Newcastle, Newcastle NSW 2300, Australia; Max Lam, Institute of Mental Health, Singapore 539747, Singapore; Nina Norgren, Human Genetics, Genome Institute of Singapore, A*STAR, Singapore 138672, Singapore; Sara A Paciga, Human Genetics and Computational Biomedicine, Pfizer Global Research and Development, Groton, Connecticut 06340, USA; Paul A Tooney, School of Biomedical Sciences and Pharmacy, University of Newcastle, Callaghan NSW 2308, Australia; Jing Qin Wu, School of Biomedical Sciences and Pharmacy, University of Newcastle, Callaghan NSW 2308, Australia
List of authors in the Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH)
Milica Pejovic-Milovancevic, Medical Faculty, University of Belgrade, Belgrade, Serbia; Diana O Perkins, Department of Psychiatry, University of North Carolina, Chapel Hill, NC, USA; Tune H Pers, Center for Biological Sequence Analysis, Department of Systems Biology, Technical University of Denmark, Lyngby, Denmark; Tracey L Petryshen, Center for Human Genetic Research and Department of Psychiatry, Massachusetts General Hospital, Boston, MA, USA; Olli Pietiläinen, Institute for Molecular Medicine Finland, FIMM, Helsinki, Finland; Jonathan Pimm, Molecular Psychiatry Laboratory, Division of Psychiatry, University College London, UK; Matti Pirinen, Wellcome Trust Centre for Human Genetics, Oxford, UK; Robert Plomin, Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King's College London, London, UK; Andrew J Pocklington, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Danielle Posthuma, Department of Functional Genomics, Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, VU University, Amsterdam, The Netherlands; Simon C Potter, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; John Powell, King's College London, UK; Alkes Price, Department of Epidemiology, Harvard University, Boston, MA, USA; Ann E Pulver, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA; Shaun M Purcell, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Digby Quested, Department of Psychiatry, University of Oxford, Oxford, UK; Henrik B Rasmussen, Institute of Biological Psychiatry, MHC Sct. Hans, Mental Health Services Copenhagen, Denmark; Anna Rautanen, Wellcome Trust Centre for Human Genetics, Oxford, UK; Radhi Ravindrarajah, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Abraham Reichenberg, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Mark A Reimers, Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA; Alexander L Richards, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Michelle Ricketts, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Marcella Rietschel, Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Heidelberg, Germany; Brien P Riley, Virginia Institute for Psychiatric and Behavioral Genetics, Departments of Psychiatry and Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, USA; Stephan Ripke, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Joshua L Roffman, Department of Psychiatry, Massachusetts General Hospital, Boston, MA, USA; Panos Roussos, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Douglas M Ruderfer, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Dan Rujescu, Department of Psychiatry, University of Halle, Halle, Germany; Veikko Salomaa, National Institute for Health and Welfare, Helsinki, Finland; Alan R Sanders, Department of Psychiatry and Behavioral Sciences, NorthShore University HealthSystem, Evanston, IL, USA; Stephen J Sawcer, Department of Clinical Neurosciences, University of Cambridge, Addenbrooke's Hospital, Cambridge, UK; Ulrich Schall, Priority Centre for Translational Neuroscience and Mental Health, University of Newcastle, Newcastle, Australia; Christian R Schubert, PharmaTherapeutics Clinical Research, Pfizer Worldwide Research and Development, Cambridge, MA, USA; Thomas G Schulze, Department of Psychiatry and Psychotherapy, University of Göttingen, Göttingen, Germany; Sibylle G Schwab, Psychiatry and Psychotherapy Clinic, University of Erlangen, Germany; Edward M Scolnick, Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Rodney J Scott, School of Biomedical Sciences, University of Newcastle, Newcastle, Australia; Larry J Seidman, Massachusetts Mental Health Center Public Psychiatry Division of the Beth Israel Deaconess Medical Center, Boston, MA, USA; Pak C Sham, Centre for Genomic Sciences, State Ket Laboratory for Brain and Cognitive Sciences, and Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Jianxin Shi, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, MD, USA; Engilbert Sigurdsson, University of Iceland, Landspitali, National University Hospital, Reykjavik, Iceland; Teimuraz Silagadze, Department of Psychiatry and Drug Addiction, Tbilisi State Medical University (TSMU), Tbilisi, Georgia; Jeremy M Silverman, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Kang Sim, Institute of Mental Health, Singapore; Pamela Sklar, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Petr Slominsky, Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, Russia; Jordan W Smoller, Psychiatric and Neurodevelopmental Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Hon-Cheong So, Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Erik Söderman, Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden; Chris C A Spencer, Wellcome Trust Centre for Human Genetics, Oxford, UK; David St Clair, University of Aberdeen, Institute of Medical Sciences, Aberdeen, Scotland, UK; Eli A Stahl, Division of Psychiatric Genomics, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Elisabeth Stogmann, Department of Clinical Neurology, Medical University of Vienna, Austria; Amy Strange, Wellcome Trust Centre for Human Genetics, Oxford, UK; Richard E Straub, Lieber Institute for Brain Development, Baltimore, MD, USA; Eric Strengman, Department of Medical Genetics, University Medical Centre, Utrecht, The Netherlands; Jana Strohmaier, Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Heidelberg, Germany; T Scott Stroup, Department of Psychiatry, Columbia University, New York, NY, USA; Zhan Su, Wellcome Trust Centre for Human Genetics, Oxford, UK; Mythily Subramaniam, Institute of Mental Health, Singapore; Patrick F Sullivan, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Jaana Suvisaari, Department of Mental Health and Substance Abuse Services, National Institute for Health and Welfare, Helsinki, Finland; Dragan M Svrakic, Department of Psychiatry, Washington University, St. Louis, MO, USA; Jin P Szatkiewicz, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Avazeh Tashakkori-Ghanbaria, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Srinivas Thirumalai, Berkshire Healthcare NHS Foundation Trust, Bracknell, UK; Draga Toncheva, Department of Medical Genetics, Medical University, Sofia, Bulgaria; Sarah Tosato, Section of Psychiatry, University of Verona, Verona, Italy; Richard C Trembath, Department of Medical and Molecular Genetics, School of Medicine, King's College London, Guy's Hospital, London, UK; Juha Veijola, Department of Psychiatry, University of Oulu, Finland; Peter M Visscher, Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; Ananth C Viswanathan, National Institute for Health Research, Biomedical Research Centre at Moorfields Eye Hospital, National Health Service Foundation Trust, London, UK; Damjan Vukcevic, Wellcome Trust Centre for Human Genetics, Oxford, UK; John Waddington, Molecular and Cellular Therapeutics, Royal College of Surgeons in Ireland, Dublin, Ireland; Matthew Waller, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Dermot Walsh, Health Research Board, Dublin, Ireland; Muriel Walshe, Institute of Psychiatry at King's College London, London, UK; James TR Walters, MRC Centre for Neuropsychiatric Genetics and Genomics, Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK; Qiang Wang, Mental Health Centre and Psychiatric Laboratory, West China Hospital, Sichuan University, Chendu, Sichuan, PR China; Dai Wang, Neuroscience Therapeutic Area, Janssen Research and Development, LLC, Raritan, NJ, USA; Bradley T Webb, Virginia Institute for Psychiatric and Behavioral Genetics, Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA; Daniel R Weinberger, Lieber Institute for Brain Development, Baltimore, MD, USA; Matthias Weisbrod, Section for Experimental Psychopathology, General Psychiatry, Heidelberg, Germany; Mark Weiser, Sheba Medical Center, Tel Hashomer, Israel; Jens R Wendland, PharmaTherapeutics Clinical Research, Pfizer Worldwide Research and Development, Cambridge, MA, USA; Paul Weston, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Pamela Whittaker, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Sara Widaa, Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Durk Wiersma, Department of Psychiatry, University Medical Center Groningen, University of Groningen, The Netherlands; Dieter B Wildenauer, Department of Psychiatry and Clinical Neurosciences, School of Psychiatry and Clinical Neurosciences, Queen Elizabeth II Medical Centre, Perth, Western Australia, Australia; Stephanie Williams, Department of Genetics, University of North Carolina, Chapel Hill, NC, USA; Nigel M Williams, Department of Psychological Medicine and Neurology, MRC Centre for Neuropsychiatric Genetics and Genomics, School of Medicine, Cardiff University, Cardiff, Wales, UK; Stephanie H Witt, Department of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Heidelberg, Germany; Aaron R Wolen, Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA; Emily HM Wong, Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, PR China; Nicholas W Wood, Department of Molecular Neuroscience, Institute of Neurology, London, UK; Brandon K Wormley, Virginia Institute for Psychiatric and Behavioral Genetics, Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA; Naomi R Wray, Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; Wellcome Trust Case-Control Consortium 2, WTCCC2; Hualin Simon Xi, Computational Sciences CoE, Pfizer Worldwide Research and Development, Cambridge, MA, USA; Clement C Zai, Campbell Family Mental Health Research Institute, Centre for Addiction and Mental Health, Toronto, Ontario, Canada; Xuebin Zheng, Human Genetics, Genome Institute of Singapore, A*STAR, Singapore; Fritz Zimprich, Department of Clinical Neurology, Medical University of Vienna, Austria
Author information
Authors and Affiliations
Consortia
Contributions
JLH, DST, MM, TAG, MTT, DC, HJE, PH, SVF, and SJG contributed to conception and design of the study. The Schizophrenia Working Group of the Psychiatric Genomics Consortium, Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH), ADB, TBA, JG, TW, PBM, OM, MN, DMH, JBG, MBH, SS, ES, HS, and KS were involved in the acquisition of data. JLH performed the statistical analyses. JLH, DST, SVF, and SJG were involved in the interpretation of data. JLH wrote the first draft of the paper and critical edits were provided by SVF and SJG Critical revisions to the paper were provided by DST, MM, TAG, DC, HJE, and PH. The Schizophrenia Working Group of the Psychiatric Genomics Consortium, Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH), ADB, TBA, JG, TW, PBM, OM, MN, DMH, JBG, MBH, SS, ES, HS, and KS provided critical review of the paper. All authors gave final approval of the version of the paper submitted for peer-review.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A full list of consortium members are listed below the Acknowledgements.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hess, J.L., Tylee, D.S., Mattheisen, M. et al. A polygenic resilience score moderates the genetic risk for schizophrenia. Mol Psychiatry 26, 800–815 (2021). https://doi.org/10.1038/s41380-019-0463-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-019-0463-8
This article is cited by
-
Blunted brain responses to neutral faces in healthy first-degree relatives of patients with schizophrenia: an image-based fMRI meta-analysis
Schizophrenia (2024)
-
Considerations, Caveats, and Suggestions for the Use of Polygenic Scores for Social and Behavioral Traits
Behavior Genetics (2024)
-
Genome-wide association studies of polygenic risk score-derived phenotypes may lead to inflated false positive rates
Scientific Reports (2023)
-
An epigenetic association analysis of childhood trauma in psychosis reveals possible overlap with methylation changes associated with PTSD
Translational Psychiatry (2022)
-
Polygenic resilience scores capture protective genetic effects for Alzheimer’s disease
Translational Psychiatry (2022)
