
Abstract
Liprins are a multifunctional family of scaffold proteins, identified by their involvement in several important neuronal functions related to signaling and organization of synaptic structures. More recently, the knowledge on the liprin family has expanded from neuronal functions to processes relevant to cancer progression, including cell adhesion, cell motility, cancer cell invasion, and signaling. These proteins consist of regions, which by prediction are intrinsically disordered, and may be involved in the assembly of supramolecular structures relevant for their functions. This review summarizes the current understanding of the functions of liprins in different cellular processes, with special emphasis on liprins in tumor progression. The available data indicate that liprins may be potential biomarkers for cancer progression and may have therapeutic importance.
Similar content being viewed by others


Introduction
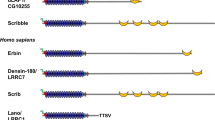
Liprins belong to the LAR (leukocyte common antigen related) protein tyrosine phosphatase interacting protein family [1]. In mammals, the liprin family consists of liprin-α (α1, α2, α3, α4), β (β1, β2) proteins, which are well conserved in evolution [1, 2], and kazrin E, a protein regulating desmosome assembly and tissue morphogenesis [3,4,5]. C. Elegans has one homolog of liprin-α (syd-2, synapse defective-2) and one homolog for liprin-β (hlb-1, liprin-β homolog) [1, 6]. In Drosophila there are single liprin-α (Dliprin-α), liprin-β and liprin-γ homologs; both liprin-β and liprin-γ interact with liprin-α [1, 7, 8]. In Drosophila, liprin-γ is thought to counteract the activity of liprin-α and liprin-β proteins in synapse formation [7]. The carboxyterminal part of liprin-α and liprin-β proteins include a highly conserved region containing three SAM (sterile-alpha motif) domains, which are important in protein–protein interactions [1, 2, 4]. Liprin-α proteins interact with the intracellular part of the transmembrane LAR and receptor tyrosine phosphatases included in this subfamily (LAR-RPTPs) with C-terminal SAM domains [1, 2, 4, 9], which are also responsible for liprin α/β binding [1, 4]. Despite forming hetero-oligomers with liprin-α, liprin-β has not been shown to bind LAR [1]. The N-terminal region of all liprins is characterized by coiled coil structures, which are responsible for homodimerization of liprin-α and liprin-β proteins, as well as for heterodimerization of liprin-α proteins with other liprin-α subfamily members [1, 2, 4].
Liprin-α1 is the only liprin-α having widely distributed mRNA and protein expression within different tissues [1, 10,11,12], whereas liprin-β’s are more broadly expressed [1] (Table 1). Liprin-α2 and -α3 are predominantly expressed in the brain tissue, whereas liprin-α4 is expressed in the heart and muscle tissues as well [1, 10, 12]. Although all four liprin-α proteins are expressed in the brain tissue, they show somewhat different brain tissue distribution and expression level [11]. Liprin-α2 and -α3 show strong expression throughout the brain, while liprin-α1 is expressed at lower levels with the exception of the olfactory bulb and the cerebellum. Liprin-α4 shows strongest expression in the cerebellum [11, 12]. Both the liprin-α1 and liprin-α3 show expression in astrocytes [11, 12], suggesting liprins may have a role in glial cells based on their expression.
Liprin-α proteins show spatiotemporal expression patterns during mouse brain development [12]. Liprin-α1 is the only member with different temporal and spatial expression pattern as compared to the other liprin-α protein members [12]. Ppfia1 is homozygously lethal, and heterozygous animals display significant changes in phenotypes including abnormalities in the vocalization and abnormal coat/hair pigmentation. On the other hand, ppfia2, ppfia3, ppfia4 and ppfibp2 KO mice are homozygously viable with ppfia2, ppfia3, and ppfibp2 KO mice showing various changes in phenotypes such as abnormal retinal vasculature morphology, vocalization, increased activity, and heart rate (www.mousephenotype.org) [13, 14]. Of note, liprins contain protein regions that are predicted to be intrinsically disordered [15, 16]. Intrinsically disordered regions are included in several proteins that are involved in the formation of multimolecular assemblies defined as biomolecular condensates, which are formed by liquid–liquid phase separation (LLPS) [17,18,19]. In addition, LLPS and biomolecular condensates may be important in cancer and in small molecule therapeutics [20, 21]. In this respect, very recent findings have shown that liprins may contribute to the assembly of biomolecular condensates [22,23,24], suggesting that both specific protein–protein interactions as well as LLPS–mediated condensates may contribute to essential physiological processes as well as play a role in different signaling pathways critical to tumor cell progression. In this review, we are focusing on recent findings on the involvement of liprins in oncogenic signaling after presenting an overview of the crucial role of liprins in neuronal cells.
The function of liprins in the nervous system and interaction with kinesins
Liprin-α proteins bind to several partners with their coiled coil and SAM domains (Fig. 1). At the presynaptic sites, liprin-α proteins display widespread functions in synapse assembly as well as in presynaptic organization. In addition to presynaptic sites, liprin-α proteins are localized in dendrites, and contribute to the maturation of the postsynaptic sites [6, 25,26,27,28,29,30,31]. Liprin-α proteins bind to MALS and CASK proteins, which are multidomain scaffolding proteins highly expressed in the mammalian nervous system [32,33,34,35]. Liprin-α proteins interact also with RIM1 [36], ERC1/ELKS [37, 38], KIF1A [26], GRIP and GIT1 [25, 27, 39] to organize presynaptic active zones and regulate neurotransmitter release. In synapses, liprin-α1 is regulated by either CAMKII or ubiquitin-proteasome mediated degradation, and liprin-α1 degradation by CAMKII is important for correct LAR distribution and dendritic development [40]. Liprin-α2 is important in promotion of protein dynamics in active zone of hippocampal synapses, and ultrastructural analysis has shown that this protein regulates the presynaptic organization and the size of the synaptic vesicle pool [35]. In mammals, liprin-α3 knockout mice show secretory impairment and defects in presynaptic active zone due to the prevalent role of liprins in synapse assembly. While liprin-α2 and -α3 may compete for positioning at the active zone, liprin-α2 cannot fully compensate the presynaptic loss of liprin-α3 [14]. Knockout of both the liprin-α2 and -α3 lead to decreased number of synaptic vesicles, reduced levels of presynaptic proteins, as well as reduction of docked vesicles in the active zone of primary mouse hippocampal neurons, which further points to the conclusion, that liprin-α2 and -α3 are necessary for presynaptic composition and normal active zone structure [23].

Liprin-α1 is shown in the drawing. Black arrows show direct interactions between the indicated proteins/protein regions (blue lines; residues in brackets). Ank ankyrin, Arf-GAP GTPase activating protein for Arf GTPases, D1 phosphatase domain, D2 phosphatase–like domain (catalytically inactive), FN-III fibronectin type III, Ig immunoglobulin, PBD paxillin binding domain, PDZ-b, PDZ binding peptide, SAM sterile-alpha motif, SHD Spa2 homology domain, TM transmembrane region. Intrinsically disordered regions were identified by the program DisEMBL [18], coiled coil regions by the program COILS [19].
In C. elegans the liprin-α homolog SYD-2 is an important player in the organization of the presynaptic active zone [6]. SYD-2 interacts with RSY-1, which negatively regulates SYD-2 and the formation of synapses [41]. SYD-2 recruits multiple synaptic components to presynaptic sites and plays a major role in the architecture of the neuromuscular junctions as well as in the assembly and stabilization of presynaptic sites [41, 42]. SYD-2 mutants display morphological changes at the presynaptic active zone and defects in presynaptic assembly that are due to problems in the oligomerization of liprin-α proteins [6, 42, 43]. Similarly, the Drosophila Dliprin-α is needed for synaptic morphogenesis and synaptic vesicular transport [8, 44]. Dliprin-α mutants display defects in synaptic morphology and photoreceptor target selection at presynaptic sites [8, 44, 45]. In C. elegans and Drosophila, the interaction between SYD-2/Liprin-alpha and SYD-1/syd-1 (RhoGAP-100F), a Rho GTPase activating protein, is important for proper localization of SYD-2/liprin-α at presynaptic sites allowing SYD-2/liprin-α to execute its active zone assembly function [30, 46, 47], as well as for proper recruitment of the synaptic proteins [43, 47, 48]. SYD-1/syd-1 acts as a positive regulator upstream of SYD-2/liprin-α. Previous data suggest, that SYD-1 and SYD-2 (liprin-alpha) are not absolutely dependent on each other to localize to presynaptic sites, but SYD-2 overexpression can overcome the defects of mutant SYD-1 [43, 47].
Several studies suggest interactions of liprins with kinesin motor proteins [26, 49, 50]. Liprin-α1 interacts with kinesin KIF1A and functions as a KIF1A receptor during transport of membrane, signaling, and cytoskeletal proteins via microtubule tracks [26]. Liprin-α1 is also important for the hedgehog signaling dependent trafficking of Kif7 and Gli proteins to the tips of cilia during the embryonic development. Liprin-α1 interacts with PP2A and promotes the dephosphorylation of Kif7. Loss of liprin-α1 leads to defective development in zebrafish embryo [49]. KIF21A suppresses microtubule growth at the cell cortex and contributes to microtubule organization at the cell edge. KIF21A is recruited to the cell cortex by KANK, which interacts with liprin-α1/β1 complex. Liprin-α1, liprin-β1, KANK1, and KIF21A co-operate with LL5β, ELKS, and CLASPs in cortical microtubule organization. LL5β and KANK1 are required for targeting of the microtubule binding proteins such as CLASPs and KIF21A, whereas liprins and ELKS play a role in scaffolds required for protein clustering. While all these proteins co-operate in microtubule organization, they have different turnover rates and dynamics at the cell cortex, which is a similar situation than that found in focal adhesions [50].
In summary, liprins act as scaffolds for assembling very large protein complexes involved in the regulation of synaptic signaling and assembly [7, 14, 29]. In addition to the established role of liprins in regulating these essential neuronal functions, liprins have recently received more attention for their role as regulators of the motility and invasion of cancer cells [4, 5, 51,52,53] (Table 2), as discussed in the next part of this review.
Genetic alterations in genes encoding liprins
Human liprin-α1 is encoded by the gene PPFIA1 (PTPRF [Protein Tyrosine Phosphatase Receptor Type F polypeptide] Interacting Protein Alpha 1), which maps to the 11q13 chromosomal region that is often amplified in multiple cancer types, such as in head and neck squamous cell carcinoma (HNSCC), breast cancer and esophageal carcinoma [54,55,56,57,58,59,60,61]. Across different studies, the 11q13 region is amplified in 30–62% of HNSCC, in 15–20% of breast cancer and in 33% of esophageal cancers [reviewed in [57]]. The minimal common region of the amplification in HNSCC, where this amplification is the most frequent, ranges from around 0.9 to 2 Mb (amplicon core 1.5 Mb), and the amplification is associated with poor survival of the patients [57,58,59,60,61,62,63,64,65]. Across several cancers, PPFIA1 is overexpressed in carcinomas including those of the head and neck and breast carcinomas (Fig. 2). In tumors, PPFIA1 is often altered by gene amplification, whereas mutations and deletions are rare. PPFIA1 has been recently reported to be nonsynonymously mutated in aggressive papillary thyroid microcarcinomas (PTMC) [66]. The functional consequences of these variants are currently unknown.

The figure shows a body-wide gene profile of the PPFIA1 gene across 15392 malignant and 3082 healthy samples. Each dot represents the expression of PPFIA1 gene in one sample. Anatomical origins of each sample are marked with colored bars below the gene plot. Sample types having higher than average expression or an outlier expression profile are additionally colored. The image has been modified from MediSapiens ISTOnline database.
In addition to gene amplifications and genetic variants, few examples of gene fusions involving liprin–encoding genes have been reported. A fusion gene PPFIBP1–ALK between liprin-β1 and anaplastic lymphoma receptor tyrosine kinase has been reported in pulmonary inflammatory myofibroblastic tumor, and it is shown to have transforming potential in vivo in mice [67]. Fusions of PPFIBP1 have also been reported in melanocytic tumors (PPFIBP1-ROS1 and PPFIBP1-MET) [68, 69]. In addition, PPFIBP2-BRAF fusion has been reported in metastatic melanoma patient [70] and PPFIBP2-RET fusion in papillary thyroid carcinoma has shown to lead to the activation of mitogen-activated protein kinase (MAPK) pathway and growth–promoting properties [71]. In general, liprin-β2 (PPFIBP2) is considered to promote tumor suppressor properties opposite to liprin-β1 (PPFIBP1) with oncogenic functions [72]. Germline loss-of-function mutations of PPFIBP2 have been associated with shorter survival in prostate cancer [73]. PPFIBP2 has been proposed as one of the possible biomarkers in endometrial cancer [74] and it has been shown to display anti-invasive properties in cancer cells [72, 75]. Thus, although PPFIA1 is so far the only gene in the liprin family which is commonly amplified in several tumor types, genetic alterations in other liprin family genes, although infrequent, have recently been found to contribute to cancer progression.
Liprin-α interaction with LAR receptor tyrosine phosphatase
The family of receptor protein tyrosine phosphatases (RPTPs) contains multiple subfamilies [76], and the LAR-RPTP subfamily include LAR, PTPσ and PTPδ receptor tyrosine phosphatases, which interact with liprin-α proteins [1, 9, 77]. LAR protein family consists of an extracellular part with immunoglobulin-like and fibronectin III domains and a cytoplasmic part with two PTPase domains, D1 and D2 [1, 9]. The D1 domain has PTPase catalytic activity, whereas the carboxy terminal D2 domain is catalytically inactive [9, 78, 79]. Liprin-α proteins bind to LAR through the SAM domains, and they co-localize at specific sites at the proximal end of focal adhesions [1, 2]. While the interaction of liprin-α proteins to LAR occur via the cytoplasmic D2 domain [2], both D1 and D2 are required for stronger interaction [80]. Liprin-β1 does not bind to LAR, but LAR can allosterically inhibit the binding of liprin-β1 to liprin-α1 providing insights into underlying mechanisms in formation of complexes of liprin proteins [80].
The cellular localization of LAR transmembrane tyrosine phosphatase is regulated by liprin-α1 and the interaction of liprin-α1 with LAR facilitates the formation of LAR clusters [1, 2, 80]. There is evidence that association between liprin-α1 and LAR is controlled by liprin-α1 autophosphorylation [1, 81]. LAR has multiple targets and it has been shown to be involved in various signaling pathways related to phosphatase regulation [82]. Liprin-α1–promoted LAR clustering attenuates phosphatase activity, showing an interesting capacity of liprin-α1 to regulate the cellular activities of target proteins through protein–protein interactions [80]. Liprin-α1 proteins promote localization of LAR family tyrosine phosphatases at specific sites on the plasma membrane, possibly regulating their interaction with the extracellular environment and their association with substrates [1, 2, 80, 81]. When positioned in the right location at the focal adhesions with the contribution of liprin-α1 proteins, LAR has the potential to dephosphorylate proteins belonging to signaling pathways involved in the regulation of adhesion [1, 2, 4]. In cancer cells, this may lead to dephosphorylation of proteins important in oncogenic signaling. Recently, PTPRF/LAR has been shown to have both the tumor suppressive and oncogenic properties, and its expression is altered in cancer. PTPRF/LAR suppresses liver cancer tumorigenesis, and reduced expression of PTPRF/LAR leads to development of tumors [83]. However, PTPRF ectodomain was upregulated in a prostate cancer mouse model [84], as well as in metastatic breast cancer suggesting that altered expression of PTPRF/LAR is tumor type dependent [85].
Liprins as regulators of cell adhesion
Focal adhesions are cellular multiprotein structures that link the actin cytoskeleton to the ECM via the transmembrane integrin receptors [86, 87]. The structures important in actin– and adhesion–dependent mesenchymal cell migration are the lamellipodia which arise at the leading edge of migrating cells [88, 89]. Several studies point to the importance of liprin-α1 in focal adhesions and in regulation of the turnover, size and shape of these structures [4, 53, 90,91,92,93], and liprin-α1 is part of the integrin adhesome network [94,95,96]. Liprin-α1 together with ERC1/ELKS and LL5 proteins (LL5α and -β) regulate focal adhesion turnover and lamellipodial dynamics near the protruding tumor cell edge [92], where these proteins are part of dynamic plasma membrane-associated platforms (PMAPs) [15]. Their dynamic behavior suggests that PMAPs are liquid–like biomolecular condensates driven by LLPS of ERC1 that can recruit liprin-α1 via specific protein–protein interactions [97].
Interestingly, recent studies have shown that among PMAP proteins, not only ERC/ELKS, but also liprin-α proteins can undergo LLPS. In Caenorhabditis elegans, both SYD-2/liprin-α and ELKS-1 undergo LLPS during presynaptic development [22]. SYD-2/liprin-α LLPS, which is driven by a central intrinsically disordered region of the protein, is important for synaptic function, and allows the recruitment of other players such as GIT1 via specific protein–protein interactions (Fig. 1). Also the mammalian family member liprin-α3 has recently been shown to undergo reversible LLPS following phosphorylation of serine 760 [23]. This study has shown that while the expression of the wildtype liprin-α3 can rescue the defect in synapses observed in neurons with double knockout of liprin-α2 and liprin-α3, expression of liprin-α3 with a mutation of serine 760 prevents both LLPS and the rescue of the synaptic defect in the double knockout neurons [23]. These studies suggest that altering the ability of liprin-α proteins to undergo LLPS may also influence the function of these proteins in tumor cell motility.
Liprin-α1 and ERC1 control active β1-integrin internalization through Rab7 positive endosomes [90]. Reduced liprin-α1 levels lead to an increase in the lamellipodia number and a decrease of their stability during migration, whereas overexpression of liprin-α1 increases the stabilization of the lamellipodia, and the turnover of focal adhesions at the protrusive front of breast cancer cells [53, 92]. Liprin-α1 stabilizes inactive β1 integrins at the cell membrane, and regulates β1-integrin internalization and recycling [90, 92, 93]. Liprin-α1 cooperates with the β1-integrin binding protein talin to promote cell spreading and integrin organization [91]. Moreover LAR and liprin-α1 association is needed for protrusions during cell spreading, but liprin-α1 has also independent functions related to focal adhesions [91]. Knockdown of LAR leads to decrease in cell spreading but not in invasion, whereas liprin-α1 knockdown itself prevents invasion of MDA-MB-231 breast cancer cells [53]. In addition, liprin-α1 interacts with GIT1 to regulate cell spreading and migration [98]. In metastatic HNSCC and breast cancer cells, liprin-α1 is localized close to the cell edge near vinculin–positive focal adhesions [51, 53]. Liprin-β1 localizes at or close to paxillin-positive focal adhesions, and binding of LAR to liprin-α1 allosterically regulates the liprin-α/liprin-β interaction [80, 99].
Liprin-α1 and -α3 regulate the formation of actin stress fibers by negatively regulating mDia1, a member of the formin protein family and a Rho effector, by displacing the protein from the plasma membrane [100]. Binding of liprin-α3 to mDia1 counteracts RhoA activation and results in reduction of the cellular actin filament formation which leads to decrease in actin filaments in cultured cells [101]. In endothelial cells, liprin-α1 is required for integrin and fibronectin recycling [102]. Liprin-α1 interacts with the cytosolic domain of the α5β1 integrins to promote fibronectin secretion and the recycling of active α5β1 integrins from the trans-Golgi network to the surface of endothelial cells, thus allowing Rab11-regulated fibronectin turnover [102].
In zebrafish, morpholino-mediated liprin-α1 knockdown causes the reduction in perivascular fibronectin deposition, and leads to severe heart morphogenesis and cardiovascular defects [102]. This finding is supported by another study in Xenopus tadpoles showing liprin-β1 to have a role in lymphatic vascular development and vessel integrity; silencing of liprin-β1 results in edema and dispersed endothelial cells when compared to the controls [103]. The available data demonstrate that liprins are important scaffold proteins regulating adhesion, integrin recycling and protrusive activity of different cell types.
Liprins and invadosomes
The term invadosome refers to actin-rich protrusions with degradative properties such as the podosomes commonly found in monocytic cells and the invadopodia present in different types of cancer cells [104,105,106,107]. Invadosomes are essential structures in cancer cell invasion, since they are required to degrade the ECM (proteases, MMPs) to promote invasion [105, 108]. Although invadosomes have been mostly studied in vitro, there is evidence supporting the existence of invadosome-like structures in vivo [109, 110]. Invadosomes in non-invasive cancer cells contain podosome-like structures opposite to the actin-based invadosome structures observed in invasive cancer cells, which have gone through EMT. Invadosomes with podosome-like structures are likely precursors of invadopodia [111, 112]. Podosomes function in adhesion as well as in degradation of ECM, and they are formed by a ring of adhesion proteins and an actin-rich core [112,113,114]. The adhesion ring of invadosomes with podosome-like structures includes the adhesion proteins β1 integrins, paxillin, talin and vinculin, whereas the core includes cortactin and actin [51, 105, 115]. Cortactin is encoded by the CTTN gene, which is located in the vicinity of PPFIA1 at the 11q13 amplicon [116]. It has been suggested that podosomes guide migration by locally anchoring protrusions [114]. Invadopodia on the other hand are irregular, less defined in shape compared to podosomes, with a cortactin– and actin–positive core, but with no adhesion ring [117]. In head and neck squamous cell carcinoma, the actin-based invadosomes differ in their protein content and turnover rate between invasive and non-invasive cells [111]. In metastatic breast cancer cells, liprin-α1 has an important role in stabilizing invadosome function, turnover and degradation of ECM, which are important functions for motile cells [53]. Invadosomes with podosome-like structures are found in poorly invasive HNSCC cells derived from primary tumor [111]. In non-invasive HNSCC cells, liprin-α1 localizes at the outer edge of the adhesion ring or behind the cell edge close to focal adhesions [51] (Fig. 3). Although liprin-α1 is recruited to the adhesion ring of the invadosomes and these structures are able to degrade the ECM, liprin-α1 is not essential for their formation nor degradation [51]. Therefore, the role of liprin-α1 may be rather in the stabilization of these structures in non-invasive cells [51]. In metastatic HNSCC and invasive breast cancer cell lines, liprin-α1 mostly localizes near the leading edge and close to focal adhesions (Fig. 3B). In these cells, liprin-α1 is required for efficient degradation of the ECM, migration, and invasive growth [51,52,53]. On the other hand, liprin-β1 does not affect ECM degradation in breast cancer cells as shown by knockdown studies [72].While liprin-α1 as well as liprin-β1 have oncogenic pro-invasive properties, liprin-β2 decreases cell migration and the capability of cancer cells to degrade ECM [53, 72].

A Simplified model of liprin-α1 contribution to invadosomes and ECM degradation in a context– and cell line–dependent manner. In specific type of cancer cells (on the left) with invadosomes capable of degrading ECM, liprin-α1 is located at the adhesion ring of the invadosome. Liprin-α1 is not necessarily required for ECM degradation per se in these cells, but rather regulates the stabilization and motility of invadosomes. On the right side of the illustration, cross section shows that liprin-α1 is recruited to the invadosome-associated compartments. Cortactin, actin, focal adhesion proteins (such as paxillin, vinculin, and talin) and integrins are recruited to the invadosome, where MMPs play a crucial role in ECM degradation. B Model for the interplay between PMAPs and focal adhesions. PMAPs are observed near dynamic focal adhesions at the front of migrating tumor cells, where PMAP proteins are required to promote focal adhesion turnover. A possible link between focal adhesions and PMAPs are represented by the KANK proteins that can interact both with the focal adhesion protein talin, and PMAP proteins liprin beta. PMAPs also link adhesions to the cytoskeleton via PMAP components interacting either with actin or with microtubules.
In Src-transformed NIH-3T3 cells, liprin-α1 is part of a membrane-less invadosome-associated compartment (IAC), near to, but distinct from both the actin-rich core of the invadosomes and the adhesion ring [118]. In these cells, liprin-α1 is not required for the formation of invadosomes, but contributes to their motility and organization: therefore liprin-α1 influences the degradation of the ECM without affecting the recruitment of the transmembrane matrix metalloprotease MT1-MMP to the structures [118]. In vivo studies in xenograft mice have revealed that liprin-α1 promotes the formation of the metastasic lesions in lungs [72]. In addition, upregulation of PPFIA1 expression indicate higher risk of metastasis relapse in breast cancer patients, specifically in estrogen positive and nodal negative group [119]. The versatile contribution of liprin-α1 on invasion is likely to be explained by the context-dependency of liprin-α1 expression, as invasive and adhesive structures differ in distinct types of cancer cells as explained above. In addition, genetic background, amplification and mutation of the PPFIA1 gene are probable contributors to the capability of liprin-α1 to modulate cellular events. It is currently obvious that the role of liprin-α1 in cell invasion and invasive growth is cell line- and context-dependent, which would explain previously reported contradicting results on the role of liprin-α1 in cell invasion as discussed below [51,52,53, 120].
Liprins in the regulation of invasion and oncogenic signaling
As discussed above, liprin-α1 is important in cell edge dynamics, cell motility, and invasion although its role in invasion has been somewhat controversial [4, 5, 51, 53, 120]. It is now known that in specific non-invasive HNSCC cells, liprin-α1 is required for expansive growth behavior of the cells, while in motile and invasive cells knockdown of liprin-α1 reduces cell invasion and invasive growth [51, 53]. Liprin-α1 knockdown leads to increased expression of vimentin in HNSCC cells, a component of intermediate filaments, as well as disturbances in organization of vimentin filament network in motile or metastatic cancer cells [51]. In support of this data, PPFIA1 has been reported to be a novel Snail1 target, which suggests that positive regulation of PPFIA1 expression by Snail1 may contribute to the invasive phenotype of breast cancer cells [121]. It is thus possible that transcriptional regulation of PPFIA1 by Snail1 may promote vimentin intermediate filament assembly, which is a hallmark of the epithelial-mesenchymal transition [121]. This data underline the importance of liprin-α1 in the regulation and interplay with the cytoskeletal elements relevant to cancer cell motility.
Liprin-α1 suppresses the expression of the transmembrane metastasis suppressor CD82 in HNSCC and breast cancer cell lines [52], and the expression of liprin-α1 and CD82 is negatively correlated in clinical breast cancer samples [122]. CD82 is a direct p53 effector, an important suppressor for metastasis in multiple cancers and an inhibitor of microprotrusions during invasion [123, 124]. CD82 can be modulated by liprin-α1 [52], it is an important player in tetraspanin-enriched membrane microdomains, and it can be activated by post-transcriptional modifications such as glycosylation and palmitoylation [123, 125, 126].
Liprin-α1 interacts with ING4 (inhibitor of growth 4), and positively regulates cell migration in RKO colon carcinoma cells in ING4-dependent manner [127]. Liprin-α1 is expressed at the protrusions, essential for movement in metastatic breast cancer cells [53, 72, 128]. Similarly, liprin-β1 co-localizes at the cell edge with liprin-α1, and contributes positively to tumor cell motility in addition to liprin-α1 [72]. Liprin-β1 interacts with the metastasis-associated protein S100A4, which may modulate liprin-α1/liprin-β1 interaction, thus acting as a component of the LAR-liprin-α1/liprin-β1 network [129].
The N-terminus of liprin-β1 co-immunoprecipitates with the adaptor proteins Kank1 and Kank2, which are suppressors of the proliferation and migration in melanoma cells [130]. Although the functional role of this interaction is not clear, liprin-β1 is likely to contribute to melanoma tumor development. While several studies suggest liprin-β1 to promote tumor progression, liprin-β2 has opposing role in the cancer invasive phenotype, and silencing of liprin‐β2 has no effects on lamellipodia density and stability in MDA-MB-231 cells [72]. Interestingly, ERK2 knockdown leads to increased expression levels of liprin-β2 in MDA-MB-231 breast cancer cells and liprin-β2 knockdown restores the invasive phenotype of ERK2-depleted cells in three-dimensional ECM [75].
Liprins are involved in a number of biological processes important in oncogenic signaling, including cell–cell or cell-substrate junctions, as well as the composition of the cell membrane [5, 51,52,53, 90, 92, 131]. For example, liprin-α1 expression associates with several pathways important in cancer cell signaling including membrane microdomains and anchoring junctions [52]. Membrane microdomains are cholesterol-containing lipid rafts, which facilitate cellular signaling. These microdomains assemble and disassemble in response to environmental factors during cellular movement, invasion and ECM remodeling [109, 132, 133].
Recently, liprins have been shown to be associated to several proteins that are involved in cancer progression and motility. Pull-down experiments in human bladder cancer cell line T24 showed that liprin-α1 binds to EphA2 and promotes progranulin-dependent motility. Liprin-α1 is abundantly expressed in several bladder cancer cell lines and it strongly co-localizes with EphA2 upon progranulin stimulation [134]. Recent findings in K562 leukemia cells have shown that PPFIA1 is a direct target of miRNA-181a, whose downregulation is associated with poor response in leukemia. In this work, liprin-α1 overexpression promoted invasive capabilities of K562 cells and transfection of PPFIA1 siRNA on CML xenograft murine model led to reduced growth of the tumor [135]. Liprin-α4 is upregulated in renal cell carcinoma cell lines, and it is directly activated by binding of the hypoxia-inducible factor 1α (HIF-1α). Liprin-α4 controls the levels of E-cadherin and β-catenin at the epithelial cell junctions [131]. All these data provide evidence of important roles of liprin proteins in oncogenic cell signaling.
Perspectives
The role of liprins in synaptic signaling in neuronal cells has been extensively studied now for a couple of decades by different in vitro and in vivo studies [1, 6,7,8, 14, 29, 35, 44] (Table 2). In contrast, the function of liprins in tumor progression has been highlighted in different tumor models only during the last decade [51,52,53, 72, 127]. While the in vivo data are still fragmentary, in the future the generation of knockout mouse models will provide deeper understanding on the role of these proteins in disease and physiology. Liprins are multifunctional scaffold proteins orchestrating the disassembly and assembly of cellular structures, such as pre- and postsynaptic sites, focal adhesions, and invadosomes [7, 15, 29, 53, 90, 118]. The increasing evidence of liprins’ contribution to tumor cell motility, signaling, recycling of membrane, and ECM proteins such as integrins and fibronectin, and localization of liprins into defined plasma membrane structures reflect their versatile functions in several physiological and pathological processes. Genetic alterations of liprins, in particular the high-level amplifications of PPFIA1 in HNSCC and breast cancer, point to the critical oncogenic role of liprins in tumor progression. The future research should focus on understanding the role of liprins in those preclinical models which take into account the tumor microenvironment and signaling between tumor cells and stroma. This would enable understanding of the cellular organization and liprin-associated protein complexes and their role in oncogenic signaling. As the interplay between LAR and liprin-α proteins in tumor development and signaling is only partially explored, it is important to find out whether liprin-α proteins contribute to the dephosphorylation activity of PTPases and thus could serve as an interesting drug target or molecular marker. The participation of liprins in LLPS and localization to plasma membrane-associated platforms may reveal in the future interesting insights of liprins in cancer cell signaling. Because the information currently is still limited, further mechanistic studies in the future are required to understand the detailed roles of liprins in oncogenic signaling, tumor progression, and drug related therapy in different tumor types.
References
Serra-Pages C, Medley QG, Tang M, Hart A, Streuli M. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem. 1998;273:15611–20.
Serra-Pages C, Kedersha NL, Fazikas L, Medley Q, Debant A, Streuli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 1995;14:2827–38.
Nachat R, Cipolat S, Sevilla LM, Chhatriwala M, Groot KR, Watt FM. KazrinE is a desmosome-associated liprin that colocalises with acetylated microtubules. J Cell Sci. 2009;122:4035–41.
de Curtis I. Function of liprins in cell motility. Exp Cell Res. 2011;317:1–8.
Chiaretti S, de Curtis I. Role of Liprins in the Regulation of Tumor Cell Motility and Invasion. Curr Cancer Drug Targets. 2016;16:238–48.
Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 1999;401:371–5.
Astigarraga S, Hofmeyer K, Farajian R, Treisman JE. Three Drosophila liprins interact to control synapse formation. J Neurosci. 2010;30:15358–68.
Kaufmann N, DeProto J, Ranjan R, Wan H, Van Vactor D. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron 2002;34:27–38.
Pulido R, Serra-Pages C, Tang M, Streuli M. The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc Natl Acad Sci USA. 1995;92:11686–90.
Zurner M, Schoch S. The mouse and human Liprin-alpha family of scaffolding proteins: genomic organization, expression profiling and regulation by alternative splicing. Genomics. 2009;93:243–53.
Spangler SA, Jaarsma D, De Graaff E, Wulf PS, Akhmanova A, Hoogenraad CC. Differential expression of liprin-alpha family proteins in the brain suggests functional diversification. J Comp Neurol. 2011;519:3040–60.
Zurner M, Mittelstaedt T. tom Dieck S, Becker A, Schoch S. Analyses of the spatiotemporal expression and subcellular localization of liprin-alpha proteins. J Comp Neurol. 2011;519:3019–39.
Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–14.
Wong MY, Liu CL, Wang SSH, Roquas ACF, Fowler SC, Kaeser PS. Liprin-alpha 3 controls vesicle docking and exocytosis at the active zone of hippocampal synapses. Proc Natl Acad Sci USA. 2018;115:2234–9.
Astro V, de Curtis I. Plasma membrane-associated platforms: dynamic scaffolds that organize membrane-associated events. Sci Signal. 2015;8:367.
Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, et al. (DP2)-P-2: database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–16.
Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98.
Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–9.
Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–4.
Boija A, Klein IA, Young RA. Biomolecular condensates and cancer. Cancer Cell. 2021;39:174–92.
Cai D, Liu Z, Lippincott-Schwartz J. Biomolecular condensates and their links to cancer progression. Trends Biochem Sci. 2021;46:535–49.
McDonald NA, Fetter RD, Shen K. Assembly of synaptic active zones requires phase separation of scaffold molecules. Nature. 2020;588:454–8.
Emperador-Melero J, Wong MY, Wang SSH, de Nola G, Nyitrai H, Kirchhausen T, et al. PKC-phosphorylation of Liprin-alpha3 triggers phase separation and controls presynaptic active zone structure. Nat Commun. 2021;12:3057.
de Curtis I. Biomolecular condensates at the front: cell migration meets phase separation. Trends Cell Biol. 2021;31:145–8.
Wyszynski M, Kim E, Dunah AW, Passafaro M, Valtschanoff JG, Serra-Pages C, et al. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34:39–52.
Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, et al. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem. 2003;278:11393–401.
Ko J, Kim S, Valtschanoff JG, Shin HW, Lee JR, Sheng M, et al. Interaction between liprin-alpha and GIT1 is required for AMPA receptor targeting. J Neurosci. 2003;23:1667–77.
Bernadzki KM, Gawor M, Pezinski M, Mazurek P, Niewiadomski P, Redowicz MJ, et al. Liprin-alpha-1 is a novel component of the murine neuromuscular junction and is involved in the organization of the postsynaptic machinery. Sci Rep. 2017;7:9116.
Spangler SA, Hoogenraad CC. Liprin-alpha proteins: scaffold molecules for synapse maturation. Biochem Soc Trans. 2007;35:1278–82.
Patel MR, Lehrman EK, Poon VY, Crump JG, Zhen M, Bargmann CI, et al. Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci. 2006;9:1488–98.
Stryker E, Johnson KGLAR. liprin alpha and the regulation of active zone morphogenesis. J Cell Sci. 2007;120:3723–8.
Olsen O, Moore KA, Nicoll RA, Bredt DS. Synaptic transmission regulated by a presynaptic MALS/Liprin-alpha protein complex. Curr Opin Cell Biol. 2006;18:223–7.
Wei Z, Zheng S, Spangler SA, Yu C, Hoogenraad CC, Zhang M. Liprin-mediated large signaling complex organization revealed by the liprin-alpha/CASK and liprin-alpha/liprin-beta complex structures. Mol Cell. 2011;43:586–98.
Olsen O, Moore KA, Fukata M, Kazuta T, Trinidad JC, Kauer FW, et al. Neurotransmitter release regulated by a MALS-liprin-alpha presynaptic complex. J Cell Biol. 2005;170:1127–34.
Spangler SA, Schmitz SK, Kevenaar JT, de Graaff E, de Wit H, Demmers J, et al. Liprin-alpha2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J Cell Biol. 2013;201:915–28.
Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, et al. RIM1 alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–6.
Dai Y, Taru H, Deken SL, Grill B, Ackley B, Nonet ML, et al. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat Neurosci. 2006;9:1479–87.
Ko J, Na M, Kim S, Lee JR, Kim E. Interaction of the ERC family of RIM-binding proteins with the liprin-alpha family of multidomain proteins. J Biol Chem. 2003;278:42377–85.
Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, et al. The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J Biol Chem. 2003;278:6291–300.
Hoogenraad CC, Feliu-Mojer MI, Spangler SA, Milstein AD, Dunah AW, Hung AY, et al. Liprin alpha 1 degradation by calcium/calmodulin-dependent protein kinase II regulates LAR receptor tyrosine phosphatase distribution and dendrite development. Dev Cell. 2007;12:587–602.
Patel MR, Shen K. RSY-1 is a local inhibitor of presynaptic assembly in C. elegans. Science. 2009;323:1500–3.
Kittelmann M, Hegermann J, Goncharov A, Taru H, Ellisman MH, Richmond JE, et al. Liprin-alpha/SYD-2 determines the size of dense projections in presynaptic active zones in C. elegans. J Cell Biol. 2013;203:849–63.
Taru H, Jin YS. The liprin homology domain is essential for the homomeric interaction of SYD-2/Liprin-alpha protein in presynaptic assembly. J Neurosci. 2011;31:16261–8.
Miller KE, DeProto J, Kaufmann N, Patel BN, Duckworth A, Van, Vactor D. Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr Biol. 2005;15:684–9.
Choe KM, Prakash S, Bright A, Clandinin TR. Liprin-alpha is required for photoreceptor target selection in Drosophila. P Natl Acad Sci USA. 2006;103:11601–6.
Hallam SJ, Goncharov A, McEwen J, Baran R, Jin Y. SYD-1, a presynaptic protein with PDZ, C2 and rhoGAP-like domains, specifies axon identity in C. elegans. Nat Neurosci. 2002;5:1137–46.
Owald D, Fouquet W, Schmidt M, Wichmann C, Mertel S, Depner H, et al. A Syd-1 homologue regulates pre- and postsynaptic maturation in Drosophila. J Cell Biol. 2010;188:565–79.
Chia PH, Patel MR, Wagner OI, Klopfenstein DR, Shen K. Intramolecular regulation of presynaptic scaffold protein SYD-2/liprin-alpha. Mol Cell Neurosci. 2013;56:76–84.
Liu YC, Couzens AL, Deshwar AR, B McBroom-Cerajewski LD, Zhang X, Puviindran V, et al. The PPFIA1-PP2A protein complex promotes trafficking of Kif7 to the ciliary tip and Hedgehog signaling. Sci Signal. 2014;7:ra117.
van der Vaart B, van Riel WE, Doodhi H, Kevenaar JT, Katrukha EA, Gumy L, et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell. 2013;27:145–60.
Pehkonen H, von Nandelstadh P, Karhemo PR, Lepikhova T, Grenman R, Lehti K, et al. Liprin-alpha1 is a regulator of vimentin intermediate filament network in the cancer cell adhesion machinery. Sci Rep. 2016;6:24486.
Pehkonen H, Lento M, von Nandelstadh P, Filippou A, Grenman R, Lehti K, et al. Liprin-alpha1 modulates cancer cell signaling by transmembrane protein CD82 in adhesive membrane domains linked to cytoskeleton. Cell Commun Signal. 2018;16:41.
Astro V, Asperti C, Cangi MG, Doglioni C, de Curtis I. Liprin-alpha1 regulates breast cancer cell invasion by affecting cell motility, invadopodia and extracellular matrix degradation. Oncogene. 2011;30:1841–9.
Jarvinen AK, Autio R, Kilpinen S, Saarela M, Leivo I, Grenman R, et al. High-resolution copy number and gene expression microarray analyses of head and neck squamous cell carcinoma cell lines of tongue and larynx. Gene Chromosome Canc. 2008;47:500–9.
Ormandy CJ, Musgrove EA, Hui R, Daly RJ, Sutherland RL. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003;78:323–35.
Carneiro A, Isinger A, Karlsson A, Johansson J, Jonsson G, Bendahl PO, et al. Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC Cancer. 2008;8:98.
Wilkerson PM, Reis-Filho JS. The 11q13-q14 amplicon: clinicopathological correlations and potential drivers. Genes Chromosomes Cancer. 2013;52:333–55.
Ramos-Garcia P, Ruiz-Avila I, Gil-Montoya JA, Ayen A, Gonzalez-Ruiz L, Navarro-Trivino FJ, et al. Relevance of chromosomal band 11q13 in oral carcinogenesis: an update of current knowledge. Oral Oncol. 2017;72:7–16.
Barros-Filho MC, Reis-Rosa LA, Hatakeyama M, Marchi FA, Chulam T, Scapulatempo-Neto C, et al. Oncogenic drivers in 11q13 associated with prognosis and response to therapy in advanced oropharyngeal carcinomas. Oral Oncol. 2018;83:81–90.
Xu C, Liu Y, Wang P, Fan W, Rue TC, Upton MP, et al. Integrative analysis of DNA copy number and gene expression in metastatic oral squamous cell carcinoma identifies genes associated with poor survival. Mol Cancer. 2010;9:143.
Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci USA. 2002;99:11369–74.
Huang X, Godfrey TE, Gooding WE, McCarty KS Jr., Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45:1058–69.
Meredith SD, Levine PA, Burns JA, Gaffey MJ, Boyd JC, Weiss LM, et al. Chromosome 11q13 amplification in head and neck squamous cell carcinoma. Association with poor prognosis. Arch Otolaryngol Head Neck Surg. 1995;121:790–4.
Akervall JA, Jin Y, Wennerberg JP, Zatterstrom UK, Kjellen E, Mertens F, et al. Chromosomal abnormalities involving 11q13 are associated with poor prognosis in patients with squamous cell carcinoma of the head and neck. Cancer. 1995;76:853–9.
Schuuring E, Verhoeven E, Vantinteren H, Peterse JL, Nunnink B, Thunnissen FBJM, et al. Amplification of genes within the chromosome-11q13 region is indicative of poor prognosis in patients with operable breast-cancer. Cancer Res. 1992;52:5229–34.
Song J, Wu S, Xia X, Wang Y, Fan Y, Yang Z. Cell adhesion-related gene somatic mutations are enriched in aggressive papillary thyroid microcarcinomas. J Transl Med. 2018;16:269.
Takeuchi K, Soda M, Togashi Y, Sugawara E, Hatano S, Asaka R, et al. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res. 2011;17:3341–8.
Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Yeh I, Botton T, Talevich E, Shain AH, Sparatta AJ, de la Fouchardiere A, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun. 2015;6:7174.
Menzies AM, Yeh I, Botton T, Bastian BC, Scolyer RA, Long GV. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res. 2015;28:607–10.
Iyama K, Matsuse M, Mitsutake N, Rogounovitch T, Saenko V, Suzuki K, et al. Identification of Three Novel fusion oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in thyroid cancers of young patients in Fukushima. Thyroid. 2017;27:811–8.
Chiaretti S, Astro V, Chiricozzi E, de Curtis I. Effects of the scaffold proteins liprin-alpha1, beta1 and beta2 on invasion by breast cancer cells. Biol Cell. 2016;108:65–75.
Wu Y, Yu H, Zheng SL, Feng B, Kapron AL, Na R, et al. Germline mutations in PPFIBP2 are associated with lethal prostate cancer. Prostate. 2018;78:1222–8.
Colas E, Perez C, Cabrera S, Pedrola N, Monge M, Castellvi J, et al. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J Cancer. 2011;129:2435–44.
von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, et al. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2. J Cell Sci. 2012;125:1465–77.
Johnson KG, Van, Vactor D. Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev. 2003;83:1–24.
Bomkamp C, Padmanabhan N, Karimi B, Ge Y, Chao JT, Loewen CJR, et al. Mechanisms of PTPsigma-mediated presynaptic differentiation. Front Synaptic Neurosci. 2019;11:17.
Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 1990;9:2399–407.
Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–57.
Xie X, Luo L, Liang M, Zhang W, Zhang T, Yu C, et al. Structural basis of liprin-alpha-promoted LAR-RPTP clustering for modulation of phosphatase activity. Nat Commun. 2020;11:169.
Serra-Pages C, Streuli M, Medley QG. Liprin phosphorylation regulates binding to LAR: evidence for liprin autophosphorylation. Biochemistry. 2005;44:15715–24.
Sarhan AR, Patel TR, Creese AJ, Tomlinson MG, Hellberg C, Heath JK, et al. Regulation of platelet derived growth factor signaling by leukocyte common antigen-related (LAR) protein tyrosine phosphatase: a quantitative phosphoproteomics study. Mol Cell Proteom. 2016;15:1823–36.
Bera R, Chiou CY, Yu MC, Peng JM, He CR, Hsu CY, et al. Functional genomics identified a novel protein tyrosine phosphatase receptor type F-mediated growth inhibition in hepatocarcinogenesis. Hepatology. 2014;59:2238–50.
Whitmore TE, Peterson A, Holzman T, Eastham A, Amon L, McIntosh M, et al. Integrative analysis of N-linked human glycoproteomic data sets reveals PTPRF ectodomain as a novel plasma biomarker candidate for prostate cancer. J Proteome Res. 2012;11:2653–65.
Levea CM, McGary CT, Symons JR, Mooney RAPTP. LAR expression compared to prognostic indices in metastatic and non-metastatic breast cancer. Breast Cancer Res Treat. 2000;64:221–8.
Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805.
Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Bio. 2009;10:21–33.
Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–49.
Innocenti M. New insights into the formation and the function of lamellipodia and ruffles in mesenchymal cell migration. Cell Adhes Migr. 2018;12:401–16.
Astro V, Tonoli D, Chiaretti S, Badanai S, Sala K, Zerial M, et al. Liprin-alpha1 and ERC1 control cell edge dynamics by promoting focal adhesion turnover. Sci Rep. 2016;6:33653.
Asperti C, Astro V, Totaro A, Paris S, de Curtis I. Liprin-alpha1 promotes cell spreading on the extracellular matrix by affecting the distribution of activated integrins. J Cell Sci. 2009;122:3225–32.
Astro V, Chiaretti S, Magistrati E, Fivaz M, de Curtis I. Liprin-alpha 1, ERC1 and LL5 define polarized and dynamic structures that are implicated in cell migration. J Cell Sci. 2014;127:3862–76.
Asperti C, Pettinato E, de Curtis I. Liprin-alpha1 affects the distribution of low-affinity beta1 integrins and stabilizes their permanence at the cell surface. Exp Cell Res. 2010;316:915–26.
Robertson J, Jacquemet G, Byron A, Jones MC, Warwood S, Selley JN, et al. Defining the phospho-adhesome through the phosphoproteomic analysis of integrin signalling. Nat Commun. 2015;6:6265.
Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol. 2015;17:1577–87.
Winograd-Katz SE, Fassler R, Geiger B, Legate KR. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol. 2014;15:273–88.
Sala K, Corbetta A, Minici C, Tonoli D, Murray DH, Cammarota E, et al. The ERC1 scaffold protein implicated in cell motility drives the assembly of a liquid phase. Sci Rep. 2019;9:13530.
Asperti C, Astro V, Pettinato E, Paris S, Bachi A, de Curtis I. Biochemical and functional characterization of the interaction between liprin-alpha1 and GIT1: implications for the regulation of cell motility. PLoS ONE. 2011;6:e20757.
Sun Z, Tseng HY, Tan S, Senger F, Kurzawa L, Dedden D, et al. Kank2 activates talin, reduces force transduction across integrins and induces central adhesion formation. Nat Cell Biol. 2016;18:941–53.
Sakamoto S, Ishizaki T, Okawa K, Watanabe S, Arakawa T, Watanabe N, et al. Liprin-alpha controls stress fiber formation by binding to mDia and regulating its membrane localization. J Cell Sci. 2012;125:108–20.
Brenig J, de Boor S, Knyphausen P, Kuhlmann N, Wroblowski S, Baldus L, et al. Structural and biochemical basis for the inhibitory effect of liprin-alpha3 on mouse diaphanous 1 (mDia1) function. J Biol Chem. 2015;290:14314–27.
Mana G, Clapero F, Panieri E, Panero V, Bottcher RT, Tseng HY, et al. PPFIA1 drives active alpha5beta1 integrin recycling and controls fibronectin fibrillogenesis and vascular morphogenesis. Nat Commun. 2016;7:13546.
Norrmen C, Vandevelde W, Ny A, Saharinen P, Gentile M, Haraldsen G, et al. Liprin (beta)1 is highly expressed in lymphatic vasculature and is important for lymphatic vessel integrity. Blood. 2010;115:906–9.
Linder S, Kopp P. Podosomes at a glance. J Cell Sci. 2005;118:2079–82.
Linder S. Invadosomes at a glance. J Cell Sci. 2009;122:3009–13.
Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–26.
Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–41.
Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105.
Di Martino J, Henriet E, Ezzoukhry Z, Goetz JG, Moreau V, Saltel F. The microenvironment controls invadosome plasticity. J Cell Sci. 2016;129:1759–68.
Gligorijevic B, Bergman A, Condeelis J. Multiparametric classification links tumor microenvironments with tumor cell phenotype. PLoS Biol. 2014;12:e1001995.
Takkunen M, Hukkanen M, Liljestrom M, Grenman R, Virtanen I. Podosome-like structures of non-invasive carcinoma cells are replaced in epithelial-mesenchymal transition by actin comet-embedded invadopodia. J Cell Mol Med. 2010;14:1569–93.
Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57.
Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–37.
Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–17.
Branch KM, Hoshino D, Weaver AM. Adhesion rings surround invadopodia and promote maturation. Biol Open. 2012;1:711–22.
Jarvinen AK, Autio R, Haapa-Paananen S, Wolf M, Saarela M, Grenman R, et al. Identification of target genes in laryngeal squamous cell carcinoma by high-resolution copy number and gene expression microarray analyses. Oncogene. 2006;25:6997–7008.
Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–52.
Sala K, Raimondi A, Tonoli D, Tacchetti C, de Curtis I. Identification of a membrane-less compartment regulating invadosome function and motility. Sci Rep. 2018;8:1164.
Yang J, Wu NN, Huang DJ, Luo YC, Huang JZ, He HY, et al. PPFIA1 is upregulated in liver metastasis of breast cancer and is a potential poor prognostic indicator of metastatic relapse. Tumour Biol. 2017;39:1010428317713492.
Tan KD, Zhu Y, Tan HK, Rajasegaran V, Aggarwal A, Wu J, et al. Amplification and overexpression of PPFIA1, a putative 11q13 invasion suppressor gene, in head and neck squamous cell carcinoma. Genes Chromosomes Cancer. 2008;47:353–62.
Maturi V, Moren A, Enroth S, Heldin CH, Moustakas A. Genomewide binding of transcription factor Snail1 in triple-negative breast cancer cells. Mol Oncol. 2018;12:1153–74.
Alfarsi LH, El Ansari R, Craze ML, Masisi BK, Ellis IO, Rakha EA, et al. PPFIA1 expression associates with poor response to endocrine treatment in luminal breast cancer. BMC Cancer. 2020;20:425.
Liu WM, Zhang XA. KAI1/CD82, a tumor metastasis suppressor. Cancer Lett. 2006;240:183–94.
Bari R, Zhang YH, Zhang F, Wang NX, Stipp CS, Zheng JJ, et al. Transmembrane interactions are needed for KAI1/CD82-mediated suppression of cancer invasion and metastasis. Am J Pathol. 2009;174:647–60.
Ono M, Handa K, Withers DA, Hakomori S. Motility inhibition and apoptosis are induced by metastasis-suppressing gene product CD82 and its analogue CD9, with concurrent glycosylation. Cancer Res. 1999;59:2335–9.
Zhou B, Liu L, Reddivari M, Zhang XA. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004;64:7455–63.
Shen JC, Unoki M, Ythier D, Duperray A, Varticovski L, Kumamoto K, et al. Inhibitor of growth 4 suppresses cell spreading and cell migration by interacting with a novel binding partner, liprin alpha1. Cancer Res. 2007;67:2552–8.
Jakobsen KR, Sorensen E, Brondum KK, Daugaard TF, Thomsen R, Nielsen AL. Direct RNA sequencing mediated identification of mRNA localized in protrusions of human MDA-MB-231 metastatic breast cancer cells. J Mol Signal. 2013;8:9.
Kriajevska M, Fischer-Larsen M, Moertz E, Vorm O, Tulchinsky E, Grigorian M, et al. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J Biol Chem. 2002;277:5229–35.
Luo M, Mengos AE, Mandarino LJ, Sekulic A. Association of liprin beta-1 with kank proteins in melanoma. Exp Dermatol. 2016;25:321–3.
Mattauch S, Sachs M, Behrens J. Liprin-alpha4 is a new hypoxia-inducible target gene required for maintenance of cell-cell contacts. Exp Cell Res. 2010;316:2883–92.
Laude AJ, Prior IA. Plasma membrane microdomains: organization, function and trafficking. Mol Membr Biol. 2004;21:193–205.
Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014;1838:532–45.
Buraschi S, Neill T, Xu SQ, Palladino C, Belfiore A, Iozzo RV, et al. Progranulin/EphA2 axis: A novel oncogenic mechanism in bladder cancer. Matrix Biol. 2020;93:10–24.
Gu C, Liu Y, Yin Z, Yang J, Huang G, Zhu X, et al. Discovery of the oncogenic Parp1, a target of bcr-abl and a potential therapeutic, in mir-181a/PPFIA1 signaling pathway. Mol Ther Nucleic Acids. 2019;16:1–14.
Acknowledgements
Figure 2 has been modified from MediSapiens ISTOnline database. Figure 3A has been created with BioRender.com software. Jane and Aatos Erkko Foundation, Academy of Finland, Cancer Foundation Finland, Luise and Henrik Kuningas Foundation, and AIRC (grant IG 20203 to IdC) are thanked for their financial support to this work.
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital.
Author information
Authors and Affiliations
Contributions
Writing, commenting, and editing of the text and content: HP, IdC, and OM. All the authors have read and agreed to the published version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pehkonen, H., de Curtis, I. & Monni, O. Liprins in oncogenic signaling and cancer cell adhesion. Oncogene 40, 6406–6416 (2021). https://doi.org/10.1038/s41388-021-02048-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-02048-1
This article is cited by
-
Polygenic risk score model for renal cell carcinoma in the Korean population and relationship with lifestyle-associated factors
BMC Genomics (2024)
-
Liprin-α1 Expression in Tumor-Infiltrating Lymphocytes Associates with Improved Survival in Patients with HPV-Positive Oropharyngeal Squamous Cell Carcinoma
Head and Neck Pathology (2023)
-
A functional interaction between liprin-α1 and B56γ regulatory subunit of protein phosphatase 2A supports tumor cell motility
Communications Biology (2022)
